

Abstract
Gefitinib is a selective inhibitor of the tyrosine kinase epidermal growth factor receptor, which inhibits tumor pathogenesis, metastasis and angiogenesis, as well as promoting apoptosis. Therefore, gefitinib presents an effective drug for the targeted therapy of lung cancer. However, the underlying mechanisms by which gefitinib induces lung cancer cell death remain unclear. To investigate the effects of gefitinib on lung cancer cells and the mechanism of such, the present study analyzed the effect of gefitinib on the autophagy, apoptosis and proliferation of the A549 and A549-gefitinib-resistant (GR) cell lines GR. The regulation of the phosphatidylinositol 3-kinase (PI3K)/protein kinase B (AKT)/ mammalian target of rapamycin (mTOR) pathway was also investigated. Acridine orange staining revealed that gefitinib induced autophagy of A549 cells but not A549-GR cells. In addition, gefitinib promoted apoptosis and inhibited proliferation of A549 cells but not A549-GR cells. Furthermore, western blot analysis demonstrated that gefitinib treatment led to the downregulation of PI3K, AKT, pAKT, mTOR and phosphorylated-mTOR protein expression in A549 cells but not A549-GR cells. LY294002 blocked the PI3K/AKT/mTOR pathway and induced autophagy and apoptosis of A549 cells, however, no synergistic effect was observed following combined treatment with gefitinib and LY294002. In conclusion, the results of the present study indicate that gefitinib promotes autophagy and apoptosis of lung cancer cells via blockade of the PI3K/AKT/mTOR pathway, which leads to lung cancer cell death.
Keywords: gefitinib, lung cancer, autophagy, apoptosis, phosphatidylinositol 3-kinase, protein kinase B, mammalian target of rapamycin
Introduction
Lung cancer is one of the most common malignant tumors worldwide, with smoking and other environmental factors considered the main risk factors of the disease (1). The disease markedly impairs patient health and quality of life (2). At present, treatments include surgical resection, radiotherapy and chemotherapy, however, molecular targeted therapy that specifically kills cancer cells with less toxicity to normal cells presents an additional treatment that may contribute to improving survival and quality of life in lung cancer patients (3–5). Gefitinib, a selective inhibitor of the tyrosine kinase, epidermal growth factor receptor (EGFR), suppresses tumor growth, metastasis and vascularization and has been demonstrated to induce tumor cell apoptosis and sensitivity to radiotherapy and chemotherapy (6–7). Gefitinib is used for the targeted therapy of lung cancer; however, the mechanism of action remains unclear. Autophagy is the stress response exhibited by eukaryotic cells to a changing internal and external environment, which maintains homeostasis via the breakdown of intracellular proteins and organelles (8).
Autophagy is considered as a double-edged sword with regard to genesis, development and the treatment of tumors as it kills tumor cells but also protect tumor cells against injury (9). Microenvironment alteration during tumor formation has been demonstrated to induce autophagy, limiting tumor metastasis, and thus modulation of the autophagic signaling pathway may present a potential therapeutic target in tumor metastasis (10). Autophagy has been identified as the early response to gefitinib in the treatment of EGFR-positive breast cancer (11), and gefitinib induces autophagy in lung cancer via activation of the AMP-activated protein kinase (AMPK) pathway (12). Furthermore, combined treatment with protein kinase B (AKT) inhibitors, chloroquine and gefitinib prevents compensatory autophagy in cells and induces EGFR-mutant lung cancer cell death (13). By contrast, autophagy may also promote lung cancer invasion and metastasis induced by Toll-like receptors, indicating that suppression of autophagy may present a novel lung cancer treatment (14). To date, no studies have confirmed whether autophagy is induced or suppressed during gefitinib-targeted therapy of lung cancer, and the association between autophagy and apoptosis remains unclear. Therefore, the present study investigated the effect of gefitinib on autophagy in the EGFR wild-type non-small cell lung cancer (NSCLC) A549 cell line and the A549-gefitinib-resistant (GR) cell line and analyzed the association between autophagy and apoptosis and the potential underlying regulatory mechanism of these processes.
