

Abstract
We and others have shown that the cystatin E/M gene is inactivated in primary human tumors, pointing to its role as a tumor suppressor gene. However, the molecular mechanism of tumor suppression is not yet understood. Using plasmid-directed cystatin E/M gene overexpression, a lentivirus-mediated tetracycline-inducible vector system, and human papillomavirus 16 (HPV 16) E6 and E7 gene-immortalized normal human epidermal keratinocytes, we demonstrated intracellular and non-cell-autonomous apoptotic growth inhibition of tumor cell lines and that growth inhibition is associated with cytoplasmic retention of NF-κB. We further demonstrated decreased phosphorylation of IκB kinase (IKKβ) and IκBα in the presence of tumor necrosis factor alpha (TNF-α), confirming the role of cystatin E/M in the regulation of the NF-κB signaling pathway. Growth suppression of nude mouse xenograft tumors carrying a tetracycline-inducible vector system was observed with the addition of doxycycline in drinking water, confirming that the cystatin E/M gene is a tumor suppressor gene. Finally, immunohistochemical analyses of cervical carcinoma in situ and primary tumors have shown a statistically significant inverse relationship between the expression of cystatin E/M and cathepsin L and a direct relationship between the loss of cystatin E/M expression and nuclear expression of NF-κB. We therefore propose that the cystatin E/M suppressor gene plays an important role in the regulation of NF-κB.
INTRODUCTION
Cervical cancer is the second-most-common cancer responsible for cancer-related death in women around the world. The incidence is increasing, with 450,000 new cases diagnosed worldwide and 12,340 diagnosed in the United States annually (1, 2). It is the third leading cause of death from cancer in women 15 to 34 years of age and the fifth leading cause of death in women 35 to 54 years of age, representing about 2% of all cancers in women in the United States. The disease is frequently found in women who have had multiple sex partners, smoking habits, and immune system dysfunctions (3). Cervical cancer is closely linked with human papillomavirus (HPV) infection, and HPVs are detected in 90% of cervical cancer lesions (4–6). While the E6 protein of HPV inactivates p53 and E7 inactivates the retinoblastoma (Rb) tumor suppressor protein, detailed studies on a large number of tumors indicate that viral infection alone is not sufficient for tumor development. Studies have also shown that 20 to 30 million Americans are infected with HPV (7). However, only those in a subset develop cervical cancer. Recently, HPVs have been implicated in the development of head and neck, lung, and breast cancers (8–10).
Cystatins are inhibitors of cysteine proteases and are classified into a large superfamily subdivided into three families based on their location, size, and complexity of polypeptide chains (11–13). Cystatin E/M belongs to the type 2 cystatin family (cystatin C, D, E/M, F, S, SA, and SN), whose members are mainly secreted, and most of them are found abundantly in body fluids and tissues. Cystatin E/M is present as an unglycosylated 14-kDa form containing 149 amino acids and a 17-kDa form that is glycosylated. Loss of this protein seems to play a significant role in abnormal skin development (14). A null mutation of the mouse cystatin E/M gene is also correlated with development of the ichq phenotype, characterized by neonatal lethality, abnormal cornification, and desquamation. Cystatin E/M was initially identified as a downregulated transcript in metastatic breast cancers (15, 16). A number of studies have implicated cystatin E/M as a human tumor suppressor gene that is inactivated in the cancers of the breast, cervix, prostate, brain, and stomach (17–23). Our studies have also shown that the inactivation of the gene is associated with homozygous deletion, promoter hypermethylation, and somatic mutations in primary tumor samples (19). Cystatin E/M gene is expressed in ductal carcinoma in situ (DCIS) but not in metastatic breast cancer, pointing to inactivation during tumor progression (16–18, 24). Finally, inactivation of cystatin E/M gene is associated with the loss of expression of estrogen and progesterone receptors and HER4, indicating an association with these proteins in the development of invasive breast cancers (25). However, the molecular mechanism of cystatin E/M-mediated tumor cell growth inhibition is not yet understood.
Cathepsins include a broad range of proteases, the serine (A and G), aspartic (D and E), and cysteine (B, K, L, S, and V) cathepsins (26). Cathepsins are present in intracellular lysosomal compartments as well as in glycosylated form in the extracellular space. The secreted forms are involved in the degradation of extracellular matrix (ECM) and basement membrane proteins. They participate in the conversion of pro-uPA (urinary plasminogen activator/urokinase) into active uPA. Overexpression of cathepsin B has been reported in aggressive human cancers, including that of the cervix (27, 28). Overexpression of cathepsin L (CTSL) is also reported in human tumors and in inflammatory arterial diseases, aortic aneurysm, and atherosclerosis (29–32). Biochemical studies have shown that cystatin E/M binds to cathepsins L and V as a noncompetitive inhibitor, thereby preventing membrane digestion by cathepsins (33). Thus, the inverse relationship of expression of cystatin E/M and expression of cathepsin L seems to play an important role in cell growth development and cancer. In the present investigation, using exogenous expression, a tetracycline-inducible lentiviral system, and immortalized human epidermal keratinocytes (HEKs), we demonstrated the existence of both intracellular and non-cell-autonomous growth-inhibitory properties of cystatin E/M. We also demonstrated that induced cystatin E/M gene expression is associated with reduced phosphorylation of IκB kinase (IKKβ) and IκBα, leading to cytoplasmic retention of NF-κB, which in turn results in reduced expression of cytokines and growth factors.
MATERIALS AND METHODS
Cell lines.
HPV-positive HeLa cells and HPV-negative C33A and HT3 cell lines representing cervical cancer and CCL23 cells representing head and neck cancer and normal human epidermal keratinocytes (HEKs) were obtained from the American Type Culture Collection (ATCC, Manassas, VA). The tumor cell lines were grown in Dulbecco's modified Eagle's medium (DMEM) containing a high level (4,500 μg/ml) of glucose, 1 mmol/liter glutamine, 100 IU/ml penicillin (Cellgro 10-013-CV; Media Tech, Grand island, NY), and 10% fetal bovine serum (FBS) (Sigma, St. Louis, MO). The HPV 16 E6 or E7 or E6/E7 immortalized HEKs (obtained from Denise Galloway, University of Washington Seattle, WA) and the normal HEKs were grown in keratinocyte culture medium supplemented with 10% FBS (Epilife MEM1500CA and 133-GS; Gibco-Life Technologies, St. Louis, MO). Two hypervariable regions (HVR1 [bp 50 to 585] and HVR2 [bp 16005 to 16503]) of the mitochondrial genome of the revised Cambridge Reference Sequence (rCRS) were used for the authentication of the cell lines (34). These sequences are commonly used for cell line authentication by other investigators (35). PCR products were sequenced at the genome sequencing core facility of the University of California, Los Angeles (UCLA), and the data showed the presence of four single nucleotide polymorphisms (SNPs) of the HVR1 region in the HeLa cells and five SNPs in the CCL23 cells. Four of the CCL23 SNPs were identical to those of the HeLa cells. Four SNPs of the HVR2 region were identical in the two cell lines.
Normal and tumor tissues.
Paraffin sections of 5 normal cervical tissues, 28 cervical carcinoma in situ (CIN) tissues, and 33 primary cervical tumors were obtained from the City of Hope National Medical Center and from the cooperative human tissue network of the NIH. Human tissues were obtained after approval from the Institutional Review Board (IRB) committees of the West Los Angeles VA Medical Center and the City of Hope National Medical Center. While information concerning the HPV status of the primary tumors was available, information concerning other clinical phenotypes was not available.
Transfection.
HeLa or C33A cells (2 × 106) were grown in 100-mm-diameter tissue culture plates overnight to achieve 70% to 80% confluence for transfection with 5 μg of pCMV control vector or the cystatin E/M gene-containing plasmid (17) using Lipofectamine 2000 reagent (Invitrogen, Inc., Carlsbad, CA) according to the manufacturer's instructions. At 24 h after transfection, cells were used for the cell viability assays at different time periods. Cotransfection of HeLa cells (2 × 106 in 100-mm-diameter tissue culture plates) was performed with 5 μg of hemagglutinin (HA)-tagged HPV 16 E6 or HPV 16 E7 plasmid vectors (Addgene, Cambridge, MA) and FLAG-tagged (Sigma Chemical Company, St. Louis, MO) cystatin E/M gene plasmid. HEK293T cells (human embryonic kidney cell line [2.5 × 105 cells]) were transfected in 6-well tissue culture plates with myc-p100 (500 ng) or cotransfected with FLAG-NIK (100 ng) and FLAG-CST6 (1.5 μg) or FLAG-cIAP1 (1.5 μg) plasmid vectors (36), and the cells were harvested 24 h after transfection and lysed with the lysis buffer (25 mM Tris-Cl [pH 7.4], 150 mM NaCl, 1 mM EDTA, 1% NP-40, 5% glycerol) on ice for 10 min. Lysates were cleared via centrifugation for 5 min and combined with 2× loading dye prior to protein separation on 4% to 20% PAGE gels.
Tetracycline-inducible lentiviral system.
An LVX tet-on advanced vector system (Clontech, San Diego, CA) consists of two plasmids: a pLVX-tet-on plasmid containing the tetracycline (Tet)-inducible transactivator element (rtTR-advanced) and a pLVX-Tight-puro plasmid that contains the minimal tetracycline response element (TRE) attached with a modified minimal cytomegalovirus (CMV) promoter driving expression of the cystatin E/M gene. The two plasmids were packaged in 293T cells with pseudotyped vesicular stomatitis virus G protein (VSV-G) particles. The lentiviral particles of high titer containing the plasmids were acquired from the UCLA core facility. The two plasmids were transfected into the HeLa cells and selected using G418 (800 μg/ml) and puromycin (2 μg/ml) for the retention of pLVX-tet-on and pLVX-Tight-puro plasmids, respectively. The selected clones (CMV or CST6 cell clones) were maintained in antibiotic selection medium.
TNF-α treatment.
HeLa or CST6 cells were grown to semiconfluence (60% confluence), grown in serum-free medium for 24 h, and then grown in the presence or absence of doxycycline (dox) (2 μg/ml) for 48 h. Cells were then treated with tumor necrosis factor alpha (TNF-α) (10 ng/ml) for 5 min, and cellular proteins were extracted using radioimmunoprecipitation assay (RIPA) lysis buffer containing a complete protease inhibitor cocktail (Roche Diagnostics, Indianapolis, IN).
Cytoplasmic/nuclear fractionation.
A cell fractionation kit (ab109719) from Abcam (Cambridge, MA) was used to obtain nuclear and cytoplasmic fractions. To validate the purity of fractions, histone H3 (ab1791; Abcam, Cambridge, MA) was used as a nuclear marker and MEK-1/2 (9122; Cell Signaling Technology, Danvers, MA) as a cytoplasmic marker.
