

ABSTRACT
SltF was identified previously as an autolysin required for the assembly of flagella in the alphaproteobacteria, but the nature of its peptidoglycan lytic activity remained unknown. Sequence alignment analyses suggest that it could function as either a muramidase, lytic transglycosylase, or β-N-acetylglucosaminidase. Recombinant SltF from Rhodobacter sphaeroides was purified to apparent homogeneity, and it was demonstrated to function as a lytic transglycosylase based on enzymatic assays involving mass spectrometric analyses. Circular dichroism (CD) analysis determined that it is composed of 83.4% α-structure and 1.48% β-structure and thus is similar to family 1A lytic transglycosylases. However, alignment of apparent SltF homologs identified in the genome database defined a new subfamily of the family 1 lytic transglycosylases. SltF was demonstrated to be endo-acting, cleaving within chains of peptidoglycan, with optimal activity at pH 7.0. Its activity is modulated by two flagellar rod proteins, FlgB and FlgF: FlgB both stabilizes and stimulates SltF activity, while FlgF inhibits it. Invariant Glu57 was confirmed as the sole catalytic acid/base residue of SltF.
IMPORTANCE The bacterial flagellum is comprised of a basal body, hook, and helical filament, which are connected by a rod structure. With a diameter of approximately 4 nm, the rod is larger than the estimated pore size within the peptidoglycan sacculus, and hence its insertion requires the localized and controlled lysis of this essential cell wall component. In many beta- and gammaproteobacteria, this lysis is catalyzed by the β-N-acetylglucosaminidase domain of FlgJ. However, FlgJ of the alphaproteobacteria lacks this activity and instead it recruits a separate enzyme, SltF, for this purpose. In this study, we demonstrate that SltF functions as a newly identified class of lytic transglycosylases and that its autolytic activity is uniquely modulated by two rod proteins, FlgB and FlgF.
INTRODUCTION
The photosynthetic bacterium Rhodobacter sphaeroides is motile through the use of a single, subpolar flagellum (1). This locomotive organelle is tightly regulated and comprised of approximately 25 different proteins arranged into three major substructures: a basal body, hook, and thin helical filament. The basal body spans the bacterial cell envelope and contains the motor, a flagellum-specific type III export apparatus and at least four ring-like structures which are all connected by a filamentous rod (2). The rod assembles into a proximal rod that lies between the MS ring and the cell wall, which is composed of nine subunits of FliE (3) and six subunits of FlgB, FlgC, and FlgF (4), as well as a distal rod that is composed of 26 subunits of FlgG (5).
During flagellum assembly, extensive modifications need to occur to the peptidoglycan (PG) sacculus to accommodate the insertion of the secretion apparatus, as well as to stabilize the function of this system by acting as an assembly scaffold (6, 7). PG is a heteropolymer of glycan strands and peptide chains forming a rigid network (sacculus) that completely surrounds bacterial cells to maintain the integrity of their cytoplasmic membranes. The glycan strands are composed of repeating N-acetylglucosamine (GlcNAc) and N-acetylmuramic acid (MurNAc) residues linked β-(1→4), and they are cross-linked together through amide linkages between the stem peptides attached to the lactyl groups of MurNAc residues. Once formed, the PG sacculus is not a static structure as it requires constant biosynthesis and reinforcement to permit cellular growth and division. PG-degrading enzymes, such as muramidases, β-N-acetylglucosaminidases, and lytic transglycosylases (LTs), play an essential role in this biosynthesis and turnover (reviewed in reference 8). Muramidases (EC 3.2.1.17; lysozymes) have the same substrate specificity as LTs (EC 4.2.2.n1/n2); however, the reaction catalyzed by a lysozyme uses a water molecule to hydrolyze the glycosidic bond between MurNAc and GlcNAc, thereby generating a reducing MurNAc product. LTs, on the other hand, are not hydrolases as they lyse PG with the concomitant formation of an intramolecular 1,6-anhydromuramoyl reaction product (9). Another site of cleavage within the carbohydrate backbone of PG is the glycosidic linkage between GlcNAc and MurNAc residues catalyzed by β-N-acetylglucosaminidases (EC 3.2.1.14; chitinase) which generates a reducing GlcNAc product (10).
An important step in basal body formation is the penetration of the rod through the PG layer. At approximately 4 nm (11), the diameter of the rod is larger than the estimated pore size of the PG sacculus (12), and thus its insertion requires the localized and controlled lysis of PG strands. FlgJ of the flagellar operon has been shown to be required for the proper assembly of the flagellum. The FlgJs produced by the beta- and gammaproteobacteria, such as Salmonella enterica serovar Typhimurium, are bifunctional enzymes, possessing an N-terminal domain responsible for proper rod assembly (13–18) and a C-terminal domain possessing β-N-acetylglucosaminidase activity (6). Various alphaproteobacteria, including R. sphaeroides, possess FlgJ homologues that lack the C-terminal PG lytic domain. In R. sphaeroides, FlgJ has the same function as the N-terminal domain of FlgJ from S. enterica, which acts as a scaffolding rod-capping protein (19). It has been shown previously that the lack of FlgJ lytic activity is compensated for by SltF, a PG-lytic enzyme encoded within the flg operon; this enzyme is responsible for flagellar rod penetration in this bacterium (20). During basal body formation, SltF is exported to the periplasm via the Sec pathway, and it is recruited to the basal body near the cell pole by interacting with FlgJ (21).
A phylogenetic analysis (21) showed that SltF is weakly related to the family 1 LTs proposed by Blackburn and Clarke (22). However, two Glu residues were tentatively identified as being essential for catalytic activity, implying the enzyme may function as a hydrolase (21); LTs use a single catalytic acid/base residue (reviewed in reference 23). No biochemical analysis has been conducted on SltF, and consequently confusion exists as to whether this enzyme functions as a hydrolase (i.e., muramidase or β-N-acetylglucosaminidase) or an LT (20, 21).
In this study, we provide the first enzymatic characterization of the PG lytic activity of SltF from R. sphaeroides as the model enzyme of the alphaproteobacteria, a diverse order that includes intracellular pathogens such as species of Brucella. In contrast to the β-N-acetylglucosaminidase activity of FlgJ from S. Typhimurium (6), SltF functions as an LT. Furthermore, two proximal rod proteins, FlgB and FlgF, are found to regulate SltF activity.
MATERIALS AND METHODS
Chemicals and reagents.
DNase I, RNase A, pronase, isopropyl β-d-1-thiogalactopyranoside (IPTG), and EDTA-free protease inhibitor tablets were purchased from Roche Diagnostics (Laval, Quebec, Canada). Restriction and other DNA-modifying enzymes were acquired from Promega (Fitchburg, WI) or New England BioLabs (Ipswich, MA). Ni2+-nitrilotriacetic acid (NTA)-agarose was obtained from Qiagen (Valencia, CA), while Source 15Q resin was purchased from GE Healthcare (Piscataway, NJ). Fisher (Nepean, Ontario, Canada) provided acrylamide, glycerol, and Luria-Bertani (LB) growth medium. All other growth media were from Difco (Detroit, MI), and unless otherwise stated, all other reagents and chemicals were from Sigma-Aldrich (Oakville, Ontario, Canada).
