

Abstract
Cyclization into closed assemblies is the most recurrent approach to realize the noncovalent synthesis of discrete, well‐defined nanostructures. This review article particularly focuses on the noncovalent synthesis of monocyclic hydrogen‐bonded systems that are self‐assembled from a single molecule with two binding‐sites. Taking advantage of intramolecular binding events, which are favored with respect to intermolecular binding in solution, can afford quantitative amounts of a given supramolecular species under thermodynamic control. The size of the assembly depends on geometric issues such as the monomer structure and the directionality of the binding interaction, whereas the fidelity achieved relies largely on structural preorganization, low degrees of conformational flexibility, and templating effects. Here, we discuss several examples described in the literature in which cycles of different sizes, from dimers to hexamers, are studied by diverse solution or surface characterization techniques.
Keywords: chelate cooperativity, hydrogen bonds, macrocycles, self-assembly, supramolecular chemistry
1. Introduction
Self‐assembly is a central area of science whose foundations lay between chemistry, with the fundamental study of molecular recognition processes; biology, with the understanding of the structure and function of complex natural systems; and materials science, with the design of advanced materials, devices, and nanostructures endowed with a particular function.1 Self‐assembly could be defined as the spontaneous generation of ordered architectures from a set of components driven by the interplay of diverse noncovalent interactions. Since these supramolecular interactions are weak in nature, self‐assembly is typically controlled by thermodynamics and thereof self‐assembled systems are formed in equilibrium conditions.
When self‐assembly is directed towards a well‐defined (uniform) and discrete (monodisperse) species, the term supramolecular synthesis or noncovalent synthesis is often employed.2, 3 To reach such aim, molecules must be carefully programmed with the required chemical information so as to associate in a given designed architecture. This process tries to mimic the “intelligent” collective behavior observed in the natural world, where the information is encoded in the covalent framework of biomolecules and determines the structure and function of complex biological machineries in the nano‐ and mesoscale.4, 5, 6, 7, 8 The noncovalent synthesis of functional nanoarchitectures with a precision analogous to that found in the biological world requires an understanding of individual noncovalent interactions (metal–ligand coordination,9 hydrogen bonding,2 ionic pairs, π–π stacking,10 van der Waals forces),11 their synergy when several of them are present in the system,12, 13 as well as the response of the system to temperature, concentration, and solvent environment changes. Of equal importance is the comprehension of cooperative and multivalent phenomena that may arise between the individual building blocks,14, 15, 16, 17 since a fine control of structure, and hence of a well‐defined function, depends largely on this issue.
A molecule with more than one binding site may, in principle, assemble into open (linear) or closed (cyclic) structures (Figure 1). The size of linear oligomers or polymers can be limited within a certain range by adjusting monomer concentration, adding end‐capping units,18, 19 controlling nucleation‐elongation processes,20 or through other approaches,21 but the supramolecular product is commonly a statistical distribution of chain lengths.22, 23, 24, 25, 26 Therefore, the goal of reaching well‐defined, discrete supramolecular structures has been normally focused on closed (multi)macrocyclic cages, where size is controlled by the geometric requirements of both the monomer and the binding interaction or by the use of particular templates, both of these approaches leading to ring closure.27, 28 The resulting closed, cyclic species may be formed quantitatively because it enjoys a thermodynamic stability that is substantially larger than the sum of the corresponding individual interactions.29 The synergistic effect that causes such increased stability is defined as chelate cooperativity and stems from the fact that an intramolecular (or intraspecies) interaction is favored over an intermolecular one, providing a series of conditions of enthalpic and entropic origin are met (see below). Although other forms of cooperativity have been described (allosteric, interannular),14, 15, 16 chelate cooperativity is mainly responsible for many of the “all‐or‐nothing” processes that are characteristic of discrete supramolecular systems: either a given supramolecular structure is formed in a certain set of conditions or nothing else can survive and only monomeric species are present.30
Figure 1.
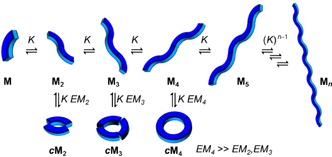
Possible supramolecular equilibria of a molecule with a given geometry and two binding sites associating with an intermolecular equilibrium constant K. Linear supramolecular oligomers (Mn) are in equilibrium with cyclic species (c M n). In this particular case, a cyclic tetramer is stabilized because the monomer and binding interaction geometric features afford a much higher effective molarity (EM) value.
A key parameter quantifying chelate cooperativity is the effective molarity (EM),31, 32 which affords an estimate of how favorable the intramolecular binding interaction is with respect to the intermolecular one. In other words, for thermodynamically controlled processes, the EM is defined by the ratio between the equilibrium constant leading to a cyclic system and the one leading to a linear system (EM=K intra/K inter).33, 34, 35, 36 Hence, the increased in stability when comparing a linear and a cyclic oligomer of a certain length (Figure 1) is given by the product K inter⋅EM, where the factor K inter considers the additional association to form the cycle, and EM takes into account that this last binding event is intramolecular.
However, cycle formation has its own enthalpic and entropic contributions, which regulate the magnitude of EM.37, 38, 39, 40, 41, 42, 43 The enthalpic component is mainly dominated by secondary interactions that may occur in the cycle due to the attractive or repulsive alignment of molecular dipoles and, primarily, by the creation of ring strain upon cyclization. Therefore, preorganization of the structure of the monomeric components towards a certain cycle size arises as a fundamental design factor to achieve high EM values and hence quantitative yields of the target self‐assembled structure. On the other hand, entropy has much to rule on supramolecular macrocyclizations. Entropy is responsible for the preference of an intramolecular binding event with respect to the intermolecular one, since in this way some rotational and the translational molecular degrees of freedom are not lost upon association. However, cyclization also involves a loss of entropy due to restriction of certain torsional motions in the closed species. This is more accused when the monomer has a flexible structure, with several rotatable bonds, so rigid monomers are preferred in order to increase EM values. Entropy also penalizes the formation of large cycles, and the EM tends to dissipate in closed assemblies formed from many molecules.
In this review article, we have assembled the efforts of many scientists directed to the noncovalent synthesis of cyclic systems from molecules that interact through two binding sites, so that the closed systems formed are two‐dimensional, or formally speaking, monocyclic.44 We are also limiting this revision to the examples found in the literature where this supramolecular interaction is the hydrogen (H)‐bond. Other excellent recent manuscripts have reviewed the self‐assembly of cyclic systems through other major supramolecular binding interactions, such as metal–ligand coordination.36, 45 H‐bonding is however considered as a powerful means to reach a high degree of control over supramolecular architectures, since it can be made highly selective and directional.2, 12, 13, 46, 47 H‐bonds are electrostatic in nature, occurring when a donor (D) group with an available acidic hydrogen atom interacts with an acceptor (A) group carrying available nonbonding electron lone pairs. The strength of this interaction depends mainly on the solvent (a competitor in the formation of H‐bonds), the chemical nature of the H‐bonding donor and acceptor functions, as well as on their number and sequence in a particular molecular fragment.23, 48, 49, 50, 51, 52
Being built from a weak interaction, when compared for instance to the covalent bond, self‐assembled H‐bonded systems in general, and cyclic systems in particular, need to be characterized through appropriate techniques.53 Due to their labile and dynamic features, H‐bonded assemblies are stable only at a well‐defined range of reactant concentrations, temperatures, and solvent environments. Spectroscopic techniques, such as NMR, Fourier transform infrared (FT‐IR), absorption, or emission spectroscopy are essential to provide structural information and to extract the main thermodynamic (equilibrium constants and enthalpic and entropic changes) and kinetic (exchange rate constants) parameters of the self‐assembled systems in solution. These studies must be accompanied by the use of other techniques that certify the size, stoichiometry, and monodispersity of the aggregates, such as gel permeation chromatography (GPC), vapor phase osmometry, NMR diffusion experiments, and mass spectrometry (MS). On the contrary, X‐ray diffraction or STM imaging, although highly valuable characterization techniques, may not always be fully representative of the architecture present in solution. After all, the intramolecular event that shifts all the equilibria to form a particular cycle is only relevant at low concentrations, whereas when the molecules are concentrated in the crystal or onto a substrate, intra‐ and intermolecular binding events are compensated. In this review article we also emphasize the most relevant techniques and methods employed by the different authors to characterize their cyclic assemblies, both in solution and onto solid substrates.
2. Hydrogen‐Bonded Macrocycles in Solution
We start this review article with diverse examples of H‐bonded monocyclic systems, formed from a given molecule with two binding sites, studied in solution. We will progress with increasing the size of the system, from dimeric to hexameric cycles. Due to the huge number of systems that dimerize in solution via H‐bonding, we are limiting our discussion to a very few selected examples that form a cycle and that are relevant for the analysis of the higher‐order systems, which are the main focus of this work and therefore will be described in further detail. In many cases, the cyclomerization constants of these assemblies were not calculated or estimated in a solvent in which the reference intermolecular association constant was not reported. Hence, for most of these systems, a comparative analysis of the EM values cannot be performed.
2.1. Two‐membered hydrogen‐bonded macrocycles
Sessler et al. designed dimeric base derivatives, containing complementary pairs, as recognition units in order to extend the degree of association of the corresponding monomers and with the objective of incorporating sequence specificity in the self‐assembly process.54, 55, 56, 57
1H NMR titration experiments of the self‐association of 1 and the monomeric species 2, 3 (Figure 2 a) in [D6]DMSO did not reveal a significant increase in the association constant in the dimer species (K b(1–1)=4.5–6.8 m −1) relative to the monomers (K b(2–3)=3.9–4.7 m −1) suggesting that just a small additional binding affinity is produced by the use of dimers. This unexpected behavior could be due to both the high conformational flexibility of the system, which implies a large entropic requirement to produce a ditopic complex (Figure 2 b) and hence a low EM value, and the poor solubility of the starting components.54 Based on this result, the authors synthesized more rigid systems by using 1,8‐diethynylanthracene as the spacer and complementary A−U base pairing entities as the recognition elements (Figure 3 a).55
Figure 2.
a) Heterodimer 1 and monomeric 2, 3 species. b) Ditopic and monotopic binding modes for self‐association of heterodimer 1 in [D6]DMSO. Adapted with permission from Ref. 54. Copyright 1992, American Chemical Society.
Figure 3.
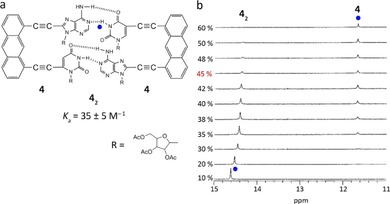
a) A−U dinucleotide system 4 and its corresponding homodimer 42. b) Changes in the 1H NMR spectra of 4 upon increasing the volume fraction of [D6]DMSO in CDCl3. Adapted with permission from Ref. 55. Copyright 1996, American Chemical Society.
1H NMR titrations of 4 in CDCl3 with DMSO showed two separate signals corresponding to the uridine N−H protons (Figure 3 b); this fact is consistent with exchange between monomer and dimer being slow on the NMR scale. Such slow exchange is usually observed in the case of very tightly bound complexes and lead the authors to infer that self‐association of 4 is favorable not only in CDCl3 but also under [D6]DMSO/CDCl3 mixed solvent conditions. The association constant of 4 in 45 % (v/v) [D6]DMSO/CDCl3 was determined to be 35±5 m −1. The strong self‐association of 4 demonstrated that the rigid homodimers of AU dinucleotide have an improved stability compared to the monomeric base pairs, whereas the use of flexible spacers (Figure 2 a) hardly improved dimer stability.54, 55
Since the appreciation that minor variations in the structure (e.g. rigidity, preorganization) and solubility are critical aspects in the self‐dimerization process, a comparative study was conducted to develop greater understanding of the effect of such factors on the association behavior.56, 57 Figures 3 and 4 display the target A−U (4–6) and G−C (7) functionalized systems chosen by the authors.57 Downfield shifts of the imino N−H protons of the uridine moieties of compounds 4–6 and of guanosine moiety of compound 7 in CDCl3 indicated the presence of strong self‐associating interactions mediated through H‐bonds (Figure 3 b and 4 d–f). Supporting this conclusion was the fact that such downfield shifts were not seen in [D6]DMSO, and under these conditions, the monomeric species predominated.
Figure 4.
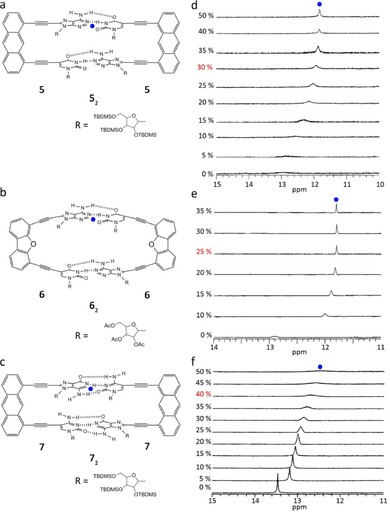
a, b) A−U dinucleoside duplexes 5 and 6. c) G−C dinucleoside duplex 7. Changes in the 1H NMR spectra of d) 5, e) 6, and f) 7 upon increasing the volume fraction of [D6]DMSO in CDCl3. Adapted with permission from Ref. 57. Copyright 1998, American Chemical Society.
As the authors demonstrated earlier, the dimeric species of 4 showed remarkable resistance toward dissociation and became fully dissociated when the relative [D6]DMSO concentration reached 60 % or higher (Figure 3 b).55 However, solutions of 52 and 62 in CDCl3 could easily be dissociated by the addition of DMSO. For instance, Figure 4 d shows that the very broad signal from the imino NH observed at 13.0 ppm in the 1H NMR spectrum of 52 became sharp when [D6]DMSO was added. Further, the signal became insensitive to [D6]DMSO concentration (11.98 ppm) when 30 % DMSO v/v was added. Such findings were consistent with species 52 being able to survive only a relatively low (<30 %) concentration of DMSO before becoming totally dissociated into monomer 5 (Figure 4 d). A similar experiment determined that 62 is even less stable, it can only survive 25 % [D6]DMSO (Figure 4 e). Despite the fact that 72 contains stronger H‐bonds than 42, the dimeric species of the GC derivative 7 was dissociated with a 40 % of [D6]DMSO (Figure 4 f). All these features indicated that either the lower structural preorganization of 6 or the presence of bulky protecting groups in 5 and 7, which causes severe steric hindrance in the dimers, leads to a significant reduction in the dimer stability. It is interesting to note, when comparing the four systems, that the kinetic behavior is also strongly affected by their different geometric features and steric hindrances. While the more stable dimer 42 exhibited a slow equilibrium with the monomer in the NMR timescale, the rest of the assemblies were in the fast exchange regime at room temperature.
