

Abstract
Studies on traditional serrated adenoma (TSA) and sessile serrated adenoma with dysplasia (SSA‐D) are rare due to the low frequency of these lesions, which are well defined by the latest WHO classification. However, introducing new morphological criteria such as intra‐epithelial lymphocytes (IELs) might facilitate colorectal polyp diagnoses. Additionally, the phenotype–genotype correlation needs to be updated as the terminology has repeatedly changed. This study analysed 516 polyps, consisting of 118 classical adenomas (CAD), 116 hyperplastic polyps (HPP), 179 SSAs, 41 SSA‐Ds, and 62 TSAs. The lesions were analysed in relation to the patients’ clinical parameters including gender, age, localisation, and size. The inflammatory background of the polyps was quantified and BRAF and KRAS mutations as well as MLH1 and CDKN2A promoter methylation were assessed. In multivariate analyses, an increase in IELs was an independent and robust new criterion for the diagnosis of SSA‐D (p < 0.001). Superficial erosions and acute neutrophil granulocytes led to reactive changes potentially resembling dysplasia. KRAS and BRAF mutations were associated with CAD/TSA and HPP/SSA, respectively. However, almost half of TSAs had a BRAF mutation and were KRAS wild type. CDKN2A seems to precede MLH1 hyper‐methylation within the serrated carcinogenesis model. The genotyping of WHO‐based entities – and especially SSA – has sharpened in comparison to previously published data. TSAs can be sub‐grouped according to their mutation status. Of note, the higher number of IELs in SSA‐D reflects their close relationship to colorectal cancers with micro‐satellite instability. Therefore, IELs might represent a new diagnostic tool for SSA‐D.
Keywords: intra‐epithelial lymphocytes, sessile serrated adenoma with dysplasia, sessile serrated adenoma, traditional serrated adenoma, BRAF, KRAS, micro‐satellite instability
Introduction
According to the 2010 WHO classification of Tumours of the Digestive System, serrated colorectal lesions can be separated from classical adenoma (CAD) and sub‐divided into hyperplastic polyps (HPP), sessile serrated adenoma (SSA) without and with dysplasia (SSA‐D), and traditional serrated adenoma (TSA) 1.
There is evidence that some of these serrated lesions lead to certain subtypes of colorectal cancer (CRC), which account for interval carcinomas found during endoscopic surveillance programmes and which are biologically different from the classical Vogelstein model for CRC 2. Recently, comprehensive molecular analysis of CRC revealed this CRC subtype to be a hyper‐mutator type with a high frequency of micro‐satellite instability (MSI) 3, 4.
However, for many practicing pathologists, definition of the different subsets of serrated precursor lesions in the colon has created considerable confusion over the years. The background of this process in terminology has been summarized by others 1, 5, 6, 7. In brief, the initial definition as ‘serrated adenoma’ from Longacre and Fenoglio‐Preiser 8 has been replaced by Torlakovic et al, who defined TSA and SSA as two different types of serrated adenoma 9. As a consequence, the term ‘serrated adenoma’ should no longer be used without this distinction.
Subsequently, the lack of cytological dysplasia in SSA caused issues for routine gastrointestinal experts, who had thus far used the term adenoma in the colon only for those lesions that showed classical features such as hyper‐chromasia, elongated pencillate nuclei and pseudo‐stratification. Thus, misleading terms were used such as ‘large hyperplastic polyps’ 10, 11, which under‐estimates the lesions as true precursors; ‘mixed polyps’ 1, 6, which evokes the impression of collision tumours rather than progressive lesions; or ‘intermediate serrated polyp’ 12, an example of individual terminology introduced by single groups. In the end, the terms SSA and TSA were retained, but controversy continues regarding the synonymy of SSA with sessile serrated polyps or sessile serrated lesions (SSL) 1, 5.
In terms of semantic precision, the latter might be the most accurate definition, as the lesion is neither a true polyp due to its sessile nature nor for many a true dysplastic adenoma. In particular, the British guidelines favour the term SSL instead of SSA 13.
However, epidemiological studies have shown that beyond true cytological dysplastic lesions (SSA‐D), large‐sized SSAs (>1.0 cm) even without cytological dysplasia behave like true precursor lesions 14. Therefore, arguments regarding the use of the integrated term ‘adenoma’ for these lesions on the basis of architectural dysplasia as in other organs were followed in the WHO classification 1, 5.
During the last 25 years of attempts to determine the most appropriate sub‐groups of serrated colorectal lesions, both molecular features and distinct morphological features have been investigated. Simply stated, if changes are made to the classification of underlying lesions, the effects on molecular distinctions have to be re‐evaluated and the phenotype–genotype correlation updated.
Hence, little is known about how the introduction of the latest WHO classification might have influenced the molecular distinction between colorectal serrated lesions. Therefore, we collected a series of polyps with more of the so far under‐represented SSA‐D and TSA lesions and asked for the first time whether the basic inflammatory response within these lesions might add new insight into the biological evolution of the serrated carcinogenesis model of CRC, with a medullary phenotype as a possible endpoint 2, 10, 15.
Materials and methods
Meta‐analysis
A Pubmed search was performed using the terms ‘Serrated adenoma’, ‘SSA’, ‘TSA’ and ‘SSA‐D’ in combination with either ‘BRAF’ or ‘KRAS’ as further classifiers. Next, studies were selected for concrete numbers of polyps investigated and attribution of numbers of BRAF or KRAS mutations. The underlying terminology was checked and if not further explicitly stated the year of the new WHO classification 2010 was set as a threshold.
Study collection
The study collection included 516 patients and was assembled by a search for serrated colorectal lesions with cytological dysplasia at four different study sites (Institutes of Pathology of the University Hospital Erlangen, University Hospital Dresden, Hospital Bayreuth and Hospital Aschaffenburg, Germany). H&E slides and corresponding formalin‐fixed, paraffin‐embedded (FFPE) material for molecular analysis were further processed at the Institute of Pathology, University Hospital Erlangen, Erlangen. The composition of the study collection is outlined in Table 1.
Table 1.