Isolation and purification of PG.
Recognizing that the PG produced by R. sphaeroides as a Gram-negative bacterium would also be chemotype A1γ, insoluble PG for use in enzymatic assays was isolated from S. Typhimurium strain LT2 for convenience using the boiling SDS protocol and purified by enzyme treatment (amylase, DNase, RNase, and pronase) as described by Clarke (24).
Bacterial strains and growth.
The sources of plasmids and bacterial strains used in this study, together with their genotypic description are listed in Table 1. Escherichia coli strains M15(pREP4), TOP10, and BL21(DE3)(pLysS) were maintained on LB broth or agar at 37°C, supplemented with ampicillin (200 μg·ml−1) and kanamycin sulfate (50 μg·ml−1) or chloramphenicol (34 μg·ml−1) as appropriate. For gene overexpression studies and overproduction of protein, E. coli strains M15(pREP4) carrying pQE-30 and BL21(DE3)(pLysS) carrying pET-19b derivative vectors were grown in LB with agitation at ambient temperature and 37°C, respectively.
TABLE 1.
Bacterial strains and plasmids used in this study
| Strain or plasmid | Genotype or description | Source or reference |
|---|---|---|
| Strains | ||
| E. coli | ||
| BL21(λDE3)(pLysS) | F− ompT hsdSB(rB− mB−) gal dcm rne131 (DE3)(pLysS) (Cmr) | Qiagen |
| M15(pREP4) | thi Δlac Δara Δgal Δmtl F′ recA+ uvr+ lon+(pREP4) Kanr | Invitrogen |
| TOP10 | F− mcrA Δ(mrr-hsdRMS-mcrBC) ϕ80lacZΔM15 ΔlacX74 recA1 araD139 Δ(ara-leu)7697 galU galK rpsL (Strr) endA1 nupG | Invitrogen |
| R. sphaeroides WS8-N | Genome sequence strain | |
| Plasmids | ||
| pQE30 | IPTG-inducible T5 expression vector, N-terminal His6 tag, Ampr | Qiagen |
| pET19b | IPTG-inducible T7 expression vector, N-terminal His10 tag, Ampr | Novagen |
| pRS1SltF | pQE30 derivative containing sltF from WS8-N with N-terminal His6 tag on ScaI/HindIII fragment, Ampr | 21 |
| pRS1SltFGlu57Ala | pRS1SltF derivative encoding SltF with Glu57Ala replacement, Ampr | 21 |
| pRSFliE | pQE30 derivative containing fliE from WS8-N with N-terminal His6 tag on SacI/HindIII fragment, Ampr | 25 |
| pRSFlgB | pQE30 derivative containing flgB from WS8-N with N-terminal His6 tag on BamHI/HindIII fragment, Ampr | 25 |
| pRSFlgC | pET19b derivative containing flgC from WS8-N with N-terminal His10 tag on NdeI/BamHI fragment, Ampr | 25 |
| pRSFlgF | pQE30 derivative containing flgF from WS8-N with N-terminal His6 tag on KpmI/HindIII fragment, Ampr | 25 |
| pRSFlgG | pET19b derivative containing flgG from WS8-N with N-terminal His10 tag on NdeI/BamHI fragment, Ampr | 25 |
Overproduction and purification of SltF.
E. coli M15(pREP4) was transformed with the respective plasmids harboring genes encoding the wild-type (21) and the Glu57Ala (20) forms of SltF (Table 1), and both gene overexpression and isolation of overproduced proteins by immobilized metal affinity chromatography (IMAC) were conducted as described previously (20). Following IMAC on Ni2+-NTA-agarose and dialysis against 50 mM sodium phosphate buffer (pH 8.0) at 4°C, SltF and SltF(Glu57Ala) were further purified by anion-exchange chromatography on a Source 15Q column. Protein was loaded onto the column previously equilibrated with dialysis buffer and then recovered with the application of a linear gradient of 0 to 1 M NaCl over 60 min at a flow rate of 0.7 ml·min−1. Under these conditions, both forms of SltF eluted in ∼5 mM NaCl. Precaution was taken to use fresh chromatographic media for the respective purifications to avoid contamination of enzyme forms with each other. Both proteins were dialyzed exhaustively against 50 mM sodium phosphate buffer (pH 6.5) with 100 mM NaCl at 4°C.
Polishing size exclusion chromatography was performed on HiLoad 16/600 Superdex 200pg. Elution was performed in 50 mM sodium phosphate buffer (pH 6.5) with 100 mM NaCl at a flow rate of 0.5 ml·min−1.
Production and purification of rod proteins.
E. coli strains M15(pREP4), carrying pQE-30, and BL21(DE3)(pLysS), carrying pET-19b derivative vectors (Table 1), were inoculated into LB supplemented with ampicillin (200 μg·ml−1) and kanamycin sulfate (50 μg·ml−1) or chloramphenicol (34 μg·ml−1), respectively, and incubated at 37 °C until early exponential phase (optical density at 600 nm [OD600] of ∼0.6). Freshly prepared IPTG (isopropyl-β-d-thiogalactopyranoside) was added to final concentrations of 0.1 and 1 mM, and expression was induced for 3 h and 2 h, respectively, at 37°C. For E. coli strain BL21(DE3)(pLysS) carrying pRSFlgC, cells were incubated at 37°C until an OD600 of 0.8 was reached, and gene overexpression was induced with 1 mM IPTG for 1 h at 37°C.
FlgF and FlgG were purified under native conditions (25). Cells were harvested by centrifugation (10,000 × g, 10 min, 4°C) and then frozen at −20°C. Thawed cell pellets were resuspended in lysis buffer (20 mM Tris-HCl, pH 8.5) containing cOmplete Mini EDTA-free protease inhibitor mixture, 10 g·ml−1 RNase A, and 5 g·ml−1 DNase I and incubated on ice for 15 min prior to disruption with an Ultrasonic liquid processor (Heat Systems, Inc., Toronto, Canada) fitted with a macroprobe. The resulting cell lysate was clarified by centrifugation (5,000 × g, 10 min, 4°C) and the soluble cell fraction was mixed with Ni2+-NTA-agarose (500 μl·liter−1 starting culture) on a Nutator for 2 h at 4°C. The resin slurry was poured into a disposable column, and contaminating proteins were removed from the resin by being washed with 20 ml of lysis buffer followed by 10 ml of wash buffer (lysis buffer containing 20 mM imidazole). Purified FlgF and FlgG were eluted in 5 to 10 ml elution buffer (lysis buffer containing 250 mM imidazole) and then dialyzed at 4°C against 50 mM sodium phosphate buffer (pH 8.0).
FliE, FlgB, and FlgC were purified under denaturing conditions as previously described (25). For FliE and FlgB, cells were harvested and lysed as described above for FlgF and FlgG. FlgC is highly susceptible to cleavage by endogenous proteases; therefore disruption of the cells was performed in denaturing buffer (6 M guanidinium hydrochloride in 20 mM Tris-HCl, pH 8.5). Following incubation in denaturing buffer for 1 h at 4°C, residual insoluble material was removed by centrifugation (10,000 × g, 10 min, 4°C). Soluble FliE, FlgB, and FlgC in denaturing buffer were isolated and purified by IMAC on Ni2+-NTA-agarose as described above, except that the Tris-HCl buffer was replaced with denaturing buffer. FliE, FlgB, and FlgC were eluted in 5 to 10 ml elution buffer (denaturing buffer containing 250 mM imidazole). Proteins were refolded by dialysis against 20 mM Tris·HCl buffer (pH 8.5), containing 250 mM NaCl at 4°C. Prior to use of FlgB and FlgF in the turbidimetric assay, both proteins were exhaustively dialyzed against 50 mM sodium phosphate (pH 7.0) at 4°C.