Taking the advantage of the strong DDAA–AADD quadruple H‐bond motifs in the 2‐ureido‐4‐pyrimidinones (UPys),58, 59 Sijbesma, Meijer and co‐workers have designed bis‐UPy scaffolds as a strategy to form stable cyclic dimers in solution.60, 61, 62, 63 For instance, UPy derivatives 8, containing methyl‐substituted alkyl linkers between the H‐bonding UPy moieties (Figure 5 a), assembled by quadruple H‐bonding to form homochiral cyclic dimers in chloroform. The preorganization of the monomers and the combined binding strength of the 8 H‐bonds resulted in a very high stability of the cyclic aggregates with pronounced selectivity between homochiral and heterochiral cyclic species.61, 62
Figure 5.
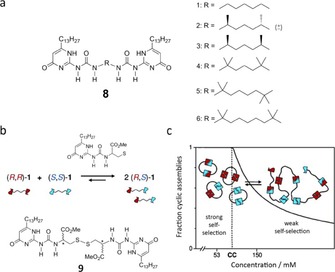
a) Structure of UPy derivative 8. Adapted with permission from Ref. 62. Copyright 2004, American Chemical Society. b) Equilibrium between diastereomers of 9 in disulfide exchange reactions. c) Correlation between self‐selection and critical concentration. Reproduced with permission from Ref. 63. Copyright 2005, American Chemical Society.
More complex studies involving disulfide exchange reactions64 demonstrated a concentration‐dependent stereochemical self‐selection process in chiral disulfides UPys (Figure 5 b).63 Using dynamic covalent chemistry (DCC) methods65 the authors established that below a critical concentration, where formation of cyclic assemblies is the dominant mode of association, the process is governed by the self‐selection of the chiral UPy groups, forming preferentially meso (R,S)‐9 cycles. Above the critical concentration the mixture was dominated by linear aggregates, due to the diminished influence of self‐selection (Figure 5 c). These results indicated the importance of strong H‐bonding in selective self‐assembly processes and how the disulfide exchange reaction can be a valuable tool to achieve pairing selectivity.
2.2. Three‐membered hydrogen‐bonded macrocycles
Zimmerman and Duerr reported the cyclotrimerization of pyrido[4,3 g]‐quinolinedione monomers 10 (Figure 6 a).66 The self‐assembly of the complementary components takes place in a cooperative way through the formation of six H‐bonds in 103. Vapor‐pressure osmometry (VPO) and concentrationdependent 1H NMR studies in chloroform confirmed the high stability of the assembly. The association constant in 10 % [D6]DMSO/CDCl3 was calculated by fitting the dilution curves to models for an n‐merization process using the Saunders–Hyne method.67 The best‐fit trimer model fit better across the entire concentration range than the best‐fit dimer and tetramer models, which deviated substantially from the experimental data (Figure 6 b). This demonstrated that the three‐membered H‐bonded cycle is the most stable species in solution (K3=20000 m −2). The EM of this trimeric cycle was recently analyzed by Ballester and de Mendoza in a review chapter as 740 m,44 which is quite high and compatible with the conditions of quantitative assembly defined by Ercolani as K inter⋅EM≫185⋅n,36 where n is the number of monomeric units in the cycle. Such a high value must originate from the low conformational flexibility of the π‐conjugated system connecting the two amide units, which preorganizes the molecule at appropriate angles for efficient cyclotrimerization.
Figure 6.
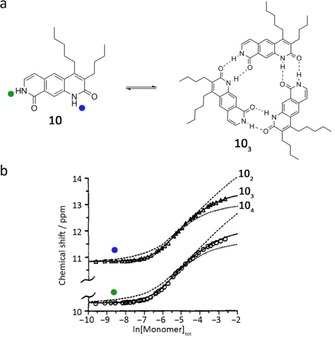
a) Cyclotrimerization of pyrido[4,3g]‐quinolinedione monomer 10. b) Saunder–Hyne analysis of 1H NMR dilution shift of 10 in 10 % [D6]DMSO/CDCl3. Adapted with permission from Ref. 66. Copyright 1992, American Chemical Society.
Another interesting motif used as a module for obtaining stable supramolecular cyclic trimers are the phthalhydrazides. These derivatives exist as an equilibrium of three tautomeric forms: the lactam‐lactam (A), the lactim‐lactam (B), and the lactim‐lactim (C), with B being the preferred form (Figure 7 a). Lactim‐lactam monomer 11 is able to self‐assemble into trimeric disks of high stability in toluene (Figure 7 b) via DA‐A′D’ H‐bonding interactions, as temperature‐dependent 1H NMR and size‐exclusion chromatography (SEC) experiments have confirmed. With suitable substituents, the authors have also proven that phthalhydrazide‐based trimers can act as a powerful self‐organizing unit and produce a stacked base triplet to form thermotropic columnar liquid crystals.68
Figure 7.
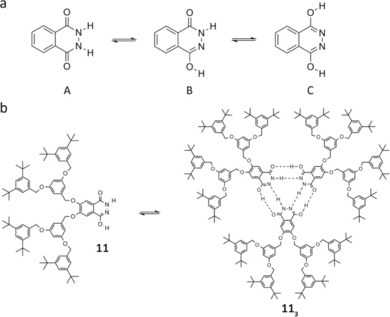
a) Phthalhydrazide tautomerization: lactam‐lactam (A), lactim‐lactam (B), and lactim‐lactim (C). b) Self‐assembly of the phthalhydrazide lactim‐lactam cyclic trimer of monomer 11. Adapted with permission from Ref. 68. Copyright 1998, American Chemical Society.
Following the previous strategy of designing artificial guanosine (G)‐cytidine (C) dinucleosides to form cohesive dimers,57 Sessler and co‐workers synthesized a novel G−C derivative (12) to extend the supramolecular cyclization process to trimers (Figure 8).69 In this case 2D‐NOESY (nuclear Overhauser effect spectroscopy) experiments showed strong cross‐peaks between the guanosine imino proton (NH‐1) and the cytidine amino proton (NH‐4) indicating a close spatial arrangement between them. These results are consistent with the Watson–Crick base pair between individual monomers of 12. Besides, electrospray ionization mass spectrometry (ESI‐MS) and SEC experiments provided the size of the resulting supramolecular species, being consistent with a trimeric structure (123).
Figure 8.
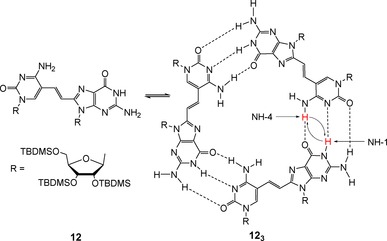
Self‐assembly of 12 into trimeric species (123). Adapted with permission from Ref. 69. Copyright 2003, American Chemical Society.
The last selected example of three‐membered H‐bonded macrocycle was reported by Ochsenfeld, Xie, and Schrader's groups.70 As shown in Figure 9, the aminopyrazole peptide hybrid 13 displays a well‐ordered structure which somehow resembles nucleic acid self‐assembly. In a strictly hierarchical process, formation of aminopyrazole “base” cyclic triplets via a H‐bond network (133) is accompanied by π‐stacking with a second rosette and final dimerization of two double rosettes (133)2 to a four‐layer superstructure 2×(133)2, stabilized by a six‐fold half‐crown alkylammonium lock. The resulting compact assembly is stable in the solid state as well as in solution. In water, the 1H NMR spectrum of 13 at 275 K along with the diffusion‐ordered spectroscopy (DOSY) measurements allowed the authors to identify two species in solution (Figure 10). The two markedly different diffusion coefficients were assigned to the monomeric oligopeptide with a calculated hydrodynamic radius of 8.4 Å and the hexameric aggregate with an Rh value of 15.6 Å (Figure 10 b). The total absence of detectable intermediate structures underlines the high degree of cooperativity of the self‐assembly process, a feature quite characteristic of cyclic species.
Figure 9.
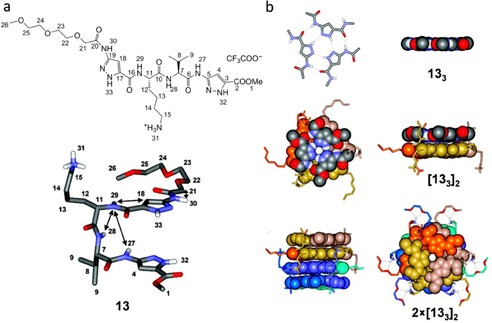
a) Peptide 13 and its conformation in the crystal. b) Segments of the crystal structure of 13, demonstrating the three hierarchical levels of self‐assembly: rosette, double rosette, and two‐double rosette. Adapted with permission from Ref. 70. Copyright 2008, American Chemical Society.
Figure 10.
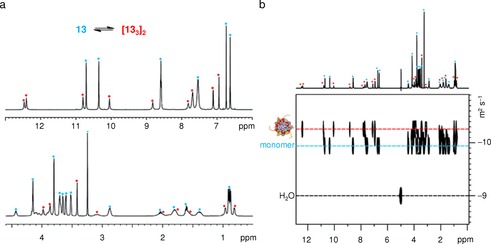
a) NMR spectrum and b) DOSY of monomer 13 and hexamer (133)2 after cooling to 275 K. Blue, monomer; red, hexamer signals. Adapted with permission from Ref. 70. Copyright 2008, American Chemical Society.
2.3. Four‐membered hydrogen‐bonded macrocycles
An especially attractive goal would be to use external stimuli to control the self‐association process between linear and cyclic species. In particular, Sleiman and co‐workers reported a new strategy to effect photochemical control over molecular self‐assembly by using photoresponsive azobenzene units as supramolecular building blocks.71 In the monomers 14 and 15 (Figure 11 a) the photoswitchable azobenzene unit is incorporated in the main chain, which can undergo a reversible trans–cis photoisomerization.72 In the trans‐isomer 14, due to the two aligned carboxylic groups, the monomer self‐assembles into linear aggregates (14 n), while in the photogenerated cis‐form 15, the carboxylic acids are oriented in a near perpendicular fashion; thus, they can self‐assemble into cyclic structures (154).73
Figure 11.
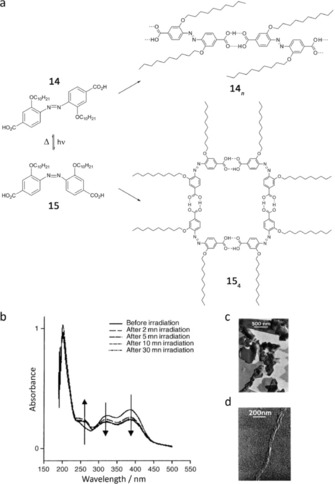
a) H‐bond self‐assembly of trans‐14 and cis‐15 azodibenzoic acid. b) UV/Vis absorption spectra recorded during the UV irradiation (λ>300 nm) of a 3.44×10−5 m solution of trans‐14 in MeOH. c) TEM image of 14. d) TEM image of cis‐15. Adapted with permission from Ref. 71. Copyright 2003, Wiley‐VCH Verlag GmbH & Co. KGaA.
The reversible photoisomerization process was followed by UV/Vis and 1H NMR spectroscopy. Irradiation of trans‐14 at λ>300 nm results in a decrease in the two absorption bands at 321 and 387 nm, with an isosbestic point at 286 nm, and the appearance of a new peak at 250 nm corresponding to cis‐15 (Figure 11 b). In [D6]DMSO, this photoisomerization results in distinct 1H NMR signals for 14 and 15, and a 1:1.3 trans–cis photostationary state can be reached after 2 h of irradiation. Kinetic studies carried out on the cis–trans thermal conversion showed that the enthalpy of activation obtained was higher in CHCl3 (ΔH≠=97.4±4.2 kJ mol−1) than in DMSO (ΔH≠=84.9±2.5 kJ mol−1), and the entropy of activation was less negative in CHCl3 (ΔS≠=−15.9±13.7 J mol−1 K−1) than in DMSO (ΔS≠=−72.8±8.0 J mol−1 K−1). This data is consistent with an additional contribution to activation energy of the cis–trans isomerization, which must be coming from the dissociation of the cis‐15 H‐bonded supramolecular structure. This result seems to confirm the presence of discrete cyclic tetramers (154) in chloroform of increased stability than linear H‐bound oligomers. Transmission electron microscopy (TEM) and dynamic light scattering (DLS) measurements also showed large differences in the self‐assembly behavior of 14 and 15 and revealed a second level of self‐organization of these molecules. Large sheet‐like aggregates were observed for trans‐14 (Figure 11 c), in agreement with the ordered arrangement of stacked linear tapes. In contrast, the cis‐15 isomer formed large bundles of 23 nm in diameter and 430 nm in length (Figure 11 d). This result suggests further organization of the supramolecular cycles of 154 through π–π stacking and/or alky‐alkyl interactions.
Again, the potent GC H‐bonding motif to direct association has been used by Perrin et al.74 to self‐assemble 16 into tetrameric rosettes. The self‐complementary G C heterocycle synthesized by these authors orients the H‐bonding faces of both guanine (ADD) and cytosine (DAA) at a 90 ° angle, which is dictated by the central pyrrole unit. Association then leads to a tetrameric cycle containing 12 H‐bonds (Figure 12 a). Variable‐temperature 1H NMR from 25 to −70 °C on a solution of 16 in [D6]DMSO/CDCl3 verified H‐bonding between the two faces of the monomer (Figure 12 b). At higher temperatures, NaH2 of the G‐face rotates rapidly on the NMR time scale, and both protons appear as a single broad coalesced resonance at 6.3 ppm. At −65 °C they resolve as two distinct peaks at 5.75 and 7.3 ppm. In contrast, the amino protons of the NbH2 of the C‐face appear as a very broad, almost undetectable resonance (6.75–7.65 ppm), which at −10 °C rapidly splits into two well‐resolved peaks at approximately 6.8 and 7.5 ppm. At −65 °C the NH2 protons present four distinct resonances. The observation of new peaks at very low temperature is consistent with a G−C pairing scheme and suggests formation of a tetrameric rosette in solution. DOSY experiments as well as ESI‐MS analysis of 16 (Figure 12 c) supported the tetrameric association, due to the fact that neither a peak for the trimer nor for any other higher‐order aggregate was detected.