Study collection
| Age | Size in cm | ||||||
|---|---|---|---|---|---|---|---|
| Polyp | n | Mean | (Range) | Gender m:f | Site r:l | Mean | (Range) |
| CAD | 118 | 66.9 | (42–89) | 66:52 | 66:52 | 0.7 | (0.2–4.0) |
| ‐TA | 61 | 66.1 | (42–84) | 35:26 | 37:24 | 0.5 | (0.2–1.5) |
| ‐TV | 54 | 66.4 | (47–89) | 28:21 | 28:26 | 0.9 | (0.4–4.0) |
| ‐VA | 3 | 72.3 | (70–74) | 1:2 | 1:2 | 1.0 | (0.3–1.4) |
| HPP | 116 | 62.2 | (22–87) | 54:62 | 45:71 | 0.5 | (0.1–2.0) |
| ‐GC | 31 | 57.7 | (22–87) | 13:18 | 12:19 | 0.5 | (0.1–1.5) |
| ‐MD | 11 | 59.3 | (44–86) | 3:8 | 7:4 | 0.5 | (0.3–0.8) |
| ‐MV | 74 | 62.7 | (32–87) | 38:36 | 26:48 | 0.5 | (0.2–2.0) |
| SSA | 179 | 62.7 | (18–93) | 82:97 | 127:52 | 0.7 | (0.2–2.5) |
| SSA‐D | 41 | 73.6 | (46–93) | 15:26 | 33:8 | 1.3 | (0.5–4.0) |
| ‐CT | 28 | 74.0 | (46–93) | 12:16 | 22:6 | 1.2 | (0.5–3.3) |
| ‐ST | 13 | 72.9 | (47–87) | 3:10 | 11:2 | 1.4 | (0.5–4.0) |
| TSA | 62 | 71.5 | (41–93) | 33:29 | 12:50 | 1.2 | (0.3–6.0) |
The Declaration of Helsinki and ethical guidelines were followed (approval numbers Re.‐No.3996 and Re.‐No. 4607).
Definition of morphological subtypes
The diagnostic criteria used followed strictly the 2010 WHO classification 1. The pre‐selection of cases excluded ambiguous lesions that might especially be found in the spectrum of TSA. So, overlaps of conventional villous adenomas with partial serration as well as goblet cell rich TSA were not part of the study. In TSA, all classical criteria such as hyper‐eosinophilia, ectopic crypt foci, narrow pencillate nuclei and high overall serration (>50%) were fulfilled. In SSA‐D, the remnant SSA was defined as strictly as in non‐dysplastic SSA.
Inflammation count
Due to the consumption of tissue for DNA extraction and molecular analysis, inflammatory cell counts relied on H&E staining alone. Therefore, only a basic distinction between granulocytes and lymphocytes was possible without subtyping. The presence of erosions was stated separately as a possible influencing factor for the cell numbers detected. Inflammatory cells were counted in a single hot spot high power field on a Zeiss Axioskop 2 microscope (Zeiss, Jena, Germany) with a see field number (SFN) of 23, aperture of 0.575 mm and area of 0.260 mm2 at a 400‐fold magnification.
Molecular analysis
DNA was extracted from two separate 10‐μm sections after macro‐dissection using the QIAamp DNA FFPE Kit (Qiagen, Hilden, Germany). DNA quantity was assessed with an ND‐1000 spectrophotometer (Thermo Scientific, Wilmington, DE, USA). DNA quality was analysed by β‐globin PCR; primers and protocols can be provided on request. KRAS and BRAF mutations were assessed simultaneously using a multiplex PCR assay, as described previously 6. MLH1 and p16 methylation analyses were performed after bisulphite treatment of DNA using the Epitect Bisulphite Kit (Qiagen, Hilden, Germany). Pyrosequencing was undertaken using the MLH1 and CDKN2A Pyrosequencing Kit each covering five CpG sites (Qiagen, Hilden, Germany) on a Q24 Pyromark System (Qiagen, Hilden, Germany). Successful KRAS and BRAF analyses were achieved for all lesions. Due to advanced DNA degradation only representative subsets of the polyp categories were available for methylation analysis after bisulphite treatment.
Statistics
The descriptive statistics included Spearman rho correlations, χ 2 tests, sensitivity, specificity calculations and, after Kolmogorov–Smirnov–Lillie test for normal distribution, Student's t‐tests with appropriate adjustments for paired or unpaired settings. The association of discriminative markers for SSA‐D diagnosis was analysed with receiver operating characteristic (ROC) curve and multivariable Cox regression analysis. The performance of ROC curve analysis was judged to be better when the area under the curve (AUC) was closer to 1. Statistical significance was assumed at p‐values < 0.05. All analyses were carried out using SPSS 21 (IBM, NY, USA).
Results
Literature study
Using a literature search, 32 studies that conducted BRAF and/or KRAS analysis on serrated colorectal polyps between 2003 and 2015 were identified, of which 31 outlined entity‐specific numbers 6, 12, 16, 17, 18, 19, 20, 21, 22, 23, 24, 25, 26, 27, 28, 29, 30, 31, 32, 33, 34, 35, 36, 37, 38, 39, 40, 41, 42, 43, 44. The underlying terminology, the date of appearance and the citations were used to segregate them into pre‐ and post‐WHO classification eras (Figure 1). Of note, some studies on TSA and SSA‐D were previously sometimes placed in the categories of serrated adenoma without classifier (SA) or mixed polyp (MP) (Supplemental Table 1). Since the introduction of the latest WHO classification, a lack of revised data regarding SSA‐D and TSA is evident. Reflecting the known frequency of approximately 0.8% for truly dysplastic serrated lesions, the underlying number of screened colorectal polyps in our series exceeded n = 5000 6.
Figure 1.
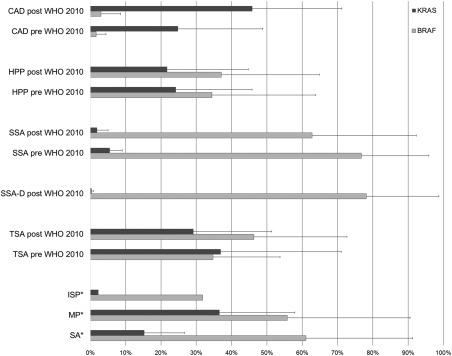
Percentage frequencies of BRAF and KRAS mutations across studies before and after the 2010 WHO classification. This graph highlights the influence of the re‐definition of colorectal polyps established by the WHO classification in 2010. No substantial differences are visible for CAD, TSA and HPP. However, SSA‐D shows a very unique and clear pattern based on KRAS and BRAF status that separates it clearly from the vague previous entities such as MP or SA. More details about the underlying studies are outlined in Supplemental Table 1.
Association with clinical parameters
SSA‐D occurred at a significantly later age than SSA (p < 0.001), with a delay of 12.0 years. This is in line with data from Fujita et al and Bettington showing a later onset of 8.2 or 17.0 years, respectively 23, 42. TSA is a later event than CAD (difference 4.7 years, p = 0.004). HPP seems to be the earliest detectable lesion in the colon, whereas singular SSA was found as early as age 18. No significant difference was observed for any lesion in terms of gender. As expected, SSA and SSA‐D were mostly located in the right side of the colon (p < 0.001), whereas TSA and HPP were mostly found on the left (p < 0.001). CAD did not show any preference for location. Each step from HPP to SSA to SSA‐D revealed a highly significant increase in diameter (each p < 0.001, Table 1). Of note, TSA showed the broadest range in size with rare small lesions and polyps from 0.3 cm up to 6.0 cm (Table 1). Hyperplastic mucosa could be found at the edges of 10 out of 61 TSAs even in large adenomas of 6.0 cm. The average serration in TSA was 83.5% (range 60–100%).