Materials and methods
Cell culture
The NSCLC A549 and A549-GR cell lines (Cancer Research Institute of Southern Medical University, Guangzhou, China) were cultured in RPMI 1640 medium supplemented with 10% fetal bovine serum and 1% mycillin (HyClone; GE Healthcare, Logan, UT, USA) at 37°C in a humidified atmosphere of 5% CO2. Cells at the exponential growth phase were then incubated with phosphate-buffered saline (PBS) (blank control group) or 50, 100 200, and 500 nmol/l gefitinib (AstraZeneca, Cambridge, UK) for 3–5 days. The levels of autophagy, proliferation and apoptosis were then analyzed.
Acridine orange (AO) staining
Cells at the exponential growth phase were suspended and seeded in a 6-well plate at a density of 1×105 cells/well. After cells became adherent, 1 ml dimethyl sulfoxide (DMSO) (Beyotime Institute of Biotechnology, Shanghai, China) was added to the first well (negative control group), 1 ml rapamycin (Sigma-Aldrich, St. Louis, MO, USA) to the second well (positive control group), and 50, 100, 200 and 500 nmol/l gefitinib was added to the third, fourth, fifth and sixth well (study group), respectively. After 48 h, the plate was stained with 50 µmol/l AO, incubated for 15 min in the dark and washed 3 times with PBS to identify the autophagic cells. Autophagic cell death was examined using a fluorescence microscope (CX41-32RFL; Olympus Corporation, Tokyo, Japan).
Western blot analysis
Cells were seeded in a 6-well plate at a density of 1×105 cells/well for 24 h. To isolate total protein, 250 µl RIPA buffer (Biosdec, Wuhan, China) was added to each well for 30 min. Expression of associated proteins was analyzed using a western blot kit (Biosdec, Wuhan, China). Protein samples (50 µg) were separated by SDS-PAGE gel electrophoresis and transferred to a 0.45 µm polyvinylidene fluoride membrane followed by incubation overnight at 4°C with rat anti-human antibodies [light chain 3B (LC3B), phosphatidylinositol 3-kinase (PI3K), Akt, phosphorylated (p)-Akt, mammalian target of rapamycin (mTOR), p-mTOR, and β-actin] (Santa Cruz Biotechnology, Inc., Santa Cruz, CA, USA) at a dilution of 1:1,000. After decoloration, the samples were incubated with horseradish peroxidase-conjugated goat anti-rabbit secondary antibody (Biosdec, Wuhan, China) at a 1:5,000 dilution for 1 h at room temperature and unbound antibody was washed away. Proteins were visualized following treatment with enhanced chemiluminescence reagent (Biosdec, Wuhan, China) for 1–2 min and exposure to X-ray film. Developed films were processed using BandScan software (Glyko Inc., Novato, CA, USA) to determine optical densities.
MTT cell proliferation/viability assay
Cells (2×103 cells/well) at the exponential phase were seeded in a 96-well plate. After cells became adherent, the medium was replaced with 5 mg/ml MTT solution (Beyotime Institute of Biotechnology) and incubated in the dark for 4 h at 37°C. The supernatant was aspirated from the plate and 150 µl DMSO (Beyotime Institute of Biotechnology) was added to each well. Next, the plate was agitated for 10 min at room temperature and absorbance was measured at a wavelength of 490 nm using a microplate reader (Bio-Rad Laboratories, Inc., Hercules, CA, USA).
Flow cytometry analysis
Cells (1×106/well) were cultured in a 6-well plate. After the cells became adherent, the medium was aspirated and cells were suspended, centrifuged (4,000 × g for 5 min) and fixed in 75% precooled ethanol overnight at −20°C. Following centrifugation, the supernatant was discarded and the sediment was washed twice with PBS, resuspended in PBS (480 µl/well) and stained with 10 µl Annexin-V and propidium iodide (PI; Beyotime Institute of Biotechnology). Cellular apoptosis was analyzed by flow cytometry (BD FACSCalibur; BD Biosciences, San Diego, CA, USA).
Inhibition of the PI3K/AKT/mTOR signaling pathway
The PI3K inhibitor, LY294002 (10 nmol/l), was added to the culture medium. Cellular autophagy, apoptosis and proliferation were analyzed 48 h after culturing.
Statistical analysis
Data were expressed as the mean ± standard deviation and analyzed by paired t-test using SPSS 16.0 software (SPSS, Inc., Chicago, IL, USA). P<0.05 was considered to indicate a statistically significant difference.