MTT assay for cell viability.
Control and transfected cells (4 × 104) were grown in 24-well tissue culture dishes for 24, 48, 72, and 96 h. The MTT [3-(4,5-dimethylthiozol-2-yl)-2,5-diphenyl tetrazolium bromide] assay was carried out following established protocols (19).
Annexin V-FITC apoptosis assay.
The apoptosis assay was carried out with an annexin V-fluorescein isothiocyanate (FITC) kit using the protocol of the manufacturer (catalog no. 556547; BD Biosciences, San Jose, CA). Briefly, 5 × 105 cells in a 0.5-ml suspension of the culture medium were centrifuged at 1,000 × g for 5 min, suspended in 0.5 ml of cold phosphate-buffered saline (PBS), centrifuged again at low speed for 5 min, and resuspended in 0.5 ml of cold annexin V binding buffer (10 mmol/liter HEPES [pH 7.4], 150 mmol/liter NaCl, 2.5 mmol/liter CaCl2, 1 mmol/liter MgCl2, 4% bovine serum albumin [BSA]). Annexin V-FITC (1.25 μl) was added, and the mixture was incubated in the dark at room temperature for 15 min. The treated cells were centrifuged at the low speed for 5 min and suspended again in 0.5 ml of cold binding buffer. Propidium iodide (10 μl) was added, and the cells were analyzed on a Becton Dickinson FACScan analytic flow cytometer. Annexin V-FITC fluorescence (FL1) was detected at 518 nm and the propidium iodide (FL2) at 620 nm.
Soft-agar colony growth assay.
Cystatin E/M gene- and CMV-transfected C33A cells were trypsinized and suspended in DMEM containing 0.1% luke-warm agar at a cell concentration of 10 × 103 cells/ml. The suspension was spread on 0.5% solidified agar 6-well tissue culture plates. The agar plates were incubated in the tissue culture incubator for 15 days, and the colonies were counted under a Zeiss microscope. Colony pictures were taken using an EVOS XL cell imaging system (Thermo Fisher Scientific, Grand Island, NY). The assay was performed in duplicate and repeated with a concentration of 20 × 103 cells/ml per well.
Cell-free supernatant growth assay.
A 50:50 mix of the cell-free supernatants from the plasmid-transfected HeLa cells was used for the growth assay in 24-well tissue culture plates. The medium (1.5 ml) was changed once in 2 days, and the MTT assay was performed on the viable cells as described.
Boyden chamber growth assay.
HeLa cells (10,000 cells) or keratinocytes (due to slow growth, 20,000 cells were used) were seeded onto the top chamber (BD Biosciences, San Diego, CA) (0.4-μm-thick inserts) of the 24-well tissue culture plates. The bottom chamber contained HeLa or CCL23 cells (15,000 cells). The top chamber received 1.0 ml of either the specialized keratinocyte medium for the HEKs or the DMEM for the HeLa cells, with each medium supplemented with 10% FBS. The bottom chamber received 1.5 ml of DMEM supplemented with 10% FBS.
Western blot analysis.
Cellular proteins were extracted as described earlier, and Western blotting was performed using 20 to 30 μg of denatured proteins on 4% to 20% SDS acrylamide gels (Invitrogen, Inc., Carlsbad, CA). Proteins transferred onto nitrocellulose were hybridized to antibodies p16 (N29) from Cal Biochem, San Diego, CA, cystatin E/M (MAB 1286) and cathepsin L (goat polyclonal AF952) from R & D Systems, Inc., Minneapolis, MN, β tubulin, (sc-9104), α actin (sc-482), and NF-κB (p65 [sc-109]) from Santa Cruz Biotechnology, Dallas, TX, FLAG (F1804) from Sigma-Aldrich Chemical Company, St. Louis, MO, and HA (3724S), Bax (2772), NF-κB (p100/p52) (3017), IκBα (9242), phospho-IκBα (ser 32) (2859), IKKβ (8943), phospho-IKKα/β (Ser176/180) (2697), Myc tag (2276), and β-actin (4970) from Cell Signaling Technology, Danvers, MA, by following the established protocol (19). For the detection of cystatin E/M expression in cell-free supernatants, immunoprecipitation of 1 ml supernatant with 1.0 μg of mouse anti-cystatin E/M antibody (MAB 1286; R & D Systems, Inc., Minneapolis, MN) followed by Western blotting with biotinylated anti-cystatin E/M antibody (BAF 286; R & D Systems, Inc., Minneapolis, MN) was performed. All Western blot analyses were performed at least twice.
Ubiquitination assay.
HeLa cells were grown in minimal essential medium (MEM) to 75% confluence and transfected with 5 μg each of HPV 16 E6, HPV 16 E7, or HPV 16 E6 plus CST6 or HPV 16 E7 plus CST6 plasmids using Lipofectamine 3000 (Invitrogen, Inc., Carlsbad, CA) transfection reagent. After growth for 4 h in Opti-MEM GlutaMAX reduced serum medium (Thermo Fisher Scientific, Waltham, MA), cells were grown in MEM–10% serum for 60 h. Cells were then treated with protease inhibitor (MG-132) (catalog no. SML1335; Sigma-Aldrich Chemical Company, St. Louis, MO) (10 μM) and deubiquitinase inhibitor (NEM) (catalog no. E3876; Sigma-Aldrich Chemical Company, St. Louis, MO) (10 μM) for 4 h, and the lysates were prepared in the ice bath by incubation with the lysis buffer (50 mM HEPES, 200 mM KCl, 1 mM EGTA, 1 mM MgCl2, 0.5 mM dithiothreitol [DTT], 0.5% NP-40 containing protease and phosphatase inhibitors) for 15 min. The lysate was spun at 15,000 rpm for 10 min, and the supernatants were used for protein concentration measurements. Immunoprecipitations were done by binding the 200 μg of protein lysates with protein G beads and anti-cystatin E/M antibody (MAB 1286; R & D Systems, Inc., Minneapolis, MN) for 1 h at 4°C and then washed 3 times with buffer containing 50 mM HEPES, 200 mM KCl, 1 mM EGTA, 1 mM MgCl2, and 0.05% NP-40 following elution performed by boiling the washed beads with 1× sample buffer for 10 min. Samples were run on 4% to 20% SDS-PAGE gels, and the blots were hybridized to the control IgG or antiubiquitin antibodies (BML-PW8810-0100; Enzo Life Sciences). After secondary antibody (IR680 and IR800; LiCOR Corp.) hybridization, membranes were scanned using a LiCOR Odyssey instrument.
Transcriptome sequencing (RNA-seq) analysis.
RNA was isolated using a PureLink RNA minikit from Ambion (12183018A; Life Technologies, Inc., Carlsbad, CA), and the libraries were made according to the well-established Illumina Ribo-Zero protocol. Six barcoded libraries were then multiplexed and sequenced on one lane of an Illumina HiSeq sequencer. Reads were unstranded and single ended, with a length of 50 nucleotides (nt). Reads with perfect tags were demultiplexed and mapped to human genome hg19 with refseq gene annotation by tophat2 (version 2.0.6) (37) together with Bowtie 1 (version 0.12.8). Only uniquely mapped reads were used by Htseq-count (38) to calculate the number of reads that aligned to each refseq gene. Genes with more than 10 reads under at least one set of conditions were used (DESeq2, version 1.4.5) (39) to perform gene differential-expression analysis.
Cytokine and growth factor analysis.
Multiplexed assays for cytokines, matrix metalloproteinases (MMPs), AKT, and growth factors were used for analysis of doxycycline-induced CST6 cells and secreted medium on a Sector Imager 6000 instrument (Meso Scale Discovery, Gaithersburg, MD). We used a Human Pro-Inflammatory 9 Plex assay kit (U-Plex Biomarker Group 1 [catalog no. K15069L-1]; MesoScale Discovery) for the measurement of interleukin-2 (IL-2), IL-8, IL-12p70, IL-1β, granulocyte-macrophage colony-stimulating factor (GM-CSF), gamma interferon (IFN-γ), IL-6, IL-10, and TNF-α levels, an AKT signaling assay kit (Akt Signaling Base kit; catalog no. K15115A-3) for the measurement of total p70S6K, glycogen synthase kinase 3b (GSK-3b), and AKT levels, an AKT phospho-signaling assay kit (Akt Signaling [Total Protein] Base kit; catalog no. K15133A-3) for the measurement of p-p70S6K, pGSK-3b, and pAKT levels, an MMP 3-Plex assay kit (Human MMP 3-Plex Base kit; catalog no. K15034A-4) for the measurement of MMP-1, MMP-3, and MMP-9 levels, an MMP 2-Plex assay kit (Human MMP 2-Plex Base kit; catalog no. K15033A-4) for the measurement of MMP-2 and MMP-10 levels, and a human growth factor assay kit (Human Growth Factor I kit; catalog no. K15029C-1) for the measurement of basic fibroblast growth factor (bFGF), soluble fms-like tyrosine kinase 1 (sFLT-1), phosphatidylinositol-glycan biosynthesis class F protein (PIGF), and vascular endothelial growth factor receptor (VEGF) levels. The samples were added to plates that were precoated with capture antibodies for the specific cytokines or growth factors. Assays were performed per the guidelines of the manufacturers, and the plates were read on a Sector 6000 imager. Samples were assayed in triplicate, and the means of the results from the triplicate assays were used for statistical analysis as described earlier (40). The assay was performed thrice.
Immunofluorescence.
Cells were grown overnight on coverslips to semiconfluence (70% to 80%) and transfected with the cystatin E/M gene or the control CMV plasmid DNAs. Immunofluorescence was performed using anti-cystatin E/M antibody (R & D Systems, Minneapolis, MN) by following the established protocol (19).
IHC.
Paraffin sections (5 μm thick) of tumor samples were dewaxed and hybridized to cystatin E/M (MAB 1286; 1:100 dilution) or cathepsin L (goat polyclonal AF952, 1:400 dilution) antibodies (R & D Systems, Inc., Minneapolis, MN) or NF-κB (p65) antibody (rabbit ab31481; 1:80 dilution) (Abcam, Cambridge, MA) by following a standard immunohistochemical (IHC) hybridization protocol (41). Normal cervix tissue was used as a control. Slides were stained with hematoxylin and eosin for pathological evaluation of normal, CIN, and tumor phenotypes. Slides were read by two pathologists using a Zeiss microscope. Percent reactivity of protein expression was scored from 0 to 100, and the intensity of expression was scored as 0, 1, 2, or 3, with 3 being the maximum level of expression.
HeLa/CST6tet-ind xenograft tumors in mice.