Inflammatory response mechanisms
As the serrated pathway in CRC is strongly linked to MSI‐high cancers with medullary features and higher rates of IELs, we tested this H&E criterion in serrated precursor lesions. Additionally, we investigated the presence of superficial erosions and intra‐epithelial granulocytes (IEGs) (Figure 2 and Supplemental Figure 1). Of note, a significant steady increase in intra‐epithelial lymphocytes (IELs) from HPP to SSA to SSA‐D was observed (p = 0.001 and p < 0.001, respectively). A number of >10 IEL/HPF seemed to be a reasonable threshold as a distinctive criterion between SSA and SSA‐D based on ROC analysis (Figure 2A). Whilst IEGs are strongly inducible by surface erosions, IELs are not influenced by this parameter (Figure 2B). Notably, CAD did not show increased numbers of IELs. The diagnostic sensitivity of IELs in SSA‐D exceeded the performance of MLH1 or p16 hyper‐methylation (Table 2). Histological details are outlined in Figure 3.
Figure 2.
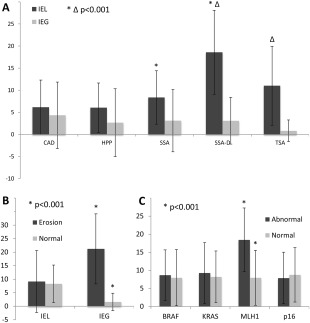
Micro‐environment of the inflammatory background in CAD; HPP; SSA, SSA‐D and TSA. (A) Hot spot single counts of IELs and IEGs per high power field. Note the steady increase in IELs from HPP to SSA to SSA‐D, whereas IEG levels remain basal. The differences between SSA and SSA‐D and between SSA‐D and TSA are highly significant. (B) To exclude an influence of superficial erosions on the results, we tested the dependency of IEG and IEL counts on erosions across all polyps. Whereas IEGs rose significantly if erosions were present, IELs remained stable. In (C), the IEL status is linked directly to molecular profiles irrespective of the underlying diagnosis. Of note, MLH1 hyper‐methylation is significantly related to the IEL response. Unpaired Student's t‐tests, values are outlined as means with error bars of one standard deviation. BRAF, BRAF mutation; KRAS, KRAS mutation; MLH1, MLH1 methylation; p16, CDKN2A methylation
Table 2.
Sensitivity and specificity of molecular parameter
| Parameter | CAD | HPP | SSA | SSA‐D | TSA |
|---|---|---|---|---|---|
| BRAF mutation | |||||
| n | (2/118) | (85/116) | (145/179) | (31/41) | (28/62) |
| Chi‐square test | p < 0.001 | p < 0.001 | p < 0.001 | p = 0.010 | p = 0.057 |
| Sensitivity | 0.017 | 0.733 | 0.810 | 0.756 | 0.452 |
| Specificity | 0.274 | 0.485 | 0.567 | 0.453 | 0.421 |
| KRAS mutation | |||||
| n | (28/118) | (16/116) | (6/179) | (1/41) | (23/62) |
| Chi‐square test | p = 0.002 | 0.747 | p < 0.001 | p = 0.021 | p < 0.001 |
| Sensitivity | 0.237 | 0.138 | 0.003 | 0.024 | 0.387 |
| Specificity | 0.879 | 0.850 | 0.792 | 0.842 | 0.885 |
| MLH1 hypermethylation | |||||
| Mean ± SD (%) | 3.0 ± 2.4 | 3.0 ± 2.7 | 4.5 ± 4.4 | 20.1 ± 21.3 | 5.7 ± 11.2 |
| n with cut‐off > 17% | (0/95) | (0/66) | (2/106) | (13/33) | (3/52) |
| Chi‐square test | p = 0.008 | p = 0.036 | p = 0.071 | p < 0.001 | p = 0.816 |
| Sensitivity | 0.000 | 0.000 | 0.019 | 0.394 | 0.057 |
| Specificity | 0.930 | 0.937 | 0.935 | 0.984 | 0.950 |
| CDKN2A hypermethylation | |||||
| Mean ± SD (%) | 8.4 ± 9.0 | 11.2 ± 14.5 | 13.7 ± 14.7 | 22.4 ± 13.3 | 17.8 ± 13.9 |
| n with cut‐off > 17% | (16/95) | (13/65) | (34/108) | (22/33) | (27/50) |
| Chi‐square test | p < 0.001 | p = 0.017 | p = 0.770 | p < 0.001 | p < 0.001 |
| Sensitivity | 0.189 | 0.200 | 0.315 | 0.667 | 0.540 |
| Specificity | 0.624 | 0.646 | 0.669 | 0.709 | 0.710 |
| Intraepithelial lymphocytes | |||||
| n with cut‐off > 10/HPF | (20/118) | (12/116) | (55/179) | (34/41) | (27/62) |
| Chi‐square test | p = 0.002 | p < 0.001 | p < 0.001 | p < 0.001 | p = 0.004 |
| Sensitivity | 0.169 | 0.103 | 0.307 | 0.829 | 0.435 |
| Specificity | 0.686 | 0.668 | 0.776 | 0.766 | 0.740 |
Figure 3.
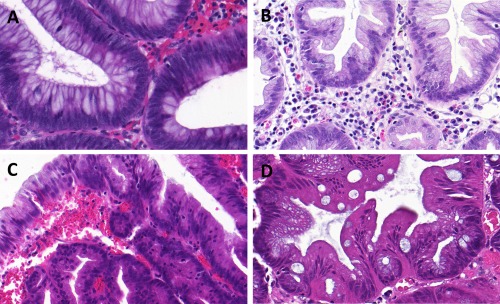
Histological details in serrated colon polyps with a focus on IELs. Detailed H&E morphology (400x magnification) showing the low number of IELs in CAD (A) and HPP (B). In direct comparison, areas within SSA‐D (C) showed a highly elevated number of IELs, whereas the number in TSAs (D) was only slightly higher than CAD and HPP and still notably lower than SSA‐D.
Molecular features of serrated lesions
Data related to the genotype of the colorectal polyps are outlined in Table 2. As expected, BRAF mutation was strongly linked to HPP and SSA. There was also a similar tendency for SSA‐D and TSA. Hence, a BRAF mutation is a strong negative marker for CAD. In contrast, KRAS mutation was strongly linked to CAD and TSA and thus is a negative marker for SSA.
Amongst the 16 KRAS mutations found in HPP, the distribution in HPP subtypes was 19.4% (6/31) in goblet cell‐rich HPP, 11.1% (1/9) in mucin‐depleted HPP and 12.2% (9/74) in micro‐vesicular HPP. The types of KRAS mutation are shown in Table 3, with a notable, but still low number of the rare Exon61 mutations in HPP, SSA and SSA‐D (Table 3).
