

Abstract
A convergent [3+2] block synthetic strategy was developed for the synthesis of the pentasaccharide repeating unit of the cell wall O‐antigen of Escherichia coli O11 strain in excellent yield in a minimum number of steps. Several suitably functionalized thioglycoside derivatives were used as glycosyl donors during the synthesis of the target compound. A thioglycoside was the glycosyl donor used to couple with another thioglycoside derivative in a highly stereoselective manner exploiting the difference of their reactivity profile. A combination of Niodosuccinimide (NIS) and perchloric acid supported over silica gel (HClO4−SiO2) was used as a thiophilic glycosylation activator system in all stereoselective glycosylation reactions. HClO4−SiO2 acted as a user‐friendly solid acid catalyst. Yields were very good in all glycosylation steps with a high stereoselective outcome. The synthetic pentasaccharide could be coupled to an appropriate protein to furnish a glycoconjugate derivative for its use in immunochemical studies.
Keywords: carbohydrates, glycosylation, oligosaccharides, polysaccharides, stereoselectivity
Diarrheal infections are frequently encountered enteric diseases all over the world, particularly in developing countries that lack adequate sanitation.1 Gastrointestinal infections are the leading cause of death in children, the elderly, and immunocompromised patients.2 Among several bacterial species associated with the enteric infections, Escherichia coli (E. coli) is predominant.3, 4 Most commonly, E. coli strains are found as the commensal microorganism in the intestine with a beneficial symbiotic relationship with the host.5 However, some strains become virulent and cause several serious infections to the host.6 In general, pathogenic E. coli strains are responsible for a) diarrheal infections;3 b) urinary tract infections,7 and c) sepsis/meningitides,8 as examples.
The diarrheagenic E. coli strains are divided into several subclasses based on the pathogenic nature of infections,3, 6 such as a) enteropathogenic E. coli (EPEC), b) enterohemorrhagic E. coli (EHEC), c) enterotoxigenic E. coli (ETEC), d) enteroaggregative E. coli (EAEC), e) enteroinvasive E. coli (EIEC), etc. Among the subclasses of diarrheagenic E. coli strains, ETEC is responsible for traveller's diarrhea9 and enteric disorders in children and the immunocompromised, as well as for causing outbreaks.10 Bacterial cell wall polysaccharide O‐antigens play a vital role in the pathogenicity of the virulent strains.11 As a consequence, several reports have appeared on the structural aspects of cell wall polysaccharides of various bacterial species.12, 13, 14 Recently, Perapelov et al. reported15 the structure of the pentasaccharide repeating unit of the cell wall O‐antigen of E. coli O11, which is a member of ETEC strains3 and associated with diarrheal outbreaks. In the current scenario, bacterial infections caused by multidrug‐resistant strains are a serious concern and require alternative approaches to conventional therapeutics for controlling the diseases.16
Cell wall polysaccharides of bacterial strains have been used to develop polysaccharide conjugate vaccines against a number of bacterial infections.17, 18 However, use of polysaccharides isolated from the natural sources suffers from several drawbacks such as a) handling of live bacterial strains for the production and collection of polysaccharides, b) batch‐to‐batch variation in the length of the polysaccharides, c) lack of adequate purity and presence of biological impurities (e.g. lipids, nucleic acids), and d) tedious isolation protocols. In the recent past, it has been reported that the use of chemically synthesized glycoconjugate derivatives could act as vaccine candidates similar or somewhat superior to the conventional polysaccharide vaccines.19, 20 Therefore, it could be useful to develop glycoconjugate derivatives for their use as therapeutics to control diarrheal infections. The first step for the development of a glycoconjugate is to develop an efficient chemical synthetic strategy for the synthesis of the oligosaccharide components. As a part of the ongoing program for the synthesis of complex oligosaccharides, a convergent synthetic strategy for the synthesis of the pentasaccharide repeating unit of the cell wall O‐antigen of E. coli O11 is reported herein.