CST6 cells were injected subcutaneously into 5-week-old NOD/SCID IL2Rγ null mice (The Jackson Laboratory, Bar Harbor, ME) for tumor formation. Animals were housed in sterile rodent microisolator caging, with a filtered cage top. Two to four animals were housed in each cage, with animals resting directly on bedding. They were given free access to sterile water and food. All cages, covers, and bedding were sterilized weekly. Tumor volume was calculated using the formula V = (4/3)πW2L, where W is half of the shorter axis diameter and L is half of the longer axis diameter, as described previously (42). Tumors were excised when the volume reached a maximum of 15 mm in one of the dimensions. All animal procedures were approved by the Institutional Animal Care and Use Committee of the West Los Angeles Veterans Affairs Medical Center, in accordance with the USPHS Policy on Humane Care and Use of Laboratory Animals.
Statistical analysis.
For the MTT optical density assay, cell viability at 570 nM was measured using a 96-well microplate reader (Power Wave XS; Biotek Instruments, Inc., Winooski, VT). To examine the differences in cell viability among groups, analysis of variance (ANOVA) modeling was used with the following covariates: group, time (measured in days), and a group-by-time interaction term. The model also allows heterogeneous variance across groups. Data are expressed as ± standard deviations (SF) of results from at least three independent MTT growth assays. P values were estimated using the slope values. For the cytokine and growth factor analysis, data points from the Meso-Scale multiplex assays were characterized on the basis of reproducible technical replicates, low percentages of coefficients of variation (CV) (<5%) present within the linear portion of the standard curve, and a value above the lower limit of detection (LLOD). Differences in the levels of expression were calculated using a two-tailed t test and were taken to be significant at a P value of ≤0.05, or at ≥2 standard deviations (SD) from the mean (SD ≥ 2.0), as appropriate. For the immunohistochemical data, statistical evaluation was performed using the two-sample Wilcoxon rank sum (Mann-Whitney) test.
RESULTS
We and others have shown that the cystatin E/M gene behaves as a human tumor suppressor gene (17–23). However, the molecular mechanism of cystatin E/M-mediated growth suppression is not yet known.
Growth inhibition in vitro with exogenous expression of cystatin E/M.
To determine the role of cystatin E/M in expression of cathepsin L, Western blot analysis of cystatin E/M gene-transfected HeLa cells confirmed the expression of cystatin E/M accompanied by reduced expression of cathepsin L (Fig. 1A). Annexin assays performed on cells grown for 48 h showed increased apoptotic cell death in cystatin E/M gene-transfected cells in comparison to that seen with plasmid-transfected or Lipofectamine-transfected or untransfected cells (Fig. 1B). The presence of the 40-kDa multimeric Bax protein in cystatin E/M-expressing cells confirmed apoptotic cell death in the transfected cells (Fig. 1C).
FIG 1.

Apoptotic cell growth inhibition of HeLa cells expressing cystatin E/M protein. (A) Transfection of cystatin E/M plasmid into HeLa cells shows the expression of both 14-kDa nonglycosylated and secreted 17-kDa glycosylated forms of cystatin E/M protein in comparison to the levels seen with control CMV plasmid-transfected cells. We could also visualize reduced expression of cathepsin L in cystatin E/M-expressing cells. (B) Annexin V-FITC assay data show enhanced apoptotic cell death in cystatin E/M (CysM)-expressing cells (27.79% [sum of data in the lower right and upper right panels]) in comparison to the HeLa cells transfected with plasmid alone (10.49%) or the Lipofectamine (Lipo)-transfected HeLa cells (7.84%) or untransfected HeLa cells (7.33%). Quad, quadrant; UL, upper left; UR, upper right; LL, lower left; LR, lower right. (C) Western blot analysis shows expression of multimeric 40-kDa Bax protein in the cystatin E/M-expressing cells and of the monomeric 20-kDa form in the control HeLa cells. (D) Western blot showing cystatin E/M expression in gene-transfected C33A cells. GAPDH, glyceraldehyde-3-phosphate dehydrogenase. (E) An MTT growth assay reveals reduced growth in gene-transfected cells in comparison to Lipofectamine-treated or CMV-transfected cells. (F) Soft-agar colony formation is also inhibited in cystatin E/M gene-transfected cells in comparison to the results seen in control cells. (G) A bar graph confirms inhibition of cystatin E/M-mediated soft-agar colony formation of C33A cells.
Growth inhibition of HPV-negative cancer cell line C33A with exogenous expression of cystatin E/M.
We have previously shown that cystatin E/M protein expression was absent in two publicly available HPV-negative cervical cancer cell lines, C33A and HT3, and in 3 of 4 primary cervical tumors (19). Two other primary tumors contained a methylated promoter of one of the alleles and a cathepsin L binding site mutation of the other allele. While C33A cells contained both methylated and unmethylated promoters, HT3 contained unmethylated promoters, indicating possible transcription in both the cell lines (19). In the present investigation, RNA-seq analysis of the two cell lines showed low but detectable expression in HT3 cells (data not shown). Expression was barely detectable in C33A cells, pointing to, as in HPV-positive tumors, inactivation of the cystatin E/M gene in the majority of HPV-negative cancer cell lines and primary tumors. To determine whether exogenous cystatin E/M gene expression leads to growth suppression of HPV-negative cancer cell lines, C33A cells were investigated after transfection with cystatin E/M or control CMV plasmids. Western blot analysis pointed to the expression of cystatin E/M in the gene-transfected cells, and the expression was proportional to the concentration of plasmid DNA used (Fig. 1D). There was transfection-related cell death and thus reduced growth in the MTT assays performed with CMV-treated cells in comparison to Lipofectamine-treated cells (Fig. 1E). However, there was increased growth inhibition in gene-transfected cells, confirming the suppressive role of the cystatin E/M gene (Fig. 1E). Furthermore, there were reductions in the size (Fig. 1F) and number (Fig. 1G) of soft-agar colonies in the gene-transfected cells, providing additional evidence for the cell growth-inhibitory function of the cystatin E/M gene in HPV-negative tumor cell lines.
Growth inhibition with lentivirus-mediated tetracycline-inducible cystatin E/M gene expression.
To rule out overexpression as the reason for growth suppression, we performed studies using a tetracycline-inducible two-plasmid lentiviral system. Stable clones (here called CST6 cells) were selected with puromycin and G418 antibiotic markers and treated with dimethyl sulfoxide (DMSO) or doxycycline (tetracycline analog) for different time periods. Cell growth assays showed growth inhibition of induced cells at both 1.0 and 2.0 μg/ml concentrations of doxycycline (Fig. 2A). Enhanced growth inhibition was seen in cells treated with 2.0 μg/ml of doxycycline. Western blot analysis showed the expression of both the 14- and 17-kDa cystatin E/M proteins in the cell extracts (Fig. 2B). To eliminate interference from fetal bovine serum proteins, cell-free supernatants were immunoprecipitated with cystatin E/M antibody prior to Western blot hybridization with cystatin E/M. The blots clearly demonstrated the expression of cystatin E/M in the cell-free supernatants, pointing to active transport of the protein into the extracellular space (Fig. 2C). As expected, intracellular cystatin E/M expression was accompanied by reduced expression of cathepsin L, specifically, the monomeric 28-kDa protein (arrow in Cat L blot in Fig. 2B). Formation of the multimeric 40-kDa Bax protein (arrow in Bax blot in Fig. 2B) was observed in induced cells, and this is in agreement with the apoptotic cell death seen in plasmid-transfected cells (Fig. 1B).
FIG 2.
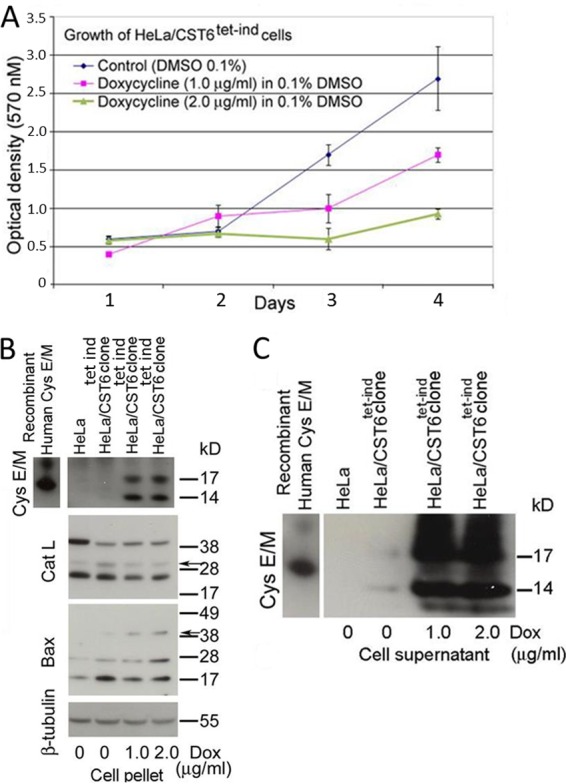
Induced cystatin E/M expression leads to cell growth inhibition. (A) Induced expression of cystatin E/M with doxycycline results in growth inhibition of CST6 cells. Slope calculations indicate P = 0.0047 for 1.0 μg/ml dox versus no dox and P = 0.0004 for 2.0 μg/ml doxycycline versus no doxycycline. (B) CST6 cells show expression of cystatin E/M that is accompanied with decreased expression of cathepsin L (arrow above 28 kDa) and the formation of multimeric Bax protein (arrow above 38 kDa). tet-ind, tetracycline induction. (C) Immunoprecipitation with mouse anti-cystatin E/M antibody followed by Western blotting with biotinylated anti-cystatin E/M antibody shows cystatin E/M expression in the cell-free supernatants, indicating secretion of the protein into the medium. Bacterial recombinant cystatin E/M proteins (R & D Systems, Inc., Minneapolis, MN) (15 and 21 kDa) were used as positive hybridization controls.
Non-cell-autonomous growth suppression of HeLa cells with cell-free supernatant containing cystatin E/M protein.
To determine whether the growth inhibition could occur due to the secreted cystatin E/M protein, HeLa cells were treated with the cell-free supernatants collected from plasmid-transfected cells. Immunofluorescence analysis showed expression of cystatin E/M in the intra- and extracellular regions, indicating expression and active transport (Fig. 3A). Western blot studies confirmed the expression of both the 14- and 17-kDa nonglycosylated and glycosylated forms of the protein (Fig. 3B). The MTT growth assay pointed to the inhibition of HeLa cell growth in the presence of cystatin E/M-containing cell-free supernatants (Fig. 3C). There was no growth inhibition in the cells grown in the presence of control CMV cell-free supernatants, confirming non-cell-autonomous growth inhibition. As shown earlier, annexin assay indicated increased cell death in cells receiving cystatin E/M-containing supernatant compared to those receiving nonexpressing supernatants in CMV-transfected or untransfected cells, confirming cystatin E/M-mediated growth inhibition through apoptotic cell death (Fig. 3D).