Results
Gefitinib induces autophagy in A549 cells
AO staining revealed that gefitinib induced the formation of autophagic vacuoles in A549 cells in a dose-dependent manner. By contrast, no significant autophagy was identified in the A549-GR cells (Fig. 1A). Furthermore, the expression of LC3B, an autophagy-related protein, varied between the two lung cancer cell lines. Following gefitinib treatment, LC3B expression exhibited a dose-dependent increase in A549 cells, however, no significant difference in LC3B expression was identified in A549-GR cells (Fig. 1B).
Figure 1.
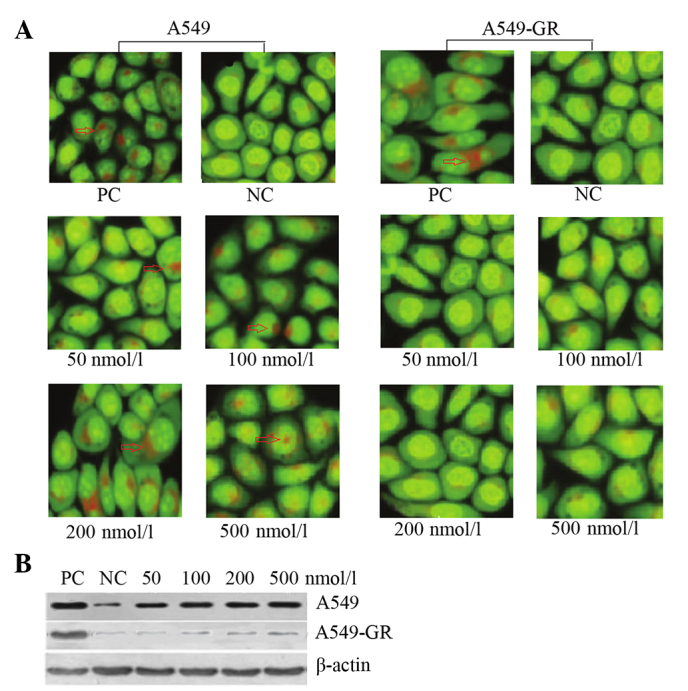
Gefitinib induces autophagy in A549 cells. (A) Autophagic vacuoles (red arrows and red staining) were observed in A549 cells following treatment with gefitinib (acridine orange staining, magnification, ×200). (B) Western blot analysis showing significant upregulation of protein light chain 3B expression in A549 cells compared with A549-GR cells following gefitinib treatment. PC, positive control; NC, negative control; GR, gefitinib-resistant.
Gefitinib induces apoptosis in A549 cells
Annexin V- and PI-positive cells were determined by flow cytometry. Apoptotic cells were located in the right upper quadrant. The results revealed that gefitinib increased the apoptotic rate of A549 cells in a dose-dependent manner. Following treatment with 500 nmol/l gefitinib, the apoptosis rate was 60.2 and <10% in A549 and A549-GR cells, respectively (Fig. 2). No significant changes in apoptosis rate were identified in A549-GR cells following gefitinib treatment therapy (Fig. 2).
Figure 2.

Gefitinib-induced apoptosis in A549 cells. (A) Flow cytometry showing enhanced apoptosis in A549 cells when compared with A549-GR cells following gefitinib treatment. (B) The apoptotic rate of A549 cells increased in a dose-dependent manner following gefitinib treatment, however, no significant differences in apoptotic rate were identified in A549-GR cells. *P<0.05 vs. 0 nmol/l. GR, gefitinib-resistant.
Gefitinib inhibits proliferation of A549 cells
MTT assay revealed gefitinib significantly attenuated A549 cell proliferation. The survival rate of A549 cells was reduced by gefitinib in a dose-dependent manner. Survival rate of A549 cells decreased to 40% following treatment with 500 nmol/l gefitinib. By contrast, cell proliferation of A549-GR cells was not affected by gefitinib treatment and no significant differences in survival rate were identified in A549-GR cells following treatment with gefitinib (Fig. 3).
Figure 3.