Table 3.
KRAS type of mutation
| Type of mutation | N = total | CAD | HPP | SSA | SSA‐D | TSA |
|---|---|---|---|---|---|---|
| pGly12Asp | 27 | 9 | 7 | 2 | 0 | 9 |
| pGly12Val | 23 | 8 | 4 | 1 | 0 | 10 |
| pGly12Cys | 9 | 5 | 2 | 2 | 0 | 0 |
| pGly12Ser | 2 | 2 | 0 | 0 | 0 | 0 |
| pGly12Ala | 2 | 0 | 0 | 0 | 0 | 2 |
| pGly12Arg | 1 | 0 | 0 | 0 | 0 | 1 |
| pGly13Asp | 6 | 4 | 0 | 0 | 0 | 2 |
| pGln61Lys | 6 | 1 | 3 | 1 | 1 | 0 |
In all polyps, BRAF and KRAS mutations were mutually exclusive. High grade dysplasia in TSA occurred with BRAF (3/7) or KRAS (4/7) mutations.
Referring to the methylation patterns of MLH1 and p16 (Table 2), we were first interested in the mean percentages detected. The 12.9% increase from SSA to SSA‐D was highly significant (p < 0.001). This difference was also maintained between SSA‐D and TSA (p < 0.001). The means of p16 methylation showed similar levels for SSA‐D and TSA, which differed significantly from CAD (p < 0.001 each) as well as HPP (p = 0.001 for SSA‐D and p = 0.01 for TSA).
To translate the mean percentages into a hyper‐methylated versus normal state, we applied the previously used cut‐off of 17% average methylation level, which was shown to cause transcriptional silencing 6. In this context, MLH1 hyper‐methylation was strongly linked to SSA‐D, but showed only moderate sensitivity. p16 hyper‐methylation was characteristics of TSA and SSA‐D with good sensitivities. Of note, MLH1 and CDKN2A hyper‐methylation were good negative markers for CAD.
Correlation of inflammatory response with methylation patterns
CDKN2A hyper‐methylation and MLH1 hyper‐methylation correlated positively across the entire study (Pearson coefficient 0.382, p < 0.001). Interestingly, there was a strong positive correlation between MLH1 methylation or CDKN2A hyper‐methylation and IELs (Pearson coefficients 0.265 and 0.113, both p < 0.001, respectively). Using ROC curve analysis the IEL counts exceeded the performance of MLH1 hyper‐methylation in the distinction of SSA‐D from SSA (Figure 4).
Figure 4.
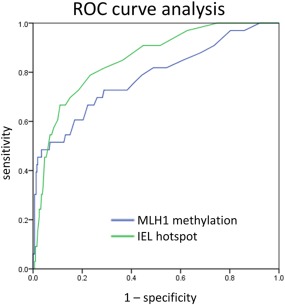
ROC curve analysis of MLH1 and IEL accuracy. Direct comparison of ROC curves of MLH1 hyper‐methylation and IEL counts and their diagnostic potential to detect SSA‐D. The ROC curve of MLH1 methylation revealed an AUC of 0.779. The ROC curve of IELs was slightly better with an area of 0.861 under the curve. Both parameters were highly significant (p < 0.001).
Dissected lesions
Twelve out of the 43 SSA‐Ds were suitable for micro‐dissection. No different mutation types were found when the SSA regions were compared to their corresponding dysplastic areas. Only one dysplastic area lacked confirmation of the BRAF mutation found in its non‐dysplastic counter‐part. This can be attributed to the very small dysplastic area in this particular SSA‐D (approximately 5% of the lesion). The paired comparison of MLH1 and CDKN2A methylation status as well as the number of IELs and IEGs are outlined in Figure 5 and revealed a tendency for an increase in MLH1 methylation and a significant and remarkable increase in IELs (p = 0.06 and p = 0.01, respectively). Of note, the dissected non‐dysplastic SSA areas showed no significant differences in these four parameters in comparison to the rest of the non‐dysplastic SSA areas (p = 0.61, 0.15, 0.60, 0.34, respectively, see also Figure 5).
Figure 5.
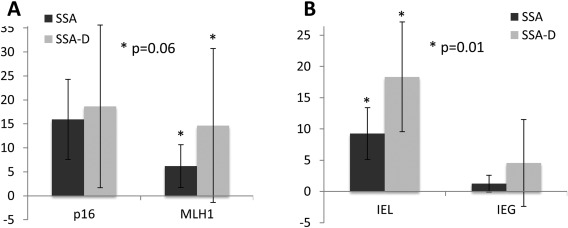
Paired analysis of SSA‐Ds with corresponding dissected areas of background SSA. (A) Methylation patterns of corresponding areas of SSA and SSA‐D within single lesions did not show differences regarding CDKN2A, but tended to show an increase in MLH1 methylation. The inflammatory response (B), however, showed significantly higher levels of IELs in dysplastic areas, whereas IEGs remained randomly distributed. Paired Student's t‐tests, values are outlined as means with error bars of one standard deviation.
Multivariate analysis
To test the independence of clinical parameters, genetics and inflammatory response on the diagnosis of SSA‐D a multivariate analysis was performed integrating all these parameters. According to this analysis, size, age, MLH1 methylation and IEL counts were independent parameters for the diagnosis of SSA‐D. As expected, granulocytes did not contribute to any significant diagnostic distinction.
Discussion
Generally tightened definition and impact of WHO classification on the phenotype and genotype of colorectal polyps
Regarding locations, the known predilection for SSAs and SSA‐Ds to occur in the proximal colon and TSAs in the distal colon was confirmed in this series with only some exceptions 6. HPP and CAD were distributed throughout the colon. The onset and size differences between SSA and SSA‐D are consistent with a recent endoscopy study 45 and indicate a form of progression in which ageing might play a crucial role 1, 35. Taking BRAF and KRAS mutations as key mutator events in CRC carcinogenesis, our study confirms a dichotomized situation. HPP, SSA and SSA‐D are more or less BRAF‐associated, whereas CAD is more prone to involve a KRAS mutation. TSA shows both events, indicating the possibility of two different subtypes of TSA 27, 46. Overall our findings confirm previous reports on the subject 1, 6, 7.
Unfortunately, a meta‐analysis regarding methylation patterns in colorectal polyps is more difficult to perform than one at the mutational level as different methylation assays have been used over time. Additionally, reports often reported higher CpG island methylator phenotype (CIMP) status in serrated lesions as a summarized form of reporting methylation results 21, 35, 47, 48. As broadly known, and depending on its definition, CIMP bundles at least five different methylation sites of different mechanistic relevance 49, 50. Only few studies highlight more biologically important gene‐related methylation patterns, allowing a more stepwise understanding of epigenetic events 19, 24. We chose to use CDKN2A hyper‐methylation as a surrogate marker for cellular senescence 51, and MLH1 hyper‐methylation as the most common reason for the development of sporadic micro‐satellite instability 24.