Figure 12.
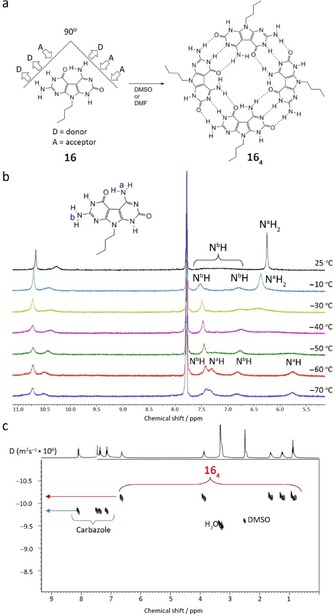
a) Monomer G C 16 and the corresponding tetrameric rosette structure 163. b) Temperature‐dependent 1H NMR experiments of 16 in [D6]DMSO/CDCl3 (60/40 %). c) DOSY of the equimolar mixture of 16 and carbazole in [D6]DMSO. Adapted with permission from Ref. 74. Copyright 2008, American Chemical Society.
In another remarkable contribution in 2011, Butkus, Wärnmark, and colleagues reported the first selective assembly by tautoleptic aggregation of an enantiomerically pure cavity, that is, a supramolecular belt, from one enantiomerically pure G C monomer containing one inherently nonself‐complementary motif.75
The demonstrated experience of de Mendoza and co‐workers in the field of UPy derivatives76, 77 allowed them to build a well‐defined cyclic tetramer like 174 (Figure 13 e).76 In their molecular design, two 2‐ureido‐4‐[1H]‐pyrimidinone (UPy) moieties were attached to a 3,6‐carbazolyl spacer with an angle of about 90 ° in order to favor cyclic arrangements of the monomer. In 17, the UPy subunits can adopt multiple conformations giving rise to cyclic oligomers of various sizes and shapes (Figure 13 a). However model optimizations for either cyclic trimers or tetramers suggested that only UPy subunits in which each monomer are pointing to opposite sides are compatible (Figure 13 a–c). Besides, in the case of the tetramer, a very favorable, sterically unhindered tubular (belt‐like) shape can be generated with both UPy subunits being mirror images pointing to the same side of the carbazole spacer (Figure 13 d). Low‐temperature NMR experiments showed unequivocally that the structure shown in Figure 13 e is the most favorable associated species. At 213 K (10 mm in CDCl3), the spectrum displays a unique set of sharp signals for the UPy subunits corresponding to a symmetric structure with a plane of symmetry bisecting the carbazole core. As expected, the methylene protons attached to the carbazolyl core split into two doublets, accounting for a rigid structure with different environments for each proton (Figure 14). These findings strongly point to a belt‐shaped tetrameric assembly. In good agreement with this geometry, the NOESY spectrum at 213 K shows contacts between H1 and the 4‐carbazolyl proton as well as with the urea protons. H2, however, is coupled with the 2‐carbazolyl proton and also to the urea protons, but to a lesser extent than H1. This indicates that both H‐bonded edges are oriented anti with respect to the carbazolyl N‐substituent (Figure 14).
Figure 13.
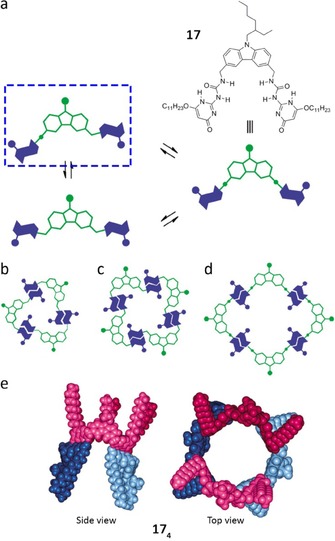
a) Disubstituted carbazole 17 and schematic representation of conformations around the UPy subunits. Schematic representation of cyclic aggregates from 17: b) trimer, c) tetramer, d) tubular‐shaped tetramer. e) Side and top views of optimized tubular‐shaped 174 aggregates. Adapted with permission from Ref. 76. Copyright 2011, American Chemical Society.
Figure 14.
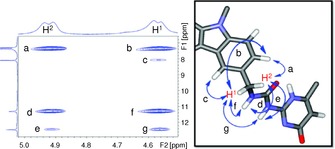
Low‐temperature (CDCl3, 213 K) NOESY spectrum of compound 17 (partial section showing contacts with methylene H1 and H2 protons). Reproduced with permission from Ref. 76. Copyright 2011, American Chemical Society.
Very recently, the group of González‐Rodríguez described H‐bonded cyclic tetramer systems that revealed high‐fidelity macrocyclization processes.78, 79 The group prepared ditopic monomers comprising a p‐diethynylbenzene ring substituted with complementary nucleosides (G and C) at both edges (Figure 15 a). The linear structure of this rigid linker, together with the 90 ° angle formed by the 5‐C position and the 8‐G position upon Watson–Crick pairing, resulted in unstrained square‐shaped assemblies (184–204). The cyclic tetramers are formed quantitatively in apolar solvents, as determined by 1 D and 2 D NMR experiments, DOSY, and ESI Q‐TOF MS. In order to dissociate them and study the underlying equilibrium processes, the authors employed three different approaches: 1) adding highly polar solvents, like DMSO or DMF, 2) decreasing the concentration to the 10−4–10−6 m range and employing highly sensitive techniques like absorption, emission, and circular dichroism (CD) spectroscopy, and 3) studying competition experiments with a mononucleoside (C).49 Tetramers 184–204 exhibit an impressive thermodynamic stability and constitute kinetically steady products in the overall self‐assembly landscape even in highly polar solvents like DMF, where H‐bonded association is typically too weak. The titration experiments with a mononucleoside competitor, were particularly revealing. As shown in Figure 15 b, upon addition of C to a solution of 184 the authors observed the gradual disappearance of the tetramer proton signals and the emergence of a new set of signals attributed to the 18⋅C complex, in fast equilibrium with excess C. This process could be equally followed by fluorescence titration experiments (Figure 15 c) monitoring the emission of the 18 molecule in the two associated states. Figure 15 d plots the fraction of 18 molecule associated as a cyclic tetramer vs. the number of equivalents of C stopper added in three solvent systems: DMF, THF, and CHCl3. In order to fully dissociate the macrocycle, about 50 C equivalents must be added, which underlines the stability of the macrocyclic assembly. The cyclotetramerization constants and EM values were determined in the different solvent systems by diverse methods. The authors point to the optimal monomer design and to the rigidity and nonrotatable nature of the multipoint G−C binding interaction as a key factor that enhances the magnitude of EM in their system (EM=102–103 m), when compared to other cyclic tetramers based on metal–ligand interactions (EM=0.1–20 m).
Figure 15.
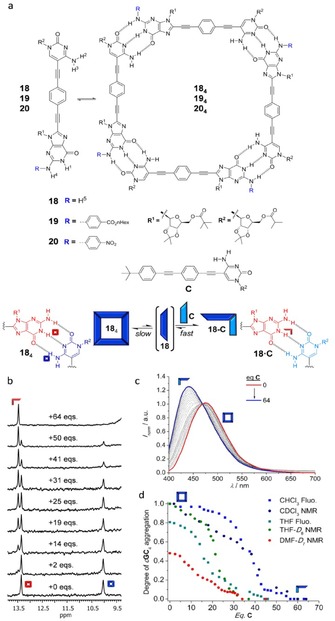
a) Chemical structure of dinucleosidic monomers 18–20, their respective cyclic tetramers and mononucleoside stopper C. Titration experiments of 184 with C at 298 K measured by b) 1H NMR (C=10−3 m) in CDCl3 or c) emission spectroscopy (C=5×10−5 m; λexc=390 nm) in CHCl3. d) Plots of the degree of 184 association, measured by 1H NMR or emission, as a function of the equivalents of C added in DMF, THF, or CHCl3. Adapted with permission from Ref. 78. Copyright, 2015, Wiley‐VCH Verlag GmbH & Co. KGaA and with permission from Ref. 79. Copyright 2015, American Chemical Society.
The guanine molecule itself has a strong tendency to associate in tetrameric macrocycles leading to the well‐known G‐quartets,80, 81 where DD–AA double H‐bonds are established with the Hoogsteen G‐edge. This species, that presents 4 carbonyl groups pointing towards the inner pore, is further stabilized by cation complexation, typically with Na+ or K+ salts (Figure 16). Complexation leads, however, to multicyclic stacked assemblies, named G‐quadruplexes, in which the cation is coordinated between pairs of quartets.82 Since the stacked quadruplexes are the thermodynamic product, the quantitative formation of isolated G‐quartets has rarely been observed.83, 84 Cation‐free or Na+‐bound quartets have been characterized in the crystal83, 84 or gas phases,85 or using covalent86, 87, 88, 89, 90 and noncovalent91 templates. A few examples where the G‐quartet structure has been imaged onto surfaces are given in the next section.
Figure 16.
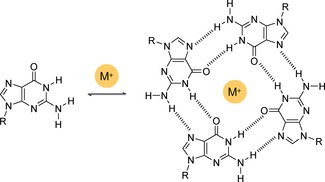
Self‐assembly of guanine derivatives into cyclic tetramers (G‐quartets) templated by alkaline salts.
2.4. Five‐membered hydrogen‐bonded macrocycles
One of the few examples of five‐membered supramolecular macrocycles was reported in 1998 by Hamilton and co‐workers.92 They previously showed that acylaminopyridine and carboxylic acid derivatives could form extended93 or cyclic aggregates94, 95 depending on the conditions and the nature of the interacting components. Molecular modeling of monomers 21 and 22 suggested that the angle between the substituents should control the structure of the final cyclic aggregate (Figure 17). For 21 (120 ° angle), both cyclic hexamers and cyclic pentamers can be formed from the planar and nonplanar disposition of the H‐bonding groups, respectively. However, with 22 (60 ° angle), cyclic trimers appear geometrically favored with no apparent distortion of the bidentate H‐bonds (Figure 17 b). According to the authors, the cyclization process in solution may be favored since 2n H‐bonds are formed from n associating particles compared to only (2n−2) for a linear type of aggregation. Concentration‐dependent 1H NMR experiments in CDCl3, [D6]DMSO:CDCl3 (1:10 and 1:5) and [D6]DMSO over a concentration range 0.1–100 mm gave rise to the corresponding dilution curves, which were analyzed by the Saunders and Hyne model.67 Dilution data for 21 fitted best to the monomer‐pentamer model and to the trimer for 22. In addition, the analysis showed that the pentameric aggregate of 21 is more stable than the trimeric association of 22 owing to the Coulombic repulsion between the carboxylate groups.
Figure 17.
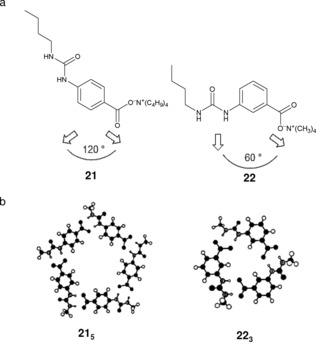
a) Monomers 21 and 22 and b) their corresponding cyclic aggregates. Adapted with permission from Ref. 92. Copyright 1998, Royal Society of Chemistry.
Monomers 23 and 24 display highly preorganized structures able to give rise to pentameric 235 and hexameric 246 cycles, respectively, mediated by either 20 or 24 H‐bonds formed between UPy subunits (Figure 18).96 To establish the structure of the cyclic aggregates, the authors used complementary methods, such as VPO, gel permeation chromatography (GPC), and ESI‐MS. All these techniques confirmed the formation of the expected aggregates but also revealed that the proposed cyclic structures 235 and 246 are not exclusive products. Additional DOSY experiments were useful to distinguish two concentration regimes for 24. At concentrations above 10 mm, the hexamers of 24 aggregate into higher‐molecular‐weight structures. Between 10 mm and 1 mm, the relative diffusion coefficient of 24 is smaller than the diffusion coefficient of 23. Therefore, at concentrations greater than 1 mm, aggregates of monomer 24 have a higher molecular weight than those of 23, which is in agreement with the pentameric and hexameric structures suggested. Despite the high binding constant of the UPy derivatives61, 62 and the highly preorganized structures of the monomers, the experimental results have shown that the formation of defined aggregates in solution is a dynamic process, in which the aggregates of interest are part of a mixed population and are the predominant species under only certain conditions. The higher stability of hexamer 246, relative to pentamer 235, may be ascribed to its, presumably, more flattened structure (Figure 18 c), which allows hierarchical aggregation into higher‐order‐molecular‐weight oligomers, as revealed by VPO and 1H NMR diffusion measurements.
Figure 18.
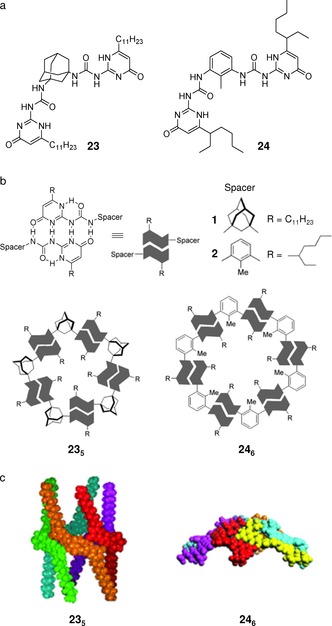
a) Structures of 1,3‐adamantane and 2‐methyl‐1,3‐phenylene bis‐ureidopyrimidinones 23 and 24. b) Schematic representation and c) front‐view representations of energy‐minimized pentameric and hexameric assemblies. Adapted with permission from Ref. 96. Copyright 2005, Wiley‐VCH Verlag GmbH & Co. KGaA.
