

Abstract
Interstitial 3p21.31 deletions have been very rarely reported. We describe a 7-year-old boy with global developmental delay, specific facial characteristics, hydronephrosis, and hypothyreosis with a de novo deletion of 3p21.31, encompassing 29 OMIM genes. Despite the wide use of microarrays, no similar case has been reported in the literature so far. Five overlapping cases are deposited in the DECIPHER database, 2 of which have significant overlapping chromosomal aberrations. They both share some phenotypic characteristics with our case, e.g. developmental delay, intellectual disability and facial dysmorphism (arched eyebrows, hypertelorism, low-set ears, and a large nose tip). In addition, loss-of-function mutations in the SETD2 gene (OMIM 612778) of the deleted region have been described in 3 patients, presenting with some similar clinical features, namely overgrowth, intellectual disability, speech delay, hypotonia, autism, and epilepsy. Therefore, SETD2 may explain part of the phenotype in our case. We focused on 3 other genes in the deleted region, based on their known functions, namely CSPG5 (OMIM 606775), PTH1R (OMIM 168468) and SMARCC1 (OMIM 601732), and assessed their potentially important role in describing the patient's phenotype. Additional cases with haploinsufficiency of this region are needed to elucidate further genotype-phenotype correlations.
Keywords: Developmental delay, Interstitial 3p21.31 deletion, Macrocephaly, 3p21.31, SETD2
Established Facts
• Reported 3p deletions involve the proximal or distal end of the short arm of chromosome 3.
• Recent reports suggest there is a third group of 3p deletions - interstitial deletions involving 3p21.31 - which have been very rarely reported.
Novel Insights
• We report on a first case of a small interstitial 3p21.31 deletion in a patient with intellectual disability, delayed speech and language development, hypotonia, hypothyreosis, and congenital anomalies of the left kidney.
• The SETD2 gene (OMIM 612778), located in the deleted region, may be partially responsible for the clinical phenotype.
Most previously reported 3p deletions involve the proximal or distal end of the short arm of chromosome 3. The more prevalent being the 3p terminal deletion syndrome, caused by 3p25pter deletions of different sizes. Its main characteristics are low birth weight, microcephaly, hypotonia, psychomotor and growth retardation, ptosis, and nonspecific dysmorphic features (telecanthus, downslanting palpebral fissures, OMIM 613792). Proximal interstitial deletions involve the 3p14 band and are associated with mild facial dysmorphism, developmental delay, intellectual disability, autism, and speech impairment [de la Hoz et al., 2015]. There are no common, well-defined breakpoints in either the proximal or distal 3p deletions.
With more recent reports, it appears there is a third group of 3p deletions - interstitial deletions involving 3p21.31 - which have been very rarely described and, again, have no common breakpoints. Three cases have been reported in the literature, none of which overlap with our patient when comparing the locations of the deletions. All reported deletions are directly adjacent to the deletion in our case [Haldeman-Englert et al., 2009; Eto et al., 2013]. In addition, 3 individuals with clinical details and different deletion sizes and breakpoints of 3p21.31 are included in the Database of Chromosomal Imbalance and Phenotype in Humans (DECIPHER) [Firth et al., 2009], and 2 cases from the DECIPHER database extend to the 3p22 band (table 1). The SETD2 gene (OMIM 612778), located in the deleted region, was recently shown to be mutated in 2 patients with overgrowth, macrocephaly, hypotonia, and speech and developmental delay [Luscan et al., 2014], and in 1 patient with intellectual disability, epilepsy, and autism [Lumish et al., 2015].
Table 1.
