

Abstract
Density functional theory (DFT) calculations were carried out to study the reaction mechanism of the Suzuki–Miyaura rhodium‐catalyzed hydroarylation of fullerene (C60) by phenylboronic acid in the presence of water. As found experimentally, our results confirm that addition of the phenyl group and the hydrogen atom in C60 occurs at the [6,6] bond. The rate‐determining step corresponds to the simultaneous transfer of a hydrogen atom from a water molecule to C60 and the recovery of the active species. The use of 2‐phenyl‐1,3,2‐dioxaborinane and the 4,4,5,5‐tetramethyl‐2‐phenyl‐1,3,2,‐dioxaborolane instead of phenylboronic acid as organoborate agents does not lead to great modifications of the energy profile. The possible higher steric hindrance of 4,4,5,5‐tetramethyl‐2‐phenyl‐1,3,2,‐dioxaborolane should not inhibit its use in the hydroarylation of C60. Overall, we show how organoboron species arylate C60 in rhodium‐based catalysis assisted by water as a source of protons.
Keywords: density functional theory, green chemistry, homogeneous catalysis, organoboron compounds, rhodium, Suzuki–Miyaura
Introduction
After thirty years since it was discovered,1 fullerene C60 is still an attractive nanocarbon‐based material due to its unique structure,2 specific properties, and numerous derivatives that enable potential applications in biology, medicine, electronics, photovoltaics, and cosmetic industries.3, 4 The reactivity of fullerenes is rich;5 the most important synthetic pathways used to functionalize C60 are not only pericyclic reactions, such as cyclopropanations,6 Diels–Alder reactions, and 1,3‐dipolar cycloadditions,7 but also the addition of free radicals or nucleophiles.8
The hydroalkylation and hydroarylation of C60 have also been fascinating areas of research.9 In this framework, organolithium and Grignard compounds are used to form intermediate anions (RC60 −), which undergo protonation to yield hydroalkylated or hydroarylated derivatives.3 Following the advance in rhodium(I) and palladium(II) catalysis of organoboron‐based hydroarylation chemistry,10, 11, 12 Itami et al. provided a catalytic pathway for the hydroalkylation and hydroarylation of C60 (Scheme 1).13, 14 Their work was inspired by the work of Miyaura et al., who discovered the rhodium‐catalyzed conjugate addition of organoboron compounds to enones in aqueous solution.11, 12, 15, 16 They also established the basis of the mechanism to carry out the addition of aryl‐ or 1‐alkenylboronic acids to enones. In this case, the catalytic cycle encompasses transmetalation between rhodium(I) enolates and arylboronic acids to produce arylrhodium(I) species followed by insertion of enones into Ar−Rh bonds.17 The mechanistic pathway of the addition of organoboron compounds to electron‐deficient alkenes such as C60 catalyzed by rhodium complexes remains rather unknown.18 Itami et al.14, 19 proposed a general catalytic cycle, based on the work of Miyaura et al.,11 for the hydroarylations of C60 fullerene, and the details of the reaction mechanism and the function of water are missing or incomplete. For this reason, here we aim to unravel the role of each agent in the Suzuki–Miyaura‐like reaction catalyzed by rhodium complexes of C60 and organoborates as cocatalysts in the presence of water. In order to do so, we explore the catalytic cycle that leads to the hydroarylation of C60 via density functional theory (DFT) methods.
Scheme 1.
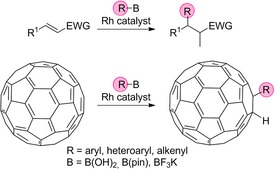
Hydroarylation of electron‐deficient alkenes (EWG: electronwithdrawing group) and fullerenes by means of organoborates and rhodium‐based catalysts.
Results and Discussion
The reaction pathway is divided into four main different parts (see Figure 1): 1) transmetalation of Rh−OH species with water and the organoborate to generate the Ar−RhB(OH)3 complex (A→C); 2) elimination of B(OH)3 to give the Rh−Ar species (C→D); 3) insertion or arylrhodation of Ar−Rh to C60 (D→F); 4) protonation of Ar−C60 through a water molecule and release of the Rh−OH species to conclude the hydroarylation process of C60.
Figure 1.