Far-Western analysis of protein-protein interactions.
Far-Western analyses were conducted according to Hall (26). Each purified rod component protein (0.05 nmol) was subjected to 17.5% SDS-PAGE and then transferred to nitrocellulose membranes. Membranes containing immobilized rod component proteins were incubated in TTBS buffer (20 mM Tris·HCl, pH 7.5, 500 mM NaCl, 0.1% Tween 20), in the presence of exogenously added purified SltF (3 μg·ml−1, final concentration) for 1 h at ambient temperature. Membranes were washed three times with the same buffer, and polyclonal anti-SltF gamma globulins (20) were added at a 1:20,000 dilution. Detection was performed by immunoblotting (see “Other analytical techniques”).
PG-lytic activity assays. (i) Turbidimetry.
The turbidimetric assay of Hash (27) was used to monitor the time course of PG solubilization by SltF. Micrococcus luteus whole cells were suspended in 50 mM sodium phosphate buffer (pH 7.0) and sonicated briefly to provide homogenous suspensions. Enzyme samples were added to 100-μl aliquots of substrate suspension, and the decrease in turbidity of the reaction mixtures was monitored continuously at OD595 for 10 to 60 min. All reactions were repeated at least in triplicate, and specific activities ± standard deviations are reported.
(ii) [18O]H2O-based assay.
The [18O]H2O-based assay developed by Herlihey et al. (6) was adapted for the characterization of the reaction specificity of SltF. This permitted differentiation between hydrolytic and LT activities, as well as facilitating direct identification of any hydrolytic reaction products associated with both soluble and insoluble fractions of PG. Freeze-dried S. Typhimurium PG was suspended in [18O]H2O to a final concentration of 1.7 mg·ml−1 and briefly sonicated to provide homogenous suspensions. Reactions were initiated by the addition of enzyme followed by incubation at 37°C overnight with gentle shaking; they were stopped by rapid freezing. The muramidase mutanolysin served as a positive control. Reaction mixtures were thawed and solubilized reaction products were separated from insoluble PG by centrifugation (15,000 × g, 15 min, 4°C) prior to analysis by LC-Q-TOF-MS. The insoluble fraction was washed four to five times with 200-μl volumes of H2O and recovered each time by centrifugation (15,000 × g, 6 min, ambient temperature). The washed PG pellet was resuspended in 0.1 mM potassium phosphate buffer (pH 6.2) and solubilized by mutanolysin prior to liquid chromatography-quadrupole time of flight mass spectrometry (LC-Q-TOF-MS) analysis. To distinguish between linear and cross-linked oligosaccharides, the reaction products were reduced with 135 mM sodium borohydride for 30 min at ambient temperature prior to the LC-MS analyses.
Mass spectrometry.
All MS analyses were conducted using instruments at the Mass Spectrometry Facility of the Advanced Analysis Center, University of Guelph. LC-Q-TOF-MS was performed by injecting samples into an Agilent 1260 Infinity liquid chromatograph interfaced to an Agilent 6540 UHD Accurate Mass Q-TOF mass spectrometer. An AdvanceBio Peptide Map C18 2.1- by 100-mm chromatographic column (Agilent) was used for separation. The initial mobile phase was 2% acetonitrile in 0.1% formic acid, and elution of molecules was achieved after a 2-min wash with the application of a multistep gradient to 60% acetonitrile in 0.1% formic acid was applied over 38 min, followed by a linear gradient to 100% acetonitrile at 50 min. The flow rate was maintained at 0.2 ml·min−1 throughout. The mass spectrometer electrospray capillary voltage was maintained at 4.0 kV and the drying temperature at 350°C with a flow rate of 5 liters·min−1. Nebulizer pressure was 15 lb/in2 gauge. Nitrogen was used as nebulizing and drying gas as well as collision gas. The mass-to-charge ratio was scanned across the m/z range 300 to 2,000, and the tandem MS (MS/MS) mass range was scanned from 50 to 3,000 m/z in positive-ion auto-MS/MS mode. The auto-MS/MS mode was set up to fragment three precursor ions per cycle (1 spectrum/s) with collision energy set 2.5-eV offset and linearly increased up to 100 eV for m/z 3,000.
Dynamic light scattering.
Dynamic light scattering measurements were recorded with a Zetasizer Nano S DLS device (Malvern Instruments). Purified proteins (2 μM) in 50 mM sodium phosphate buffer (pH 8.0) with 100 mM NaCl were filtered through 0.22-μm-pore membranes prior to data collection. At least 11 valid readings were recorded for each temperature, with an increase of 1°C per measurement over a range of 25 to 90°C.
Other analytical techniques.
Identification of isoelectric points (pI) was performed using ProtParam (28). Integrated Microbial Genome (IMG) (29) searches for SltF homologs were performed using the amino acid sequence of R. sphaeroides WS8-N SltF as the probe. Analyses of sequence data were performed using the MUSCLE software (30), whereas both secondary and tertiary structure predictions were made using Phyre2 (31). Protein concentrations were determined using the Pierce bicinchoninic acid (BCA) protein assay kit, with bovine serum albumin (BSA) serving as the standard (Pierce Biotechnology, Rockford, IL). SDS-PAGE on 15% acrylamide gels was conducted by the method of Laemmli (32) with Coomassie brilliant blue staining. Western immunoblotting for the detection of SltF was performed using a 1:20,000 dilution of polyclonal anti-SltF gamma globulins (20) and a 1:10,000 dilution of the secondary antibody, donkey anti-rabbit IgG peroxidase-linked antibody (GE Healthcare Life Sciences). Detection was performed using SuperSignal West Pico (Thermo Scientific). Circular dichroism (CD) spectroscopy was performed as described previously (6), using a 0.1-cm path-length cell at an internal temperature of 25°C. The spectra were recorded as an average of four data accumulations, with a scan speed of 50 nm·min−1, bandwidth of 1 nm, 1 s, data pitch of 1 nm, and range of 190 to 250 nm. Analyses of the spectra were performed using the web server K2D3 (33).
RESULTS
In silico analysis of SltF.