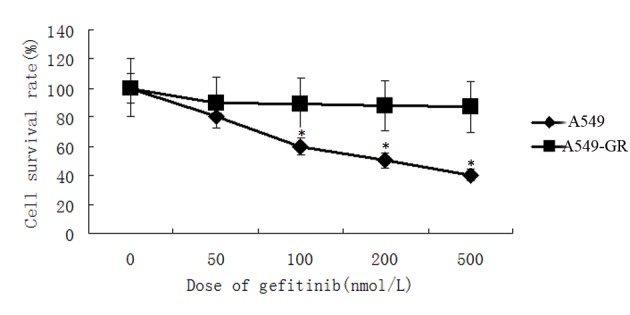
MTT assay revealed that gefitinib decreased A549 cell survival rate in a dose-dependent manner, however, no significant differences in cell survival were identified following gefitinib treatment in A549-GR cells. *P<0.05 vs. A549-GR. GR, gefitinib-resistant.
Gefitinib mediates autophagy and apoptosis of A549 cells via the PI3K/AKT/mTOR pathway
To elucidate the underlying molecular mechanism of gefitinib-mediated autophagy and apoptosis, the expression of proteins associated with the PI3K/AKT/mTOR pathway was measured using western blot analysis prior to and following gefitinib treatment in A549 and A549-GR cells. The results demonstrated that PI3K, AKT, pAKT, mTOR and p-mTOR protein expression was significantly downregulated in A549 cells following treatment with 500 nmol/l gefitinib. By contrast, no significant differences in PI3K, AKT, pAKT, mTOR and p-mTOR protein expression were identified in A549-GR cells following treatment with gefitinib (Fig. 4).
Figure 4.

Gefitinib mediates autophagy and apoptosis of A549 cells via suppression of the PI3K/AKT/mTOR pathway. Western blot analysis showing reduced protein expression of PI3K, AKT, pAKT, mTOR and p-mTOR in A549 cells following gefitinib treatment, however, no differences in protein expression were identified in A549-GR cells. GR, gefitinib-resistant; PI3K, phosphatidylinositol 3-kinase; AKT, protein kinase B; mTOR, mammalian target of rapamycin; p, phosphorylated.
Effect of gefitinib on autophagy and apoptosis in A549 cells following blockade of the PI3K/AKT/mTOR pathway
AO staining indicated that LY294002 treatment alone activated autophagy in A549 cells. Although LY294002 inhibited the PI3K/AKT/mTOR pathway, no significant differences in autophagic cell death were identified in A549 cells following combined treatment with gefitinib and LY294002 when compared with LY294002 alone (Fig. 5A). Flow cytometry revealed that LY294002 activated A549 cell apoptosis, however, no synergistic effect was observed in LY294002-induced apoptosis following combined treatment with gefitinib (Fig. 5B).
Figure 5.
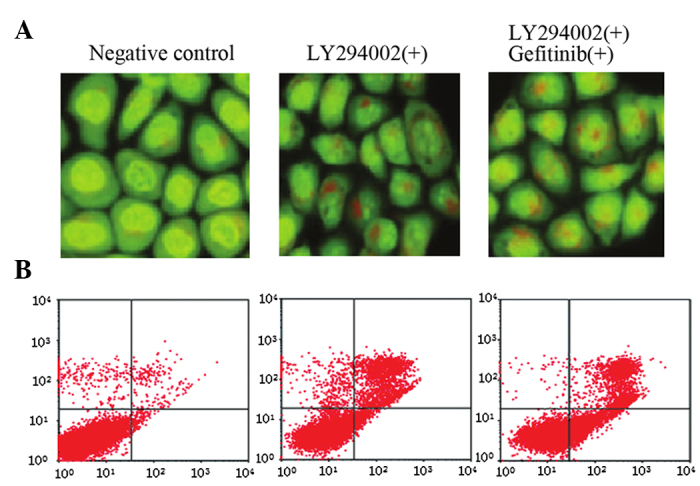
Effect of gefitinib on autophagy and apoptosis in A549 cells following suppression of the PI3K/AKT/mTOR pathway by LY294002. (A) Acridine orange staining revealing LY294002 induces autophagy in A549 cells, however no synergism was observed following combined treatment with gefitinib and LY294002. (B) Flow cytometry showing that LY294002 promotes apoptosis in A549 cells, however no synergism was observed following combined treatment with gefitinib and LY294002.