FIG 3.

Non-cell-autonomous growth inhibition of HeLa cells. (A) Immunofluorescence shows cytoplasmic expression of cystatin E/M in 50% to 60% of cystatin E/M gene-transfected HeLa cells. (B) Western blot analysis indicates protein expression beyond 12 days in the transfected cells. (C) HeLa cells grown in the presence of secreted medium (50:50 with MEM) showed statistically significant (P value < 0.0001) growth inhibition in cystatin E/M-expressing supernatants. Growth inhibition was not seen in cells grown with cell-free supernatants collected from the parental HeLa or CMV-transfected cells. (D) Annexin V-FITC assay shows enhanced apoptotic cell death in HeLa cells grown in the presence of cystatin E/M-expressing supernatants (59.48%) in comparison to cell death in CMV (47.20%) or untransfected (2.73%) supernatants.
We then tested the supernatant collected from the inducible system for the growth-inhibitory effect of cystatin E/M. Since the expression level would be lower than that seen with plasmid-transfected cells, we employed Boyden chamber studies for continuous expression of cystatin E/M in the presence of 2.0 μg/ml of doxycycline. The top chamber contained HeLa or CST6 cells or the CMV cells as a source of cystatin E/M, and the bottom chamber contained HeLa cells as test cells. Analysis of the top chamber cells after 48 h of induction with doxycycline clearly demonstrated expression of cystatin E/M from the induced CST6 cells (Fig. 4A). MTT growth assays of cells in the bottom chamber performed at different time periods showed statistically significant growth inhibition (P < 0.0001) when cells were grown with doxycycline-induced CST6 cells in the top chamber (Fig. 4B). Growth inhibition was not observed in the control HeLa cells or doxycycline-treated HeLa/CMV- or DMSO-treated CST6 cells, indicating a clear association between growth inhibition and cystatin E/M expression. Growth inhibition was also studied using a head and neck cancer cell line and the bottom Boyden chamber. The CST6 cells grown in the top chamber again showed growth inhibition with the addition of doxycycline (Fig. 4C). CCL23 cells in the bottom chamber also showed growth inhibition with induction of cystatin E/M from CST6 cells (Fig. 4D). CCL23 cells grown without induction did not show growth inhibition, confirming the non-cell-autonomous growth-suppressive function of cystatin E/M. Although it is possible that doxycycline could have diffused through the Boyden chamber membrane and inhibited cell growth (antibiotic effect), the absence of cell growth inhibition with doxycycline-treated lentiviral control CMV cells in the top chamber (see Fig. 4B) clearly indicated that the growth inhibition was not due to the antibiotic effect of doxycycline but was instead due to the secretion of cystatin E/M protein from the induced CST6 cells.
FIG 4.

Non-cell-autonomous growth inhibition of HeLa and CCL23 cells with induced cystatin E/M expression. (A) Induced cystatin E/M expression (in both the 17-kDa and 14-kDa proteins in the Western blot) was seen in CST6 cells collected from the Boyden system top chamber. (B) Induction of cystatin E/M led to statistically significant (P < 0.0001) inhibition of growth of HeLa cells grown in the bottom chamber. Growth inhibition was not seen in HeLa cells grown with HeLa cells or CMV cells in the presence of doxycycline or with CST6 cells in the absence of doxycycline in the top chamber. (C) Statistically significant growth inhibition (<0.05) of CST6 cells (in the top chamber) was seen when the cells were grown in the presence of doxycycline (2 μg/ml). (D) CCL23 cells grown in the bottom chamber showed growth inhibition when the cells were grown in the presence of doxycycline-treated CST6 cells (P < 0.05).
Non-cell-autonomous HeLa cell growth inhibition mediated by cystatin E/M protein secreted by HEKs.
To find out whether growth inhibition could be demonstrated with the physiological concentration of cystatin E/M protein secreted by nonneoplastic cells, we used the HPV 16 E6 or E7 or E6 and E7 immortalized human epidermal keratinocyte (HEK) cell line system. This study would also resolve the issue of growth inhibition mediated by doxycycline versus cystatin E/M in the inducible system. While the normal and E6 immortalized HEK cell lines showed appreciable levels of cystatin E/M expression, there was reduced expression in E7 and E6/E7 immortalized HEK cell lines (Fig. 5A). Immunofluorescence analysis confirmed reduced expression of cystatin E/M in HEK/HPV 16 E7 cells in comparison to that seen in HEK/HPV 16 E6 cells (Fig. 5B). While uniform expression was noticed in HEK/HPV 16 E6 cells, a punctated pattern indicating decreased expression was seen in HEK/HPV 16 E7 cells. Western blot analysis of cell-free supernatant samples confirmed secretion of cystatin E/M in the immortalized HEK cell lines, with reduced expression again seen in HPV 16 E7-containing HEK cells (Fig. 5C). To determine the reason behind decreased cystatin E/M expression in HPV 16 E7-containing cells, cotransfections were carried out with HA-tagged HPV 16 E6 or E7 plasmid and FLAG-tagged cystatin E/M in the HeLa cells. Hybridization to the FLAG antibody confirmed the expression of cystatin E/M in the HeLa cells transfected with the cystatin E/M plasmid or in cotransfection with HPV 16 E6 or E7 plasmids (Fig. 5D). Cystatin E/M expression was not observed in HeLa cells transfected with HPV 16 E6 or E7 alone. Immunoprecipitation with HA antibody followed by Western blot analysis using the FLAG antibody demonstrated binding of cystatin E/M to the HPV 16 E7 protein but not to HPV 16 E6, indicating a specific interaction between HPV 16 E7 and cystatin E/M (Fig. 5E).
FIG 5.

Reduced expression of cystatin E/M in HPV 16 E7 immortalized HEK cells. (A) Western blot analysis reveals expression of cystatin E/M in normal human epidermal keratinocytes (HEK cells) and HEK cells immortalized with the HPV 16 E6 gene or E7 gene or with the E6 gene plus the E7 gene. However, reduced expression was seen in the HEK/HPV 16 E7 cell line in comparison to the normal cell line or the HEK/HPV 16 E6 cell line. (B) Immunofluorescence staining confirms reduced expression in HEK/HPV 16 E7 cells (×100 magnification). The inset for a single cell shows uniform expression in HEK/HPV 16 E6 cells and reduced punctuated staining in HEK/HPV 16 E7 cells. (C) Western blot analysis of the supernatants (100 μl) again shows cystatin E/M expression in the immortalized HEK cells with reduced expression in HEK/HPV 16 E7cells. (D) Hybridization with the FLAG antibody shows cystatin E/M expression in the HeLa cells transfected with the FLAG tag cystatin E/M plasmid. Expression was not seen in HeLa cells transfected with the E6 gene or the E7 gene alone. (E) Hybridization of HA (HPV 16 E6 or E7 gene tag)-immunoprecipitated samples with the FLAG (cystatin E/M tag) antibody shows the presence of cystatin E/M protein in cells cotransfected with the HPV 16 E7 gene, pointing to an interaction between HPV 16 E7 and cystatin E/M proteins. (F) Hybridization of anti-cystatin E/M antibody-immunoprecipitated lysates with the antiubiquitin (Ubq) antibody shows higher-intensity hybridization of high-molecular-mass proteins in HPV 16 E7- and cystatin E/M-cotransfected HeLa cells, indicating cystatin E/M degradation by HPV 16 E7. This interpretation is also supported by the absence of a visible cystatin E/M band in the cell-free supernatants of HPV 16 E7- and cystatin E/M-cotransfected cells. β-Tubulin and GAPDH (glyceraldehyde-3-phosphate dehydrogenase) were used as the input controls.
To find out whether there was ubiquitination in HPV 16 E7-containing cells, HeLa cell lysates prepared from HPV 16 E6 or HPV 16 E7 alone or cotransfected with the cystatin E/M DNA were immunoprecipitated with the anti-cystatin E/M antibody and Western blots of the immunoprecipitates were hybridized to the antiubiquitin antibodies. There was increased hybridization to higher-molecular-mass bands in HPV 16 E7- and cystatin E/M-cotransfected cells, pointing to the ubiquitination of cystatin E/M by HPV 16 E7 (Fig. 5F). Western blot analysis of anti-cystatin E/M antibody immunoprecipitates of the supernatants collected prior to inhibitor addition showed the presence of the cystatin E/M protein in HPV 16 E6-cystatin E/M-cotransfected cells. This protein was again not visualized in HPV 16 E7-cystatin E/M-cotransfected cells, pointing to the degradation of expression of cystatin E/M mediated by HPV 16 E7.
To determine whether expression of cystatin E/M in the immortalized keratinocytes is related to a lower growth rate, MTT growth assays were performed. The investigation showed significantly reduced growth in the immortalized HEK cells in comparison to the HeLa cells (P value < 0.0001) (Fig. 6A). Further, the HEK/HPV 16 E6 cells showing higher levels of cystatin E/M expression grew more slowly than the HEK/HPV 16 E7 cells, demonstrating a direct relationship between the level of cystatin E/M expression and cell growth inhibition.
FIG 6.

Inhibition of HeLa cell growth by HEK/HPV 16 E6 and HEK/HPV 16 E7 keratinocytes. (A) Both the HEK/HPV 16 E6 cells and HEK/HPV 16 E7 cells expressing cystatin E/M grew more slowly than the nonexpressing HeLa cells (P value < 0.0001). (B to D) Coculture studies show inhibition of HeLa cell growth in the presence of HEK/HPV 16 E6 or HEK/HPV 16 E7 keratinocytes. Statistically significant growth inhibition is seen for HeLa:HEK/HPV 16 E6 cells in comparison to HeLa:HEK/HPV 16 E7 cells (P value < 0.037). (E) Boyden chamber assay showed statistically significant HeLa cell growth inhibition with HEK/HPV 16 E6 or HEK/HPV 16 E7 keratinocytes (P value for HEK/HPV 16 E6 versus HeLa cells, <0.0001; P value for HEK/HPV 16 E7 versus HeLa cells, <0.027). Statistically significant (P value < 0.05) growth inhibition was also seen with HEK/HPV 16 E6 cells in comparison to that seen with HEK/HPV 16 E7 cells, again indicating a direct relationship between cystatin E/M expression and cell growth inhibition.