Alignments of the amino acid sequence of R. sphaeroides SltF with known and hypothetical homologues present in the flagellar operons of alphaproteobacteria found in genome database and the IMG (29) led to the identification of four consensus motifs (Fig. 1 A; see Fig. S1 in the supplemental material). All of these motifs are located within the PG-lytic N-terminal domain of SltF (20). The putative catalytic residue of R. sphaeroides SltF, Glu57, comprises motif I. Despite minimal overall sequence similarity, this motif aligns with the ESGR signature motif of the family 1A and 1C LTs, such as Slt70, and MltE (EmtA) from E. coli (22), suggesting that SltF functions as an LT. If so, however, then it would represent a new subclass of the family 1 enzymes because its other consensus motifs are either unique or localized differently within the enzyme. Thus, for example, the GCMQ sequence of motif III, where the Cys (and Gly) is invariant (boldface), resembles the GLMQ of motif II of the family 1A and -E LTs and goose-type lysozymes. Similarly, the AAAAYHS sequence of SltF motif V resembles the AAYN of motif III of these LTs. Significantly, these motifs of the family 1 LTs are involved in substrate binding (34, 35). Nonetheless, the differences involving invariant residues are significant. Moreover, a closer analysis of the aligned SltF sequences permitted their separation into two distinct subsets, distinguished primarily on the presence in motif I of either a Ser or Thr residue immediately following the putative catalytic Glu. This separation revealed a close evolutionary relationship among the enzymes aligned but a clear divergence into the two consistently distinct groups, as depicted in Fig. 1.
FIG 1.

Consensus sequence motifs and predicted structure of SltF. (A) The five consensus motifs (labeled I to V) identified in the aligned apparent homologs of R. sphaeroides SltF (see Fig. S1 in the supplemental material) are separated into two subfamilies (green and blue boxes). Motifs I, III, and V align with consensus motifs I, II, and III of family 1A and 1C LTs (yellow boxes). Boldface lettering denotes greater than 80% identity among the respective aligned sequences, while letters in red are invariant. (B and C) The schematic and surface models of the predicted three-dimensional structure of R. sphaeroides SltF. Residues Asp29 to Ala169 were threaded by Phyre2 onto family 1A Slt70 from E. coli (PDB accession no. 1SLY). The putative catalytic residue Glu57 is in yellow, while residues Tyr152 and Tyr163 are in blue.
To date there is no crystallographic structure of SltF from R. sphaeriodes or its close homologs. As noted above, it and its homologs appear to be distinct from other PG lytic enzymes. Comparison to the structures known, R. sphaeroides SltF appears to be most similar to the LT domain of E. coli Slt70: its amino acid sequence is 17.8% identical and 24.9% similar. Recognizing this limited similarity, the structure of R. sphaeroides SltF involving residues Asp29 to Glu169 was predicted by Phyre using E. coli Slt70 (PDB accession no. 1SLY) as the template. The resulting structure consists of mainly α-helical folds (α, 66%; β, 2%), which are separated by a deep cleft that is proposed to contain the active site. Glu57 of motif I is located at the end of an α-helix, and it is positioned appropriately within the middle of the putative active site cleft to potentially serve as a catalytic acid/base (Fig. 1B and C). Two invariant Tyr residues, Tyr152 and Tyr163, present in motif V, are located near the proposed catalytic residue Glu57 and thus may participate in stabilizing the protonated charge state of the carboxylate group of Glu57 (34, 35).
Secondary structure of SltF.
Recombinant SltF (involving residues Ala27 to Pro265) was isolated and purified to apparent homogeneity, as determined by SDS-PAGE analysis, using a combination of affinity and anion-exchange chromatographies (see Fig. S2 in the supplemental material). The enzyme is subject to limited digestion in its noncatalytic, C-terminal domain, and so a polishing size exclusion chromatography on Superdex was performed when a single form of the enzyme was required. The overall yield of protein was similar to that reported previously (20, 21).
The secondary structure of SltF was determined by CD spectroscopy involving a constrained least-squares analysis. This analysis indicated the protein to be comprised of 83.4% α-structure and 1.48% β-structure (see Fig. S3 in the supplemental material). Recognizing that the recombinant protein is longer by an additional 96 C-terminal amino acids, these data are consistent with the structure predicted by Phyre and thus enhance confidence in the modeled structure.
Lytic activity of SltF.
The turbidimetric assay of Hash (27) was used to confirm the PG lytic activity of purified SltF with M. luteus whole cells as the substrate. As seen from the representative plot presented in Fig. 2, incubation of the whole cells with SltF led to the time-dependent loss of turbidity expected with the cleavage of the cell wall into soluble products by lytic enzymes. This loss of turbidity was continuous with time such that prolonged incubation led to eventual clearing of the cells, activity indicative of endo-acting lytic enzymes (23). The activity of SltF was very sensitive to slight pH changes, and the optimum was established to be pH 7.0 (Fig. 2). The specific activity of SltF at pH 7.0 was determined to be 60.0 ± 5.83 ΔA600 units·min−1·mg protein−1.
FIG 2.

Lytic activity of SltF. (A) SltF (2 μM) was incubated at ambient temperature with M. luteus cells suspended in 50 mM sodium phosphate buffer (pH 7.0) alone (●) or in the presence of 2 μM FlgB (■) or 2 μM FlgF (▲). Shown are representative curves for the progress of lysis as monitored turbidimetrically at OD595. ◆, 2 μM SltF(Glu57Ala) assayed in the absence of other added protein. Negative controls: ○, no enzyme added; □, FlgB; △, FlgF alone. (B) pH activity profile of SltF (error bars denote standard errors [SE]; n = 3).
Reaction specificity of SltF.
The heavy water assay previously developed by Herlihey et al. (6) was used to identify the type of lytic activity that SltF from R. sphaeroides catalyzes. S. Typhimurium PG in 50 mM sodium phosphate buffer (pH 7.0), prepared in [18O]H2O, was incubated with SltF (and mutanolysin as a positive control for hydrolytic activity). Following 16 h of reaction, soluble products were separated from insoluble material by centrifugation and subjected to LC-MS analysis. Digestion with mutanolysin led to complete solubilization of the PG and the typical production of a variety of muropeptide products (Fig. 3). MS/MS analysis of these revealed their enrichment with 18O, as expected from the introduction of [18O]H2O across the former β-(1→4) glycosidic linkages. The analysis of one of the major muropeptide products, GlcNAc-MurNAc-(tetrapeptide), is presented in Fig. 4 as an example of this protocol. With SltF, very few soluble products were detected (Fig. 3), and MS analysis indicated that most did not contain higher proportions of 18O than would be naturally present (Fig. 4). These data suggested that SltF does not function as a hydrolase. Tandem MS analysis of the SltF reaction products confirmed that the soluble muropeptides contained GlcNAc-1,6-anhydroMurNAc-(peptides), both cross-linked and un-cross-linked (Table 2), thus confirming that SltF indeed functions as an LT. However, a low proportion of hydrolytic products (viz. those containing 18O) were observed in the soluble reaction products of SltF (Table 2). This observation is not unexpected given the reported secondary (minor) muramidase-like activity of LTs, such as MltD, Slt70, MltF from E. coli (36), and CwlQ from Bacillus subtilis (37).
FIG 3.