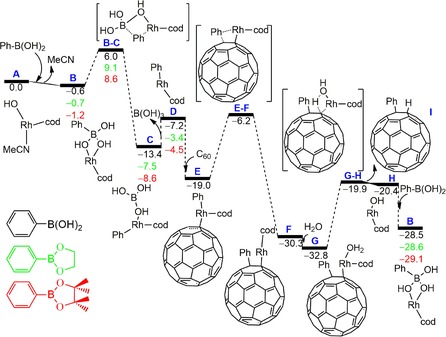
Gibbs energy reaction profiles for the rhodium‐catalyzed addition to fullerene (C60) of phenylboronic acid (black), 2‐phenyl‐1,3,2‐dioxaborinane (green), and 4,4,5,5‐tetramethyl‐2‐phenyl‐1,3,2,‐dioxaborolane (red) in ortho‐dichlorobenzene solution. Values for the Gibbs energy are given in kcal mol−1. Schematic drawings of molecules refer to the reaction with phenylboronic acid.
The catalytic species A is obtained from the precatalytic species [Rh(cod)(MeCN)2]BF4 (cod=1,5‐cyclooctadiene) through the dissociation of an acetonitrile ligand followed by the coordination of the hydroxy group from a water molecule releasing a proton that, with the initial counter anion BF4 −, yields HBF4. This step has an energetic cost of 24.3 kcal mol−1,20 and is a common step for all organoborates.14, 19 First, we discuss the reaction mechanism of the Suzuki–Miyaura rhodium‐catalyzed hydroarylation of C60 by phenylboronic acid in the presence of water. The neutral species [Rh(cod)(MeCN)(OH)] (A) can release an acetonitrile ligand and coordinate Ph−B(OH)2 in a slightly exergonic process (0.6 kcal mol−1) to give B. Then species B overcomes a barrier of 6.6 kcal mol−1 to transfer the phenyl group from boron to rhodium to yield Ph(cod)RhB(OH)3 (complex C).
Release of B(OH)3 from C results in D with an energetic cost of only 6.2 kcal mol−1. In D, rhodium is surrounded by the cod and phenyl ligands. η2‐Coordination of D to C60 to form E is favorable by 11.8 kcal mol−1, in agreement with previous experimental21 and computational insights,22 with slightly longer Rh−C bonds (2.28 and 2.31 Å) to be compared with 2.20 Å for RhH(CO)(PH3)2(C60) in part due to the sterically demanding cod ligand. From E, the transfer of the phenyl group to the adjacent carbon atom requires to overcome a relatively low barrier of 12.8 kcal mol−1, driving to the next intermediate F, which is quite stable (11.3 kcal mol−1 more stable than its precursor E). The resulting attacked bond in F is a [6,6] bond as experimentally found.14, 19 We also examined the attack to a [5,6] bond of C60 in E and confirmed that the transition state and the resulting complex are less stable by 3.6 and 7.5 kcal mol−1, respectively. Moreover, we were not able to obtain an optimized structure with η 5 and η 6 coordination of the PhRh(cod) in E or Rh(cod) in F to C60.
Finally, in order to get the organic substituted C60 product and regenerate the catalyst, a coordination of a water molecule to rhodium occurs to yield G. This complex (G) is transformed into H via the transfer of a hydrogen atom from water to the carbon atom of C60 bonded to rhodium and concomitant release of (cod)Rh−OH. The role of water is then to provide the hydrogen atom for the formation of the phenyl(hydro)[60]fullerene and the OH group to recover the (cod)Rh−OH species.14, 19 The structure, acidity, and aromaticity of this phenyl(hydro)[60]fullerene species were discussed previously by Geerlings and co‐workers.23 The complete G→H transformation has a Gibbs energy barrier of 12.9 kcal mol−1. In the next step, Rh in H coordinates a new entering Ph−B(OH)2 molecule, which is a reactant in excess, to recover the catalytic species B again and close the catalytic cycle. The energy difference between species B at the beginning and at the end of the catalytic cycle in Figure 1 corresponds to the Gibbs reaction energy of the transformation of C60, H2O, and PhB(OH)2 into B(OH)3 and C60PhH in a process that is exergonic by 27.9 kcal mol−1.