To verify whether cystatin E/M protein secreted from the immortalized HEK cells plays a role in cell growth inhibition (i.e., non-cell-autonomous growth inhibition), we performed coculture studies with HeLa cells grown in the presence of HEK/HPV 16 E6 or HEK/HPV 16 E7 cells. The results showed a decrease in growth of HeLa cells in the presence of HEK/HPV 16 E6 cells with different HeLa:HEK/HPV 16 E6 cell ratios in comparison to the growth seen with different HeLa:HEK/HPV 16 E7 cell ratios (Fig. 6B to D). Correlating to the higher expression of cystatin E/M, statistically significant growth inhibition of HeLa cells was seen when the cells were grown with HEK/HPV 16 E6 cells in comparison to the results seen with HEK/HPV 16 E7 cells (P value < 0.037 [at a 50:50 ratio]) (Fig. 6C). To further confirm the non-cell-autonomous growth suppression mediated by secreted cystatin E/M, Boyden chamber assays were performed with HeLa cells or the HEK/HPV 16 E6 or HEK/HPV 16 E7 cells in the top chamber. MTT growth assay of the HeLa cells plated in the bottom chamber showed statistically significant growth inhibition of HeLa cells under conditions of growth with the HEK/HPV 16 E6 (P value < 0.0001) or HEK/HPV 16 E7 (P value < 0.027) cells in comparison to that seen with the HeLa cells in the top chamber (Fig. 6E). These results further demonstrated a growth-inhibitory effect of cystatin E/M protein present in the secreted medium of the immortalized HEK cells. Enhanced growth suppression in the presence of HPV 16 E6 immortalized HEKs expressing and secreting higher levels of cystatin E/M pointed to a direct relationship between the level of cystatin E/M expression and tumor cell growth inhibition.
Doxycycline treatment leads to cystatin E/M expression and minimal change in the expression of other proteins.
As a step toward understanding the molecular mechanism of tumor cell growth suppression, we determined the effect of doxycycline in the parental HeLa cells versus the CST6 cells. There was statistically significant (P = 0.0477) growth inhibition in CST6 cells compared to HeLa cells at a 2 μg/ml concentration of doxycycline, pointing to an additional growth-inhibitory effect of the cystatin E/M protein produced and secreted in doxycycline-treated CST6 cells (Fig. 7A and B). Both cell lines showed >75% growth inhibition in 4 days with 4 μg/ml doxycycline treatment, indicating drug-induced toxicity at higher concentrations. Thus, we restricted our molecular mechanistic studies to treatments using 2 μg/ml doxycycline.
FIG 7.

Minimal expression changes in doxycycline-treated HeLa or CST6 cells. (A) Enhanced growth inhibition is seen in CST6 cells treated with doxycycline (2 μg/ml) in comparison to doxycycline-treated HeLa cells (P = 0.0477). Treatment with 4 μg/ml doxycycline led to >80% cell death in both cell lines, pointing to antibiotic-induced toxicity. wrt, with respect to. (B) Western blot analysis of cell supernatants showing cystatin E/M expression in doxycycline-treated CST6 cells. (C) Representative RNA expression profile showing that differential expression changes in doxycycline-treated CST6 cells were limited to increased expression of cystatin E/M (150-fold) and GALNT16 (2.5-fold) and decreased expression of Jun (50%) and CTSL (10%). Due to low basal levels, differential levels of expression in doxycycline-treated HeLa cells may not be significant. (D) Multiplex growth factor assays showing statistically significant (P < 0.05) reduced expression of IL-10 and MMP-10 in doxycycline-treated CST6 cells and MMP-1 and MMP-10 in doxycycline-treated CST6 cell supernatants (Sup).
Of the 20,000 genes analyzed from the RNA-seq data of parental and doxycycline-treated cell lines (HeLa versus HeLa dox and CST6 versus CST6 dox), differential expression levels were seen for only a few after doxycycline treatment (Fig. 7C). Although doxycycline-treated HeLa cells showed 50%-decreased expression of CDH1 (e-cadherin) and CDH2 (n-cadherin) genes, expression levels were low in the untreated HeLa cells, indicating that these differences may not be significant. CST6 cells had higher differential levels of expression of several genes than HeLa cells in the basal state, reflecting genetic changes induced by the lentiviral vector system. However, the differential expression of CST6 and doxycycline-treated CST6 cells was restricted to increased expression of cystatin E/M and GALNT16 (polypeptide N-acetylgalactosaminyltransferase 16, an enzyme involved in protein glycosylation) and decreased expression of c-Jun and CTSL (cathepsin L). Although expression of a few other genes was also affected after doxycycline treatment, basal expression levels in the untreated CST6 cells were low, indicating that the differences were most likely nonsignificant. These results therefore indicated that the major difference in the gene expression patterns of doxycycline-treated HeLa versus CST6 cells was primarily limited to increased expression of cystatin E/M, confirming a direct relationship between cystatin E/M expression and growth inhibition in doxycycline-treated CST6 cells.
To identify the effect on cytokines and growth factors associated with cell growth inhibition at the protein level, the cell lysates and cell-free supernatants were analyzed using the Human 9-plex Electrochemiluminescent Multi-Spot enzyme-linked immunosorbent assay (ELISA) system in the Meso Scale discovery platform (38). Statistically significant (P < 0.05) inhibition was seen in the expression of IL-10 and of matrix metalloproteinases MMP-1 and -10, again confirming minimal expression changes induced by doxycycline treatment of CST6 cells (Fig. 7D).
Cystatin E/M expression correlates to the inhibition of the canonical NF-κB signaling pathway.
Apoptotic cell death and formation of multimeric Bax protein (Fig. 1C and 2B) and inhibitory effects on MMPs (Fig. 7D) in cells with induced expression of cystatin E/M suggested that cystatin E/M was targeting the transcription factor NF-κB. To verify this hypothesis, we determined the expression of NF-κB and p16, the tumor suppressor protein that we have previously shown to be associated with NF-κB ubiquitination, in doxycycline-treated CST6 cells. We found expression levels of both NF-κB and p16 to be increased after doxycycline treatment (Fig. 8A). Cytoplasmic and nuclear fractions isolated and verified using MEK and histone H3 markers showed higher cytoplasmic retention of NF-κB in doxycycline-treated CST6 cells (Fig. 8B and C). Additionally, we observed decreased expression of phospho-IκBα in the cell lysate and in the cytoplasmic fraction of doxycycline-treated CST6 cells, indicating inhibition of IκBα phosphorylation by cystatin E/M (Fig. 8D).
FIG 8.

Induced cystatin E/M expression leads to cytoplasmic retention of NF-κB. (A) Western blot analysis shows increased expression of NF-κB and p16 in doxycycline-treated CST6 cells. (B) Cell fractionation shows hybridization of MEK primarily to the cytoplasmic fraction and histone H3 primarily to the nuclear fraction. (C) Analysis of the cellular fractions showing increased cytoplasmic (Cyt) expression of NF-κB but not p16 in comparison to the control cytoplasmic protein MEK in doxycycline-treated CST6 cells. Nuc, nucleus. (D) Expression of phospho-IκBα visualized in total cell lysate and the cytoplasmic fraction of CST6 cells; the expression was reduced after doxycycline treatment.
To determine whether decreased phospho-IκBα expression was associated with increased expression of IκBα, control and doxycycline-treated HeLa and CST6 cells were analyzed. The Western blot studies did not reveal an appreciable difference in the levels of expression of IκBα in the untreated and doxycycline-treated HeLa cells (Fig. 9A). However, there was increased IκBα expression in doxycycline-treated CST6 cells in comparison to the untreated control CST6 cells (Fig. 9A). Higher IκBα expression was also associated with decreased phospho-IκBα expression, confirming the inhibitory effect of doxycycline on the phosphorylation of IκBα in CST6 cells.
FIG 9.

Inhibition of canonical NF-κB signaling pathway by cystatin E/M protein. (A) Basal expression of nonphosphorylated IκBα was seen in HeLa cells and was not altered after doxycycline treatment. However, IκBα expression increased in CST6 cells after doxycycline treatment, correlating to decreased phosphor lation of IκBα. Treatment with TNF-α enhanced phospho-IκBα expression in both the HeLa and CST6 cell lines, and the expression was not altered in HeLa cells after doxycycline treatment. On the other hand, treatment with doxycycline led to reduced expression of phospho-IκBα in CST6 cells. (B) The basal IKKβ expression level was high in the control CST6 cells, phospho-IKKβ expression was visualized only with the addition of TNF-α, and the expression was reduced in doxycycline-treated cells. (C) Induced cystatin E/M expression did not inhibit endogenous activation of p100 (NF-κB2) to produce p52 in CST6 cells. In HEK 293T cells, exogenous NIK expression led to activation of myc-tag p100 to produce myc-p52. While this activation was inhibited by cIAP1/2 expression, inhibition was not observed with the expression of cystatin E/M. (D) Mechanistic representation showing inhibition of the IKKβ-mediated canonical and not the NIK-mediated noncanonical NF-κB signaling pathway by cystatin E/M. TNFR, tumor necrosis factor receptor; TNFSFR, tumor necrosis factor superfamily receptor.
Tumor necrosis factor alpha (TNF-α) is a known inducer of NF-κB activity through the activation of the IKK complex for the phosphorylation and ubiquitination of IκBα, the inhibitor protein of NF-κB. We therefore treated HeLa and CST6 cells with TNF-α for 5 min in the presence or absence of doxycycline for 48 h. Treatment with TNF-α alone increased the expression of phospho-IκBα in both cell lines in comparison to that seen in the untreated control cell lines (Fig. 9A). Addition of TNF-α to doxycycline-treated HeLa cells did not affect the expression of phospho-IκBα. However, phosphorylation of IκBα was reduced in doxycycline-treated CST6 cells, pointing to a cystatin E/M-mediated inhibitory effect on the TNF receptor signaling pathway. To confirm this phenomenon, we studied the activation of the upstream protein IKKβ in the presence and absence of doxycycline. While there was high expression of IKKβ, phosphorylation of this protein was undetectable at the basal level in both the HeLa and CST6 cell lines (data for CST6 cells are shown in Fig. 9B). Treatment with TNF-α-induced phosphorylation of IKKβ in CST6 cells and doxycycline treatment for 48 h followed by TNF-α treatment showed reduced phospho-IKKβ expression, clearly indicating that cystatin E/M inhibits TNF receptor-mediated signaling to NF-κB.
Commercially available phospho-IKKβ antibodies hybridize to both phospho-IKKα and IKKβ, raising the possibility that IKKα phosphorylation would also be inhibited. While IKKα/IKKβ dimerization is observed in the IKK complex of the canonical NF-κB pathway, IKKα homodimerization is seen in the noncanonical pathway. Therefore, to determine whether the noncanonical NF-κB signaling pathway was inhibited, we investigated the effect of cystatin E/M on the expression of p52, the active form of the NF-κB2, from its precursor protein, p100. There was high endogenous expression of p100 and p52 in CST6 cells, and the expression was not inhibited with doxycycline-induced expression of cystatin E/M (Fig. 9C). We therefore studied the effect on exogenously activated p52 using myc-tag p100 and FLAG tag NIK cotransfected into HEK 293T cells. While inhibition of p52 expression was observed with exogenous expression of cIAP1/2, a known inhibitor of the NF-κB2 pathway, inhibition was not observed with expression of cystatin E/M in the cotransfected cells. These results clearly indicated that cystatin E/M inhibits IKKβ phosphorylation of the canonical NF-κB signaling pathway and not IKKα phosphorylation of the noncanonical NF-κB signaling pathway (Fig. 9D).