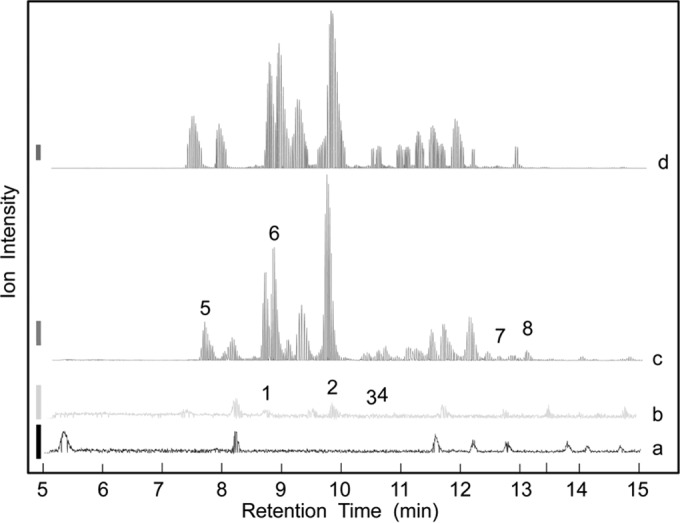
Characterization of SltF as an endo-lytic transglycosylase by LC-Q-TOF MS analysis of its reaction products. S. Typhimurium PG was suspended in 50 mM sodium phosphate buffer (pH 7.0) made with [18O]H2O to a final concentration of 1.76 mg·ml−1 and treated with either 4.0 μM SltF or 1.1 μM mutanolysin (positive control). After incubation at 30°C overnight, soluble reaction products were separated from insoluble material by centrifugation. The insoluble PG pellet from the SltF digestion was washed exhaustively with H2O and then resuspended in 0.1 mM potassium phosphate buffer (pH 6.2) for solubilization by 1.1 μM mutanolysin. The soluble muropeptides of this secondary digestion were recovered by centrifugation. Each fraction was subjected to LC-Q-TOF MS analysis. (a) PG control, no enzyme added; (b) SltF soluble fraction; (c) SltF insoluble fraction digested with mutanolysin; (d) mutanolysin control digestion. The identities of the numbered muropeptide fractions are listed in Table 2. The vertical bars to the left denote 20,000 (a and b) and 200,000 (c and d) intensity units.
FIG 4.

Tandem Q-TOF MS analysis of select muropeptides. Example of muropeptides recovered from (a) mutanolysin (positive control) and (b) soluble SltF digests of S. Typhimurium PG by LC-MS as described in the legend to Fig. 3. The muropeptides were subjected to tandem Q-TOF MS analysis (c and d, respectively). The blue spectral line in the MS spectrum of panel a denotes the 18O-containing isotope of the respective muropeptide. The monoisotopic masses (M + 2H)2+ are presented for each of the identified fragments.
TABLE 2.
ESI MS analysis of select muropeptides released from insoluble PG by SltF
| Fraction no.a | Annotationb |
m/z |
z | ||
|---|---|---|---|---|---|
| Observed | Expected | Δ | |||
| SltF soluble reaction products | |||||
| 1 | G-AnhM(Tetra) | 461.6960 | 461.6960 | 0 | 2 |
| 2 | G-[18O]M(Tetra) | 471.7054 | 471.7050 | −0.0004 | 2 |
| 3 | G-AnhM(Gly-Penta) | 490.2275 | 490.2015 | −0.0260 | 2 |
| 4 | G-AnhM(Tri) | 26.1818 | 426.1722 | −0.0096 | 2 |
| SltF insoluble reaction products | |||||
| 5 | G-AnhM(Tri) | 426.1779 | 426.1722 | −0.0057 | 2 |
| 6 | G-AnhM(Tetra) | 461.6961 | 461.6960 | −0.0001 | 2 |
| 7 | G-M(Tetra)-G-AnhM(Tetra)-(Tetra)M-G | 922.3848 | 922.3815 | −0.0033 | 3 |
| 7(R)c | G-M(Tetra)-G-AnhM(Tetra)-(Tetra)M-G | 923.0609 | 923.0533 | −0.0076 | 3 |
| 8 | G-M(Tetra)-(Tetra)AnhM-G | 922.3847 | 922.3815 | −0.0032 | 2 |
| 8(R) | G-M(Tetra)-(Tetra)AnhM-G | 923.3951 | 923.3895 | −0.0056 | 2 |
The muropeptide fractions correspond to those of the reverse-phase high-performance liquid chromatography (RP-HPLC) separation presented in Fig. 3.
Identification of each muropeptide was made by tandem Q-TOF-MS analysis of each parent ion (data not shown). G, GlcNAc; M, MurNAc; AnhM, 1,6-anhydroMurNAc; [18O]M, 18O-containing MurNAc; Tri, tripeptide l-Ala-d-Glu-DAP; Tetra, tetrapeptide l-Ala-d-Glu-DAP-d-Ala; Penta, pentapeptide l-Ala-d-Glu-DAP-d-Ala-d-Ala; Gly, glycine.
R denotes reduction of the sample with NaBH4 prior to analysis to quantify the number of reducing sugar ends (m/z of starting material + 1 = 1 reducing end).
The low concentration of muropeptides in the soluble fraction of SltF digests suggested endo-type activity. If so, lytic products would be either large and hence remain insoluble or still cross-linked to the remaining PG saccular material. To investigate if SltF functions as an endo-enzyme, the insoluble material from the above-mentioned digest of S. Typhimurium PG was washed extensively with Milli-Q H2O prior to a secondary solubilization by mutanolysin. With this treatment, any original hydrolytic products within the insoluble fraction would retain their 18O label, while muropeptides would be enriched with 1,6-anydroMurNAc if SltF indeed functions as an endo-LT. As seen in Fig. 3, the muropeptide profile of PG digested with SltF followed by mutanolysin (trace d) is distinct from the control reaction with mutanolysin alone (trace c). Tandem MS analysis of the SltF/mutanolysin digest of the insoluble reaction product revealed that, as with the soluble fraction described earlier, most of the muropeptides did not contain high proportions of 18O. Moreover, the majority of the reaction products were linear oligomeric sugars terminating with one anhydromuramoyl moiety. The product mixture exhibited multiple chromatographic peaks at m/z 922.3847, and positive-ion-mode electrospray ionization (ESI) generated ions with charged states of +1, +2, and +3, which correspond to neutral molecular masses of 921, 1,843, and 2,764 Da, respectively (Table 2). The m/z of 922.3847 with a charged state of +1 is consistent with GlcNAc-1,6-anhydroMurNAc-tetrapeptide, whereas the additional charged states of +2 and +3 are either linear oligosaccharides terminating in 1,6-anhydro-MurNAc or (GlcNAc-MurNAc-tetrapeptide)-(GlcNAc-1,6-anhydro-MurNAc-tetrapeptide) products cross-linked with (GlcNAc-MurNAc-tetrapeptide)n. To distinguish between the linear or cross-linked oligosaccharides, the reaction products were fragmented by tandem MS with or without prior reduction by sodium borohydride. These results indicated that SltF produces linear oligosaccharides terminating in 1,6-anhydro-MurNAc with a single cross-link (Table 2).
Identification of the catalytic residue(s) of SltF.
The mechanism of action of LTs involves a single acidic residue to be positioned in the active site cleft/pocket (reviewed in reference 23). In an earlier study, both Glu57 and Glu83 were identified as being potentially essential residues in R. sphaeroides SltF. Replacement of each led to a diminution of activity as determined by the semiquantitative methods of zymo-dot and lyso-plate analyses (20). Glu57 is an invariant residue among the aligned sequences of identified alphaproteobacterial SltFs (Fig. 1), and it comprises consensus motif I. In contrast, Glu83 is not conserved in other SltF homologues, and it would appear to be positioned to the side of the active site cleft and to be too far to serve in concert with Glu57. Based on this understanding of the Phyre model structure and alignment of SltF to family 1A/C LTs (21), Glu57 is proposed to function as the sole catalytic acid/base residue in SltF.