2.5. Six‐membered hydrogen‐bonded macrocycles
The groups of Lehn97 and Mascal98, 99, 100 have pioneered the synthesis of Janus‐type molecules like 25, 26, and 27, which contain H‐bonding codes of both cytosine (AAD) and guanine (DDA) that mediates their self‐organization into a hexameric supramolecular macrocylic structure (Figure 19). The quantitative estimation of the association constant of 25 and 26 in apolar solvents was hampered by solubility problems, but 1H NMR spectroscopic and VPO measurements in chloroform indicated that the desired hexamer is formed in solution.96 Moreover, X‐ray crystallographic studies confirmed the presence of the cyclic hexamer 276 in the solid state (Figure 19 c).98, 99 In this structure, 276 hexamers overlapped each other to describe a highly porous solid with an interwoven network of channels (Figure 19 d). This work represents one of the few examples where suitable monocrystals for X‐ray analysis have been obtained in which the structure resolved faithfully represents the assembly found in solution. The rigid nature and full preorganization of the molecule towards cyclization must inhibit the formation of linear oligomers and other H‐bonded species and clearly helps in the formation of a well‐ordered network at the solid state.
Figure 19.
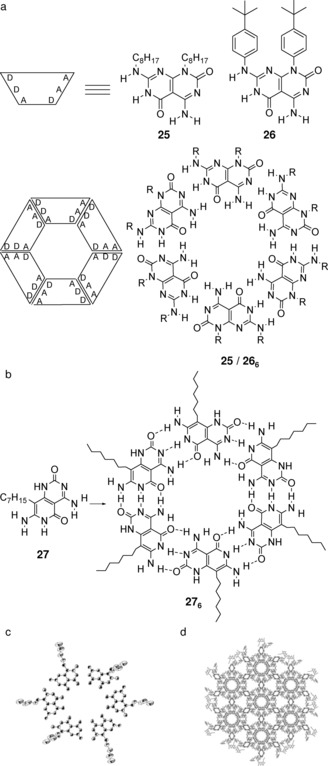
a) Proposed self‐assembly of the self‐complementary heterocycles 25 and 26 into a supramolecular macrocycle; schematic and structural representation (AD: hydrogen acceptor‐donor site). Adapted with permission from Ref. 97. Copyright 1996, Royal Society of Chemistry. b) Self‐assembled hexameric structure from monomer 27. Adapted with permission from Ref. 98. Copyright 1996, Wiley‐VCH Verlag GmbH & Co. KGaA. c) Oblique and d) cell view of the hexagonal packing of 27 in the crystal. Adapted with permission from Ref. 99. Copyright 1999, American Chemical Society.
Using the complementary DDA–AAD H‐bonding motif, Kolotuchin and Zimmerman designed a more robust hexameric aggregate (Figure 20).101 Additionally to the 18 H‐bonds formed by the pairing of self‐complementary sites, six secondary H‐bonds are also present involving the 2‐NH groups. Solventdependent 1H NMR experiments showed that the cyclic aggregate 286 is stable in 15 % aqueous THF. The authors demonstrated that this unit can effectively self‐assemble dendrimers.102, 103, 104 To this aim the 2,8‐diamino‐2‐ethylpyrimido‐(4,5‐b)(1,8)naphthyridine‐3H‐4‐one subunit (29) was synthesized with a first (29 a), second (29 b), and third generation (29 c) Fréchet‐type dendron attached to the 8‐amino group (Figure 21 a). Studies using 1H NMR, SEC, and DLS support the cooperative formation of cyclic hexamers in apolar solvents. The stability of the self‐assembled dendrimers is dependent on the size of the attached dendron. Thus, CDCl3 solutions of 29 a–c were titrated with [D6]DMSO until the hexamer signal was no longer visible. Figure 21 b shows representative data for hexamer (29 a)6 whose tert‐butyl and NH group signal intensity regularly decreases upon addition of [D6]DMSO and then fully disappears at 35 % (v/v) [D6]DMSO/CDCl3. The same experiments were carried out for 29 b and 29 c, but in these cases the amount of [D6]DMSO required to fully dissociate the hexamer in CDCl3 was 19 and 9 % (v/v), respectively. These results indicated that the stability of the hexameric aggregates decreases with increasing dendrimer size: (29 a)6>(29 b)6>(29 c)6, which suggests important steric effects between the peripheral bulky groups.
Figure 20.
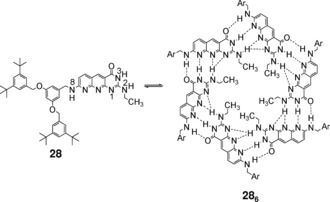
Monomer 28 and the corresponding hexameric aggregate 286. Adapted with permission from Ref. 101. Copyright 1998, American Chemical Society.
Figure 21.
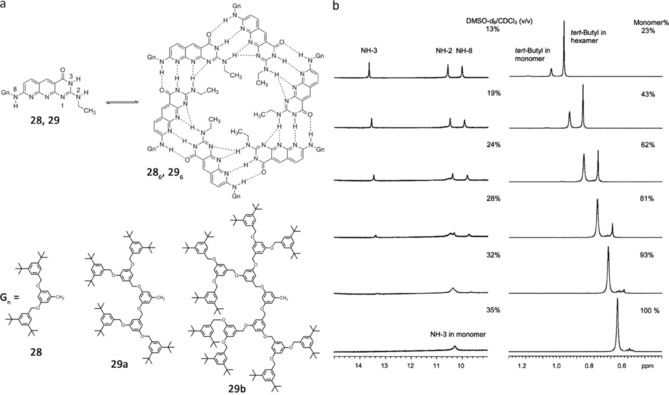
a) Molecular structure of monomers 28 and 29 and the corresponding hexameric assembly. b) 1H NMR titration of 286 in CDCl3 by [D6]DMSO. Adapted with permission from Ref. 104. Copyright 2002, American Chemical Society.
Chen and co‐workers used hydrazide‐based supramolecular synthons linked through a 120 ° spacer, with properly encoded recognition sites, to favor a spontaneous self‐assembly process into well‐defined hexameric rosettes, as shown in Figure 22 a.105 However, the conformational flexibility of the monomer, which can also adopt a conformation with a C2 axis, allows the formation of linear polymers in solution, which may compete with macrocyclization. First evidence supporting the formation of cyclic supramolecular associates of 30–32 was provided by 1H NMR spectra in CDCl3, (Figure 22 b). Dilution 1H NMR experiments within a concentration range of 100–1 mm showed no prominent changes suggesting a high stability of the assembly. However, when a 10 mm solution of 32 in CDCl3 was titrated with [D6]DMSO, in addition to the signals corresponding to the hexamer, new signals corresponding to the monomer also appeared when the [D6]DMSO content reached about 4 %. No other signals were found during the whole titration process (Figure 22 c). This phenomenon along with the high association constant calculated for the hexameric species (between ∼1016 and 106 m −5 in CDCl3–[D6]DMSO mixtures) suggests that the self‐assembly/disassembly of hexamer (32)6 is a highly cooperative process (Ka=3.67×1016 m −5 in 4 % [D6]DMSO/CDCl3) and rules out the formation of linear species.
Figure 22.
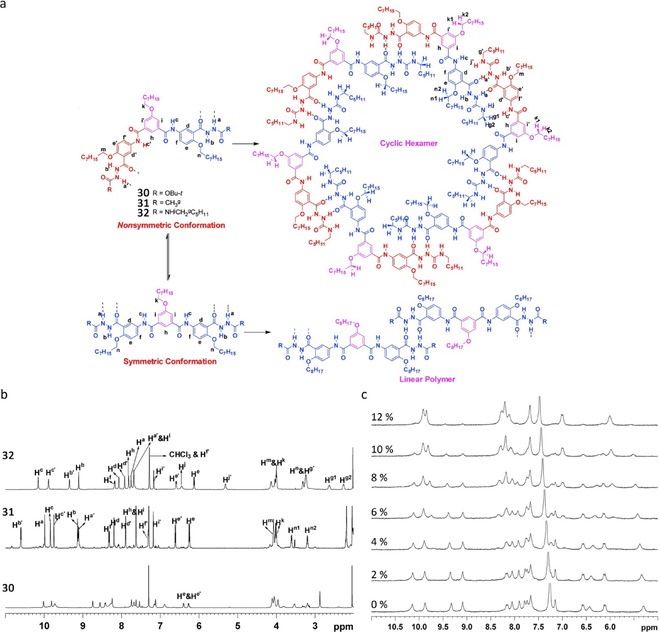
a) Conformation and aggregation analysis of 120 ° spacer linked hydrazide‐based ditopic monomers 30–32: nonsymmetric conformation vs. symmetric conformation; cyclic hexamer vs. linear polymer, with hydrogen labeling scheme indicated. b) Stacked partial 1H NMR spectra of 30–32 in CDCl3 (298 K, 600 MHz). c) 1H NMR spectra of 32 in CDCl3 titrated with [D6]DMSO (10 mm, 298 K, 300 MHz). Adapted with permission from Ref. 105. Copyright 2009, American Chemical Society.
Because of the dynamically and kinetically stable nature of this kind of assemblies, the assembly from monomer 33 with chiral auxiliary terminal groups also displayed supramolecular chirality in solution, which was confirmed by concentration‐dependent CD spectra in chloroform (Figure 23).
Figure 23.
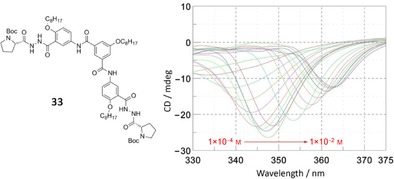
Chemical structure of 33 with chiral auxiliary terminative group and CD spectra at different concentrations in CHCl3. Adapted with permission from Ref. 105. Copyright 2009, American Chemical Society.
By changing the location of the alkoxy group on the spacer of the hydrazide‐based monomer (from position 5 to position 4) the authors were able to fix a syn–anti conformation (Figure 24 a). As a result, a high regioselectivity for the self‐assembly process was achieved (Figure 24 b).106 1H NMR experiments of compounds 34 and 35 in CDCl3 suggest the formation of well‐defined shape‐persistent cyclic hexamers in solution, where the exchange of hydrazide units on the inner and outer sides is slow on the NMR time scale, and two sets of signals were observed for both parts as well (Figure 24 c). These results, along with the evidence of intermolecular contacts among the molecules in the cyclic species observed by NOESY and COSY (correlation spectroscopy) analysis, clearly show a precise control of the relative positions of the hydrazide units in the resulting cyclic hexamers.
Figure 24.
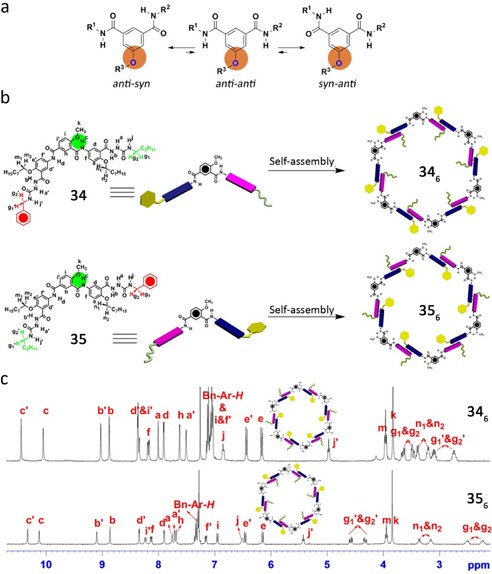
a) Representation of conformation equilibrium of the isophthalamide spacer. b) Self‐assembly of compounds 34 and 35. c) 1H NMR spectra for compounds 34 and 35 (CDCl3, 298 K, 10 mm). Adapted with permission from Ref. 106. Copyright 2013, Nature Publishing Group.
Yagai et al. have demonstrated that the barbiturate unit is a powerful functional group for the organization of π‐conjugated units into unique nanostructures.107, 108, 109, 110 For instance, monomers 36, 37, and 38 self‐assemble into H‐bonded hexameric rosettes in apolar solvents, which can further stack to afford highly ordered nanostructures such as nanorings107, 108, 109 or nanorods.109 These nanostructures were clearly visualized by atomic force microscopy (AFM) studies (Figures 25 and 26).
Figure 25.
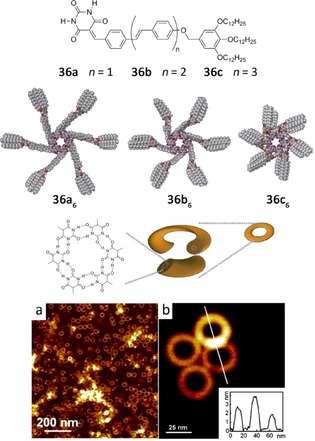
Chemical structures of 36 and proposed mechanism for the formation of nanorings and nanofibers by hydrogen‐bonded macrocycles. AFM height images of nanostructures of a) 36 b at 1×10−4 m and b) 36 a at 2×10−5 m spin‐coated from MCH solutions onto HOPG (z scale: 20 nm). Adapted with permission from Ref. 108. Copyright, 2011, Wiley‐VCH Verlag GmbH & Co. KGaA and with permission from Ref. 107. Copyright 2009, American Chemical Society.
Figure 26.
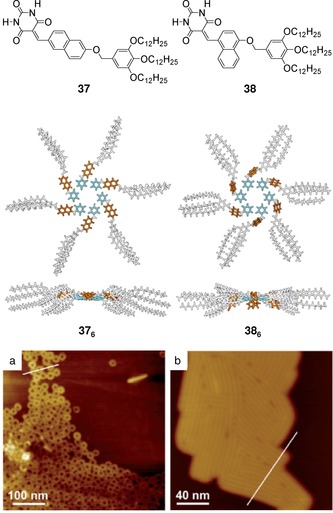
Chemical structures of 37 and 38 and their corresponding hexameric rosettes. AFM height images of nanostructures of a) 37 at 5×10−5 m and b) 38 at 1×10−4 m spin‐coated from MCH solutions onto HOPG. Adapted with permission from Ref. 109. Copyright 2012, Wiley‐VCH Verlag GmbH & Co. KGaA.