Clinical characteristics of previously reported cases with overlapping deletions
| Characteristics | Case |
|||||
|---|---|---|---|---|---|---|
| our case | 248943 | 258176 | 284462 | 293473 | 253934 | |
| Coordinates of reported deletions (hg19) | chr3: 45,881,062–48,009,576 | chr3: 45,773,561–48,068,283 | chr3: 44,251,270–47,936,671 | chr3: 46,852,723–50,824,570 | chr3: 42,167,061–46,303,826 | chr3: 42,671,667–47,995,168 |
| Chromosome band | 3p21.31 | 3p21.31 | 3p21.31 | 3p21.31p21.2 | 3p22.1p21.31 | 3p22.1p21.31 |
| Gender | M | F | M | F | M | M |
| Developmental delay | + | – | – | – | + | – |
| Speech delay | + | + | – | – | – | – |
| Intellectual disability | + | + | + | – | – | – |
| Autism | + | – | + | – | – | – |
| Craniofacial characteristics | ||||||
| Prominent forehead | – | – | + | – | – | – |
| Hypertelorism | + | – | – | + | – | + |
| Downslanting | ||||||
| palpebral fissure | + | – | + | – | – | – |
| Nose | bulbous tip | – | – | – | – | depressed nasal bridge |
| Ears | low-set | – | – | low-set, posteriorly rotated ears | – | low-set, abnormal external ear |
| Palate | – | – | – | – | – | |
| Other | ptosis | cleft palate, blepharophimosis, micrognathia | ||||
| Cardiovascular characteristics | – | – | – | transposition of great arteries, atrial septum defect | – | hypoplastic left heart |
| Hypotonia | + | + | – | – | – | – |
| Other | clinodactyly of the 5th finger | clinodactyly of the 5th finger | visual impairment | anal atresia | – | hydrocephalus, supernumerary ribs, renal hypoplasia, hypospadias, abnormality of the sacrum, anal atresia |
M = Male; F = female; + = present; - = not reported.
We report on a first case of a specific 3p21.31 deletion in a 7-year-old boy and review the above-mentioned cases with respect to genotype-phenotype overlap with our case.
Patient and Methods
Patient
Our patient, a 7-year-old boy, born as the first child to healthy nonconsanguineous parents was re-evaluated. The family history is unremarkable. The boy has a younger healthy sister. In the third trimester of the pregnancy, less fetal movement was reported. An elective cesarean section due to a uterine myoma was performed in the 37th week of gestation. The boy's birth weight was 2,320 g (3rd-5th percentile), birth length 48 cm (50th percentile) and his head circumference was 34 cm (50th percentile). Muscular hypotonia and hypoglycemia were noted and treated immediately after birth. An abdominal ultrasound revealed hydronephrosis, pyeloureteral stenosis and a dysplastic left kidney. Latent hypothyreosis was diagnosed when he was 5 months old. In addition, he was described as having facial stigmata (no details available). At the age of 4, a clinical geneticist diagnosed a long face, downslanting palpebral fissures, large nose tip, deep philtrum, and low-set, dysmorphic ears (fig. 1). A MRI of his head, at the age of 7, showed a frontal parasagittal arachnoid cyst located under the vertex, which slightly displaced the superior frontal gyrus. It was stable and remained unchanged in size (2.8 × 1.3 × 2.9 cm) compared to a previous MRI done 3 years earlier. Also, a mild atrophy of the posterior corpus callosum, punctiform high-intensity changes in deep white matter around both the lateral ventricles posteriorly and the Chiari malformation type 1 were described. All changes have been stable for the past 3 years. Currently, the boy is on regular follow-up by a nephrologist for his hydronephrosis and pyeloureteral stenosis and an orthopedist for his flat feet. He receives hormone replacement therapy due to latent hypothyreosis. At the age of 7, his growth parameters were as follows: height 127.3 cm (86th percentile), weight 28 kg (91st percentile), and head circumference 55.2 cm (98th percentile).
Fig. 1.
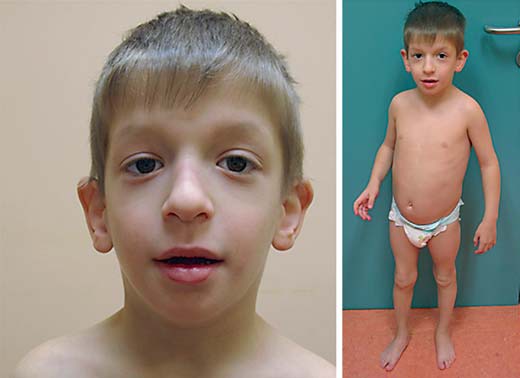
Craniofacial characteristics in our patient with a 3p21.31 microdeletion. A long face, downslanting palpebral fissures, large nose tip, deep philtrum, and low-set, dysmorphic ears are shown.