To confirm the participation of Glu57 in the catalytic mechanism of R. sphaeroides SltF, SltF(Glu57Ala) was purified to apparent homogeneity using the same protocol used for the wild-type enzyme (see Fig. S2 in the supplemental material), and CD spectroscopy was used to confirm that the secondary structure was not altered by the amino acid replacement. The resulting spectrum was found to be indistinguishable from that of the wild-type enzyme (see Fig. S3 in the supplemental material). SltF(Glu57Ala) was devoid of detectable activity using the turbidimetric assay (Fig. 2), where detection limits are 0.04 ΔA600 units·min−1 under the conditions employed, and no increase in 1,6-anhydromuro-peptide products was observed in reaction mixtures (data not shown).
Interaction of SltF with flagellar rod proteins.
A far-Western experiment was performed to investigate possible interactions between the rod component proteins FliE, FlgB, FlgC, FlgF, and FlgG on SltF and their effect, if any, on its activity. The respective recombinant proteins were overproduced and purified to apparent homogeneity by affinity chromatography. Whereas both recombinant FlgF and FlgG remained stable and therefore could be purified under native conditions, FliE, FlgB, and FlgC had to be denatured in guanidinium hydrochloride to prevent either proteolytic digestion or aggregation (31).
The far-Western experiment involved the loading and separation of the five proteins in an SDS-PAGE gel and then probing with SltF followed by detection of the latter with an anti-SltF antibody (20). Despite the overall structural similarities between these proteins as predicted earlier by in silico modeling based on amino acid sequence alignments (25), SltF appeared to bind to only the two proximal rod proteins FlgB (15 kDa) and FlgF (27 kDa) under the conditions used (Fig. 5). Both interactions were confirmed by reversing the far-Western experiment using the rod proteins as the probes (data not shown).
FIG 5.
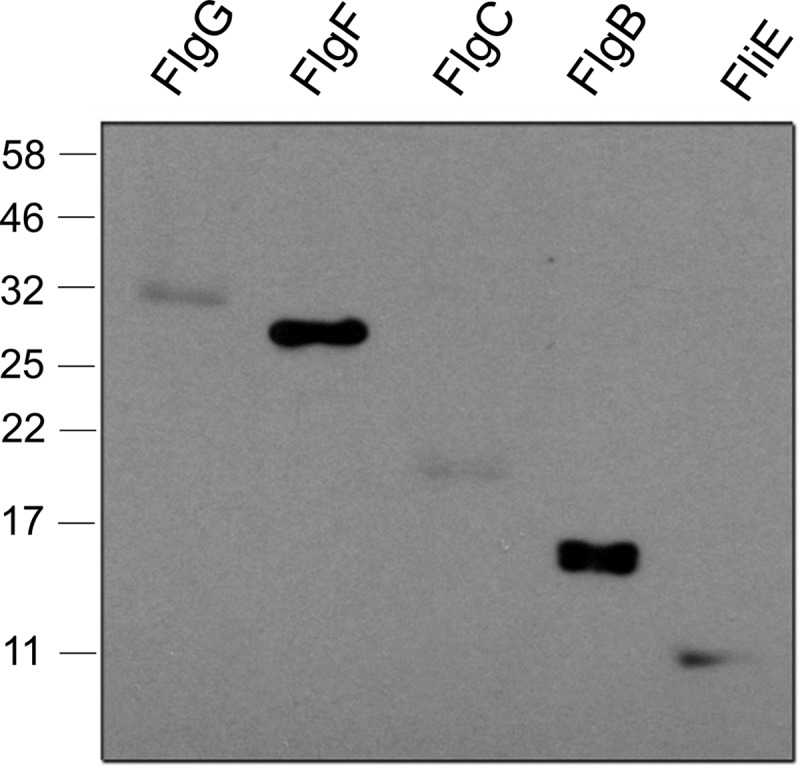
Interaction of SltF with flagellar rod proteins. Shown is a far-Western blot of flagellar rod proteins from R. sphaeroides using SltF as the probe. A total of 0.05 nmol of each purified rod protein was loaded, and following electrophoresis and blotting onto nitrocellulose, SltF was added to a final concentration of 3 μg·ml−1. The interaction was detected using a 1:20,000 dilution of anti-SltF gamma globulins.
The turbidimetric assay was used to determine if these interactions affect the PG-lytic activity of SltF. Enzyme (2 μM) was incubated for 10 min at ambient temperature in the absence and presence of equimolar concentrations of the respective proteins prior to assay using M. luteus whole cells as the substrate in 50 mM sodium phosphate buffer (pH 7.0). FlgF was observed to totally inhibit SltF activity, while, conversely, FlgB appeared to stimulate SltF lytic activity (Fig. 2). Thus, at 266 ± 27.4 ΔA600 units·min−1·mg protein−1, the specific activity of the SltF·FlgB complex was over 4-fold higher than that of the free enzyme. As a control, FlgB alone did not have any PG-lytic activity under the conditions employed. These results thus not only confirm the interactions detected but also suggest a regulatory mechanism for SltF's activity.
To determine if the protein-protein interaction with FlgB may have a stabilizing effect on SltF, equimolar concentrations of SltF and FlgB were incubated overnight at 4°C, and residual enzymatic activity was determined using the turbidimetric assay. SltF incubated alone under these conditions was totally devoid of lytic activity, whereas the activity of the SltF-FlgB complex (57.7 ± 6.73 ΔA600 units·min−1·mg protein−1) remained similar to that of freshly purified SltF when assayed alone. Thus, the inactivation of SltF was minimized when stored together with FlgB.
An additional experiment was performed to compare the stability of the SltF-FlgB complex to that of SltF alone using dynamic light scattering. The thermal aggregation temperature of SltF alone was 51°C, and this increased to 55°C with the SltF-FlgB complex. Taken together, these data suggest that the increase in SltF activity observed in the presence of FlgB may be a result of the stabilization of an active form of SltF by this proximal rod protein.
DISCUSSION
This study presents the first biochemical analysis of the enzymatic activity of SltF, an autolysin recruited for flagellar insertion in many alphaproteobacteria, including important bacterial pathogens such as species of Brucella. In beta- and gammaproteobacteria, penetration of flagellar rods through the cell wall is accomplished by the bifunctional enzyme FlgJ, which possesses a β-N-acetylglucosaminidase domain at its C terminus (13, 16). Based on our previous (20, 21) and current findings, we propose a different mechanism for rod penetration that occurs in alphaproteobacteria in which a specialized LT, SltF, is responsible for localized PG degradation. Moreover, we identified two flagellar rod proteins, FlgB and FlgF, with which SltF interacts directly, one of which both stabilizes the enzyme and stimulates its lytic activity.