Another remarkable six‐membered system was provided by Fenniri and colleagues in a system related to the one of Lehn and Mascal. They created a heterobicyclic G C monomer containing the Watson–Crick DDA H‐bond array of guanine and the AAD array of cytosine (Figure 27 a). Because of the asymmetry of its H‐bonding edges, the G C monomer undergoes self‐assembly in water111, 112, 113, 114 as well as in polar solvents115, 116, 117 to form a six‐membered macrocycle maintained by 18 H‐bonds. The resulting hexameric rosette then self‐organizes hierarchically to produce tubular stacks (named rosette nanotubes, RNs) with tunable dimensions and properties (Figure 27 b). The peripheral diameter and its properties are dictated by the choice of the functional groups conjugated to this motif whereas the inner space is directly related to the distance separating the H‐bonding arrays within G C.118, 119 For instance, the authors designed a heterotricyclic self‐complementary molecule, which possesses the same H‐bonding code, but separated by an internally fused pyridine ring (Figure 28 a).118, 119 Compared to 41, the assembled rosette nanotube 40 shows a higher diameter and a larger π system that can allow electronic transport along the main axis (Figure 28 b). AFM, TEM, and SEM images of randomly oriented RNs obtained from 40 featured an outer diameter of 4.3 nm, the inner and outer diameters of 40 increasing by 0.4 and 0.9 nm relative to 41, respectively (Figure 28 b–e).119
Figure 27.
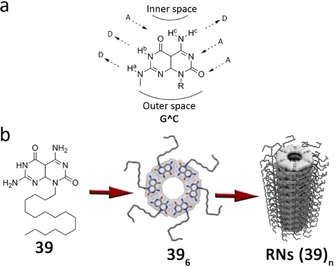
a) G C motif. Reproduced with permission from Ref. 113. Copyright 2002, American Chemical Society. b) Self‐assembly of monomer 39 into hexameric rosettes and RNs. Reproduced with permission from Ref. 117. Copyright 2013, American Chemical Society.
Figure 28.
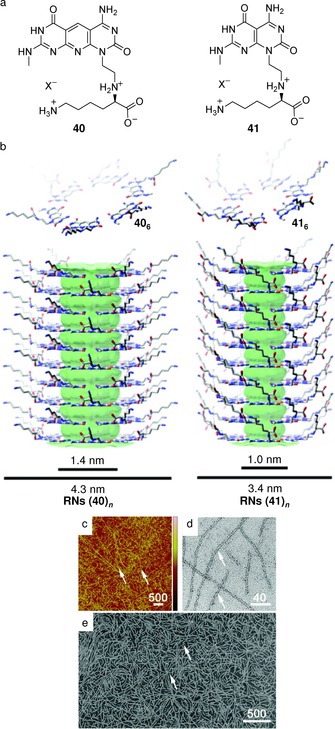
a) Monomers 40 and 41 and b) their corresponding hexameric rosettes (middle), and RNs (lower) (X=CF3CO2 −). Imaging of 40 (0.1 mg mL−1 in water) by c) TM‐AFM (5 μm scan, height scale=0–10 nm), d) TEM, and e) SEM. White arrows point to individual RNs. Reproduced with permission from Ref. 119. Copyright, 2010, American Chemical Society.
One of the latest developments of the group consists of the generation of related systems with chiroptical behavior.120, 121 They rely on the ability of a single chiral molecule to express multiple supramolecular chirality outputs as a result of preferred conformational states under a set of physical conditions (e.g. different solvents). These chiromers are supramolecular conformational isomers that a) are thermodynamically stable, b) can memorize their chirality, and c) can amplify their chirality in an achiral environment.
3. Hydrogen‐Bonded Macrocycles Studied onto Surfaces
We thought it would be convenient to include in this review a discussion on H‐bonded cyclic systems studied on diverse surfaces, like highly oriented pyrolytic graphite (HOPG) or gold substrates, by scanning tunneling microscopy (STM). We will focus again on planar monocyclic systems, the typical motifs stabilized in two dimensions, in which the discrete macrocycle constitutes the primary repeating unit. This unit then forms a network by means of secondary interactions that are different from the H‐bonds that hold the cycle together. Compared with the previous examples studied in solution, most of these cyclic systems deposited onto surfaces are bound by relatively weak H‐bonding interactions, which would be too labile to afford the same macrocycles in solution. Cyclic systems are still formed due to the stabilizing molecule–substrate interactions and the fact that the molecules are concentrated on a surface. Upon physisorption, several degrees of translational, rotational, and vibrational freedom are lost, and as a result, molecules that display very weak and ill‐defined binding in solution can form well‐ordered assemblies when confined in two dimensions. This concentration effect, however, eliminates the preference for intramolecular vs. intermolecular binding observed in solution. Therefore, nothing can assure that closed H‐bonded species are going to dominate over open oligomers onto surfaces, even if the molecule is preorganized for ring‐closure. Macrocycles are either observed at low monolayer coverages or because the overall network is more stable when the monomer assembles in its cyclic form. As a matter of fact and to the best of our knowledge, none of the monocyclic systems reported so far have been quantitatively obtained in solution and then transferred to a solid substrate with total fidelity and structural integrity.
Dipole–dipole interactions occur when the electronegative portion of one molecule is attracted to the electropositive portion of a second molecule. This interaction can also be seen as a weak and single H‐bond. Using low‐temperature STM, Mashiko and co‐workers122 showed that cyano‐substituted porphyrins, like 42, adsorbed on a gold surface, could form cyclic trimers and tetramers endowed with a small central pore (Figure 29 a–e). The estimated length of the CH⋅⋅⋅NC bond is around 2.6 Å and 2.5 Å, suggesting weak H‐bonding interactions. They anticipate useful electronic and optoelectrical functions for their systems, as electron or energy transfer could occur across hydrogen‐bonded interfaces. Furthermore, the group of Mashiko points out that the central porphyrins and cyanophenyl substituents within their clusters are decoupled from both the surface and surrounding clusters, since the bulky tert‐buthylphenyl insulators attached to the surface prevent any direct interaction. Later, Diederich and co‐workers123 were able to self‐assemble a larger ZnII porphyrin derivative 43 into discrete macrocyclic‐polymorphs on the Cu (111) surface, growing in size. At submonolayer coverage, the cis‐porphyrin 43 forms cyclic trimers, tetramers, pentamers, and hexamers held together by CN⋅⋅⋅H−C(sp2) H‐bonding (Figure 29 f–p), involving the positive polarized hydrogen ortho to the CN group of the neighboring molecule. Also, antiparallel CN⋅⋅⋅CN dipolar interactions help in the stabilization of the oligomeric macrocycles. The authors argued that the very strong adsorbate–Cu (111) substrate interactions, due to the extended π‐system of the porphyrins, compensate the loss in antiparallel CN⋅⋅⋅CN forces and are the origin of the diversity of the obtained porous macrocycles, compared with the earlier work of Mashiko.122
Figure 29.
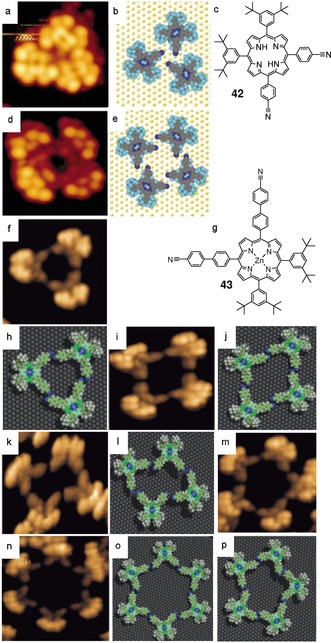
a,d) STM images at 63 K of the supramolecular aggregation of compound 42 on Au (111). c) Chemical structure of porphyrin 42. b,e) Corresponding molecular models. Reproduced with permission from Ref. 122. Copyright 2001, Nature Publishing Group. f–p) STM images of porphyrin 43 on Cu (111) with corresponding molecular models. g) Chemical structure of 43. Reproduced with permission from Ref. 123. Copyright 2009, Wiley‐VCH Verlag GmbH & Co. KGaA.
These H⋅⋅⋅N≡C interactions featured by associating cyanophenyl rings are substantially weaker than classic H‐bonds involving more acidic O−H or N−H H‐bond donors. The H‐bond formed between two carboxylic acids is the most widely used form by researchers for preprogramming 2 D networks. The benzene ring solely meta‐substituted with two carboxylic acid substituents (isophtahic acid) cannot form a porous nanostructure and prefers a densely‐packed network with zigzag linear chains. However, there are ways to circumvent this and promote cycle formation. For instance, Hecht, Grill, and colleagues124 made use of peripheral azobenzene derivatives equipped with −COOH groups to direct the supramolecular self‐assembly of highly stable discrete hexameric rosettes (Figure 30 a,b). In the rosette, each molecule is connected to both of its neighbors and forms the maximum number of nondistorted carboxylic acid dimer motifs. Therefore, the binding energy per molecule is maximized. They measured a distance of 3.6±0.7 Å between oxygen atoms, so the pore diameter is around 1.7 nm. They failed to assure molecular switching, as the isomerization of the azobenzene derivatives was suppressed on the surface, probably due to the lack of bulky tert‐butyl groups on the inner H‐bonding phenyl fragment, leading to an increased coupling to the surface. On the other hand, De Feyter and co‐workers125 proved how coronene (COR) could act as a molecular template directing the assembly of isophthalic acid 44 and afford a heteromolecular cluster (Figure 30 c,d). Six molecules are held together by 12 H‐bonds, leaving a cavity of ∼1 nm in diameter, where COR fits perfectly and further stabilizes this 2 D crystal via van der Waals interactions with a force of −13 kcal mol−1, according to their molecular mechanics calculations. Larger pores able to host a CuII‐phthalocyanine were also obtained by Bai and colleagues126 with the triple‐armed amphiphile 45 (Figure 30 e,f). They believe their approach may allow profiting from the properties of molecular semiconductors, such as phthalocyanines, in potential applications like photoconductors, electronic devices and gas sensors.
Figure 30.
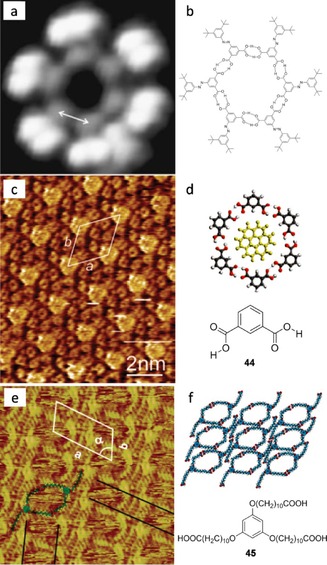
2 D nanoporous self‐assembled systems based on carboxylic acids. a) STM image (5.2×5.2 nm2; I=0.11 nA; V=1 V) on Au (111) and b) self‐assembled model. Reproduced with permission from Ref. 124. Copyright 2008, Springer. c) STM image at the 1‐octanoic acid/HOPG interface of the self‐assembled pattern of 44 and COR (I set=530 pA, V set=−1.00 V). d) Corresponding molecular model. Reproduced with permission from Ref. 125. Copyright 2008, American Chemical Society. e) STM image of the network formed by 45 on HOPG and f) model. Reproduced with permission from Ref. 126. Copyright 2004, American Chemical Society.
Other molecular families carrying O⋅⋅⋅H H‐bonding moieties can be used. Mamdouh et al.127 imaged at the solid–liquid interface porous discrete cyclic tetramers from an alkylated monodendron 46 (Figure 31 a,b). The formation of tetramers is due to H‐bonding between the carboxylic group of one monodendron and the hydroxyl group of another one. The alkyl chains help in reducing the mobility of the molecules when physisorbed on graphite.
Figure 31.
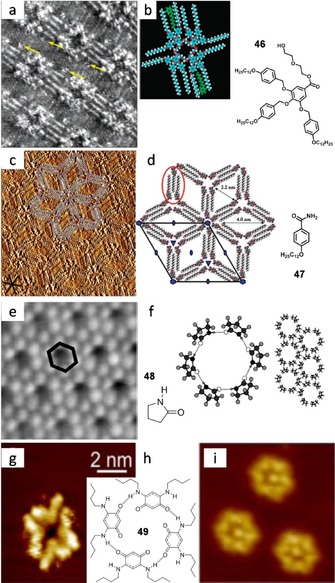
a) STM image of 46 at the 1‐phenyloctane/HOPG interface (I t=0.5 nA, V bias=−0.518 V). b) Tentative molecular model and chemical structure. Reproduced with permission from Ref. 127. Copyright 2004, American Chemical Society. c) STM image at the 1‐phenyloctane/HOPG interface (I t=300 pA, V s=0.80 V). d) Computed model. Reproduced with permission from Ref. 128. Copyright 2009, American Chemical Society. e) STM image of 48 on Au (111) and f) structure and model. Reproduced with permission from Ref. 129. Copyright 2000, Elsevier. g, i) STM images of 49 on Ag (111). h) Molecular structure of 49 and association model. Adapted with permission from Ref. 130. Copyright 2015, AIP Publishing LLC.
Matzger and co‐workers128 formed hexamers based on an amphiphilic amide 47, further stabilized by van der Waals interactions between the long alkyl chains (Figure 31 c,d). The authors described how, depending on the conformation of the amide, 3‐ and 6‐fold symmetric H‐bonding patterns can be seen. In the 3‐fold rotation axis, a typical arrangement of three dimers, united by a single H‐bond each, takes place. Hexameric rings involving 12 H‐bonding donors and six acceptors exist as well at the sixfold axis. In this case, the two oxygen lone pairs make H‐bonds with two hydrogen atoms in an amide. An amide was also used as H‐bonding carrier by Cousty and co‐workers,129 who were able to stabilize hexamers of 2‐pyrrolidone 48 on Au (111) (Figure 31 e,f). They observed a periodic arrangement of hexamers (unit cell parameters: a=1.33±0.05 nm, b=1.33±0.05 nm, and γ=120±1°) corresponding to cyclic pyrrolidone funnel‐like hexamers. They proposed a model where each molecule establishes one H‐bond with each one of its two nearest neighbors and the plane of each molecule is tilted with respect to the gold substrate. The distance measured between neighboring residues is 0.5 nm and fits the funnel‐like arrangement. Theoretical calculations showed that this macrocycle was only slightly more stable than the pyrrolidone dimer. However, the hexameric shape fits better the Au (111) lattice, and the molecule–substrate interactions are, therefore, stronger than for a dimer, explaining the absence of polymorphism.