For 2‐phenyl‐1,3,2.dioxaborinane and 4,4,5,5‐tetramethyl‐2‐phenyl‐1,3,2‐dioxaborolane, the Gibbs reaction energies are the same: −27.9 kcal mol−1. Apart from the relatively high energy cost to generate the catalytic species A from the precatalytic species [Rh(cod)(MeCN)2]BF4, for the catalytic cycle, the G to H conversion is the rate determining step. However, the phenyl transfer in E→F is in competition, being the corresponding barrier of the transition state just 0.1 kcal mol−1 lower in energy (see Figure 2).24
Figure 2.
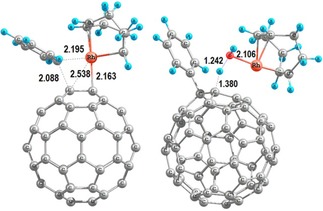
Density functional theory (DFT)‐optimized geometries for the transition states of the transformations E→F (left) and G→H (right); key distances are given in Å.
On the other hand, considering the non‐stoichiometric amount of C60 with respect to the metal catalyst, the coordination of a second C60 molecule substituting the cod ligand in the intermediate species displayed in Figure 1 was also investigated (see Figure S1 in the Supporting Information). This alternative path for the transformation of A to H implies the loss of acetonitrile and cod, the attack of RhOH to C60, and subsequent addition of PhB(OH)2. Overall, this path to H has to surmount a Gibbs energy barrier higher than 40 kcal mol−1 and, therefore, it is not operative and can be ruled out.
The Gibbs energy profiles corresponding to alternative organoborates, such as 2‐phenyl‐1,3,2,‐dioxaborinane and 4,4,5,5‐tetramethyl‐2‐phenyl‐1,3,2,‐dioxaborolane, are similar to that obtained with the phenylboronic acid (Figure 1). The fact that 2‐phenyl‐1,3,2,‐dioxaborinane gives a nearly identical energy profile to that of phenylboronic acid concurs with the experimental result that this organoborate also results in the formation of the corresponding aryl(hydro)fullerene. However, for the 4,4,5,5‐tetramethyl‐2‐phenyl‐1,3,2,‐dioxaborolane, no yield is observed experimentally. This is unexpected because the different organoborates share the same rate‐determining step. Maybe it could be related to the somewhat more energetically difficult B to C transformation. What is clear is that steric hindrance seems not to be the cause of the decreased reactivity of 4,4,5,5‐tetramethyl‐2‐phenyl‐1,3,2,‐dioxaborolane.
Conclusions
We have investigated by DFT calculations the reaction mechanism of the Suzuki–Miyaura rhodium‐catalyzed hydroarylation of C60 by phenylboronic acid in the presence of water. Our results show that the until now unknown role of water is to coordinate the PhC60Rh(cod) complex to provide the hydrogen atom for the formation of the phenyl(hidro)[60]fullerene and the OH group to recover the (cod)Rh−OH species. This step is the rate‐determining step of the whole catalytic cycle.
Our results with alternative organoborates, such as dioxaborinane and 4,4,5,5‐tetramethyl‐2‐phenyl‐1,3,2,‐dioxaborolane, showed no remarkable differences for the phenyl transfer to the metal center, thus excluding the steric hindrance of the organoborate as a key factor to tune and improve the yield of the Suzuki–Miyaura reaction of C60 as a substrate, rhodium as a catalyst, and organoborate as cocatalysts. Water turns out to be a key reactant.
Experimental Section
All DFT calculations were completed with the Gaussian09 set of programs.25 For geometry optimizations, the well‐established and computationally fast hybrid‐GGA functional B3LYP was used.26 Geometry optimizations were performed without symmetry constraints, and the located stationary points were characterized by analytical frequency calculations. The electronic configuration of the molecular systems was described with the Gaussian 6‐31G(d) basis set with a polarization function for H, C, N, B, and O.27
For rhodium, we used the small‐core, quasi‐relativistic Stuttgart/Dresden (SDD) effective core potential with an associated valence contracted basis set (standard SDD keywords in Gaussian 09).28 Zero‐point energies (ZPEs) and thermal corrections were calculated at the B3LYP level. Single‐point energy calculations with the M06 functional29 in solution were performed with the 6‐311G(d,p) basis set for main group atoms,30 and again the same SDD pseudopotential for rhodium.31 Solvent effects were included with the polarizable continuous solvation model polarizable continuum model (PCM) using ortho‐dichlorobenzene (o‐DCB) as a solvent (the solvent employed in experimental studies is a 9:1 mixture of o‐DCB and water). The Gibbs energy profiles in water are given in the Supporting Information. Relative Gibbs energy differences in water and in o‐DCB are lower than 2.0 kcal mol−1.