Growth inhibition of in vivo CST6 mouse xenograft tumors with induced cystatin E/M expression.
To determine whether growth of mouse xenograft tumors could be inhibited by induced cystatin E/M expression, CST6 cells were injected subcutaneously into 5-week-old NOD/SCID IL2Rγ null mice for tumor formation. Once small tumors were visible (7 days), mice received 5% sucrose water (control group—8 mice) or 5% sucrose water containing doxycycline (2 mg/ml) (experimental group—12 mice). Tumor volumes were measured weekly, and the tumors were excised when the size reached the animal protocol-approved limit. The investigation showed statistically significant (P = 0.0047) inhibition of tumor cell growth in the doxycycline-treated mice in comparison to the results seen with the control mice (Fig. 10A). Reflecting tumor growth inhibition, a significant decrease in the size of the tumors excised from the experimental group was seen (three each of representative control and experimental-group tumors are shown in Fig. 10B). To demonstrate that the growth inhibition was associated with the expression of cystatin E/M, tumor cells were sonicated in a lysis buffer containing protease inhibitors. Due to blood cell contamination in the tumors, direct Western blotting could not be carried out. However, blotting analysis of cystatin E/M immunoprecipitated proteins showed expression of cystatin E/M in the doxycycline-treated tumors (Fig. 10C). Hybridization with the control actin probe indicated the presence of comparable levels in the control and the experimental tumor lysates, indicating a direct correlation between the expression of cystatin E/M and tumor cell growth inhibition.
FIG 10.

Xenograft tumor growth inhibition of CST6 cells with induced cystatin E/M expression. (A) The experimental group (n = 12) receiving doxycycline water showed tumor growth reduction in comparison to the control group (n = 8) (P value = 0.0047). (B) Representative photographs confirm decreased tumor growth in the experimental animals. (C) Immunoprecipitated proteins showing expression of cystatin E/M in doxycycline-treated animals correlating with tumor growth suppression. α-Actin was used as the positive hybridization for the input control.
Inverse relationship between cystatin E/M expression and cathepsin L expression in normal cervix tissue.
To verify the inverse relationship of the levels of expression of cystatin E/M (inhibitor) and cathepsin L (substrate) in normal human tissues, immunohistochemical (IHC) analysis was performed on normal cervix tissue, CIN (carcinoma in situ; also known as cervical intraneoplasia) tissue, and primary cervical tumors. The IHC studies performed on normal skin, sebaceous glands, and cervix and endocervix (EN) tissues showed expression of cystatin E/M in the cytoplasm and in the perinuclear region (Fig. 11A to D). Nuclear expression was also seen in some areas of the tissue sections. While normal epithelial cells did not show expression of cathepsin L, expression was observed in endocervical glands near the secreting lumens (Fig. 11E and F). Thus, an inverse relationship of the levels of expression of cystatin E/M and cathepsin L was observed in normal cervical tissues.
FIG 11.

Cystatin E/M expression in normal skin, normal cervix, and CIN tissues. (A and B) Pictures were taken of skin from a healthy subject (C and D) Photomicrographs were taken of a healthy cervix with epithelium (C) and endocervical gland (D). Panels A to D all show expression of cystatin E/M. Expression in perinuclear localizations is shown by arrows. (E) Normal epithelial cells do not show expression of cathepsin L. (F) Cathepsin L expression is seen in endocervical glands as a fine granular cytoplasmic reaction concentrated near secretory lumens (arrow); none of the results showed nuclear presentation. (G) Cystatin E/M is expressed in epithelial cells (EP) and in endocervix (EN) (arrow) in CIN tissue. (H) Cathepsin L expression is seen in secretory granules of endocervix (EN). Neither the epithelium nor the endocervix showed cathepsin L expression. All photographs were obtained with a 60× oil objective lens.
Loss of cystatin E/M expression in primary cervical tumors.
To verify whether loss of cystatin E/M expression was related to tumor development, preneoplastic CIN specimens and primary cervical tumors were analyzed. IHC investigation of CIN specimens (1 CIN I sample and 27 CIN III samples) showed cystatin E/M expression in all 28 samples. Expression was seen in both the epithelium and endocervix (Fig. 11G; the arrow points to the endocervix). Cathepsin L expression was seen in secretory granules of endocervix (see expression in the EN space indicated in Fig. 11H). The epithelium and endocervix did not express cathepsin L, and the endocervical secretory granules did not express cystatin E/M, indicating an inverse relationship between cystatin E/M and cathepsin L in the CIN specimens such as was seen in the normal cervix tissue.
Analysis of 33 primary cervical tumors showed the loss of or reduced (5% to 10% of cells with 1+ intensity) expression of cystatin E/M in 26 tumors. Seven tumors had positive cystatin E/M expression (higher percentage of cells with 2+ intensity) that was previously reported by our group to represent cathepsin L binding site mutations. Loss of cystatin E/M expression in the tumors (representative data are shown for tumor 55 in Fig. 12A) was accompanied by overexpression of cathepsin L. Expression of cathepsin L was also seen in cystatin E/M-expressing tumors that contained binding site mutations (data are shown for tumor 59 containing a cathepsin L binding site M34T cystatin E/M gene mutation in Fig. 12A).
FIG 12.

Loss of cystatin E/M expression in primary cervical tumors and inverse relationship to nuclear NF-κB expression. (A) Composite pictures of tumors 55 and 59 with cystatin E/M and cathepsin L immunohistochemical stains show minimal or no expression of cystatin E/M but high expression of cathepsin L in tumor 55 (×40 magnification). High expression of cystatin E/M is seen in the superficial epithelium with CIN III pathology (inset) but not in the invasive tumor (T). The ×40 images of the tumor show cathepsin L expression extending into the interstitial tissues. Tumor 59, containing a cathepsin L binding site mutation, showed high nuclear expression of cystatin E/M, but expression of cathepsin L was also seen in the invasive cancerous cells. (B) Box plot representation of cystatin E/M and cathepsin L expression in 28 CIN specimens versus 33 primary tumors showing statistically significant decrease in the percentage of cells expressing cystatin E/M (P value = 0.0003), a joint score of percent reactivity × intensity of cystatin E/M (P value = 0.003), and an increase in the joint score of percent reactivity × intensity of cathepsin L expression (P value = 0.04) in primary tumors. (C) Normal endocervix tissue and a CIN III specimen showing cytoplasmic NF-κB expression. Occasional nuclear staining is seen in these cells. Invasive primary tumors contained increased nuclear NF-κB expression.
Two-sample Wilcoxon rank sum (Mann-Whitney) test results showed a statistically significant (P value = 0.0003) difference in the percentages of reactivity of cystatin E/M in CIN specimens versus primary tumors (Fig. 12B). Statistical significance (P value = 0.003) was also observed for results of comparisons of the joint score of reactivity × intensity of cystatin E/M in CIN specimens versus the tumors. Statistical significance (P value = 0.04) was also observed for results of comparisons of the joint reactivity × intensity of cathepsin L expression in the stroma of CIN specimens versus the primary tumors. In this case, the levels of expression of cathepsin L in the stroma of CIN specimens were lower than those observed in the stroma of tumors. Thus, the results indicated that loss of cystatin E/M expression was accompanied by enhanced cathepsin L expression during the development of CIN specimens to invasive cervical tumors.
Overexpression of nuclear NF-κB in primary cervical tumors.
To determine whether the inhibitory effect of cystatin E/M on NF-κB observed in the in vitro studies could be related to cytoplasmic localization of transcription factors in the normal and CIN tissues and nuclear localization in primary tumors, IHC analysis was performed. While the normal cervix and CIN specimens (five of each examined) showed cytoplasmic expression of NF-κB (p65; rel A protein), nuclear expression of NF-κB was seen in all the 20 primary tumors (Fig. 12C). These results therefore pointed to a strong association between the loss of cystatin E/M expression and nuclear expression of NF-κB in the primary cervical tumors.
DISCUSSION
Numerous studies have shown inactivation of the cystatin E/M gene in human tumors (16–23). However, all the functional studies relied on the exogenous expression of cystatin E/M using plasmid constructs. As such, there could be potential phenotypic changes that could affect the validity of conclusions based on analysis of plasmid-transfected cells. In the present investigation, three model systems were utilized, namely, transient exogenous transfection of the cystatin E/M gene, a tetracycline-inducible lentiviral system, and HPV 16 E6, HPV 16 E7, or HPV 16 E6 and E7 immortalized human epidermal keratinocytes, the tissue of origin for cervical cancer. In contrast to analyses of exogenous expression, the inducible system allows the same cells to be compared with and without the addition of doxycycline. The parental HeLa cells were also studied with and without doxycycline treatment as controls for the effect of doxycycline on growth suppression. Similarly, in comparison to nonphysiological expression of cystatin E/M in mouse xenograft tumors of plasmid-transfected cell lines, the cells containing the inducible vector system are more suitable to test cystatin E/M-mediated tumor growth suppression. We have further used HPV 16 E6- and E7-transfected normal human epidermal keratinocytes to demonstrate intracellular and non-cell-autonomous growth-inhibitory properties of cystatin E/M. We have presented evidence that growth inhibition is related to apoptosis and cytoplasmic retention and therefore to inactivation of NF-κB.
The extracellular matrix (ECM) plays a significant role in cell growth and migration, as a number of growth-signaling inputs, including Wnt, Notch, and hormone and integrin signaling, emanate from the cytokines and growth factors secreted into the ECM (43, 44). The matrix metalloproteinases (MMPs) are also integral components of the signaling process in normal cell migration and tumor cell invasion and metastasis. Further, tumors with metastatic potential themselves secrete cytokines and growth factors for cancer cell self-renewal, as proposed for the self-renewal of cancer stem cells (CSCs) (45, 46). For example, fibroblasts from normal mammary tissues do not support growth of slowly growing tumor cell lines; however, fibroblasts from a breast cancer would support faster growth of the same tumor cell line, indicating the production of growth factors by breast cancer fibroblasts (47)
Cancer cells whose growth has been inhibited by chemotherapy-radiation therapy secrete cytokines and chemokines, resulting in a paracrine effect on the neighboring tumor cells (48). The growth signaling then leads to the development of chemotherapy- and radiation-resistant cancer stem cells. For example, IL-6 activates the JAK/STAT pathway and IL-8 activates the AKT pathway. Both cytokines could also activate the NF-κB pathway, which in turn could activate the production of cytokines and growth factors. Thus, there is a feedback loop between cytokines and growth factors and transcription factors leading to aggressive cancer cell growth. Inhibition of this loop through the inhibition of growth factors is required for the control of cancer cell growth. In addition to its inhibitory role with respect to cathepsin L, our studies show that cystatin E/M inhibits expression of growth factors mediated by the cytoplasmic retention of NF-κB, which could be related to the cathepsin L- and legumain-independent inhibitory function of cystatin E/M correlating to tumor cell growth inhibition.