Discussion
Advances in molecular biological techniques have resulted in the development of targeted therapies, which are important for the treatment of cancer. Gefitinib is a selective tyrosine kinase inhibitor that interrupts EGFR activation via binding to ATP-competitive binding sites of the extracellular domain, which further inhibits cell proliferation and neovascularization and stimulates cell apoptosis (15–18). In May 2003, the Food and Drug Administration (USA) approved gefitinib for late stage platinum and paclitaxel-resistant NSCLC. Currently, gefitinib is used for the treatment of NSCLC (19–21), however, the mechanism of action remains unclear. In the present study, gefitinib-sensitive (A549) and GR (A549-GR) lung cancer cells were used to examine the effect of gefitinib on NSCLC and the underlying mechanism of action.
The results of the present study demonstrated that gefitinib stimulates autophagy in lung cancer cells (Fig. 1). Autophagy, also known as type II programmed cell death, is closely associated with tumorigenicity and tumor development. Autophagy is able to promote or inhibit cell apoptosis, suggesting that autophagy exhibits different functions in a variety of histocytes. For example, in previous studies autophagy inhibited apoptosis in breast carcinoma and the suppression of autophagy promoted apoptosis in breast carcinoma (22,23). However, the stimulation of autophagy promoted apoptosis in colon carcinoma (24). Mimulone was reported to induce autophagic cell death in lung cancer A549 cells (25), indicating that interference with autophagy in lung cancer cells may present a potential treatment for lung cancer (26,27). Bortezomib-induced apoptosis in lung cancer cells is promoted by the suppression of autophagy (28) and autophagy in lung cancer cells is regulated via interference with the AMPK pathway (29). In the present study, gefitinib induced autophagy in lung cancer A549 cells while enhancing apoptosis and suppressing proliferation. By contrast, A549-GR cells rarely exhibited autophagy or apoptosis and cell proliferation was not suppressed in response to gefitinib therapy. These findings suggest that gefitinib exhibits antitumor activity in lung cancer via the induction of autophagy and apoptosis.
The PI3K/AKT/mTOR pathway is one of the most important downstream signaling pathways activated following EGFR activation with multiple biological functions that are involved in cell cycle regulation (30,31). The PI3K/AKT/mTOR pathway exhibits an important function in the regulation of autophagy (32,33). Therefore, autophagy is promoted by blockade of the PI3K/AKT/mTOR pathway (34) and impaired by activation of the pathway (35). In the present study, gefitinib decreased the expression of PI3K, AKT, pAKT, mTOR and p-mTOR in A549 lung cancer cells and blockade of the PI3K/AKT pathway following treatment with LY294002 induced autophagy and apoptosis, indicating that gefitinib induces autophagy and apoptosis in lung cancer cells via inhibition of the PI3K/AKT/mTOR pathway. Furthermore, the results of the present study demonstrated that combined gefitinib treatment did not exhibit a synergistic effect on the autophagy and apoptosis induced in lung cancer cells following the inactivation of PI3K/AKT/mTOR pathway by LY294002. It is hypothesized that blockade of the PI3K/AKT/mTOR pathway interrupts targets of gefitinib, which in turn affect the activity of gefitinib. Previous studies have reported that gefitinib suppressed EGF-induced cellular migration (36) and induced apoptosis in breast cancer cells (37) via PI3K pathway interference. Furthermore, the PI3K pathway affects lung cancer cell sensitivity to gefitinib (38,39).
Gefitinib contributes to tumor suppression by inducing autophagy and apoptosis and inhibiting proliferation in lung cancer cells via the blockade of PI3K/AKT/mTOR pathway. However, the association between gefitinib-induced autophagy and apoptosis in lung cancer cells and the underlying mechanism of such requires further study.