The M06 energy calculations were carried out with the scf=tight and integral(grid=ultrafinegrid) keywords. Reported energies are M06/6‐311G(d,p)∼SDD//B3LYP/6‐31G(d)∼SDD electronic energies corrected with ZPEs, thermal energies, and entropy effects calculated at 298 K using the B3LYP/6‐31G(d)∼SDD method. We also corrected the energy for solvation effects present in an o‐DCB solution that were calculated at the M06/6‐311G(d,p)∼SDD//B3LYP/6‐31G(d)∼SDD level with the PCM method.32 Finally, we applied a concentration correction of 1.89 kcal mol−1 for the Gibbs energies in solution to account for the condition change from 1 atm to 1 m concentration when going from gas phase to solution.33 For water, the correction was 4.27 kcal mol−1. Furthermore, for the aqueous solvation free energy of the proton, we assumed the value of −262.2 kcal mol−1 from the literature.34
Supporting information
As a service to our authors and readers, this journal provides supporting information supplied by the authors. Such materials are peer reviewed and may be re‐organized for online delivery, but are not copy‐edited or typeset. Technical support issues arising from supporting information (other than missing files) should be addressed to the authors.
Supplementary
Acknowledgements
A.P. thanks the Spanish Ministry of Economy and Competitiveness (MINECO) for a Ramón y Cajal contract (RYC‐2009‐05226) and grant no. CTQ2014‐59832‐JIN, and a European Commission Career Integration Grant (CIG09‐GA‐2011‐293900). J.P.M. gratefully acknowledges a Ph.D. fellowship (register/application no. 217067/312543) from the Mexican National Council of Science and Technology (CONACYT). M.S. acknowledges funding through a European Union (EU) FEDER fund (UNGI08‐4E‐003 and UNGI10‐4E‐801), the Generalitat de Catalunya (Spain) (project 2014SGR931), a Catalan Institution for Research and Advanced Studies (ICREA) Academia prize (2014), and MINECO project CTQ2014‐54306‐P.
J. P. Martínez, M. Solà, A. Poater, ChemistryOpen 2015, 4, 774.
References
- 1. Kroto H. W., Heath J. R., O'Brien S. C., Curl R. F., Smalley R. E., Nature 1985, 318, 162–163. [Google Scholar]
- 2.
- 2a. Fullerenes: Chemistry, Physics, and Technology, (Eds.: K. M. Kadish, R. S. Ruoff), Wiley, New York, 2000; [Google Scholar]
- 2b. Fullerenes: Principles and Applications, (Eds.: F. Langa, J.-F. Nierengarten), RSC, Cambridge, 2007; [Google Scholar]
- 2c. Martín N., Chem. Commun. 2006, 2093–2104; [DOI] [PubMed] [Google Scholar]
- 2d. Martín N., Altable M., Filippone S., Martín-Domenech A., Synlett 2007, 3077–3095. [Google Scholar]
- 3. Tong J., Zimmerman M. C., Li S., Yi X., Luxenhofer R., Jordan R., Kabanov A. V., Biomaterials 2011, 32, 3654–3665. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.
- 4a. Kato S., Taira H., Aoshima H., Saitoh Y., Miwa N., J. Nanosci. Nanotechnol. 2010, 10, 6769–6774; [DOI] [PubMed] [Google Scholar]
- 4b. Jennepalli S., Pyne S. G., Keller P. A., RSC Adv. 2014, 4, 46383–46398; [Google Scholar]
- 4c. Hosseini M., Rivera-Nazario D. M., Echegoyen L. A., ACS Appl. Mater. Interfaces 2014, 6, 3712–3720; [DOI] [PubMed] [Google Scholar]
- 4d. Cho S. W., Piper L. F. J., DeMasi A., Preston A. R. H., Smith K. E., Chauhan K. V., Sullivan P., Hatton R. A., Jones T. S., J. Phys. Chem. C 2010, 114, 1928–1933. [Google Scholar]
- 5.