H‐bonded assemblies in which the individual components carry opposite charges have higher stabilities than assemblies consisting of neutral components and can be seen as ion‐pair‐reinforced motifs.2 Recently, Kunkel et al.130 prepared a strongly dipolar p‐benzoquinonemonoimines to study the competition between chemical interactions and substrate effects in the self‐assembly on metal surfaces. When sublimating the butyl zwitterionic molecule 49 and making the deposition on the Au (111) surface, ring‐shaped tetramers stabilized by four H‐bonds were obtained (Figure 31 g,h). In this case the geometrical hindrance of the alkyl substituents is an important factor to limit the size of the cluster. For instance, in the case of the parent p‐benzoquinone‐monoimine, with no alkyl substituent and therefore smaller constraints, larger rings consisting of six molecules are the most frequently observed. For this bigger self‐assembled macrocycle, the pore is large enough to host a seventh molecule (Figure 31 i).
Stöhr et al.131 also used classic H‐bonds, involving in this case N−H moieties. They stabilized discrete trimers of the perylene derivative 50 on the Cu (111) surface by intermolecular resonance‐assisted H‐bonding. Upon annealing a sample with a coverage of 0.85 mL 50 at 300 °C, trimeric structures in the form of rings are found (Figure 32 a–b). The authors argue whether their molecular surface assembly, which is highly robust, may be suitable for the construction of self‐organizing structures able to host deposited material at lower temperatures. The reported energy per single acceptor‐donor pair in solution quantified by typical methods like NMR or UV/Vis is close to 1.0–1.4 kcal mol−1.132 However, since H‐bonding is a strong dipole‐dipole attraction, highly dependent on the molecular environment, these energy values in solution are to be taken on surfaces as upper limits. Even so, successive H‐bond motifs allow the stabilization of more complex macrocycles. Melamine 51 appears here as a very useful synthon for the construction of self‐assembled monolayers, as it presents three DAD recognition sites. When increasing the number of H‐bonds to three parallel ones in a ADA–DAD pair, the association energy rises to very high values. In 2009, Walch et al.133 were able to obtain stable monolayers of melamine as six‐membered rings, stabilized by 12 internal N…H−N H‐bonds at the liquid‐graphite interface. In these systems, fatty acids, such as nonanoic acid, were used as solvent. It is interesting to see that nonanoic acid not only serves as a medium for the solute molecules, but it also stabilizes and isolates the macrocycles on the surface. Twelve solvent molecules arrange radially around the melamine molecules, establishing a total of 36 H‐bonds and allowing the formation of crystalline hexameric cycles (Figure 32 c,d). 51 forms very tight complexes with carboxylic acids as a result of proton transfer. Through selective adsorption of appropriately functionalized nanoparticles, Walch and co‐workers believe their isotopological networks could serve to investigate particle‐particle interaction as a function of the distance.
Figure 32.
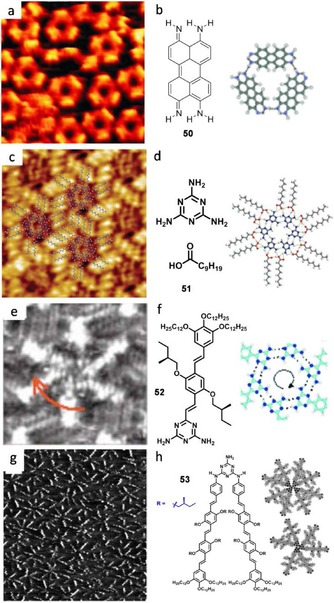
a) STM image of 50 on Cu (111) and b) model. Adapted with permission from Ref. 131. Copyright 2005, Wiley‐VCH Verlag GmbH & Co. KGaA. c) STM image of 51 at the nonanoic acid/HOPG interface (I t=110 pA, V s=1.0 V). d) Association model and chemical structures. Reproduced with permission from Ref. 133. Copyright 2009, American Chemical Society. e) STM image of 52 at the 1‐phenyloctane/HOPG interface (I t=0.50 nA, V s=−0.50 V). f) Chemical structure and association modes. Adapted with permission from Ref. 134. Copyright 2004, Wiley‐VCH Verlag GmbH & Co. KGaA. g) STM image of 53 at the 1‐phenyloctane/HOPG interface (I t=0.48 nA, V s=−0.486 V). h) Chemical structure and association modes. Adapted with permission from Ref. 135. Copyright 2008, Wiley‐VCH Verlag GmbH & Co. KGaA.
Using a related H‐bonding motif, the diaminotriazine, Jonkheijm et al.134 stabilized at the solid‐liquid interface hexameric π‐conjugated rosettes from molecule 52, which bears a oligo(p‐phenylenevinylene) (OPV) fragment (Figure 32 e,f). They bet on the ability of their molecule to function as an ideal channel for transportation, as it self‐assembles into a soluble tubular aggregate, by virtue of its apolar shell. Later, Hoeben et al.135 saw how the true ADA–DAD motif of melanine could stabilize other self‐assembled macrocycles at the 1‐phenyloctane/HOPG interface. By applying a drop of the V‐shaped‐molecule 53 solution into the freshly cleaved HOPG substrate, trimers and tetramers were observed (Figure 32 g,h). In the first example,134 each molecule establishes four H‐bonds for stabilizing the six‐membered rosette (∼7 Å pore diameter) with six hydrogen donor–acceptor pairs. In the second one,133 the bifunctional molecules with two H‐bonding arrays available for complementary binding allow the formation of smaller discrete macrocycles.
Some research groups have combined N⋅⋅⋅H and O⋅⋅⋅H H‐bonding in order to get more interesting self‐assembled macrocycles. For example, Ziener and co‐workers136 obtained trimers from an interplay of both H‐bonds and van der Waals interactions of lactim/lactam moieties (Figure 33 a–b). The synthesized phthalhydrazide 54 undergoes tautomerism between three forms, and discrete cyclic units are formed if neighboring molecules establish double H‐bonds in both sides of the molecule, through their two N−C−O H‐bonding moieties. The trimer is formed by six H‐bonds in the lactim/lactam tautomeric form. This adaptive nanostructure may serve, in combination with metal complexation, as a template to achieve addressable functional devices for molecular or optical electronics.
Figure 33.
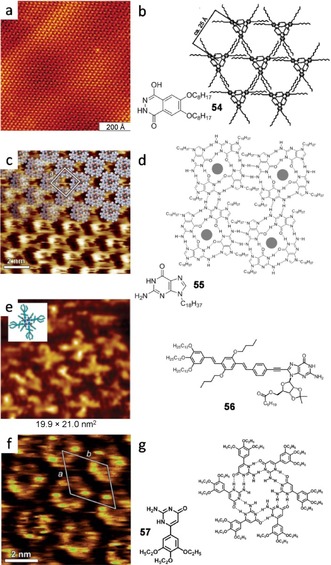
a) STM image of 54 at the 1‐chloronaphtalene/HOPG interface (I t=27 pA, V s=251 mV). b) Chemical structure and model. Reproduced with permission from Ref. 136. Copyright 2006, American Chemical Society. c) STM image of 55 at the TCB/HOPG interface (I t=5 pA, V s=200 mV). d) Association mode and chemical structure. Adapted with permission from Ref. 137. Copyright 2010, Wiley‐VCH Verlag GmbH and Co. KGaA. e) STM image of 56 at the 1‐phenyloctane/HOPG interface of 56 (It=28 pA, Vs=−0.600 mV) along with model and chemical structure. Reproduced with permission from Ref. 145. Copyright 2010, American Chemical Society. f) STM image of 57 at the TCB/HOPG interface (I t=17 pA, V s=450 mV). g) Chemical structure and model. Reproduced with permission from Ref. 146. Copyright 2011, Royal Society of Chemistry.
Ciesielski et al. explored the self‐assembly process of nucleobase derivatives such as the naturally occurring guanine.137 The G quartet (G4) is an H‐bonded macrocycle first identified in 1962 as the basis for the aggregation of 5′‐guanosine monophosphate,138 and it can be seen as an ideal scaffold to construct functional assemblies.82, 139, 140, 141, 142 However it was observed to self‐assemble on the HOPG surface as ribbons unless it forms a cation‐templated assembly with K+ when adding 100 eq of potassium picrate (Figure 33 c,d). The cation plays a crucial role in stabilizing the guanine tetramer through ion–dipole interactions with the inner carbonyl moieties. The unit cell contains four molecules and lead to an area of 5.21±1.35 nm2. The authors attribute the high‐contrast circular feature to the K+ ions not being located in the same plane as the G4, as it is also the case in solution.143 The observed packing motif differs also from the one observed in UHV without templating ions144 Ciesielski and co‐workers present a first prototype in the generation of nanopatterned responsive architectures with their in situ reversible assembly and reassembly between two highly ordered supramolecular structures. Another interesting approach for the stabilization of the G4 on the surface was published by González–Rodríguez et al. that same year.145 They capped a guanosine derivative with an OPV oligomer and observed how compound 56 formed a monolayer of individual quartets on the HOPG substrate, without any cation‐templated stabilization of the assembly (Figure 33 e). The authors argue that the bulky ribose group blocks the NH2 proton and the formation of G‐ribbons is prevented because of steric hindrance, resulting in the stabilization of the circular tetramers. These authors also succeeded in the construction of large disk‐shaped organic nanoparticles and aim at progressively enhance the fluorescence of their dynamic self‐assembled nanoobjects. Later, inspired by Watson–Crick base pairing, again Ciesielski et al.146 prepared an isocytosine (iC) derivative 57 bearing two different faces: one with an AA H‐bonding pattern and the other presenting a DD motif. In that way, they crystallized a heterohexameric macrocyle on the HOPG/TCB interface, further stabilized by intermolecular van der Waals interactions between the six‐membered assemblies (Figure 33 f–g). The measured unit cell parameters lead to an area of 6.54±0.27 nm2 containing six iC derivatives, a single molecule occupying an area of 1.09±0.05 nm2, and the pore size is around 6 Å. The comparison of the heterocycles shown in Figure 33 constitutes a nice picture to demonstrate the influence of the H‐bonding pattern and molecular geometry on the size of the cyclic associated motif obtained, from trimers to hexamers.
4. Conclusion
Cyclization into closed assemblies is the most recurrent approach to realize the noncovalent synthesis of discrete, well‐defined nanostructures. The advantage that an intramolecular binding interaction offers, in comparison with the intermolecular association, is a convenient approach to afford quantitative amounts of a given supramolecular species under thermodynamic control. This review article provides numerous examples of H‐bonded monocyclic systems and their characterization by solution or surface techniques. It is clear that the size of the assembly will depend on geometric issues such as the monomer structure and the directionality of the binding interaction, whereas the fidelity achieved relies largely on structural preorganization, low degrees of conformational flexibility, and templating effects.
Employing reversible interactions between programmed monomers to build complex nanostructures is clearly more appealing than the fully covalent approach.147, 148, 149, 150 The latter has the main advantage of robust, nondynamic stability, but the disadvantage of tedious, often inefficient synthetic routes. Therefore, in order to realize the full potential of noncovalent synthesis, we must develop both characterization techniques and supramolecular strategies that allow us to convincingly determine the structure of a given aggregate, on one hand, and to “freeze” its dynamic behavior, on the other.
Biographical Information
David González‐Rodríguez obtained his bachelor′s degree in chemistry at the Universidad Complutense de Madrid in 1999. He then joined the Nanoscience and Molecular Materials research group of Prof. Tomas Torres at the Universidad Autónoma de Madrid, where he obtained his PhD in Organic Chemistry in 2003. Between 2005 and 2008, he worked in the laboratories of Profs. E. W. Meijer and A. P. H. J. Schenning at the Eindhoven University of Technology as a Marie Curie postdoctoral fellow. Since 2011, he has led the Nanostructured Molecular Systems and Materials (MSMn) group at the Universidad Autónoma de Madrid, where he is Associate Professor. His research interests focus on the development of new, versatile strategies to improve or create novel functions in organic materials by rationally ordering molecules at the nanoscale using the tools of self‐assembly. The potential applications of such molecular nanostructured systems range from optoelectronic π‐functional devices to custom‐tailored nanoporous networks.

Acknowledgements
The authors gratefully acknowledge funding from the European Research Council (ERC‐StG 279548) and the Spanish Ministry of Economy and Competitiveness (MINECO) (CTQ2011‐23659 and CTQ2014‐57729‐P).
M. J. Mayoral, N. Bilbao, D. González-Rodríguez, ChemistryOpen 2016, 5, 10.
References
- 1.Special Issue on the Status of Self-assembly at the Beginning of the XXI Century, Science 2002, 295.
- 2. Prins L. J., Reinhoudt D. N., Timmerman P., Angew. Chem. Int. Ed. 2001, 40, 2382–2426; [DOI] [PubMed] [Google Scholar]; Angew. Chem. 2001, 113, 2446–2492. [Google Scholar]
- 3. Reinhoudt D. N., Crego-Calama M., Science 2002, 295, 2403–2407. [DOI] [PubMed] [Google Scholar]
- 4. Gregorio C. C., Weber A., Bondad M., Pennise C. R., Fowler V. M., Nature 1995, 377, 83–86. [DOI] [PubMed] [Google Scholar]
- 5. Berger B., Shor P. W., J. Struct. Biol. 1998, 121, 285–294. [DOI] [PubMed] [Google Scholar]
- 6. Marshall W. F., Rosenbaum J. L., J. Cell Biol. 2001, 155, 405–414. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7. Seeman N. C., Nature 2003, 421, 427–431. [DOI] [PubMed] [Google Scholar]
- 8. Chworos A., Severcan I., Koyfman A. Y., Weinkam P., Oroudjev E., Hansma H. G., Jaeger L., Science 2004, 306, 2068–2072. [DOI] [PubMed] [Google Scholar]
- 9. Sun W.-Y., Yoshizawa M., Kusukawa T., Fujita M., Curr. Opin. Chem. Biol. 2002, 6, 757–764. [DOI] [PubMed] [Google Scholar]
- 10. Adams H., Hunter C. A., Lawson K. R., Perkins J., Spey S. E., Urch C. J., Sanderson J. M., Chem. Eur. J. 2001, 7, 4863–4877. [DOI] [PubMed] [Google Scholar]
- 11. Müller-Dethlefs K., Hobza P., Chem. Rev. 2000, 100, 143–167. [DOI] [PubMed] [Google Scholar]
- 12. González-Rodríguez D., Schenning A. P. H. J., Chem. Mater. 2011, 23, 310–325. [Google Scholar]
- 13. Schenning A. P. H. J., González-Rodríguez D. in Organic Nanomaterials: Synthesis Characterization, and Device Applications (Eds.: T. Torres, G. Bottari), John Wiley & Sons, Inc., Hoboken, New Jersey, 2015, pp. 33–58. [Google Scholar]
- 14. Hunter C. A., Anderson H. L., Angew. Chem. Int. Ed. 2009, 48, 7488–7499; [DOI] [PubMed] [Google Scholar]; Angew. Chem. 2009, 121, 7624–7636. [Google Scholar]
- 15. Ercolani G., Schiaffino L., Angew. Chem. Int. Ed. 2011, 50, 1762–1768; [DOI] [PubMed] [Google Scholar]; Angew. Chem. 2011, 123, 1800–1807. [Google Scholar]
- 16.Focus Issue on Cooperativity, Nat. Chem. Biol 2008, 4, 433–507.