References
- 1.Hernández-Hernández JR, de Moreno Vega-Herrero MB, Iglesias-Heras M, García-García R, Hernández-Terciado F, Celdrán-Gil J. Lung cancer in avila province, Spain. Incidence rates, epidemiolgy of the year 2012 and trends in the last 20 years. Semergen. 2015;41:362–369. doi: 10.1016/j.semerg.2014.07.003. (In Spanish) [DOI] [PubMed] [Google Scholar]
- 2.Ostroff JS. Quality lung cancer screening protects quality of life: No harm, no foul. Cancer. 2014;120:3275–3276. doi: 10.1002/cncr.28835. [DOI] [PubMed] [Google Scholar]
- 3.Maemondo M. Lung cancer: Progress in diagnosis and treatments. Topics: iii. Treatment; 3. Chemotherapy for patients with non-small cell lung cancer, 2) Driver mutation and molecular-targeted therapy in lung cancer. Nihon Naika Gakkai Zasshi. 2014;103:1314–1321. doi: 10.2169/naika.103.1314. (In Japanese) [DOI] [PubMed] [Google Scholar]
- 4.Martinez P, Martinez-Marti A, Navarro A, Cedrés S, Felip E. Molecular targeted therapy for early-stage non-small-cell lung cancer: Will it increase the cure rate? Lung Cancer. 2014;84:97–100. doi: 10.1016/j.lungcan.2014.01.018. [DOI] [PubMed] [Google Scholar]
- 5.Printz C. Targeted therapy in lung cancer: Survival, quality of life improved for some patients. Cancer. 2014;120:2625–2626. doi: 10.1002/cncr.28943. [DOI] [PubMed] [Google Scholar]
- 6.Zhao H, Fan Y, Ma S, Song X, Han B, Cheng Y, Huang C, Yang S, Liu X, Liu Y, et al. Final overall survival results from a phase iii, randomised, placebo-controlled, parallel-group study of gefitinib versus placebo as maintenance therapy in patients with locally advanced or metastatic non-small-cell lung cancer (INFORM; C-tong 0804) J Thorac Oncol. 2015;10:655–664. doi: 10.1097/JTO.0000000000000445. [DOI] [PubMed] [Google Scholar]
- 7.Yu H, Zhang J, Wu X, Luo Z, Wang H, Sun S, Peng W, Qiao J, Feng Y, Wang J, Chang J. A phase II randomized trial evaluating gefitinib intercalated with pemetrexed/platinum chemotherapy or pemetrexed/platinum chemotherapy alone in unselected patients with advanced non-squamous non-small cell lung cancer. Cancer Biol Ther. 2014;15:832–839. doi: 10.4161/cbt.28874. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Yang Z, Klionsky DJ. Eaten alive: A history of macroautophagy. Nat Cell Biol. 2010;12:814–822. doi: 10.1038/ncb0910-814. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Apel A, Zentgraf H, Büchler MW, Herr I. Autophagy-A double-edged sword in oncology. Int J Cancer. 2009;125:991–995. doi: 10.1002/ijc.24500. [DOI] [PubMed] [Google Scholar]
- 10.Xu Y, Xia X, Pan H. Active autophagy in the tumor microenvironment: A novel mechanism for cancer metastasis. Oncol Lett. 2013;5:411–416. doi: 10.3892/ol.2012.1015. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Dragowska WH, Weppler SA, Wang JC, Wong LY, Kapanen AI, Rawji JS, Warburton C, Qadir MA, Donohue E, Roberge M, et al. Induction of autophagy is an early response to gefitinib and a potential therapeutic target in breast cancer. PLoS One. 2013;8:e76503. doi: 10.1371/journal.pone.0076503. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Xu Z, Hang J, Hu J, Gao B. Gefitinib, an egfr tyrosine kinase inhibitor, activates autophagy through AMPK in human lung cancer cells. J BUON. 2014;19:466–473. [PubMed] [Google Scholar]