- 5a. Bingel C., Chem. Ber. 1993, 126, 1957–1959; [Google Scholar]
- 5b. Nierengarten J.-F., Gramlich V., Cardullo F., Diederich F., Angew. Chem. Int. Ed. Engl. 1996, 35, 2101–2103; [Google Scholar]; Angew. Chem. 1996, 108, 2242–2244; [Google Scholar]
- 5c. Camps X., Hirsch A., J. Chem. Soc. Perkin Trans. 1 1997, 1595–1596; [Google Scholar]
- 5d. Nierengarten J.-F., Felder D., Nicoud J.-F., Tetrahedron Lett. 1998, 39, 2747–2750; [Google Scholar]
- 5e. Cheng F., Yang X., Zhu H., Song Y., Tetrahedron Lett. 2000, 41, 3947–3950; [Google Scholar]
- 5f. Martín N., Altable M., Filippone S., Martin-Domenech A., Poater A., Solà M., Chem. Eur. J. 2005, 11, 2716–2729; [DOI] [PubMed] [Google Scholar]
- 5g. Alegret N., Rodriguez-Fortea A., Poblet J. M., Chem. Eur. J. 2013, 19, 5061–5069; [DOI] [PubMed] [Google Scholar]
- 5h. Maroto E. E., Filippone S., Suarez M., Martinez-Alvarez R., de Cozar A., Cossio F. P., Martin N., J. Am. Chem. Soc. 2014, 136, 705–712; [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5i. Maroto E. E., Mateos J., García-Borràs M., Osuna S., Filippone S., Herranz M. A., Solà M., Martín N., J. Am. Chem. Soc. 2014, 137, 1190–1197. [DOI] [PubMed] [Google Scholar]
- 6.
- 6a. Rotello V. M., Howard J. B., Yadav T., Conn M. M., Viani E., Giovane L. M., Lafleur A. L., Tetrahedron Lett. 1993, 34, 1561–1562; [Google Scholar]
- 6b. Tsuda M., Ishida T., Nogami T., Kurono S., Ohashi M., J. Chem. Soc. Chem. Commun. 1993, 1296–1297; [Google Scholar]
- 6c. Komatsu K., Murata Y., Sugita N., Takeuchi K., Wan T. S. M., Tetrahedron Lett. 1993, 34, 8473–8476; [Google Scholar]
- 6d. Lamparth I., Maichle-Mössmer C., Hirsch A., Angew. Chem. Int. Ed. Engl. 1995, 34, 1607–1609; [Google Scholar]; Angew. Chem. 1995, 107, 1755–1757; [Google Scholar]
- 6e. Murata Y., Kato N., Fujiwara K., Komatsu K., J. Org. Chem. 1999, 64, 3483–3488; [DOI] [PubMed] [Google Scholar]
- 6f. Mikami K., Matsumoto S., Tonoi T., Okubo Y., Suenobu T., Fukuzumi S., Tetrahedron Lett. 1998, 39, 3733–3736; [Google Scholar]
- 6g. Segura J. L., Martín N., Chem. Rev. 1999, 99, 3199–3246; [DOI] [PubMed] [Google Scholar]
- 6h. Takaguchi Y., Tajima T., Ohta K., Motoyoshiya J., Aoyama H., Wakahara T., Akasaka T., Fujitsuka M., Ito O., Angew. Chem. Int. Ed. 2002, 41, 817–819; [DOI] [PubMed] [Google Scholar]; Angew. Chem. 2002, 114, 845–847. [Google Scholar]
- 7.