- 17. Badjic J. D., Nelson A., Cantrill S. J., Turnbull W. B., Stoddart J. F., Acc. Chem. Res. 2005, 38, 723–732. [DOI] [PubMed] [Google Scholar]
- 18. Dudek S. P., Pouderoijen M., Abbel R., Schenning A. P. H. J., Meijer E. W., J. Am. Chem. Soc. 2005, 127, 11763–11768. [DOI] [PubMed] [Google Scholar]
- 19. Schmid S. A., Abbel R., Schenning A. P. H. J., Meijer E. W., Sijbesma R. P., Herz L. M., J. Am. Chem. Soc. 2009, 131, 17696–17704. [DOI] [PubMed] [Google Scholar]
- 20. De Greef T. F. A., Smulders M. M. J., Wolffs M., Schenning A. P. H. J., Sijbesma R. P., Meijer E. W., Chem. Rev. 2009, 109, 5687–5754. [DOI] [PubMed] [Google Scholar]
- 21. Hunter C. A., Tomas S., J. Am. Chem. Soc. 2006, 128, 8975–8979. [DOI] [PubMed] [Google Scholar]
- 22. Moore J. S., Curr. Opin. Colloid Interface Sci. 1999, 4, 108–116. [Google Scholar]
- 23. Brunsveld L., Folmer B. J. B., Meijer E. W., Sijbesma R. P., Chem. Rev. 2001, 101, 4071–4097. [DOI] [PubMed] [Google Scholar]
- 24. Zeng F. W., Zimmerman S. C., Kolotuchin S. V., Reichert D. E. C., Ma Y. G., Tetrahedron 2002, 58, 825–843. [Google Scholar]
- 25. Lehn J. M., Polym. Int. 2002, 51, 825–839. [Google Scholar]
- 26. Michelsen U., Hunter C. A., Angew. Chem. Int. Ed. 2000, 39, 764–767; [PubMed] [Google Scholar]; Angew. Chem. 2000, 112, 780–783. [Google Scholar]
- 27. Seidel S. R., Stang P. J., Acc. Chem. Res. 2002, 35, 972–983. [DOI] [PubMed] [Google Scholar]
- 28. Sato S., Iida J., Suzuki K., Kawano M., Ozeki T., Fujita M., Science 2006, 313, 1273–1276. [DOI] [PubMed] [Google Scholar]
- 29. Hogben H. J., Sprafke J. K., Hoffmann M., Pawlicki M., Anderson H. L., J. Am. Chem. Soc. 2011, 133, 20962–20969. [DOI] [PubMed] [Google Scholar]
- 30. Sprafke J. K., Odell B., Claridge T. D. W., Anderson H. L., Angew. Chem. Int. Ed. 2011, 50, 5572–5575; [DOI] [PubMed] [Google Scholar]; Angew. Chem. 2011, 123, 5687–5690. [Google Scholar]
- 31. Kirby A. J., Adv. Phys. Org. Chem. 1980, 17, 183–278. [Google Scholar]
- 32. Mandolini L., Adv. Phys. Org. Chem. 1986, 22, 1–111. [Google Scholar]
- 33. Chi X., Guerin A. J., Haycock R. A., Hunter C. A., Sarson L. D., J. Chem. Soc. Chem. Commun. 1995, 2563–2565. [Google Scholar]
- 34. Ercolani G., J. Phys. Chem. B 1998, 102, 5699–5703. [Google Scholar]
- 35. Ercolani G., J. Phys. Chem. B 2003, 107, 5052–5057. [Google Scholar]
- 36. Ercolani G., Struct. Bonding (Berlin) 2006, 121, 167–215. [Google Scholar]
- 37. Hunter C. A., Misuraca M. C., Turega S. M., J. Am. Chem. Soc. 2011, 133, 20416–20425. [DOI] [PubMed] [Google Scholar]
- 38. Misuraca M. C., Grecu T., Freixa Z., Garavini V., Hunter C. A., van Leeuwen P., Segarra-Maset M. D., Turega S. M., J. Org. Chem. 2011, 76, 2723–2732. [DOI] [PubMed] [Google Scholar]
- 39.Refer to Ref. 29.
- 40. Hunter C. A., Misuraca M. C., Turega S. M., Chem. Sci. 2012, 3, 589–601. [Google Scholar]
- 41. Hunter C. A., Misuraca M. C., Turega S. M., Chem. Sci. 2012, 3, 2462–2469. [Google Scholar]
- 42. Adams H., Chekmeneva E., Hunter C. A., Misuraca M. C., Navarro C., Turega S. M., J. Am. Chem. Soc. 2013, 135, 1853–1863. [DOI] [PubMed] [Google Scholar]
- 43. Sun H., Hunter C. A., Navarro C., Turega S., J. Am. Chem. Soc. 2013, 135, 13129–13141. [DOI] [PubMed] [Google Scholar]
- 44. Ballester P., de Mendoza J. in Modern Supramolecular Chemistry (Eds.: F. Diederich, P. J. Stang, R. R. Tykwinski), Wiley-VCH, Weinheim, 2008, pp. 69–111. [Google Scholar]
- 45. Würthner F., You C.-C., Saha-Möller C. R., Chem. Soc. Rev. 2004, 33, 133–146. [DOI] [PubMed] [Google Scholar]
- 46. Steiner T., Angew. Chem. Int. Ed. 2002, 41, 48–76; [Google Scholar]; Angew. Chem. 2002, 114, 50–80. [Google Scholar]
- 47. Hydrogen Bonded Supramolecular Structures (Eds.: Z.-T. Li, L.-Z. Wu), Springer-Verlag, Berlin, Heidelberg, 2015. [Google Scholar]
- 48. Sijbesma R. P., Meijer E. W., Chem. Commun. 2003, 5–16. [DOI] [PubMed] [Google Scholar]
- 49. Camacho-García J., Montoro-García C., López-Pérez A. M., Bilbao N., Romero-Pérez S., González-Rodríguez D., Org. Biomol. Chem. 2015, 13, 4506–4513. [DOI] [PubMed] [Google Scholar]
- 50. Sessler J. L., Lawrence C. M., Jayawickramarajah J., Chem. Soc. Rev. 2007, 36, 314–325. [DOI] [PubMed] [Google Scholar]
- 51. Sivakova S., Rowan S. J., Chem. Soc. Rev. 2005, 34, 9–21. [DOI] [PubMed] [Google Scholar]
- 52. Yang S. K., Zimmerman S. C., Isr. J. Chem. 2013, 53, 511–520. [Google Scholar]
- 53. Analytical Methods in Supramolecular Chemistry (Eds.: C. A. Schalley), Wiley-VCH, Weinheim, 2012. [Google Scholar]
- 54. Sessler J. L., Magda D., Furuta H., J. Org. Chem. 1992, 57, 818–826. [Google Scholar]
- 55. Sessler J. L., Wang R., J. Am. Chem. Soc. 1996, 118, 9808–9809. [Google Scholar]
- 56. Sessler J. L., Wang R., Angew. Chem. Int. Ed. 1998, 37, 1726–1729; [DOI] [PubMed] [Google Scholar]; Angew. Chem. 1998, 110, 1818–1821. [Google Scholar]
- 57. Sessler J. L., Wang R., J. Org. Chem. 1998, 63, 4079–4091. [Google Scholar]
- 58. Beijer F. H., Sijbesma R. P., Kooijman H., Spek A. L., Meijer E. W., J. Am. Chem. Soc. 1998, 120, 6761–6769. [Google Scholar]
- 59. Söntjens S. H. M., Sijbesma R. P., van Genderen M. H. P., Meijer E. W., J. Am. Chem. Soc. 2000, 122, 7487–7493. [Google Scholar]
- 60. Folmer B. J. B., Sijbesma R. P., Kooijman H., Spek A. L., Meijer E. W., J. Am. Chem. Soc. 1999, 121, 9001–9007. [Google Scholar]
- 61. Tessa ten Cate A., Dankers P. Y. W., Kooijman H., Spek A. L., Sijbesma R. P., Meijer E. W., J. Am. Chem. Soc. 2003, 125, 6860–6861. [DOI] [PubMed] [Google Scholar]
- 62. Tessa ten Cate A., Kooijman H., Spek A. L., Sijbesma R. P., Meijer E. W., J. Am. Chem. Soc. 2004, 126, 3801–3808. [DOI] [PubMed] [Google Scholar]
- 63. Tessa ten Cate A., Dankers P. Y. W., Sijbesma R. P., Meijer E. W., J. Org. Chem. 2005, 70, 5799–5803. [DOI] [PubMed] [Google Scholar]
- 64. Black S. P., Sanders J. K. M., Stefankiewicz A. R., Chem. Soc. Rev. 2014, 43, 1861–1872. [DOI] [PubMed] [Google Scholar]
- 65. Corbett P. T., Leclaire J., Vial L., West K. R., Wietor J.-L., Sanders J. K. M., Otto S., Chem. Rev. 2006, 106, 3652–3711. [DOI] [PubMed] [Google Scholar]
- 66. Zimmerman S. C., Duerr B. F., J. Org. Chem. 1992, 57, 2215–2217. [Google Scholar]
- 67. Saunders M., Hyne J. B., J. Chem. Phys. 1958, 29, 1319–1323. [Google Scholar]
- 68. Suárez M., Lehn J.-M., Zimmerman S. C., Skoulios A., Heinrich B., J. Am. Chem. Soc. 1998, 120, 9526–9532. [Google Scholar]
- 69. Sessler J. L., Jayawickramarajah J., Sathiosatham M., Sherman C. L., Brodbelt J. S., Org. Lett. 2003, 5, 2627–2630. [DOI] [PubMed] [Google Scholar]
- 70. Rzepecki P., Hochdörffer K., Schaller T., Zienau J., Harms K., Ochsenfeld C., Xie X., Schrader T., J. Am. Chem. Soc. 2008, 130, 586–591. [DOI] [PubMed] [Google Scholar]
- 71. Rakotondradany F., Whitehead M. A., Lebuis A.-M., Sleiman H. F., Chem. Eur. J. 2003, 9, 4771–4780. [DOI] [PubMed] [Google Scholar]
- 72. Dhammika Bandara H. M., Burdette S. C., Chem. Soc. Rev. 2012, 41, 1809–1825. [DOI] [PubMed] [Google Scholar]
- 73. Rakotondradany F., Sleiman H. F., Whitehead M. A., J. Mol. Struct. Theochem. 2007, 806, 39–50. [Google Scholar]
- 74. Asadi A., Patrick B. O., Perrin D. M., J. Am. Chem. Soc. 2008, 130, 12860–12861. [DOI] [PubMed] [Google Scholar]
- 75. Orentas E., Wallentin C.-J., Bergquist K.-E., Lund M., Butkus E., Wärnmark K., Angew. Chem. Int. Ed. 2011, 50, 2071–2074; [DOI] [PubMed] [Google Scholar]; Angew. Chem. 2011, 123, 2119–2122. [Google Scholar]
- 76. Yang Y., Xue M., Marshall L. J., de Mendoza J., Org. Lett. 2011, 13, 3186–3189. [DOI] [PubMed] [Google Scholar]
- 77. Marshall L. J., de Mendoza J., Org. Lett. 2013, 15, 1548–1551. [DOI] [PubMed] [Google Scholar]
- 78. Montoro-García C., Camacho-García J., López-Pérez A. M., Bilbao N., Romero-Pérez S., Mayoral M. J., González-Rodríguez D., Angew. Chem. Int. Ed. 2015, 54, 6780–6784; [DOI] [PubMed] [Google Scholar]; Angew. Chem. 2015, 127, 6884–6888. [Google Scholar]
- 79. Romero-Pérez S., Camacho-García J., Montoro-García C., López-Pérez A. M., Sanz A., Mayoral M. J., González-Rodríguez D., Org. Lett. 2015, 17, 2664–2667. [DOI] [PubMed] [Google Scholar]
- 80. Davis J. T., Angew. Chem. Int. Ed. 2004, 43, 668–698; [DOI] [PubMed] [Google Scholar]; Angew. Chem. 2004, 116, 684–716. [Google Scholar]
- 81. Davis J. T., Spada G. P., Chem. Soc. Rev. 2007, 36, 296–313. [DOI] [PubMed] [Google Scholar]
- 82. González-Rodríguez D., van Dongen J. L. J., Lutz M., Spek A. L., Schenning A. P. H. J., Meijer E. W., Nat. Chem. 2009, 1, 151–155. [DOI] [PubMed] [Google Scholar]
- 83. Sessler J. L., Sathiosatham M., Doerr K., Lynch V., Abboud K. A., Angew. Chem. Int. Ed. 2000, 39, 1300–1303; [DOI] [PubMed] [Google Scholar]; Angew. Chem. 2000, 112, 1356–1359. [Google Scholar]
- 84. Inui Y., Shiro M., Fukuzumi S., Kojima T., Org. Biomol. Chem. 2013, 11, 758–764. [DOI] [PubMed] [Google Scholar]
- 85. Fraschetti C., Montagna M., Guarcini L., Guidoni L., Filippi A., Chem. Commun. 2014, 50, 14767–14770. [DOI] [PubMed] [Google Scholar]
- 86. Kotch F. W., Sidorov V., Lam Y.-F., Kayser K. J., Li H., Kaucher M. S., Davis J. T., J. Am. Chem. Soc. 2003, 125, 15140–15150. [DOI] [PubMed] [Google Scholar]
- 87. Nikan M., Sherman J. C., Angew. Chem. Int. Ed. 2008, 47, 4900–4902; [DOI] [PubMed] [Google Scholar]; Angew. Chem. 2008, 120, 4978–4980. [Google Scholar]
- 88. Nikan M., Sherman J. C., J. Org. Chem. 2009, 74, 5211–5218. [DOI] [PubMed] [Google Scholar]