- 13.Bokobza SM, Jiang Y, Weber AM, Devery AM, Ryan AJ. Combining AKT inhibition with chloroquine and gefitinib prevents compensatory autophagy and induces cell death in EGFR mutated NSCLC cells. Oncotarget. 2014;5:4765–4778. doi: 10.18632/oncotarget.2017. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Zhan Z, Xie X, Cao H, Zhou X, Zhang XD, Fan H, Liu Z. Autophagy facilitates TLR4- and TLR3-triggered migration and invasion of lung cancer cells through the promotion of TRAF6 ubiquitination. Autophagy. 2014;10:257–268. doi: 10.4161/auto.27162. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Lee CH. A mechanistic study on gefitinib-induced apoptosis reveals a new link between EGFR and hTERT in breast cancers. Arch Pharm Res. 2009;32:1333–1334. doi: 10.1007/s12272-009-1920-8. [DOI] [PubMed] [Google Scholar]
- 16.Chang CY, Shen CC, Su HL, Chen CJ. Gefitinib induces apoptosis in human glioma cells by targeting bad phosphorylation. J Neurooncol. 2011;105:507–522. doi: 10.1007/s11060-011-0632-3. [DOI] [PubMed] [Google Scholar]
- 17.Wu J, Min R, Wu M, Chen W. Gefitinib induces mitochondrial-dependent apoptosis in Saccharomyces cerevisiae. Mol Med Rep. 2011;4:357–362. doi: 10.3892/mmr.2011.427. [DOI] [PubMed] [Google Scholar]
- 18.Lu PH, Kuo TC, Chang KC, Chang CH, Chu CY. Gefitinib-induced epidermal growth factor receptor-independent keratinocyte apoptosis is mediated by the JNK activation pathway. Br J Dermatol. 2011;164:38–46. doi: 10.1111/j.1365-2133.2010.10038.x. [DOI] [PubMed] [Google Scholar]
- 19.Karim NA, Musaad S, Zarzour A, Patil S, Jazieh AR. Phase II clinical trial of gefitinib for the treatment of chemonaive patients with advanced non-small cell lung cancer with poor performance status. Clin Med Insights Oncol. 2014;8:121–128. doi: 10.4137/CMO.S15172. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Tamiya A, Tamiya M, Shiroyama T, Saijo N, Nakatani T, Minomo S, Tsuji T, Takeuchi N, Omachi N, Kurata K, et al. Phase II trial of carboplatin, S-1 and gefitinib as first-line triplet chemotherapy for advanced non-small cell lung cancer patients with activating epidermal growth factor receptor mutations. Med Oncol. 2015;32:40. doi: 10.1007/s12032-014-0474-x. [DOI] [PubMed] [Google Scholar]
- 21.Ko R, Kenmotsu H, Hisamatsu Y, Akamatsu H, Omori S, Nakashima K, Oyakawa T, Wakuda K, Shukuya T, Ono A, et al. The effect of gefitinib in patients with postoperative recurrent non-small cell lung cancer harboring mutations of the epidermal growth factor receptor. Int J Clin Oncol. 2015;20:668–673. doi: 10.1007/s10147-014-0761-8. [DOI] [PubMed] [Google Scholar]
- 22.Sun WL, Lan D, Gan TQ, Cai ZW. Autophagy facilitates multidrug resistance development through inhibition of apoptosis in breast cancer cells. Neoplasma. 2015;62:199–208. doi: 10.4149/neo_2015_025. [DOI] [PubMed] [Google Scholar]
- 23.Alayev A, Berger SM, Kramer MY, Schwartz NS, Holz MK. The combination of rapamycin and resveratrol blocks autophagy and induces apoptosis in breast cancer cells. J Cell Biochem. 2015;116:450–457. doi: 10.1002/jcb.24997. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Song X, Kim SY, Zhang L, Tang D, Bartlett DL, Kwon YT, Lee YJ. Role of AMP-activated protein kinase in cross-talk between apoptosis and autophagy in human colon cancer. Cell Death Dis. 2014;5:e1504. doi: 10.1038/cddis.2014.463. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.An HK, Kim KS, Lee JW, Park MH, Moon HI, Park SJ, Baik JS, Kim CH, Lee YC. Mimulone-induced autophagy through p53-mediated AMPK/mTOR pathway increases caspase-mediated apoptotic cell death in A549 human lung cancer cells. PLoS One. 2014;9:e114607. doi: 10.1371/journal.pone.0114607. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Lu HY, Chang YJ, Fan NC, Wang LS, Lai NC, Yang CM, Wu LC, Ho JA. Synergism through combination of chemotherapy and oxidative stress-induced autophagy in A549 lung cancer cells using redox-responsive nanohybrids: A new strategy for cancer therapy. Biomaterials. 2015;42:30–41. doi: 10.1016/j.biomaterials.2014.11.029. [DOI] [PubMed] [Google Scholar]