Molecular Karyotyping
Postnatal cytogenetic analysis showed a normal male karyotype. Microarray analysis (180K CGH array, Agilent Technologies, Santa Clara, Calif., USA) (fig. 2A) revealed a de novo microdeletion of 2.18 Mb in chromosome 3, region 3p21.31 (arr[hg19] 3p21.31(45,881,062-48,009,576)×1). The region contains 29 OMIM genes.
Fig. 2.

A Array-CGH results in our case with complete chromosome 3 (left) and the magnified 3p21.31 region (right). B Sizes and locations of our case and 5 reported overlapping cases (red bars) using the UCSC Genome Browser. Gene content (OMIM genes) is presented under the reported cases.
Discussion
A search through several databases - ECARUCA (http://umcecaruca01.extern.umcn.nl:8080/ecaruca/ecaruca.jsp), DECIPHER (https://decipher.sanger.ac.uk/index), ClinGen (https://www.clinicalgenome.org/), and scientific literature indexed in PubMed (http://www.ncbi.nlm.nih.gov/pubmed) was performed. The Database of Genomic Variants (DGV; http://dgv.tcag.ca/) was checked for the presence of similar CNVs in control populations and none were revealed. According to all cases reported in the literature or deposited in databases with phenotype description (fig. 2B; table 1), there were 5 cases with overlapping deletions. Case 253934 was reported to have 3 CNVs altogether. One was 5.3 Mb in size and included the reported region herein, detailed in table 1 and figure 2B. Additionally, 2 CNVs of much smaller size were classified as likely benign (492.64-kb large deletion at 12p11.23) and as a variant of unknown significance (17.78-kb large deletion at 13q34), according to the above-mentioned databases and the DGV. All other cases mentioned in the DECIPHER database have only one CNV.
One of the previously diagnosed cases (DECIPHER case 248943) strikingly resembles our patient, not only in reported genomic rearrangement, but in sharing clinical findings as well. Both patients have intellectual disability, delayed speech and language development and muscular hypotonia. Another case with a significant deletion location and size overlap (DECIPHER case 258176) shares some phenotypic features, facial dysmorphism, intellectual impairment and autism. The case with the smallest joined region of overlap (DECIPHER case 284462) presented with a transposition of the great arteries and an atrial septum defect. Since there is no structural heart defect in our case or in the above-mentioned similar cases, the responsible gene may be located in the region between chr3: 48,068,283-50,824,570. Two previously reported cases with the adjacently deleted regions [Eto et al., 2013] had congenital heart anomalies (ventricular septal defect in patient 1 and atrial septum defect and patent ductus arteriosus in patient 2), supporting the suggestion that the responsible gene is located proximally to the deletion discovered in our patient.
With extensive use of the exome sequencing approach, SETD2 loss-of-function mutations were recently reported in 3 patients [Luscan et al., 2014; Lumish et al., 2015]. Patient 1 has a mutation that introduced a premature stop codon [Luscan et al., 2014]. The main reported clinical features are macrocephaly with a long face, prominent forehead and downslanting palpebral fissures, hypotonia, mild developmental delay and intellectual disability. Patient 2, described with macrocephaly, a long face, developmental delay, intellectual disability, and significant speech problems, has a missense mutation predicted to result in a loss-of-function allele. The third patient [Lumish et al., 2015] has a frameshift mutation and developmental delay, macrocephaly with a Chiari I malformation and ventriculomegaly, hypotonia, autism and seizures. With so many overlapping clinical features, we speculate that SETD2 haploinsufficiency could be an important cause of some clinical characteristics in our patient. The DGV database cites one sample with partial SETD2 deletion from the 1000 Genomes Consortium project. There are no data on this sample apart from the fact that it is a male individual.
PTH1R (OMIM 168468) has been linked to 2 distinctive phenotypes in humans, which are inherited in an autosomal dominant manner: Murk Jansen metaphyseal chondrodysplasia and primary failure of tooth eruption (PFE). Murk Jansen metaphyseal chondrodysplasia is a consequence of activating mutations in PTH1R; therefore, the phenotype is not expected in our patient. PFE is suggested to be caused by a haploinsufficiency mechanism and would be expected in our case. So far, no teeth abnormalities have been seen in our patient. Moreover, there are some deletions reported in DGV, which include the PTH1R gene. Two possible explanations are plausible: first and more likely, gene deletions may not be related to PFE. Second, since the phenotype of PFE predominantly affects posterior teeth and causes posterior open bite, this might not be recognized as a relevant health condition; therefore, samples/individuals have been included in DGV.