Since the target compound 1 contains four α‐glycosidic linkages and one β‐glycosidic linkage in the molecule, judiciously functionalized monosaccharide intermediates were chosen to couple them together with an appropriate stereochemical outcome at the glycosidic linkages. Application of a series of sequential glycosylation reactions in a multistep synthetic scheme often requires a large number of reaction steps resulting in a low overall yield. Alternatively, the number of steps can be decreased with the significant improvement of the overall yield by using a block glycosylation strategy. Therefore, in the present study, a convergent [3+2] block glycosylation approach was adopted to achieve the target compound. A set of suitably functionalized monosaccharide intermediates 4, 5,21 8,22 11,23 and 12 24 were prepared from the reducing sugars, applying the reaction conditions reported in the literature (Figure 1). The trisaccharide acceptor 10 was synthesized using sequential glycosylations and protecting group manipulations using the monosaccharide intermediates 4, 5, and 8. The disaccharide thioglycoside 13 was prepared by the glycosylation of two thioglycoside derivatives (11 and 12) in which compound 11 acted as glycosyl donor, allowing compound 12 to act as glycosyl acceptor by tuning their relative reactivity profile. The trisaccharide acceptor 10 was allowed to couple stereoselectively with the disaccharide thioglycoside 13 under a recently reported glycosylation condition25 to give the pentasaccharide derivative 14 in fairly good yield. The 2‐aminoethyl group at the reducing terminal of the pentasaccharide 1 could be used to conjugate the glycan moiety with an appropriate protein.
Figure 1.
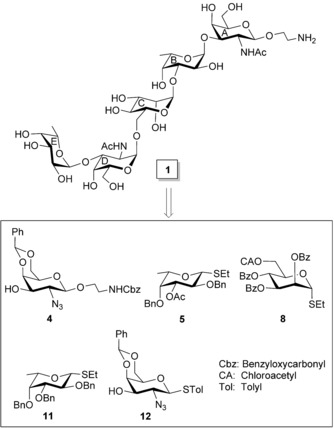
Structure of the synthesized pentasaccharide corresponding to the cell wall O‐antigen of E. coli O11 strain as its 2‐aminoethyl glycoside.
Stereoselective 1,2‐trans glycosylation of 1,3,4,6‐tetra‐O‐acetyl‐2‐azido‐2‐deoxy‐α/β‐d‐galactopyranose (2)26 with 2‐(N‐benzyloxycarbonyl)amino ethanol in the presence of borontrifluoride diethyletharate in acetonitrile utilizing the concept of nitrile effect27 of the solvent in β‐glycosylation furnished compound 3 in 69 % yield. A small quantity (∼10 %) of the 1,2‐cis glycoside was also formed, which was separated by repeated column chromatography. Appearance of a doublet at δ=4.30 ppm (J=8.0 Hz, H‐1) in the 1H NMR spectrum of compound 3 confirmed the formation of the 1,2‐trans glycoside. Treatment of compound 3 with sodium methoxide28 gave the de‐O‐acetylated product, which on treatment with benzaldehyde dimethyl acetal in the presence of p‐toluenesulfonic acid29 led to the formation of compound 4 in 76 % overall yield. Iodonium‐ion‐mediated stereoselective 1,2‐cis glycosylation of compound 4 with l‐fucosyl thioglycoside donor 5 in the presence of a combination25 of N‐iodosuccinimide (NIS) and perchloric acid supported over SiO2 (HClO4−SiO2)30 in a mixed solvent (CH2Cl2−Et2O; 1:2) resulted in the formation of the disaccharide derivative 6 in 70 % yield. The spectral analysis of compound 6 confirmed its formation: signals at δ=5.05 (d, J=3.5 Hz, H‐1B) and 4.28 ppm (d, J=9.0 Hz, H‐1A) in the 1H NMR and at 102.7 (C‐1A) and 100.3 ppm (C‐1B) in the 13C NMR spectra. Treatment of compound 6 with sodium methoxide furnished the disaccharide acceptor 7 in 90 % yield. Following the similar reaction condition as in the preparation of compound 6, compound 7 was stereoselectively coupled with d‐mannose‐derived thioglycoside donor 8 in the presence of a combination25 of NIS and HClO4−SiO2 in CH2Cl2−Et2O (1:2) to produce trisaccharide derivative 9 in 71 % yield. Exclusive formation of compound 9 was confirmed from its spectral analysis: signals at δ=5.71 (br s, H‐1C), 5.10 (d, J=3.5 Hz, H‐1B), and 4.42 ppm (d, J=8.5 Hz, H‐1A) in the 1H NMR and at 102.7 (C‐1A), 100.1 (C‐1B), and 99.1 ppm (C‐1C) in the 13C NMR spectra. The chloroacetyl group in compound 9 was selectively removed31 by the treatment with thiourea to give the trisaccharide acceptor 10 in 70 % yield without affecting the benzoyl group present in it (Scheme 1).