Due to the technical limitations associated with studying PG-degrading enzymes, such as their limited solubility and the fact that their substrate PG is totally insoluble, the nature of the lytic activity of SltF had not been determined. With the lack of a soluble and defined substrate, previous researchers had based their assessment and interpretations of SltF activity on zymo-dot and lyso-plate assays coupled with amino acid sequence alignments and structural homologies (21). However, many of the PG lytic enzymes share the same lysozyme fold, which has led to assumptions of activity and/or misidentification. Such was the case for FlgJ of the beta- and gammaproteobacteria, where it was described as either a muramidase or LT. This issue was recently resolved with the development of a novel assay to differentiate between hydrolases and LTs (6). This assay permits the direct labeling (or not in the case of LTs) of reaction products with the stable 18O isotope from [18O]H2O coupled with MS and compositional analyses. Application of this assay to the characterization of FlgJ from S. Typhimurium led to the surprising discovery that it functions as neither a muramidase nor an LT, but rather as a β-N-acetylglucosaminidase (6). In the present study, the observed increase in production of linear oligomeric chains terminating in 1,6-anhydromuramic acid residues coupled with minimal introduction of 18O into the reaction products facilitated the identification of SltF as being an endo-acting LT.
The finding of endo activity is not unexpected given that the function of SltF is to generate a pore within the existing PG sacculus for the assembly of a single subpolar flagellum in R. sphaeroides (1). Nonetheless, most LTs characterized to date are exo-acting; the only other LTs demonstrated experimentally to be endo-acting are MltC (EmtA) from E. coli and MltD from Helicobacter pylori (reviewed in references 10 and 22). That a lytic enzyme would need to be recruited for this function is consistent with our understanding of PG metabolism around the poles of rod- and coccus-shaped bacteria. It is generally accepted that these regions of the bacterial sacculus contain inert PG and lytic enzymes would not be readily present (reviewed in references 8 and 38). The insertion of a flagellum would require a localized rearrangement of the PG layer involving endo-activity rather than the processive release of 1,6-anhydroMurNAc products that accompany its turnover during biosynthesis. This type of functional specificity involving the rearrangement of the sacculus has been ascribed to H. pylori MltD (39).
The consensus motifs identified in SltF and its known and predicted homologs loosely resembled those found in family 1 LTs. Likewise, the Phyre algorithm selected E. coli Slt70, a family 1A enzyme, as the best fit among the proteins in the Protein Structure Database for structural prediction. Notwithstanding the limitations of protein structure predictions based on the threading of amino acid sequences onto known tertiary structures (31), the catalytic domain of SltF is expected to adopt a lysozyme-like fold mostly composed of α-helices, a prediction supported by CD analysis. However, the distinct differences between the consensus motifs found in SltF homologs and those associated with other family 1 LTs indicate that SltF would be the prototype for a new subfamily, which we name family 1F. Moreover, this subfamily could be further subdivided based on whether a Ser or Thr follows the catalytic Glu in signature motif I. Indeed, separation of the aligned amino acid sequences on this basis revealed the close evolutionary relatedness of the hypothetical enzymes but their divergence into distinctive subsets, as reflected in each of the five consensus motifs.
The catalytic activity of autolysins is tightly regulated both temporally and spatially to prevent catastrophic autolysis (22, 40). With S. enterica, FlgJ is exported to the periplasm via the flagellum-specific type III export apparatus (13). Once the lytic domain of FlgJ has bored a hole in the PG, rod assembly continues through the wall and reaches the extracellular space. FlgJ remains associated with the growing rod, and consequently it is secreted extracellularly (41), thereby precluding any indiscriminate autolysis. R. sphaeroides SltF, on the other hand, is exported to the periplasm via the Sec pathway (21), where it was assumed to remain after rod completion. If left uncontrolled in the periplasm, continued random SltF activity would be deleterious to the bacterium. In this study, however, we found that SltF's activity is modulated by two proximal rod proteins, FlgB and FlgF. Thus, association with FlgB both stabilizes and enhances SltF activity, while FlgF inhibits it. FlgB, together with FliE, constitutes the most proximal portion of the rod (3, 42), which is consistent with enhancing SltF activity at the appropriate site for boring the required hole within the PG sacculus. Presumably once done, FlgF binds to preclude further lysis. That SltF forms complexes with both FlgB and FlgF would ensure that its localization remains fixed to the flagellum and not be rampant within the periplasm. This would be especially appropriate as the bacterium produces only a single subpolar flagellum, and hence no further endo-lytic activity of this form would be required.
At this juncture, it is not known whether or not the FlgB and FlgF homologs in S. Typhimurium and related species modulate the autolytic activity of their bifunctional FlgJ in the same manner. However, the use of physical association to control and modulate SltF activity is analogous to that observed with EtgA, a dedicated LT associated with the injectisome-type III secretion system (43). Indeed, the bacterial flagellum is evolutionarily related to this secretion apparatus as many proteins associated with both systems are conserved in sequence and function (44, 45). With EtgA from enteropathogenic E. coli, its localization is fixed (46) and its activity enhanced (47) by physical association with the inner rod protein EscI. The observation that R. sphaeroides SltF's activity is both fixed and enhanced by one structural protein and inhibited by another is novel.
Bacterial motility is widespread in nature and provides an evolutionary advantage. The assembly of the cell wall-spanning flagellum is tightly regulated, which results in a hierarchical pattern of gene expression (48). sltF is localized within the flgG operon (19), and homologues of SltF are widespread in flagellated alphaproteobacteria that lack a bimodular FlgJ. An exception among the alphaproteobacteria is Caulobacter crescentus, which appears to lack an SltF homolog. Instead, PleA is proposed to provide the requisite PG lytic activity for assembly of flagella and pili in this bacterium (49). PleA and SltF do not appear to be evolutionarily related as they share only 13.3% amino acid identity (20.7% similarity) and, unlike SltF, PleA is not encoded in a flagellar genetic context. Finally, and unfortunately, despite being referred to as an LT, it is not known whether PleA in fact functions as an LT, muramidase, or β-N-acetylglucosaminidase.
The observation that SltF is both endo-acting and stimulated by FlgB supports the earlier proposal that it is a specialized PG-degrading enzyme with a defined role in locally degrading the PG sacculus to permit the insertion of the flagellar rod (20). It is believed that the ancestral FlgJ contained only the single domain as found in alphaproteobacteria and that the bimodular FlgJ emerged in the common ancestor of beta- and gammaproteobacteria by fusing the PG-lytic domain to the C terminus of FlgJ (50).
The original organization of the LTs was based on the identification of homologs of the enzymes identified and characterized from E. coli and Pseudomonas aeruginosa (22). Thus, E. coli was thought originally to produce six LTs, each representing the prototype for the respective LT families. We discovered a seventh LT in E. coli (51), which became the prototype for family 1E, and more recently, an eighth has been reported (52). Each of these LTs is involved in the general biosynthesis of PG and cell division. However, it is becoming apparent that LTs are even more prevalent within bacteria as homologs and paralogs appear to be produced for specific tasks, such as the SltF for flagellum insertion described in this study. Indeed, the interaction of specialized LTs with components of secretion apparatuses appears to be a common theme, as others, such as EtgA from the injectisome type III secretion system (46, 47) and VirB from the type IV secretion system (53), have been described. Such apparent redundancy reflects their important role for cell growth and survival, thus making them an attractive new target for the discovery of novel antibiotics (23). A compound that inhibits the mechanism of action of these enzymes would indiscriminately block a variety of LT-catalyzed physiological events in a bacterium which may prove to be as effective as the β-lactam antibiotics, compounds that inhibit each of the redundant penicillin-binding proteins of a bacterium with lethal effect. However, progress toward this end will only occur with the development of a facile assay for this interesting and unique class of bacterial enzymes.