Another interesting gene from the region is CSPG5 (OMIM 606775) that spans the cell membrane. It is only expressed in the central nervous system, and it is primarily involved in neuritogenesis [Nakanishi et al., 2006]. Haploinsufficiency might have an impact on the developing brain, but further cases and studies are needed to show a direct correlation.
Conclusion
This is the first clinical report of a relatively small interstitial 3p21.31 deletion in a patient with intellectual disability, delayed speech and language development, hypotonia, hypothyreosis, and congenital anomalies of the left kidney. Only one case with very similar deletion size and location was previously reported, according to current literature and all available data in public databases (DECIPHER case 248943). Moreover, the DECIPHER case mentioned above, shares important phenotypic characteristics with our patient. In addition, loss-of-function mutations in the SETD2 gene in the 3p21.31 region were reported in 3 patients with overlapping clinical features, suggesting that SETD2 may be partially responsible for the clinical phenotype. On the other hand, there is one case with a partial SETD2 deletion deposited in DGV; therefore, more data is needed in order to clarify the mechanism and pathogenicity of SETD2 gene deletions.
Nevertheless, the 3p21.31 region may contain additional genes with an important influence on cognitive function as well as speech development and also renal development. Reports on additional cases will elucidate the common phenotype and provide insights into the functional knowledge of all the genes involved.
Statement of Ethics
The authors have no ethical conflicts to disclose. Written informed consent was obtained from the patient for publication of this case report and any accompanying images.
Disclosure Statement
The authors have no conflicts of interest to declare.
Acknowledgments
We thank the family for their generous collaboration. This study uses data generated by the DECIPHER community. A full list of centers which contributed to the generation of the data is available at http://decipher.sanger.ac.uk and via email, decipher@sanger.ac.uk. This study was funded by the Wellcome Trust.
References
- 1.de la Hoz AB, Maortua H, García-Rives A, Martínez-González MJ, Ezquerra M, Tejada M. 3p14 de novo interstitial microdeletion in a patient with intellectual disability and autistic features with language impairment: a comparison with similar cases. Case Rep Genet. 2015;2015:876348. doi: 10.1155/2015/876348. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Eto K, Sakai N, Shimada S, Shioda M, Ishigaki K, et al. Microdeletions of 3p21.31 characterized by developmental delay, distinctive features, elevated serum creatine kinase levels, and white matter involvement. Am J Med Genet A. 2013;161A:3049–3056. doi: 10.1002/ajmg.a.36156. [DOI] [PubMed] [Google Scholar]
- 3.Firth HV, Richards SM, Bevan AP, Clayton S, Corpas M, et al. DECIPHER: Database of Chromosomal Imbalance and Phenotype in Humans Using Ensembl Resources. Am J Hum Genet. 2009;84:524–533. doi: 10.1016/j.ajhg.2009.03.010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Haldeman-Englert CR, Gai X, Perin JC, Ciano M, Halbach SS, et al. A 3.1-Mb microdeletion of 3p21.31 associated with cortical blindness, cleft lip, CNS abnormalities, and developmental delay. Eur J Med Genet. 2009;52:265–268. doi: 10.1016/j.ejmg.2008.11.005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Luscan A, Laurendeau I, Malan V, Francannet C, Odent S, et al. Mutations in SETD2 cause a novel overgrowth condition. J Med Genet. 2014;51:512–517. doi: 10.1136/jmedgenet-2014-102402. [DOI] [PubMed] [Google Scholar]
- 6.Lumish HS, Wynn J, Devinsky O, Chung WK. Brief report: SETD2 mutation in a child with autism, intellectual disabilities and epilepsy. J Autism Dev Disord. 2015;45:3764–3770. doi: 10.1007/s10803-015-2484-8. [DOI] [PubMed] [Google Scholar]
- 7.Nakanishi K, Aono S, Hirano K, Kuroda Y, Ida M, et al. Identification of neurite outgrowth-promoting domains of neuroglycan C, a brain-specific chondroitin sulfate proteoglycan, and involvement of phosphatidylinositol 3-kinase and protein kinase C signaling pathways in neuritogenesis. J Biol Chem. 2006;25:24970–24978. doi: 10.1074/jbc.M601498200. [DOI] [PubMed] [Google Scholar]