- 7a. Maggini M., Scorrano G., Prato M., J. Am. Chem. Soc. 1993, 115, 9798–9799; [Google Scholar]
- 7b. Prato M., Maggini M., Acc. Chem. Res. 1998, 31, 519–526; [Google Scholar]
- 7c. Imahori H., Norteda H., Yamada H., Nishimura Y., Yamazaki I., Sakata Y., Fukuzumi S., J. Am. Chem. Soc. 2001, 123, 100–110; [DOI] [PubMed] [Google Scholar]
- 7d. Wang G.-W., Zhang T.-H., Hao E.-H., Jiao L.-J., Murata Y., Komatsu K., Tetrahedron 2003, 59, 55–60; [Google Scholar]
- 7e. Wang G.-W., Li J.-X., Li Y.-J., Liu Y.-C., J. Org. Chem. 2006, 71, 680–684; [DOI] [PubMed] [Google Scholar]
- 7f. Alvarez A., Ochoa E., Verdecia Y., Suárez M., Solà M., Martín N., J. Org. Chem. 2005, 70, 3256–3262. [DOI] [PubMed] [Google Scholar]
- 8.
- 8a. Hirsch A., Brettreich M., Fullerenes: Chemistry and Reactions, Wiley-VCH, Weinheim, 2005; [Google Scholar]
- 8b. Izquierdo M., Osuna S., Filippone S., Martín-Domenech A., Solà M., Martín N., J. Org. Chem. 2009, 74, 1480–1487; [DOI] [PubMed] [Google Scholar]
- 8c. Izquierdo M., Osuna S., Filippone S., Martín-Domenech N., Solà M., Martín N., J. Org. Chem. 2009, 74, 6253–6259; [DOI] [PubMed] [Google Scholar]
- 8d. Izquierdo M., Osuna S., Filippone S., Martín-Domenech A., Solà M., Martín N., Eur. J. Org. Chem. 2009, 6231–6238; [DOI] [PubMed] [Google Scholar]
- 8e. Keshavarz-K K. M., Knight B., Srdanov G., Wudl F., J. Am. Chem. Soc. 1995, 117, 11371–11372; [Google Scholar]
- 8f. Nambo M., Wakamiya A., Yamaguchi S., Itami K., J. Am. Chem. Soc. 2009, 131, 15112–15113; [DOI] [PubMed] [Google Scholar]
- 8g. Hirsch A., Li Q., Wudl F., Angew. Chem. Int. Ed. Engl. 1991, 30, 1309–1310; [Google Scholar]; Angew. Chem. 1991, 103, 1339–1341. [Google Scholar]
- 9. Tzirakis M. D., Orfanopoulos M., Chem. Rev. 2013, 113, 5262–5321. [DOI] [PubMed] [Google Scholar]
- 10.
- 10a. Cho C. S., Motofusa S., Ohe K., Uemura S., Shim S. C., J. Org. Chem. 1995, 60, 883–888; [Google Scholar]
- 10b. Zhang T., Shi M., Chem. Eur. J. 2008, 14, 3759–3764; [DOI] [PubMed] [Google Scholar]
- 10c. Nambo M., Wakamiya A., Itami K., Chem. Sci. 2012, 3, 3474–3481. [Google Scholar]
- 11. Takaya Y., Ogasawara M., Hayashi T., Sakai M., Miyaura N., J. Am. Chem. Soc. 1998, 120, 5579–5580. [Google Scholar]
- 12. Hayashi T., Yamasaki K., Chem. Rev. 2003, 103, 2829–2844. [DOI] [PubMed] [Google Scholar]
- 13. Mori S., Nambo M., Chi L. C., Bouffard J., Itami K., Org. Lett. 2008, 10, 4609–4612. [DOI] [PubMed] [Google Scholar]
- 14. Nambo M., Noyori R., Itami K., J. Am. Chem. Soc. 2007, 129, 8080–8081. [DOI] [PubMed] [Google Scholar]
- 15.