Consensus meetings that resulted in the introduction of the WHO classification concluded that SSA is a very distinct lesion. It shares molecular similarities with its advanced counterpart SSA‐D, which therefore can now be taken out of the pooled entities formerly considered as mixed polyps or serrated adenoma. The mutual exclusivity of BRAF and KRAS mutation status can be used as a strong argument against the theoretical possibility of a mixed ‘collision’ polyp 6. Mixed BRAF/KRAS status was not seen, either in our study or in the 32 studies reported previously. Beyond this basic knowledge, it should be noted which aspects of each lesion need further attention (overview given in Figure 6).
Figure 6.
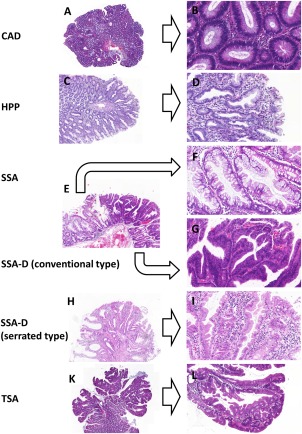
Histology overview. Histological overview of samples diagnosed according to the latest WHO classification: CAD (A, B), HPP (C, D), SSA (E, F) corresponding SSA‐D areas (E, G) and TSA (K, L). Of note, some authors suggest that SSA‐Ds are separated into conventional type SSA‐D (G) or serrated type SSA‐D (H, I). Left 40x, right 200x magnification.
Classical adenoma
Higher degrees of villosity in CAD have been associated with higher rates of KRAS mutation 39. Our series showed a similar tendency, but the association could not be statistically confirmed due to the very small number of cases of pure villous adenoma. Some villous CADs show an overlap with TSA. These subtypes have been recently presented as (low) serrated tubulovillous adenomas of the large intestine 41. Cut‐offs for serration patterns, eg the proposal of 20% serration 52 or predominant serration (meaning > 50%) 41, are open for discussion. Additionally, secondary serration patterns in high grade dysplasia due to general elevated proliferation should be taken into account (Supplemental Figure 1). In general, more studies dissecting the morphologically distinct regions, as we did in SSA‐D, are necessary in this field.
Hyperplastic polyps
In contrast to statements from early investigations conducted long before the latest WHO classification, BRAF mutation is rare in HPP as BRAF is the main initiating mutation event. Comparison of HPP in micro‐vesicular, mucin‐depleted and goblet cell‐rich HPP revealed a slight enrichment of KRAS mutation in the latter. Therefore, as suggested previously 1, micro‐vesicular and mucin‐depleted HPP may be the progenitors of SSA rather than goblet cell‐rich HPP.
Sessile serrated adenoma
SSA showed the highest incidence of BRAF mutations, which seem to be essential for this lesion. SSA later showed a slow increase in CDKN2A methylation preceding MLH1 methylation. This indicates that altered senescence could be an earlier biological event in the development of this lesion 51.
Traditional serrated adenoma
In the morphological analysis, TSAs showed an exophytic growth pattern, hyper‐eosinophilia and the presence of ectopic crypt foci. At the molecular level, either BRAF or KRAS mutations were present in more than 80% of TSAs. Although molecularly different the lesions showed the same morphology 27, 37. CDKN2A methylation levels in TSA were also elevated, showing disturbed senescence within these lesions. However, the MLH1 methylation level was normal in TSA. Even though the ‘dysplastic’ senescent cells in TSA more or less resemble duodenal mucosa, this high rate of mutations argues against a presumably metaplastic concept 53, but favour a truly adenomatous lesion. It has recently been shown by Chetty et al 46 that TSAs contain adjacent areas of CAD, HP or SSA in almost half of all polyps. As no molecular data were collected in this study, this morphological phenomenon should be discussed with care. There is a well‐known overlap between CAD and TSA, which has led to different controversial thresholds for the amount of serration. Long ago, the term ‘substantial serration’ was sufficient 9, but this has been replaced by thresholds of 20% 52 or the recent predominant serration model (>50%) 41, 46. Therefore, we outlined the mean serration and its range in TSA.
HPP‐like areas adjacent to TSA were found in approximately one‐sixth of the lesions. These areas were regarded by us as a rather hyperplastic edge phenomenon as they also occurred in large lesions. Unless dissection studies are performed, it remains unknown whether these HPP areas are the soil for TSA growth or are hyperplastic reactive changes 46.
In contrast to the proposal of Chetty et al, the presence of an SSA as a background lesion in a true dysplastic lesion justifies the term SSA‐D according to the WHO 1. At first glance, attempts to separate TSA from an SSA‐D with a serrated pattern seem confusing (see below). However, clear molecular differences, eg a KRAS mutation would favour TSA. The same is true for inflammatory changes, as in our study higher rates of IELs indicated a serrated type dysplasia within an advanced SSA‐D instead of a TSA. The remnant SSA concept proposed by the WHO only applies for ‘caught in the act’ lesions and tumour overgrowth might lead to the diagnosis of a TSA due to the vanished background SSA. This might be one reason for the two pathways of TSAs distinguished by BRAF or KRAS mutation 34, 53.
Sessile serrated adenoma with dysplasia
To date, the diagnosis of SSA‐D has been based on having an SSA as the background lesion. The dysplastic areas could occur in a conventional type dysplasia resembling classical adenomatous regions. As stated above, a second type of morphology has been shown in SSA‐D, involving a serrated structure 45. Both types of dysplastic area were present in the study and showed no distinct molecular features. Of note, reactive inflammatory changes other than erosions and acute neutrophil granulocyte infiltration should be excluded (Supplemental Figure 1). SSA‐D often shows a high number of BRAF mutations, like its regular SSA counter‐part. Exceptionally, we found one SSA‐D with a rare exon 61 KRAS mutation but this was combined with MLH1 hyper‐methylation and high IEL counts. Micro‐dissection of the SSA part of this polyp was not feasible and the complete lesion was analysed as one. However, in this and all other SSA‐D there was no contradictory mutational status in the pooled or micro‐dissected situation. As reported previously, this contradicts the concept of a mixed polyp 6. The loss of MLH1 seems to be exclusively restricted to SSA‐D. Additionally, the evidence from this molecular analysis for a progression model from SSA towards SSA‐D supports the conventional as well as the serrated type of dysplasia in SSA‐D.