Doxycycline treatment has been shown to inhibit hepatic tumor cell growth through inhibition of epithelial-to-mesenchymal transition (EMT) and by enhanced expression of E-cadherin and reduced expression of N-cadherin, vimentin, twist, and snail (49). Since our studies show that there is >75% HeLa cell growth inhibition with 4 μg/ml of doxycycline treatment, the results seen with the higher doxycycline concentrations (5 and 10 μg/ml) used by Meng et al. (49) suggest a possible doxycycline-induced toxic effect on hepatic tumor cells. Our RNA-seq data from the parental HeLa cells have further indicated very low expression of cadherins and MMPs and only modestly decreased expression in doxycycline-treated cells (data not shown). Additionally, we did not see reduced expression of the mesenchymal markers vimentin, twist, and snail in doxycycline-treated HeLa cells, pointing to the absence of EMT transformation in our cell system. Among the 20,000 genes analyzed using the RNA-seq data, we observed elevated expression of cystatin E/M (150-fold) and GALNT16 (2.5-fold) and reduced expression of c-Jun (50%) in doxycycline-treated CST6 cells as the only significant changes, underscoring the utility of our inducible system in the investigation of the molecular mechanism of tumor cell growth inhibition.
We have demonstrated a growth-suppressive effect with secreted proteins of immortalized human epidermal keratinocytes, which are precursor cells of cervical cancer. Although slower growth in comparison to that of the HeLa cells was seen in both HEK/HPV 16 E6 and HEK/HPV 16 E7 immortalized cells, the HEK/HPV 16 E6 cells with higher expression of cystatin E/M grew more slowly than the HEK/HPV 16 E7 immortalized cells. Further, coculture and Boyden chamber studies confirmed reduced growth of HeLa cells with HEK/HPV 16 E6 cells in comparison to growth with HEK/HPV 16 E7 cells, pointing to an enhanced growth-inhibitory effect mediated by the increased expression of cystatin E/M protein in HEK/HPV 16 E6 cells. We have also shown that the reduced cystatin E/M expression in HEK/HPV 16 E7 cells is related to the interaction between E7 and cystatin E/M proteins. Our results therefore suggest that cystatin E/M is one of the targets of oncogenic HPV.
Vast literature is available on the inactivation of tumor suppressor genes in the development of human tumors. However, gene therapeutic interventions are highly difficult due to development of somatic mutations over time in gene-transfected cells. Thus, it will be beneficial to approach cell growth inhibition through the use of non-cell-autonomous growth inhibitors. Our results indicate that we could induce cystatin E/M expression from CST6 cells and test its growth-inhibitory activity in animal metastatic tumor model systems. Other proteins with non-cell-autonomous growth-inhibitory properties include STAT and SMAD-4, which acts through TGF-β signaling pathways (50, 51). Thus, there may be avenues for combined therapeutic studies performed with cystatin E/M, STAT, and SMAD-4 proteins, for the control of cancer cell growth in in vitro and in vivo animal model systems.
ACKNOWLEDGMENTS
We thank Ofer Eidelman and Catherine Joswik for helpful discussions. We gratefully acknowledge cell growth assay support from Jenna Chatoff and Kimberly Hwang and technical support for the ubiquitination study from Ankur Gholkar and for cytokine and growth factor assays from Yvonne Eudy.
This work was supported by the VA Greater Los Angeles Healthcare System, West Los Angeles Surgical Education Research program, and by a VA Merit Grant to E.S.S. Support for this work from the Cystic Fibrosis Foundation and Center for Neurology and Regenerative Medicine to M.S. is also acknowledged.
H.S., N.V., M.S.V., S.R., A.Z., S.K.B., K.P., M.S., J.Z.T., and E.S.S. performed the biological, biochemical, and animal studies. N.A.M. and E.S.S. performed immunohistochemical studies, and L.-J.L. and D.W.G. performed statistical analysis. All of us read the manuscript and approved its content.
None of us have financial conflicts of interest.
REFERENCES
- 1.Siegel R, Naishadham D, Jemal A. 2013. Cancer Statistics, 2013. CA Cancer J Clin 63:11–30. doi: 10.3322/caac.21166. [DOI] [PubMed] [Google Scholar]
- 2.GLOBOCAN International Agency for Research on Cancer (IARC). 2013. Cancer fact sheet no. 397. World Health Organization, Geneva, Switzerland: http://www.who.int/mediacentre/factsheets/fs297/en/. [Google Scholar]
- 3.Couture MC, Page K, Stein ES, Sansothy N, Sichan K, Kaldor J, Evans JL, Maher L, Palefsky J. 2012. Cervical human papillomavirus infection among young women engaged in sex work in Phnom Penh, Cambodia: prevalence, genotypes, risk factors and association with HIV infection. BMC Infect Dis 12:166–176. doi: 10.1186/1471-2334-12-166. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.de Freitas AC, Coimbra EC, Leitão MD. 2014. Molecular targets of HPV oncoproteins: potential biomarkers for cervical carcinogenesis. Biochim Biophys Acta 1845:91–103. [DOI] [PubMed] [Google Scholar]
- 5.Elfström KM, Herweijer E, Sundström K, Arnheim-Dahlström L. 2014. Current cervical cancer prevention strategies including cervical screening and prophylactic human papillomavirus vaccination: a review. Curr Opin Oncol 26:120–129. doi: 10.1097/CCO.0000000000000034. [DOI] [PubMed] [Google Scholar]
- 6.Poljak M, Seme K, Mave PJ, Kocjan BJ, Cuschieri KS, Rogovskaya SI, Arbyn M, Syrjänen S. 2013. Human papillomavirus prevalence and type-distribution, cervical cancer screening practices and current status of vaccination implementation in Central and Eastern Europe. Vaccine 31(Suppl 7):H59–H70. doi: 10.1016/j.vaccine.2013.03.029. [DOI] [PubMed] [Google Scholar]
- 7.Vedham V, Divi RL, Starks VL, Verma M. 2014. Multiple infections and cancer: implications in epidemiology. Technol Cancer Res Treat 13:177–194. [DOI] [PubMed] [Google Scholar]
- 8.Smith EM, Pawlita M, Rubenstein LM, Haugen TH, Hamsikova E, Turek LP. 2010. Risk factors and survival by HPV-16 E6 and E7 antibody status in human papillomavirus positive head and neck cancer. Int J Cancer 127:111–117. doi: 10.1002/ijc.25015. [DOI] [PubMed] [Google Scholar]
- 9.Sarchianaki E, Derdas SP, Ntaoukakis M, Vakonaki E, Lagoudaki ED, Lasithiotaki I, Sarchianaki A, Koutsopoulos A, Symvoulakis EK, Spandidos DA, Antoniou KM, Sourvinos G. 2014. Detection and genotype analysis of human papillomavirus in non-small cell lung cancer patients. Tumour Biol 35:3203–3209. doi: 10.1007/s13277-013-1419-2. [DOI] [PubMed] [Google Scholar]
- 10.Herrera-Goepfert R, Vela-Chávez T, Carrillo-García A, Lizano-Soberón M, Amador-Molina A, Oñate-Ocaña LF, Hallmann RS. 2013. High-risk human papillomavirus (HPV) DNA sequences in metaplastic breast carcinomas of Mexican women. BMC Cancer 13:445. doi: 10.1186/1471-2407-13-445. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Anastasi A, Brown MS, Kembhavi AA, Nicklin MJH, Sayers CA, Sunter DC, Barrett AJ. 1983. Cystatin, a protein inhibitor of cysteine proteinases. Biochem J 211:129–138. doi: 10.1042/bj2110129. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Rawlings ND, Barrett AJ. 1990. Evolution of proteins of the cystatin superfamily. J Mol Evol 30:60–71. doi: 10.1007/BF02102453. [DOI] [PubMed] [Google Scholar]
- 13.Barrett AJ, Fritz H, Grubb A, Isemura S, Jarvinen M, Katunuma N, Machleidt W, Muller-Esterl W, Sasaki M, Turk V. 1986. Nomenclature and classification of the proteins homologous with the cysteine-proteinase inhibitor chicken cystatin. Biochem J 236:312. doi: 10.1042/bj2360312. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Zeeuwen PL, van Vlijmen-Willems IM, Hendriks W, Merkx GF, Schalkwijk J. 2002. A null mutation in the cystatin M/E gene of ichq mice causes juvenile lethality and defects in epidermal cornification. Hum Mol Genet 11:2867–2875. doi: 10.1093/hmg/11.23.2867. [DOI] [PubMed] [Google Scholar]
- 15.Sotiropoulou G, Anisowicz A, Sager R. 1997. Identification, cloning, and characterization of cystatin M, a novel cysteine proteinase inhibitor, down-regulated in breast cancer. J Biol Chem 272:903–910. doi: 10.1074/jbc.272.2.903. [DOI] [PubMed] [Google Scholar]
- 16.Zhang J, Shridhar R, Dai Q, Song J, Barlow SC, Yin L, Sloane BF, Miller FR, Meschonat C, Li BD, Abreo F, Keppler D. 2004. Cystatin m: a novel candidate tumor suppressor gene for breast cancer. Cancer Res 64:6957–6964. doi: 10.1158/0008-5472.CAN-04-0819. [DOI] [PubMed] [Google Scholar]
- 17.Shridhar R, Zhang J, Song J, Booth BA, Kevil CG, Sotiropoulou G, Sloane BF, Keppler D. 2004. Cystatin M suppresses the malignant phenotype of human MDA-MB-435S cells. Oncogene 23:2206–2215. doi: 10.1038/sj.onc.1207340. [DOI] [PubMed] [Google Scholar]
- 18.Kioulafa M, Balkouranidou I, Sotiropoulou G, Kaklamanis L, Mavroudis D, Georgoulias V, Lianidou ES. 2009. Methylation of cystatin M promoter is associated with unfavorable prognosis in operable breast cancer. Int J Cancer 125:2887–2892. doi: 10.1002/ijc.24686. [DOI] [PubMed] [Google Scholar]