- 15a. Tseng N.-W., Mancuso J., Lautens M., J. Am. Chem. Soc. 2006, 128, 5338–5338; [DOI] [PubMed] [Google Scholar]
- 15b. Tonogaki K., Itami K., Yoshida J., Org. Lett. 2006, 8, 1419–1422; [DOI] [PubMed] [Google Scholar]
- 15c. Miura T., Murakami M., Chem. Commun. 2007, 217–224; [DOI] [PubMed] [Google Scholar]
- 15d. Kurahashi T., Shinokubo H., Osuka A., Angew. Chem. Int. Ed. 2006, 45, 6336–6338; [DOI] [PubMed] [Google Scholar]; Angew. Chem. 2006, 118, 6484–6486; [Google Scholar]
- 15e. Ueura K., Miyamura S., Satoh T., Miura M., J. Organomet. Chem. 2006, 691, 2821–286; [Google Scholar]
- 15f. Ukai K., Aoki M., Takaya J., Iwasawa N., J. Am. Chem. Soc. 2006, 128, 8706–8707. [DOI] [PubMed] [Google Scholar]
- 16.Recent examples of rhodium-catalyzed 1,4-addition of arylboronic acids to α,β-unsaturated carbonyl compounds:
- 16a. Paquin J.-F., Defieber C., Stephenson C. R. J., Carreira E. M., J. Am. Chem. Soc. 2005, 127, 10850–10851; [DOI] [PubMed] [Google Scholar]
- 16b. Walter C., Auer G., Oestreich M., Angew. Chem. Int. Ed. 2006, 45, 5675–5677; [DOI] [PubMed] [Google Scholar]; Angew. Chem. 2006, 118, 5803–5805; [Google Scholar]
- 16c. Duan W.-L., Iwamura H., Shintani R., Hayashi T., J. Am. Chem. Soc. 2007, 129, 2130–2138; [DOI] [PubMed] [Google Scholar]
- 16d. Mariz R., Luan X., Gatti M., Linden A., Dorta R., J. Am. Chem. Soc. 2008, 130, 2172–2173; [DOI] [PubMed] [Google Scholar]
- 16e. Nishimura T., Wang J., Nagaosa M., Okamoto K., Shintani R., Kwong F.-Y., Yu W.-Y., Chan A. S. C., Hayashi T., J. Am. Chem. Soc. 2010, 132, 464–465. [DOI] [PubMed] [Google Scholar]
- 17.
- 17a. Sakai M., Hayashi H., Miyaura N., Organometallics 1997, 16, 4229–4231; [Google Scholar]
- 17b. Poater A., Ragone F., Mariz R., Dorta R., Cavallo L., Chem. Eur. J. 2010, 16, 14348–14353. [DOI] [PubMed] [Google Scholar]
- 18. Fagnou K., Lautens M., Chem. Rev. 2003, 103, 169–196. [DOI] [PubMed] [Google Scholar]
- 19. Nambo M., Segawa Y., Wakamiya A., Itami K., Chem. Asian J. 2011, 6, 590–598. [DOI] [PubMed] [Google Scholar]
- 20. Poater A., Vummaleti S. V. C., Cavallo L., Organometallics 2013, 32, 6330–6336. [Google Scholar]
- 21.
- 21a. Ishii Y., Hoshi H., Hamada Y., Hidai M., Chem. Lett. 1994, 23, 801–804; [Google Scholar]
- 21b. Douthwaite R. E., Green M. L. H., Stephens A. H. H., Turner J. F. C., J. Chem. Soc. Chem. Commun. 1993, 1522–1523. [Google Scholar]
- 22. Ikeda A., Kameno Y., Nakao Y., Sato H., Sakaki S., J. Organomet. Chem. 2007, 692, 299–306. [Google Scholar]
- 23.
- 23a. Van Lier G., Safi B., Geerlings P., J. Phys. Chem. Solids 1997, 58, 1719–1727; [Google Scholar]
- 23b. Amat M. C., Van Lier G., Solà M., Duran M., Geerlings P., J. Org. Chem. 2004, 69, 2374–2380. [DOI] [PubMed] [Google Scholar]
- 24. Kozuch S., Shaik S., Acc. Chem. Res. 2011, 44, 101–110. [DOI] [PubMed] [Google Scholar]
- 25.Gaussian 09, Revision C.01, M. J. Frisch, G. W. Trucks, H. B. Schlegel, G. E. Scuseria, M. A. Robb, J. R. Cheeseman, G. Scalmani, V. Barone, B. Mennucci, G. A. Petersson, H. Nakatsuji, M. Caricato, X. Li, H. P. Hratchian, A. F. Izmaylov, J. Bloino, G. Zheng, J. L. Sonnenberg, M. Hada, M. Ehara, K. Toyota, R. Fukuda, J. Hasegawa, M. Ishida, T. Nakajima, Y. Honda, O. Kitao, H. Nakai, T. Vreven, J. A. Montgomery, Jr., J. E. Peralta, F. Ogliaro, M. Bearpark, J. J. Heyd, E. Brothers, K. N. Kudin, V. N. Staroverov, R. Kobayashi, J. Normand, K. Raghavachari, A. Rendell, J. C. Burant, S. S. Iyengar, J. Tomasi, M. Cossi, N. Rega, J. M. Millam, M. Klene, J. E. Knox, J. B. Cross, V. Bakken, C. Adamo, J. Jaramillo, R. Gomperts, R. E. Stratmann, O. Yazyev, A. J. Austin, R. Cammi, C. Pomelli, J. W. Ochterski, R. L. Martin, K. Morokuma, V. G. Zakrzewski, G. A. Voth, P. Salvador, J. J. Dannenberg, S. Dapprich, A. D. Daniels, Ö. Farkas, J. B. Foresman, J. V. Ortiz, J. Cioslowski, D. J. Fox, Gaussian, Inc., Wallingford, CT, USA, 2009.