Scheme 1.
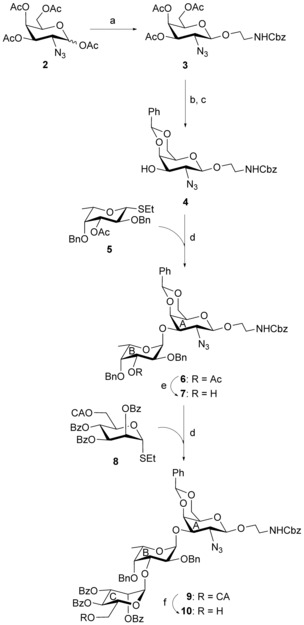
Reagents and conditions: a) 2‐(N‐benzyloxycarbonyl)amino ethanol, BF3⋅Et2O, CH3CN, 0 °C → rt, 36 h, 69 %; b) 0.1 m CH3ONa, CH3OH, rt, 3 h; c) PhCH(OCH3)2, p‐TsOH, mol. sieve 3 Å, CH3CN, rt, 8 h, overall 76 %; d) NIS, HClO4−SiO2, mol. sieve 4 Å, CH2Cl2−Et2O (1:2), −15 °C, 25 min, 6: 70 % and 9: 71 %; e) 0.1 m CH3ONa, CH3OH, rt, 1 h, 90 %; f) thiourea, CH2Cl2−CH3OH (1:1), 50 °C, 2 h, 70 %.
In another experiment, stereoselective orthogonal glycosylation32 of thioglycoside 11 and thioglycoside 12 in the presence of a combination25 of NIS and HClO4−SiO2 in a mixed solvent (CH2Cl2−Et2O; 1:4) afforded the disaccharide thioglycoside derivative 13 in 68 % yield. The presence of a benzyl group at the C‐2 position of compound 11 improved the nucleophilicity of the anomeric sulfur atom, whereas the azido group at the C‐2 position of compound 12 together with the tolyl moiety linked to the sulfur atom deactivated the nucleophilicity of the anomeric sulfur atom. As a result, compound 11 was activated faster than compound 12 in the presence of the thiophilic activator and behaved as a glycosyl donor. Having a hydroxy group present in the molecule, compound 12 played the role of a glycosyl acceptor. The spectral analysis of compound 13 confirmed its formation: signals at δ=4.91 (d, J=3.5 Hz, H‐1E) and 4.31 ppm (d, J=9.5 Hz, H‐1D) in the 1H NMR and 100.8 (C‐1E) and 85.4 ppm (C‐1D) in the 13C NMR spectra. Finally, glycosylation of the trisaccharide acceptor 10 with the disaccharide thioglycoside donor 13 in the presence of a combination25 of NIS and HClO4−SiO2 in a mixed solvent (CH2Cl2−Et2O; 1:4) resulted in the formation of the pentasaccharide derivative 14 in 70 % yield. The stereochemistry at the glycosidic linkages in compound 14 was confirmed from its spectral analysis: signals at δ=5.64 (br s, 1 H, H‐1C), 5.14 (d, J=3.5 H, H‐1D), 5.05 (d, J=3.5 Hz, H‐1B), 5.01 (d, J=3.5 Hz, H‐1E), and 4.25 ppm (d, J=8.5 Hz, H‐1A) in the 1H NMR and 102.8 (C‐1A), 100.6 (C‐1D), 99.6 (C‐1C), 99.4 (C‐1E), and 98.9 ppm (C‐1B) in the 13C NMR spectra. Compound 14 was subjected to a sequence of functional‐group transformations consisting of a) direct conversion of azido group into acetamido group using thioacetic acid,33 b) removal of O‐benzoyl group using sodium methoxide, and c) removal of benzyl ethers and benzylidene acetal by catalytic transfer hydrogenation34 using triethylsilane in the presence of Pd(OH)2−C to give the target pentasaccharide 1 as its 2‐aminoethyl glycoside in 54 % overall yield. The structure of compound 1 was unambiguously confirmed from its spectral analysis: signals at δ=5.10 (br s, H‐1C), 5.04 (d, J=3.0 Hz, H‐1B), 5.03 (d, J=3.0 Hz, H‐1E), 4.92 (d, J=3.5 Hz, H‐1D), and 4.59 ppm (d, J=8.5 Hz, H‐1A) in the 1H NMR and 102.6 (C‐1C), 101.2 (C‐1B), 101.0 (C‐1E), 100.9 (C‐1A), and 97.2 ppm (C‐1D) in the 13C NMR spectra (Scheme 2).