Introducing the immune response in colorectal polyps to the serrated pathway to CRC
Taking a closer look at the assumed model of progression from certain HPP to SSA to SSA‐D, the initiating mutator event, which is already present in HPP, is BRAF mutation, as is proposed in the WHO model. During tumour progression, CDKN2A hyper‐methylation increases, leading to loss of p16. This process can be seen as proof of the concept of oncogenically induced cell senescence 51. In SSA‐D, the loss of MLH1 leads to the accumulation of mutations and, therefore, the occurrence of true dysplastic areas. This step is believed to be the turning point towards sporadic MSI high tumours 48.
This hyper‐mutated genotype induces immunogenic missense proteins, which stimulate a lymphocyte‐rich immune response as seen in medullary colorectal carcinoma.
Although right‐sided MSI‐high CRC subtypes share several morphological features of SSA‐D, IELs in different serrated colorectal polyps have never been comprehensively investigated. This includes several inter‐observer studies searching for the best criteria for HPP, SSA and TSA 1, 5, 6, 7. Studies on IELs in regular CADs are rare too. In Lynch syndrome patients, elevated numbers of IELs were found in polyps with mismatch repair (MMR) loss, but not in the control group with maintained MMR 54. Three other studies focussed on immunogenic events mainly in CADs. One showed an elevation of cytotoxic T‐cells in high grade dysplasia 55. McLean et al presented a very precise profile of macrophages in CAD 56. Pai et al used IELs as one of the criteria to define different subtypes of CAD in patients with and without synchronous SSA 57. However, the different subtypes of serrated polyps have never previously been segregated using this feature.
The strong evidence for high IEL rates in the dysplastic part of SSA‐D indicates that these areas are not the same as CAD areas, but biologically distinct as an early reaction to a hyper‐mutant genotype. The WHO classification currently has no positive criterion for SSA‐D. In our series, high numbers of IELs were highly specific for SSA‐D areas and exceeded the performance of detectable MLH1 hyper‐methylation as a diagnostic marker.
Perspectives and limitations for use in daily routine diagnostics
We chose to rely on molecular features of the polyps for diagnosis. Therefore, this study is limited by the use of H&E staining alone to judge the immune response occurring in the polyps. We assume that the infiltrates were T‐lymphocytes rather than B‐lymphocytes as there was no visible increase in plasma cells. However, an immuno‐histochemical sub‐classification and increased numbers of investigated SSA‐D are warranted for confirmation.
It should also be noted that higher levels of IELs could occur in different settings of immunogenic events. High grade dysplasia can show an increase in IELs 55 and hot spots of IELs may occur in advanced gigantic CADs with an accumulation of mutations. All other causes of a possibly altered immune response, such as inflammatory bowel diseases, bowel obstruction and disturbed microbial flora should be considered during careful differential diagnosis. However, this series was representatively taken from general colorectal surveillance and contained polyps of similar small size (mostly <2.0 cm) in as yet untreated patients. Of note, SSA‐D shows the potential for highly accelerated progression towards carcinoma 14. Therefore, larger lesions are extraordinarily rare.
As an advantage, counting IELs is a well‐established H&E technique in routine pathology. Taken together, we have shown that higher rates of IELs distinguish the dysplastic areas in an SSA‐D from CAD. This morphological difference can be used as a further argument against the concept of a ‘mixed polyp’ 6 and fit very well into the serrated carcinogenesis model with phenotypic features of medullary carcinoma.
Author contributions
TTR and TK conceived the study design and analysed data; DA, GB, AH, KE, AL, ME, MV and AW gathered specimen, clinical data and reviewed cases; AW and IZ carried out statistics; TTR, RS and AH performed and analysed molecular pathology. RA evaluated clinical and endoscopic data. All authors were involved in writing the paper and had final approval of the submitted and published versions.
Supporting information
The following supplementary material may be found in the online version of this article:
Supplemental Table 1. Study overview
Supplemental Figure 1. Pitfalls in the diagnosis of serrated lesions. As is known in other fields of gastrointestinal pathology, eg Barrett's mucosa etc, severe inflammatory changes and erosions should not be overestimated as dysplasia in HPP and SSA (A). Be aware of focal secondary serrations in CAD due to higher proliferation in areas of high grade dysplasia (B).
Supplemental Figure 2. Endoscopic appearance of SSA, SSA‐D and TSA: The endoscopic appearance of SSA‐D and TSA could only be assessed in a few samples (SSA, SSA‐D and TSA, n = 3 for each). For a systematic endoscopic analysis, we refer to a recent study by Burgess et al 45. However, an endoscopic comparison of SSA versus SSA‐D and TSA could be made. SSA (A) showed the typical flat appearance with diffuse margins. SSA‐D (B) gave the impression of a ‘polyp in a polyp’. The completely flat lesion is marked by black arrow‐heads and corresponds to a typical SSA. A more polypoid structure with altered pit‐pattern is visible on the left side of the white arrowhead, presumably reflecting the dysplastic parts in SSA‐D. TSA (C) shows a predominantly exophytic growth pattern.
Acknowledgement
Special thanks to Madeleine Demleitner and Angela Neumann for excellent technical assistance.
Contract/grant details: The study was funded by the Bavarian Gastroenterology Society All authors disclose no conflict of interest.