- 26.
- 26a. Becke A. D., J. Chem. Phys. 1993, 98, 5648–5652; [Google Scholar]
- 26b. Lee C., Yang W., Parr R. G., Phys. Rev. B 1988, 37, 785–789; [DOI] [PubMed] [Google Scholar]
- 26c. Stephens P., Devlin F. J., Chabalowski C. F., Frisch M. J., J. Phys. Chem. 1994, 98, 11623–11627. [Google Scholar]
- 27.
- 27a. Hehre W. J., Ditchfield R., Pople J. A., J. Chem. Phys. 1972, 56, 2257–2261; [Google Scholar]
- 27b. Hehre W. J., Radom L., Schleyer P. v. R., Pople J. A., Ab Initio Molecular Orbital Theory; Wiley, New York, 1986. [Google Scholar]
- 28.
- 28a.U. Häussermann, M. Dolg, H. Stoll, H. Preuss, P: Schwerdtfeger, R. M. Pitzer, Mol. Phys 1993, 78, 1211–1224;
- 28b. Küchle W., Dolg M., Stoll H., Preuss H., J. Chem. Phys. 1994, 100, 7535–7542; [Google Scholar]
- 28c. Leininger T., Nicklass A., Stoll H., Dolg M., Schwerdtfeger P., J. Chem. Phys. 1996, 105, 1052–1059. [Google Scholar]
- 29. Zhao Y., Truhlar D. G., Theor. Chem. Acc. 2008, 120, 215–241. [Google Scholar]
- 30. Wachters A. J. H., J. Chem. Phys. 1970, 52, 1033–1036. [Google Scholar]
- 31. Mariz R., Luan X., Poater A., Gatti M., Blumentritt S., Drinkel E., Linden A., Bürgi J. J., Cavallo L., Dorta R., Chem. Eur. J. 2010, 16, 14335–14347. [DOI] [PubMed] [Google Scholar]
- 32.
- 32a. Barone V., Cossi M., J. Phys. Chem. A 1998, 102, 1995–2001; [Google Scholar]
- 32b. Tomasi T., Persico M., Chem. Rev. 1994, 94, 2027–2094. [Google Scholar]
- 33.
- 33a. Kelly C. P., Cramer C. J., Truhlar D. G., J. Chem. Theory Comput. 2005, 1, 1133–1152; [DOI] [PubMed] [Google Scholar]
- 33b. Kelly C. P., Cramer C. J., Truhlar D. G., J. Phys. Chem. B 2006, 110, 16066–16081; [DOI] [PubMed] [Google Scholar]
- 33c. Bryantsev V. S., Diallo M. S., W. A. Goddard III , J. Phys. Chem. B 2008, 112, 9709–9719. [DOI] [PubMed] [Google Scholar]
- 34. Tawa G. J., Topol I. A., Burt S. K., Caldwell R. A., Rashin A. A., J. Chem. Phys. 1998, 109, 4852–4963. [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
As a service to our authors and readers, this journal provides supporting information supplied by the authors. Such materials are peer reviewed and may be re‐organized for online delivery, but are not copy‐edited or typeset. Technical support issues arising from supporting information (other than missing files) should be addressed to the authors.
Supplementary