Scheme 2.
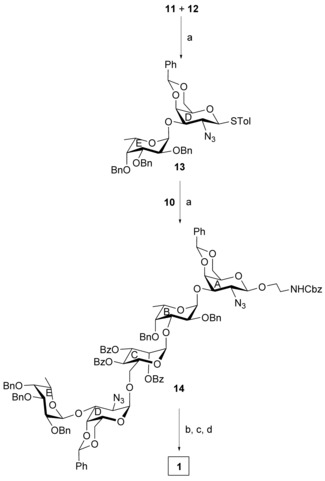
Reagents and conditions: a) NIS, HClO4−SiO2, CH2Cl2−Et2O (1:4), −10 °C, 30 min, 13: 68 % 14: 70 %; b) CH3COSH, pyridine, rt, 16 h; c) 0.1 m CH3ONa, CH3OH, rt, 6 h; d) Et3SiH, Pd(OH)2−C (20 %), CH3OH, rt, 18 h, overall 54 %.
In summary, a pentasaccharide corresponding to the repeating unit of the cell wall polysaccharide of E. coli O11 was synthesized in good yield using a convergent [3+2] block glycosylation approach. During the synthesis of the target molecule, similar reaction conditions were used in all intermediate glycosylation reaction steps with satisfactory yield and stereoselectivity. A stereoselective glycosylation was carried out between thioglycoside donor and thioglycoside acceptor by tuning their relative reactivity profile.
Experimental Section
For details of the experimental conditions and characterization data for the compounds reported, see the Supporting Information.
Supporting information
As a service to our authors and readers, this journal provides supporting information supplied by the authors. Such materials are peer reviewed and may be re‐organized for online delivery, but are not copy‐edited or typeset. Technical support issues arising from supporting information (other than missing files) should be addressed to the authors.
Supplementary
Acknowledgements
A. S. thanks the Council of Scientific and Industrial Research (CSIR), New Delhi, India for providing a Junior Research Fellowship. This work was supported by CSIR, New Delhi, India (A. K. M., Project No. 02(0038)/11/EMR‐II). Mr. Barun Majumdar of the Biophysics Department, Bose Institute is thankfully acknowledged for recording NMR spectra.
A. Si, A. K. Misra, ChemistryOpen 2016, 5, 47.
References
- 1. Pawlowski S. W., Warren C. A., Guerrant R., Gastroenterology 2009, 136, 1874–1886. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2. Lund B. M., O'Brien S. J., Foodborne Pathog. Dis. 2011, 8, 961–973. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3. Nataro J. P., Kaper J. B., Clin. Microbiol. Rev. 1998, 11, 142–201. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4. Garcia M.-I., Le Bouguenéc C., Clin. Microbiol. Infect. 1998, 4, 38–43. [DOI] [PubMed] [Google Scholar]
- 5. Clements A., Young J. C., Constantinou N., Frankel G., Gut Microbes 2012, 3, 71–87. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6. Kaper J. B., Nataro J. P., Mobley H. L., Nat. Rev. Microbiol. 2004, 2, 123–140. [DOI] [PubMed] [Google Scholar]
- 7. Manges A. R., Johnson J. R., Foxman B., O'Bryan T. T., Fullerton K. E., Riley L. W., N. Engl. J. Med. 2001, 345, 1007–1013. [DOI] [PubMed] [Google Scholar]
- 8. Johnson J. R., Gajewski A., Lesse A. J., Russo T. A., J. Clin. Microbiol. 2003, 41, 5798–5802. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9. Shah N., DuPont H. L., Ramsey D. J., Am. J. Trop. Med. Hyg. 2009, 80, 609–614. [PubMed] [Google Scholar]
- 10. Qadri F., Svennerholm A. M., Faruque A. S., Sack R. B., Clin. Microbiol. Rev. 2005, 18, 465–483. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11. Lerouge I., Vanderleyden J., FEMS Microbiol. Rev. 2002, 26, 17–47. [DOI] [PubMed] [Google Scholar]