- 89. Bare G. A. L., Liu B., Sherman J. C., J. Am. Chem. Soc. 2013, 135, 11985–11989. [DOI] [PubMed] [Google Scholar]
- 90. Laguerre A., Stefan L., Larrouy M., Genest D., Novotna J., Pirrotta M., Monchaud D., J. Am. Chem. Soc. 2014, 136, 12406–12414. [DOI] [PubMed] [Google Scholar]
- 91. Inui Y., Fukuzumi S., Kojima T., Dalton Trans. 2013, 42, 3779–3782. [DOI] [PubMed] [Google Scholar]
- 92. Zafar A., Geib S. J., Hamuro Y., Hamilton A. D., New J. Chem. 1998, 22, 137–141. [Google Scholar]
- 93. Fan E., Vicent C., Geib S. J., Hamilton A. D., Chem. Mater. 1994, 6, 1113–1117. [Google Scholar]
- 94. Yang J., Fan E., Gieb S. J., Hamilton A. D., J. Am. Chem. Soc. 1993, 115, 5314–5315. [Google Scholar]
- 95. Yang J., Marendaz J.-L., Geib S. J., Hamilton A. D., Tetrahedron Lett. 1994, 35, 3665–3668. [Google Scholar]
- 96. Keizer H. M., Gonzalez J. J., Segura M., Prados P., Sijbesma R. P., Meijer E. W., de Mendoza J., Chem. Eur. J. 2005, 11, 4602–4608. [DOI] [PubMed] [Google Scholar]
- 97. Marsh A., Silvestri M., Lehn J.-M., Chem. Commun. 1996, 1527–1528. [Google Scholar]
- 98. Mascal M., Hext N. M., Warmuth R., Moore M. H., Turkenburg J. P., Angew. Chem. Int. Ed. Engl. 1996, 35, 2204–2206; [Google Scholar]; Angew. Chem. 1996, 108, 2348–2350. [Google Scholar]
- 99. Mascal M., Hext N. M., Warmuth R., Arnall-Culliford J. R., Moore M. H., Turkenburg J. P., J. Org. Chem. 1999, 64, 8479–8484. [Google Scholar]
- 100. Mascal M., Farmer S. C., Arnall-Culliford J. R., J. Org. Chem. 2006, 71, 8146–8150. [DOI] [PubMed] [Google Scholar]
- 101. Kolotuchin S. V., Zimmerman S. C., J. Am. Chem. Soc. 1998, 120, 9092–9093. [Google Scholar]
- 102. Zimmerman S. C., Zeng F., Reichert D. E. C., Kolotuchin S. V., Science 1996, 271, 1095–11098. [DOI] [PubMed] [Google Scholar]
- 103. Corbin P. S., Lawless L. J., Li Z., Ma Y., Witmer M. J., Zimmerman S. C., Proc. Natl. Acad. Sci. USA 2002, 99, 5099–5104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 104. Ma Y., Kolotuchin S. V., Zimmerman S. C., J. Am. Chem. Soc. 2002, 124, 13757–13769. [DOI] [PubMed] [Google Scholar]
- 105. Yang Y., Xue M., Xiang J.-F., Chen C.-F., J. Am. Chem. Soc. 2009, 131, 12657–12663. [DOI] [PubMed] [Google Scholar]
- 106. Yang Y., Huang F., Chen C.-F., Xia M., Cai Q., Qian F.-J., Xiang J., Sci. Rep. 2013, 3, 1059. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 107. Yagai S., Kubota S., Saito H., Unoike K., Karatsu T., Kitamura A., Ajayaghosh A., Kanesato M., Kikkawa Y., J. Am. Chem. Soc. 2009, 131, 5408–5410. [DOI] [PubMed] [Google Scholar]
- 108. Yagai S., Goto Y., Karatsu T., Kitamura A., Kikkawa Y., Chem. Eur. J. 2011, 17, 13657–13660. [DOI] [PubMed] [Google Scholar]
- 109. Yagai S., Goto Y., Lin X., Karatsu T., Kitamura A., Kuzuhara D., Yamada H., Kikkawa Y., Saeki A., Seki S., Angew. Chem. Int. Ed. 2012, 51, 6643–6647; [DOI] [PubMed] [Google Scholar]; Angew. Chem. 2012, 124, 6747–6751. [Google Scholar]
- 110. Yagai S., Suzuki M., Lin X., Gushiken M., Noguchi T., Karatsu T., Kitamura A., Saeki A., Seki S., Kikkawa Y., Tani Y., Nakayama K.-i., Chem. Eur. J. 2014, 20, 16128–16137. [DOI] [PubMed] [Google Scholar]
- 111. Fenniri H., Mathivanan P., Vidale K. L., Sherman D. M., Hallenga K., Wood K. V., Stowell J. G., J. Am. Chem. Soc. 2001, 123, 3854–3855. [DOI] [PubMed] [Google Scholar]
- 112. Fenniri H., Deng B.-L., Ribbe A. E., Hallenga K., Jacob J., Thiyagarajan P., Proc. Natl. Acad. Sci. USA 2002, 99, 6487–6492. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 113. Fenniri H., Deng B.-L., Ribbe A. E., J. Am. Chem. Soc. 2002, 124, 11064–11072. [DOI] [PubMed] [Google Scholar]
- 114. Deng B.-L., Beingessner R. L., Johnson R. S., Girdhar N. K., Danumah C., Yamazaki T., Fenniri H., Macromolecules 2012, 45, 7157–7162. [Google Scholar]
- 115. Tikhomirov G., Oderinde M., Makeiff D., Mansouri A., Lu W., Heirtzler F., Kwok D. Y., Fenniri H., J. Org. Chem. 2008, 73, 4248–4251. [DOI] [PubMed] [Google Scholar]
- 116. Tikhomirov G., Yamazaki T., Kovalenko A., Fenniri H., Langmuir 2008, 24, 4447–4450. [DOI] [PubMed] [Google Scholar]
- 117. Durmus A., Gunbas G., Farmer S. C., Olmstead M. M., Mascal M., Legese B., Cho J.-Y., Beingessner R. L., Yamazaki T., Fenniri H., J. Org. Chem. 2013, 78, 11421–11426. [DOI] [PubMed] [Google Scholar]
- 118. Borzsonyi G., Johnson R. S., Myles A. J., Cho J.-Y., Yamazaki T., Beingessner R. L., Kovalenko A., Fenniri H., Chem. Commun. 2010, 46, 6527–6529. [DOI] [PubMed] [Google Scholar]
- 119. Borzsonyi G., Beingessner R. L., Yamazaki T., Cho J.-Y., Myles A. J., Malac M., Egerton R., Kawasaki M., Ishizuka K., Kovalenko A., Fenniri H., J. Am. Chem. Soc. 2010, 132, 15136–15139. [DOI] [PubMed] [Google Scholar]
- 120. Johnson R. S., Yamazaki T., Kovalenko A., Fenniri H., J. Am. Chem. Soc. 2007, 129, 5735–5743. [DOI] [PubMed] [Google Scholar]
- 121. Hemraz U. D., El-Bakkari M., Yamazaki T., Cho J.-Y., Beingessner R. L., Fenniri H., Nanoscale 2014, 6, 9421–9427. [DOI] [PubMed] [Google Scholar]
- 122. Yokoyama T., Yokoyama S., Okuno Y., Mashiko S., Nature 2001, 413, 619–621. [DOI] [PubMed] [Google Scholar]
- 123. Fendt L.-A., Stöhr M., Wintjes N., Enache M., Jung T. A., Diederich F., Chem. Eur. J. 2009, 15, 11139–11150. [DOI] [PubMed] [Google Scholar]
- 124. Selvanathan S., Peters M. V., Schwarz J., Hecht S., Grill L., Appl. Phys. A 2008, 93, 247–252. [Google Scholar]
- 125. Lei S., Surin M., Tahara K., Adisoejoso J., Lazzaroni R., Tobe Y., De Feyter S., Nano Lett. 2008, 8, 2541–2546. [DOI] [PubMed] [Google Scholar]
- 126. Lu J., Lei S.-b., Zeng Q.-d., Kang S.-z., Wang C., Wan I.-j., Bai C.-l., J. Phys. Chem. B 2004, 108, 5161–5165. [Google Scholar]
- 127. Mamdouh W., Uji-i H., Dulcey A. E., Percec V., De Feyter S., De Schryver F. C., Langmuir 2004, 20, 7678–7685. [DOI] [PubMed] [Google Scholar]
- 128. Ahn S., Morrison C. N., Matzger A. J., J. Am. Chem. Soc. 2009, 131, 7946–7947. [DOI] [PubMed] [Google Scholar]
- 129. Xie Z. X., Charlier J., Cousty J., Surf. Sci. 2000, 448, 201–211. [Google Scholar]
- 130. Kunkel D. A., Hooper J., Simpson S., Miller D. P., Routaboul L., Braunstein P., Doudin B., Beniwal S., Dowben P., Skomski R., Zurek E., Enders A., J. Chem. Phys. 2015, 142, 101921. [DOI] [PubMed] [Google Scholar]
- 131. Stöhr M., Wahl M., Galka C. H., Riehm T., Jung T. A., Gade L. H., Angew. Chem. Int. Ed. 2005, 44, 7394–7398; [DOI] [PubMed] [Google Scholar]; Angew. Chem. 2005, 117, 7560–7564. [Google Scholar]
- 132. Jeffrey G. A., An Introduction to Hydrogen Bonding, Oxford University Press, Oxford, New York, 1997. [Google Scholar]
- 133. Walch H., Maier A- K., Heckl W. M., Lackinger M., J. Phys. Chem. C 2009, 113, 1014–1019. [Google Scholar]
- 134. Jonkheijm P., Miura A., Zdanowska M., Hoeben F. J. M., De Feyter S., Schenning A. P. H. J., De Schryver F. C., Meijer E. W., Angew. Chem. Int. Ed. 2004, 43, 74–78; [DOI] [PubMed] [Google Scholar]; Angew. Chem. 2004, 116, 76–80. [Google Scholar]
- 135. Hoeben F. J. M., Zhang J., Lee C. C., Pouderoijen M. J., Wolffs M., Würthner F., Schenning A. P. H. J., Meijer E. W., De Feyter S., Chem. Eur. J. 2008, 14, 8579–8589. [DOI] [PubMed] [Google Scholar]
- 136. Mourran A., Ziener U., Möller M., Suarez M., Lehn J. M., Langmuir 2006, 22, 7579–7586. [DOI] [PubMed] [Google Scholar]
- 137. Ciesielski A., Lena S., Masiero S., Spada G. P., Samori P., Angew. Chem. Int. Ed. 2010, 49, 1963–1966; [DOI] [PubMed] [Google Scholar]; Angew. Chem. 2010, 122, 2007–2010. [Google Scholar]
- 138. Gellert M., Lipsett M. N., Davies D. R., Proc. Natl. Acad. Sci. USA 1962, 48, 2013–2018. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 139. Lena S., Masiero S., Pieraccini S., Spada G. P., Chem. Eur. J. 2009, 15, 7792–7806. [DOI] [PubMed] [Google Scholar]
- 140. Masiero S., Gottarelli G., Pieraccini S., Chem. Commun. 2000, 1995–1996. [DOI] [PubMed] [Google Scholar]
- 141. Martic S., Liu X., Wang S., Wu G., Chem. Eur. J. 2008, 14, 1196–1204. [DOI] [PubMed] [Google Scholar]
- 142. Spada G. P., Lena S., Masiero S., Pieraccini S., Surin M., Samori P., Adv. Mater. 2008, 20, 2433–2439. [Google Scholar]
- 143. Marlow A. L., Mezzina E., Spada G. P., Masiero S., Davis J. T., Gottarelli G., J. Org. Chem. 1999, 64, 5116–5123. [DOI] [PubMed] [Google Scholar]
- 144. Otero R., Schock M., Molina L. M., Laegsgaard E., Stensgaard I., Hammer B., Besenbacher F., Angew. Chem. Int. Ed. 2005, 44, 2270–2275; [DOI] [PubMed] [Google Scholar]; Angew. Chem. 2005, 117, 2310–2315. [Google Scholar]
- 145. González-Rodríguez D., Janssen P. G. A., Martín-Rapún R., De Cat I., De Feyter S., Schenning A. P. H. J., Meijer E. W., J. Am. Chem. Soc. 2010, 132, 4710–4719. [DOI] [PubMed] [Google Scholar]
- 146. Ciesielski A., Colella S., Zalewski L., Bruchmann B., Samori P., Cryts. Eng. Comm. 2011, 13, 5535–5537. [Google Scholar]
- 147. Iyoda M., Yamakawa J., Rahman M. J., Angew. Chem. Int. Ed. 2011, 50, 10522–10553; [DOI] [PubMed] [Google Scholar]; Angew. Chem. 2011, 123, 10708–10740. [Google Scholar]
- 148. Shimizu L. S., Salpage S. R., Korous A. A., Acc. Chem. Res. 2014, 47, 2116–2127. [DOI] [PubMed] [Google Scholar]
- 149. Modern Supramolecular Chemistry. Strategies for Macrocycle Synthesis (Eds.: F. Diederich, P. J. Stang, R. R. Tykwinski), Wiley-VCH, Weinheim, 2008. [Google Scholar]
- 150. Höger S., Angew. Chem. Int. Ed. 2005, 44, 3806–3808; [DOI] [PubMed] [Google Scholar]; Angew. Chem. 2005, 117, 3872–3875. [Google Scholar]