- 19.Veena MS, Lee G, Keppler D, Mendonca M, Redpath JL, Standbridge EJ, Wilczynsky SP, Srivatasan ES. 2008. Inactivation of the cystatin E/M tumor suppressor gene in cervical cancer. Genes Chrom Cancer 47:740–754. doi: 10.1002/gcc.20576. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Pulukuri SM, Gorantla B, Knost JA, Rao JS. 2009. Frequent loss of cystatin E/M expression implicated in the progression of prostate cancer. Oncogene 28:2829–2838. doi: 10.1038/onc.2009.134. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Qiu J, Ai L, Ramachandran C, Yao B, Gopalakrishnan S, Fields CR, Delmas AL, Dyer LM, Melnick SJ, Yachnis AT, Schwartz PH, Fine HA, Brown KD, Robertson KD. 2008. Invasion suppressor cystatin E/M (CST6): high-level cell type-specific expression in normal brain and epigenetic silencing in gliomas. Lab Invest 88:910–925. doi: 10.1038/labinvest.2008.66. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Chen X, Cao X, Dong W, Xia M, Luo S, Fan Q, Xie J. 2010. Cystatin M expression is reduced in gastric carcinoma and is associated with promoter hypermethylation. Biochem Biophys Res Commun 391:1070–1074. doi: 10.1016/j.bbrc.2009.12.022. [DOI] [PubMed] [Google Scholar]
- 23.Rivenbark AG, Coleman WB. 2009. Epigenetic regulation of cystatins in cancer. Front Biosci (Landmark Ed) 14:453–462. [DOI] [PubMed] [Google Scholar]
- 24.Jin L, Zhang Y, Li H, Yao L, Fu D, Yao X, Xu LX, Hu X, Hu G. 2012. Differential secretome analysis reveals CST6 as a suppressor of breast cancer bone metastasis. Cell Res 22:1356–1373. doi: 10.1038/cr.2012.90. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Ko E, Park SE, Cho EY, Kim Y, Hwang JA, Lee YS, Nam SJ, Bang S, Park J, Kim DH. 2010. Cystatin M loss is associated with the losses of estrogen receptor, progesterone receptor, and HER4 in invasive breast cancer. Breast Cancer Res 12:R100. doi: 10.1186/bcr2783. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Hashimoto Y, Kondo C, Kojima T, Nagata H, Moriyama A, Hayakawa T, Katunuma N. 2006. Significance of 32-kDa cathepsin L secreted from cancer cells. Cancer Biother Radiopharm 21:217–224. doi: 10.1089/cbr.2006.21.217. [DOI] [PubMed] [Google Scholar]
- 27.Häckel CG, Krueger S, Grote HJ, Oshiro Y, Hodges S, Johnston DA, Johnson ME, Roessner A, Ayala AG, Czerniak B. 2000. Overexpression of cathepsin B and urokinase plasminogen activator is associated with increased risk of recurrence and metastasis in patients with chondrosarcoma. Cancer 89:995–1003. [PubMed] [Google Scholar]
- 28.Kruszewski WJ, Rzepko R, Wojtacki J, Skokowski J, Kopacz A, Jaœkiewicz K, Drucis K. 2004. Overexpression of cathepsin B correlates with angiogenesis in colon adenocarcinoma. Neoplasma 51:38–43. [PubMed] [Google Scholar]
- 29.Wei DH, Jia XY, Liu YH, Guo FX, Tang ZH, Li XH, Wang Z, Liu LS, Wang GX, Jian ZS, Ruan CG. 2013. Cathepsin L stimulates autophagy and inhibits apoptosis of ox-LDL-induced endothelial cells: potential role in atherosclerosis. Int J Mol Med 31:400–406. [DOI] [PubMed] [Google Scholar]
- 30.Lv BJ, Lindholt JS, Wang J, Cheng X, Shi GP. 2013. Plasma levels of cathepsins L, K, and V and risks of abdominal aortic aneurysms: a randomized population-based study. Atherosclerosis 230:100–105. doi: 10.1016/j.atherosclerosis.2013.05.018. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Potts W, Bowyer J, Jones H, Tucker D, Freemont AJ, Millest A, Martin C, Vernon W, Neerunjun D, Slynn G, Harper F, Maciewicz R. 2004. Cathepsin L-deficient mice exhibit abnormal skin and bone development and show increased resistance to osteoporosis following ovariectomy. Int J Exp Pathol 85:85–96. doi: 10.1111/j.0959-9673.2004.00373.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Stabuc B, Mrevlje Z, Markovic J, Stabuc-Silih M. 2006. Expression and prognostic significance of Cathepsin L in early cutaneous malignant melanoma. Neoplasma 53:259–262. [PubMed] [Google Scholar]
- 33.Cheng T, Hitomi K, Van Vlijmen-Willems IM, de Jongh GJ, Yamamoto K, Nishi K, Watts C, Reinheckel T, Schalkwijk J, Zeeuwen PL. 2006. Cystatin M is a high affinity inhibitor of cathepsin V and cathepsin L by a reactive site that is distinct from the legumain-binding site. A novel clue for the role of cystatin M in epidermal cornification. J Biol Chem 281:15893–15899. [DOI] [PubMed] [Google Scholar]
- 34.Andrews RM, Kubacka I, Chinnery PF, Lightowlers RN, Turnbull DM, Howell N. 1999. Reanalysis and revision of the Cambridge reference sequence for human mitochondrial DNA. Nat Genet 23:147. doi: 10.1038/13779. [DOI] [PubMed] [Google Scholar]
- 35.Kalous O, Conklin D, Desai AJ, O'Brien NA, Ginther C, Anderson L, Cohen DJ, Britten CD, Taylor I, Christensen JG, Slamon DJ, Finn RS. 2012. Dacomitinib (PF-00299804), an irreversible Pan-HER inhibitor, inhibits proliferation of HER2-amplified breast cancer cell lines resistant to trastuzumab and lapatinib. Mol Cancer Ther 11:1978–1987. doi: 10.1158/1535-7163.MCT-11-0730. [DOI] [PubMed] [Google Scholar]
- 36.Zarnegar BJ, Wang Y, Mahoney DJ, Dempsey PW, Cheung HH, He J, Shiba T, Yang X, Yeh WC, Mak TW, Korneluk RG, Cheng G. 2008. Noncanonical NF-kappaB activation requires coordinated assembly of a regulatory complex of the adaptors cIAP1, cIAP2, TRAF2 and TRAF3 and the kinase NIK. Nat Immunol 9:1371–1378. doi: 10.1038/ni.1676. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Trapnell C, Pachter L, Salzberg SL. 2009. TopHat: discovering splice junctions with RNA-Seq. Bioinformatics 25:1105–1111. doi: 10.1093/bioinformatics/btp120. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Anders S, Pyl TP, Huber W. 2015. HTSeq—a Python framework to work with high-throughput sequencing data. Bioinformatics 31:166–169. doi: 10.1093/bioinformatics/btu638. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Love MI, Huber W, Anders S. 2014. Moderated estimation of fold change and dispersion for RNA-Seq data with DESeq2. Genome Biol 15:550. doi: 10.1186/s13059-014-0550-8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Kim SG, Veena MS, Basak SK, Han E, Tajima T, Gjertson DW, Starr J, Eidelman O, Pollard HB, Srivastava M, Srivatsan ES, Wang MB. 2011. Curcumin treatment suppresses IKKβ kinase activity of salivary cells of patients with head and neck cancer: a pilot study. Clin Cancer Res 17:5953–5961. doi: 10.1158/1078-0432.CCR-11-1272. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Basak SK, Veena MS, Oh S, Lai C, Vangala S, Elashoff D, Fishbein MC, Sharma S, Rao NP, Rao D, Phan R, Srivatsan ES, Batra RK. 2013. The CD44high tumorigenic subsets in lung cancer biospecimens are enriched for low miR-34a expression. PLoS One 8:e73195. doi: 10.1371/journal.pone.0073195. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Duarte VM, Han E, Veena MS, Salvado A, Suh J, Liang L-J, Faull KF, Srivatsan ES, Wang MB. 2010. Curcumin enhances the effect of cisplatin in suppression of head and neck squamous cell carcinoma via inhibition of IKKβ protein of the nuclear factor kB pathway. Mol Cancer Ther 9:2665–2675. doi: 10.1158/1535-7163.MCT-10-0064. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Wood SL, Pernemalm M, Crosbie PA, Whetton AD. 2014. The role of the tumor-microenvironment in lung cancer-metastasis and its relationship to potential therapeutic targets. Cancer Treat Rev 40:558–566. [DOI] [PubMed] [Google Scholar]
- 44.Hecht M, von Metzler I, Sack K, Kaiser M, Sezer O. 2008. Interactions of myeloma cells with osteoclasts promote tumour expansion and bone degradation through activation of a complex signalling network and upregulation of cathepsin K, matrix metalloproteinases (MMPs) and urokinase plasminogen activator (uPA). Exp Cell Res 314:1082–1093. doi: 10.1016/j.yexcr.2007.10.021. [DOI] [PubMed] [Google Scholar]
- 45.Allen M, Louise Jones J. 2011. Jekyll and Hyde: the role of the microenvironment on the progression of cancer. J Pathol 223:162–176. [DOI] [PubMed] [Google Scholar]
- 46.Korkaya H, Liu S, Wicha MS. 2011. Breast cancer stem cells, cytokine networks, and the tumor microenvironment. J Clin Invest 121:3804–3809. doi: 10.1172/JCI57099. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Yu Y, Xiao CH, Tan LD, Wang QS, Li XQ, Feng YM. 2014. Cancer-associated fibroblasts induce epithelial-mesenchymal transition of breast cancer cells through paracrine TGF-β signalling. Br J Cancer 110:724–732. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Orjalo AV, Bhaumik D, Gengler BK, Scot GK, Campisi J. 2009. Cell surface-bound IL-1alpha is an upstream regulator of the senescence-associated IL-6/IL-8 cytokine network. Proc Natl Acad Sci U S A 106:17031–17036. doi: 10.1073/pnas.0905299106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Meng J, Sun B, Zhao X, Zhang D, Zhao X, Gu Q, Dong X, Zhao N, Liu P, Liu Y. 2014. Doxycycline as an inhibitor of the epithelial-to-mesenchymal transition and vasculogenic mimicry in hepatocellular carcinoma. Mol Cancer Ther 13:3107–3122. doi: 10.1158/1535-7163.MCT-13-1060. [DOI] [PubMed] [Google Scholar]
- 50.Schroeder MC, Chen C-L, Gajewski K, Halder G. 2013. A non-cell-autonomous tumor suppressor role for Stat in eliminating oncogenic scribble cells. Oncogene 32:4471–4479. doi: 10.1038/onc.2012.476. [DOI] [PubMed] [Google Scholar]
- 51.Yang G, Yang X. 2010. Smad4-mediated TGF-beta signaling in tumorigenesis. Int J Biol Sci 6:1–8. [DOI] [PMC free article] [PubMed] [Google Scholar]