References
- 1. Snover DC, Ahnen DJ, Burt RW, et al Serrated polyps of the colon and rectum and serrated polyposis In WHO Classification of Tumours of the Digestive System (4th edn), Bosman FT, Carneiro F, Hruban RH, et al (eds). IARC Press: Lyon, 2010; 160–166. [Google Scholar]
- 2. Vogelstein B, Fearon ER, Hamilton SR, et al Genetic alterations during colorectal‐tumor development. N Engl J Med 1988; 319: 525–532. [DOI] [PubMed] [Google Scholar]
- 3. De Sousa EMF, Wang X, Jansen M, et al Poor‐prognosis colon cancer is defined by a molecularly distinct subtype and develops from serrated precursor lesions. Nat Med 2013; 19: 614–618. [DOI] [PubMed] [Google Scholar]
- 4. Cancer Genome Atlas Network . Comprehensive molecular characterization of human colon and rectal cancer. Nature 2012; 487: 330–337. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5. Ensari A, Bilezikci B, Carneiro F, et al Serrated polyps of the colon: how reproducible is their classification? Virchows Arch 2012; 461: 495–504. [DOI] [PubMed] [Google Scholar]
- 6. Rau TT, Agaimy A, Gehoff A, et al Defined morphological criteria allow reliable diagnosis of colorectal serrated polyps and predict polyp genetics. Virchows Arch 2014; 464: 663–672. [DOI] [PubMed] [Google Scholar]
- 7. Rex DK, Ahnen DJ, Baron JA, et al Serrated lesions of the colorectum: review and recommendations from an expert panel. Am J Gastroenterol 2012; 107: 1315–1329; quiz 1314, 1330. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8. Longacre TA, Fenoglio‐Preiser CM. Mixed hyperplastic adenomatous polyps/serrated adenomas. A distinct form of colorectal neoplasia. Am J Surg Pathol 1990; 14: 524–537. [DOI] [PubMed] [Google Scholar]
- 9. Torlakovic EE, Gomez JD, Driman DK, et al Sessile serrated adenoma (SSA) vs. traditional serrated adenoma (TSA). Am J Surg Pathol 2008; 32: 21–29. [DOI] [PubMed] [Google Scholar]
- 10. Burt R, Jass JR. Hyperplastic polyposis In WHO Classification of Tumours Pathology and Genetics of Tumours of the Digestive System (3rd series), Hamilton SR, Aaltonen LA. (eds). IARC Press: Lyon, 1999; 135–137. [Google Scholar]
- 11. Dell'Abate P, Iosca A, Galimberti A, et al Large hyperplastic polyps of the colon. Surg Endosc 2000; 14: 865. [DOI] [PubMed] [Google Scholar]
- 12. Kwon HJ, Cho NY, Chang MS, et al Intermediate serrated polyp as an intermediate lesion of hyperplastic polyp and sessile serrated polyp/adenoma in terms of morphological and molecular features. Hum Pathol 2014; 45: 1759–1765. [DOI] [PubMed] [Google Scholar]
- 13. Bateman AC, Shepherd NA. UK guidance for the pathological reporting of serrated lesions of the colorectum. J Clin Pathol 2015; 68: 585–591. [DOI] [PubMed] [Google Scholar]
- 14. Buda A, De Bona M, Dotti I, et al Prevalence of different subtypes of serrated polyps and risk of synchronous advanced colorectal neoplasia in average‐risk population undergoing first‐time colonoscopy. Clin Transl Gastroenterol 2012; 3: e6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15. Galon J, Mlecnik B, Bindea G, et al Towards the introduction of the ‘Immunoscore’ in the classification of malignant tumours. J Pathol 2014; 232: 199–209. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16. Burnett‐Hartman AN, Newcomb PA, Potter JD, et al Genomic aberrations occurring in subsets of serrated colorectal lesions but not conventional adenomas. Cancer Res 2013; 73: 2863–2872. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17. Carr NJ, Mahajan H, Tan KL, et al Serrated and non‐serrated polyps of the colorectum: their prevalence in an unselected case series and correlation of BRAF mutation analysis with the diagnosis of sessile serrated adenoma. J Clin Pathol 2009; 62: 516–518. [DOI] [PubMed] [Google Scholar]
- 18. Chan TL, Zhao W, Leung SY, et al BRAF and KRAS mutations in colorectal hyperplastic polyps and serrated adenomas. Cancer Res 2003; 63: 4878–4881. [PubMed] [Google Scholar]
- 19. Dhir M, Yachida S, Van Neste L, et al Sessile serrated adenomas and classical adenomas: an epigenetic perspective on premalignant neoplastic lesions of the gastrointestinal tract. Int J Cancer 2011; 129: 1889–1898. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20. Feng Y, Bommer GT, Zhao J, et al Mutant KRAS promotes hyperplasia and alters differentiation in the colon epithelium but does not expand the presumptive stem cell pool. Gastroenterology 2011; 141: 1003–1013.e1‐10. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21. Fernando WC, Miranda MS, Worthley DL, et al The CIMP phenotype in BRAF mutant serrated polyps from a prospective colonoscopy patient cohort. Gastroenterol Res Pract 2014; 2014: 374926. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22. Fu B, Yachida S, Morgan R, et al Clinicopathologic and genetic characterization of traditional serrated adenomas of the colon. Am J Clin Pathol 2012; 138: 356–366. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23. Fujita K, Yamamoto H, Matsumoto T, et al Sessile serrated adenoma with early neoplastic progression: a clinicopathologic and molecular study. Am J Surg Pathol 2011; 35: 295–304. [DOI] [PubMed] [Google Scholar]
- 24. Gaiser T, Meinhardt S, Hirsch D, et al Molecular patterns in the evolution of serrated lesion of the colorectum. Int J Cancer 2013; 132: 1800–1810. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25. Glatz K, Pritt B, Glatz D, et al A multinational, internet‐based assessment of observer variability in the diagnosis of serrated colorectal polyps. Am J Clin Pathol 2007; 127: 938–945. [DOI] [PubMed] [Google Scholar]
- 26. Han Y, Zhou ZY. Clinical features and molecular alterations of traditional serrated adenoma in sporadic colorectal carcinogenesis. J Dig Dis 2011; 12: 193–198. [DOI] [PubMed] [Google Scholar]
- 27. Jass JR, Baker K, Zlobec I, et al Advanced colorectal polyps with the molecular and morphological features of serrated polyps and adenomas: concept of a ‘fusion’ pathway to colorectal cancer. Histopathology 2006; 49: 121–131. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28. Kambara T, Simms LA, Whitehall VL, et al BRAF mutation is associated with DNA methylation in serrated polyps and cancers of the colorectum. Gut 2004; 53: 1137–1144. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29. Kim KM, Lee EJ, Ha S, et al Molecular features of colorectal hyperplastic polyps and sessile serrated adenoma/polyps from Korea. Am J Surg Pathol 2011; 35: 1274–1286. [DOI] [PubMed] [Google Scholar]
- 30. Kim KM, Lee EJ, Kim YH, et al KRAS mutations in traditional serrated adenomas from Korea herald an aggressive phenotype. Am J Surg Pathol 2010; 34: 667–675. [DOI] [PubMed] [Google Scholar]
- 31. Kim MJ, Lee EJ, Suh JP, et al Traditional serrated adenoma of the colorectum: clinicopathologic implications and endoscopic findings of the precursor lesions. Am J Clin Pathol 2013; 140: 898–911. [DOI] [PubMed] [Google Scholar]
- 32. Konishi K, Yamochi T, Makino R, et al Molecular differences between sporadic serrated and conventional colorectal adenomas. Clin Cancer Res 2004; 10: 3082–3090. [DOI] [PubMed] [Google Scholar]