- 12. Stenutz R., Weintraib A., Widmalm G., FEMS Microbiol. Rev. 2006, 30, 382–403. [DOI] [PubMed] [Google Scholar]
- 13. Liu B., Knirel Y. A., Feng L., Perepelov A. V., Senchenkova S. N., Wang Q., Reeves P. R., Wang L., FEMS Microbiol. Rev. 2008, 32, 627–653. [DOI] [PubMed] [Google Scholar]
- 14. Liu B., Knirel Y. A., Feng L., Perepelov A. V., Senchenkova S. N., Reeves P. R., Wang L., FEMS Microbiol. Rev. 2014, 38, 56–89. [DOI] [PubMed] [Google Scholar]
- 15. Li Y., Perepelov A. V., Guo D., Shevelev S. D., Senchenkova S. N., Shahskov A. S., Liu B., Wang L., Knirel Y. A., FEMS Immunol. Med. Microbiol. 2011, 61, 258–268. [DOI] [PubMed] [Google Scholar]
- 16. Byarugaba D. K., Int. J. Antimicrob. Agents 2004, 24, 105–110. [DOI] [PubMed] [Google Scholar]
- 17. Butler J. C., Breiman R. F., Campbell J. F., Lipman H. B., Broome C. V., Facklam R. R., JAMA 1993, 270, 1826–1831. [PubMed] [Google Scholar]
- 18. Ada G., Isaacs D., Clin. Microbiol. Infect. 2003, 9, 79–85. [DOI] [PubMed] [Google Scholar]
- 19. Roy R., Carbohydrate-based Vaccines, Vol. 989, ACS Symposium Ser., Washington, DC, 2008, pp. 1–379. [Google Scholar]
- 20. Galonić D. P., Gin D. Y., Nature 2007, 446, 1000–1007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21. Ziegler T., Carbohydr. Res. 1994, 262, 195–212. [DOI] [PubMed] [Google Scholar]
- 22. Fyrner T., Svensson S. C. T., Konradsson P., Tetrahedron 2012, 68, 6712–6720. [Google Scholar]
- 23. Lönn H., Carbohydr. Res. 1985, 139, 105–113. [DOI] [PubMed] [Google Scholar]
- 24. Zhang Z., Ollmann I. R., Ye X.-S., Wischnat R., Wischnat R., Baasov T., Wong C.-H., J. Am. Chem. Soc. 1999, 121, 734–753. [Google Scholar]
- 25. Mukherjee C., Misra A. K., Synthesis 2007, 683–692. [Google Scholar]
- 26. Guazzelli L., Catelani G., D′Andrea F., Giannarelli A., Carbohydr. Res. 2009, 344, 298–303. [DOI] [PubMed] [Google Scholar]
- 27. Toepfer A., Kinzy W., Schmidt R. R., Liebigs Ann. Chem. 1994, 449–464. [Google Scholar]
- 28. Zemplén G., Ber. Dtsch. Chem. Ges. 1926, 59, 1254–1266. [Google Scholar]
- 29. Hanessian S., Preparative Carbohydrate Chemistry, Marcel Dekker, New York, 1997, pp. 53–67. [Google Scholar]
- 30. Chakraborti A. K., Gulhane R., Chem. Commun. 2003, 1896–1897. [DOI] [PubMed] [Google Scholar]
- 31. Masaki M., Kitahara T., Kurita H., Ohta M., J. Am. Chem. Soc. 1968, 90, 4508–4509. [Google Scholar]
- 32. Kanie O., Ito Y., Ogawa T., J. Am. Chem. Soc. 1994, 116, 12073–12074. [Google Scholar]
- 33. Rosen T., Lico I. M., Chu D. T. W., J. Org. Chem. 1988, 53, 1580–1582. [Google Scholar]
- 34. Santra A., Ghosh T., Misra A. K., Beilstein J. Org. Chem. 2013, 9, 74–78. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
As a service to our authors and readers, this journal provides supporting information supplied by the authors. Such materials are peer reviewed and may be re‐organized for online delivery, but are not copy‐edited or typeset. Technical support issues arising from supporting information (other than missing files) should be addressed to the authors.
Supplementary