- 27.Lee TG, Jeong EH, Kim SY, Kim HR, Kim CH. The combination of irreversible EGFR TKIs and SAHA induces apoptosis and autophagy-mediated cell death to overcome acquired resistance in EGFR T790m-mutated lung cancer. Int J Cancer. 2015;136:2717–2729. doi: 10.1002/ijc.29320. [DOI] [PubMed] [Google Scholar]
- 28.Wu G, Li H, Ji Z, Jiang X, Lei Y, Sun M. Inhibition of autophagy by autophagic inhibitors enhances apoptosis induced by bortezomib in non-small cell lung cancer cells. Biotechnol Lett. 2014;36:1171–1178. doi: 10.1007/s10529-014-1470-0. [DOI] [PubMed] [Google Scholar]
- 29.Xu Z, Hang J, Hu J, Gao B. Gefitinib, an EGFR tyrosine kinase inhibitor, activates autophagy through AMPK in human lung cancer cells. J BUON. 2014;19:466–473. [PubMed] [Google Scholar]
- 30.Davis NM, Sokolosky M, Stadelman K, Abrams SL, Libra M, Candido S, Nicoletti F, Polesel J, Maestro R, D'Assoro A, et al. Deregulation of the EGFR/PI3K/PTEN/AKT/mTORC1 pathway in breast cancer: Possibilities for therapeutic intervention. Oncotarget. 2014;5:4603–4650. doi: 10.18632/oncotarget.2209. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Duan H, Qu L, Shou C. Activation of EGFR-PI3K-AKT signaling is required for mycoplasma hyorhinis-promoted gastric cancer cell migration. Cancer Cell Int. 2014;14:135. doi: 10.1186/s12935-014-0135-3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Heras-Sandoval D, Pérez-Rojas JM, Hernández-Damián J, Pedraza-Chaverri J. The role of PI3K/AKT/mTOR pathway in the modulation of autophagy and the clearance of protein aggregates in neurodegeneration. Cell Signal. 2014;26:2694–2701. doi: 10.1016/j.cellsig.2014.08.019. [DOI] [PubMed] [Google Scholar]
- 33.Liu M, Li CM, Chen ZF, Ji R, Guo QH, Li Q, Zhang HL, Zhou YN. Celecoxib regulates apoptosis and autophagy via the PI3K/Akt signaling pathway in SGC-7901 gastric cancer cells. Int J Mol Med. 2014;33:1451–1458. doi: 10.3892/ijmm.2014.1713. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Wang C, Zhang X, Teng Z, Zhang T, Li Y. Downregulation of PI3K/Akt/mTOR signaling pathway in curcumin-induced autophagy in APP/PS1 double transgenic mice. Eur J Pharmacol. 2014;740:312–320. doi: 10.1016/j.ejphar.2014.06.051. [DOI] [PubMed] [Google Scholar]
- 35.Datta K, Suman S, Fornace AJ. Radiation persistently promoted oxidative stress, activated mTOR via PI3K/AKT, and downregulated autophagy pathway in mouse intestine. Int J Biochem Cell Biol. 2014;57:167–176. doi: 10.1016/j.biocel.2014.10.022. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Shien T, Doihara H, Hara H, Takahashi H, Yoshitomi S, Taira N, Ishibe Y, Teramoto J, Aoe M, Shimizu N. PLC and PI3K pathways are important in the inhibition of EGF-induced cell migration by gefitinib (‘Iressa’, ZD1839) Breast Cancer. 2004;11:367–373. doi: 10.1007/BF02968044. [DOI] [PubMed] [Google Scholar]
- 37.Deng M, Wang J, Chen Y, Zhang L, Liu D. Combination of SF1126 and gefitinib induces apoptosis of triple-negative breast cancer cells through the PI3K/AKT-mTOR pathway. Anticancer Drugs. 2015;26:422–427. doi: 10.1097/CAD.0000000000000202. [DOI] [PubMed] [Google Scholar]
- 38.Jeannot V, Busser B, Brambilla E, Wislez M, Robin B, Cadranel J, Coll JL, Hurbin A. The PI3K/AKT pathway promotes gefitinib resistance in mutant KRAS lung adenocarcinoma by a deacetylase-dependent mechanism. Int J Cancer. 2014;134:2560–2571. doi: 10.1002/ijc.28594. [DOI] [PubMed] [Google Scholar]
- 39.Li H, Schmid-Bindert G, Wang D, Zhao Y, Yang X, Su B, Zhou C. Blocking the PI3K/AKT and MEK/ERK signaling pathways can overcome gefitinib-resistance in non-small cell lung cancer cell lines. Adv Med Sci. 2011;56:275–284. doi: 10.2478/v10039-011-0043-x. [DOI] [PubMed] [Google Scholar]