- 33. Lee EJ, Choi C, Park CK, et al Tracing origin of serrated adenomas with BRAF and KRAS mutations. Virchows Arch 2005; 447: 597–602. [DOI] [PubMed] [Google Scholar]
- 34. Morimoto T, Mitomi H, Saito T, et al Distinct profile of HIF1alpha, PTCH, EphB2, or DNA repair protein expression and BRAF mutation in colorectal serrated adenoma. J Gastroenterol Hepatol 2014; 29: 1192–1199. [DOI] [PubMed] [Google Scholar]
- 35. O'Brien MJ, Yang S, Mack C, et al Comparison of microsatellite instability, CpG island methylation phenotype, BRAF and KRAS status in serrated polyps and traditional adenomas indicates separate pathways to distinct colorectal carcinoma end points. Am J Surg Pathol 2006; 30: 1491–1501. [DOI] [PubMed] [Google Scholar]
- 36. Spring KJ, Zhao ZZ, Karamatic R, et al High prevalence of sessile serrated adenomas with BRAF mutations: a prospective study of patients undergoing colonoscopy. Gastroenterology 2006; 131: 1400–1407. [DOI] [PubMed] [Google Scholar]
- 37. Tsai JH, Liau JY, Lin YL, et al Traditional serrated adenoma has two pathways of neoplastic progression that are distinct from the sessile serrated pathway of colorectal carcinogenesis. Mod Pathol 2014; 27: 1375–1385. [DOI] [PubMed] [Google Scholar]
- 38. Velho S, Moutinho C, Cirnes L, et al BRAF, KRAS and PIK3CA mutations in colorectal serrated polyps and cancer: primary or secondary genetic events in colorectal carcinogenesis? BMC Cancer 2008; 8: 255. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39. Yamada M, Sekine S, Ogawa R, et al Frequent activating GNAS mutations in villous adenoma of the colorectum. J Pathol 2012; 228: 113–118. [DOI] [PubMed] [Google Scholar]
- 40. Yang S, Farraye FA, Mack C, et al BRAF and KRAS mutations in hyperplastic polyps and serrated adenomas of the colorectum: relationship to histology and CpG island methylation status. Am J Surg Pathol 2004; 28: 1452–1459. [DOI] [PubMed] [Google Scholar]
- 41. Bettington M, Walker N, Rosty C, et al Serrated tubulovillous adenoma of the large intestine. Histopathology 2016; 68: 578–587. [DOI] [PubMed] [Google Scholar]
- 42. Bettington M, Walker N, Rosty C, et al Clinicopathological and molecular features of sessile serrated adenomas with dysplasia or carcinoma. Gut 2015, epub ahead of print [DOI] [PubMed] [Google Scholar]
- 43. Inoue A, Okamoto K, Fujino Y, et al B‐RAF mutation and accumulated gene methylation in aberrant crypt foci (ACF), sessile serrated adenoma/polyp (SSA/P) and cancer in SSA/P. Br J Cancer 2015; 112: 403–412. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44. Ito M, Mitsuhashi K, Igarashi H, et al MicroRNA‐31 expression in relation to BRAF mutation, CpG island methylation and colorectal continuum in serrated lesions. Int J Cancer 2014; 135: 2507–2515. [DOI] [PubMed] [Google Scholar]
- 45. Burgess NG, Pellise M, Nanda KS, et al Clinical and endoscopic predictors of cytological dysplasia or cancer in a prospective multicentre study of large sessile serrated adenomas/polyps. Gut 2015, in press. [DOI] [PubMed] [Google Scholar]
- 46. Chetty R, Hafezi‐Bakhtiari S, Serra S, et al Traditional serrated adenomas (TSAs) admixed with other serrated (so‐called precursor) polyps and conventional adenomas: a frequent occurrence. J Clin Pathol 2015; 68: 270–273. [DOI] [PubMed] [Google Scholar]
- 47. Jass JR. Classification of colorectal cancer based on correlation of clinical, morphological and molecular features. Histopathology 2007; 50: 113–130. [DOI] [PubMed] [Google Scholar]
- 48. Maeda T, Suzuki K, Togashi K, et al Sessile serrated adenoma shares similar genetic and epigenetic features with microsatellite unstable colon cancer in a location‐dependent manner. Exp Ther Med 2011; 2: 695–700. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49. Ogino S, Cantor M, Kawasaki T, et al CpG island methylator phenotype (CIMP) of colorectal cancer is best characterised by quantitative DNA methylation analysis and prospective cohort studies. Gut 2006; 55: 1000–1006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50. Zlobec I, Bihl M, Foerster A, et al Comprehensive analysis of CpG island methylator phenotype (CIMP)‐high, ‐low, and ‐negative colorectal cancers based on protein marker expression and molecular features. J Pathol 2011; 225: 336–343. [DOI] [PubMed] [Google Scholar]
- 51. Kriegl L, Neumann J, Vieth M, et al Up and downregulation of p16(Ink4a) expression in BRAF‐mutated polyps/adenomas indicates a senescence barrier in the serrated route to colon cancer. Mod Pathol 2011; 24: 1015–1022. [DOI] [PubMed] [Google Scholar]
- 52. Bariol C, Hawkins NJ, Turner JJ, et al Histopathological and clinical evaluation of serrated adenomas of the colon and rectum. Mod Pathol 2003; 16: 417–423. [DOI] [PubMed] [Google Scholar]
- 53. Bettington ML, Chetty R. Traditional serrated adenoma: an update. Hum Pathol 2015; 46: 933–938. [DOI] [PubMed] [Google Scholar]
- 54. Meijer TW, Hoogerbrugge N, Nagengast FM, et al In Lynch syndrome adenomas, loss of mismatch repair proteins is related to an enhanced lymphocytic response. Histopathology 2009; 55: 414–422. [DOI] [PubMed] [Google Scholar]
- 55. Rubio CA, Jacobsson B, Castanos‐Velez E. Cytotoxic intraepithelial lymphocytes in colorectal polyps and carcinomas. Anticancer Res 1999; 19: 3221–3227. [PubMed] [Google Scholar]
- 56. McLean MH, Murray GI, Stewart KN, et al The inflammatory microenvironment in colorectal neoplasia. PLoS One 2011; 6: e15366. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57. Pai RK, Mackinnon AC, Joseph L, et al Identification of histologically distinct conventional adenomas that arise predominately in patients with sessile serrated adenomas. Am J Surg Pathol 2010; 34: 355–363. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
The following supplementary material may be found in the online version of this article:
Supplemental Table 1. Study overview
Supplemental Figure 1. Pitfalls in the diagnosis of serrated lesions. As is known in other fields of gastrointestinal pathology, eg Barrett's mucosa etc, severe inflammatory changes and erosions should not be overestimated as dysplasia in HPP and SSA (A). Be aware of focal secondary serrations in CAD due to higher proliferation in areas of high grade dysplasia (B).
Supplemental Figure 2. Endoscopic appearance of SSA, SSA‐D and TSA: The endoscopic appearance of SSA‐D and TSA could only be assessed in a few samples (SSA, SSA‐D and TSA, n = 3 for each). For a systematic endoscopic analysis, we refer to a recent study by Burgess et al 45. However, an endoscopic comparison of SSA versus SSA‐D and TSA could be made. SSA (A) showed the typical flat appearance with diffuse margins. SSA‐D (B) gave the impression of a ‘polyp in a polyp’. The completely flat lesion is marked by black arrow‐heads and corresponds to a typical SSA. A more polypoid structure with altered pit‐pattern is visible on the left side of the white arrowhead, presumably reflecting the dysplastic parts in SSA‐D. TSA (C) shows a predominantly exophytic growth pattern.