Supplementary Material
ACKNOWLEDGMENTS
We thank Dyanne Brewer and Armen Charchoglyan of the Mass Spectrometry Facility (Advanced Analysis Centre, University of Guelph) for expert technical assistance and advice and both Javier de la Mora and Teresa Ballado (Instituto de Fisiología Celular) for technical assistance.
Footnotes
Supplemental material for this article may be found at http://dx.doi.org/10.1128/JB.00203-16.
REFERENCES
- 1.Armitage JP, Macnab RM. 1987. Unidirectional, intermittent rotation of the flagellum of Rhodobacter sphaeroides. J Bacteriol 169:514–518. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Young GM, Schmiel DH, Miller VL. 1999. A new pathway for the secretion of virulence factors by bacteria: the flagellar export apparatus functions as a protein-secretion system. Proc Natl Acad Sci U S A 96:6456–6461. doi: 10.1073/pnas.96.11.6456. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Müller V, Jones CJ, Kawagishi I, Aizawa S, Macnab RM. 1992. Characterization of the fliE genes of Escherichia coli and Salmonella typhimurium and identification of the FliE protein as a component of the flagellar hook-basal body complex. J Bacteriol 17:2298–2304. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Jones CJ, Macnab RM, Okino H, Aizawa S. 1990. Stoichiometric analysis of the flagellar hook-(basal-body) complex of Salmonella typhimurium. J Mol Biol 212:377–387. doi: 10.1016/0022-2836(90)90132-6. [DOI] [PubMed] [Google Scholar]
- 5.Okino H, Isomura M, Yamaguchi S, Magariyama Y, Kudo S, Aizawa SI. 1989. Release of flagellar filament-hook-rod complex by a Salmonella typhimurium mutant defective in the M ring of the basal body. J Bacteriol 171:2075–2082. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Herlihey FA, Moynihan PJ, Clarke AJ. 2014. The essential protein for bacterial flagella formation FlgJ functions as a β-N-acetylglucosaminidase. J Biol Chem 289:31029–31042. doi: 10.1074/jbc.M114.603944. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Roure S, Bonis M, Chaput C, Ecobichon C, Mattox A, Barrière C, Geldmacher N, Guadagnini S, Schmitt C, Prévost MC, Labigne A, Backert S, Ferrero RL, Boneca IG. 2012. Peptidoglycan maturation enzymes affect flagellar functionality in bacteria. Mol Microbiol 86:845–856. doi: 10.1111/mmi.12019. [DOI] [PubMed] [Google Scholar]
- 8.Vollmer W, Bertsche U. 2008. Murein (peptidoglycan) structure, architecture and biosynthesis in Escherichia coli. Biochim Biophys Acta 1778:1714–1734. doi: 10.1016/j.bbamem.2007.06.007. [DOI] [PubMed] [Google Scholar]
- 9.Höltje JV, Mirelman D, Sharon N, Schwarz U. 1975. Novel type of murein transglycosylase in Escherichia coli. J Bacteriol 124:1067–1076. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Vollmer W, Joris B, Charlier P, Foster S. 2008. Bacterial peptidoglycan (murein) hydrolases. FEMS Microbiol Rev 32:259–286. doi: 10.1111/j.1574-6976.2007.00099.x. [DOI] [PubMed] [Google Scholar]
- 11.Suzuki H, Yonekura K, Murata K, Hirai T, Oosawa K, Namba K. 1998. A structural feature in the central channel of the bacterial flagellar FliF ring complex is implicated in type III protein export. J Struct Biol 124:104–114. doi: 10.1006/jsbi.1998.4048. [DOI] [PubMed] [Google Scholar]
- 12.Demchick P, Koch AL. 1996. The permeability of the wall fabric of Escherichia coli and Bacillus subtilis. J Bacteriol 178:768–773. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Nambu T, Minamino T, Macnab RM, Kutsukake K. 1999. Peptidoglycan-hydrolyzing activity of the FlgJ protein, essential for flagellar rod formation in Salmonella typhimurium. J Bacteriol 181:1555–1561. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Dijkstra AJ, Keck W. 1996. Peptidoglycan as a barrier to transenvelope transport. J Bacteriol 178:5555–5562. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Das M, Chopra AK, Wood T, Peterson JW. 1998. Cloning, sequencing and expression of the flagellin core protein and other genes encoding structural proteins of the Vibrio cholerae flagellum. FEMS Microbiol Lett 165:239–246. [DOI] [PubMed] [Google Scholar]
- 16.Hirano T, Minamino T, Macnab RM. 2001. The role in flagellar rod assembly of the N-terminal domain of Salmonella FlgJ, a flagellum specific muramidase. J Mol Biol 312:359–369. doi: 10.1006/jmbi.2001.4963. [DOI] [PubMed] [Google Scholar]
- 17.Hashimoto W, Ochiai A, Momma K, Itoh T, Mikami B, Maruyama Y, Murata K. 2009. Crystal structure of the glycosidase family 73 peptidoglycan hydrolase FlgJ. Biochem Biophys Res Commun 381:16–21. doi: 10.1016/j.bbrc.2009.01.186. [DOI] [PubMed] [Google Scholar]
- 18.Maruyama Y, Ochiai A, Itoh T, Mikami B, Hashimoto W, Murata K. 2010. Mutational studies of the peptidoglycan hydrolase FlgJ of Sphingomonas sp. strain A1. J Basic Microbiol 50:311–317. doi: 10.1002/jobm.200900249. [DOI] [PubMed] [Google Scholar]
- 19.González-Pedrajo B, de la Mora J, Ballado T, Camarena L, Dreyfus G. 2002. Characterization of the flgG operon of Rhodobacter sphaeroides WS8 and its role in flagellum biosynthesis. Biochim Biophys Acta 1579:55–63. doi: 10.1016/S0167-4781(02)00504-3. [DOI] [PubMed] [Google Scholar]
- 20.de la Mora J, Ballado T, González-Pedrajo B, Camarena L, Dreyfus G. 2007. The flagellar muramidase from the photosynthetic bacterium Rhodobacter sphaeroides. J Bacteriol 189:7998–8004. doi: 10.1128/JB.01073-07. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.de la Mora J, Osorio-Valeriano M, González-Pedrajo B, Ballado T, Camarena L, Dreyfus G. 2012. The C terminus of the flagellar muramidase SltF modulates the interaction with FlgJ in Rhodobacter sphaeroides. J Bacteriol 194:4513–4520. doi: 10.1128/JB.00460-12. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Blackburn NT, Clarke AJ. 2001. Identification of four families of peptidoglycan lytic transglycosylases. J Mol Evol 52:78–84. doi: 10.1007/s002390010136. [DOI] [PubMed] [Google Scholar]
- 23.Scheurwater E, Reid CW, Clarke AJ. 2008. Lytic transglycosylases: bacterial space-making autolysins. Int J Biochem Cell Biol 40:586–591. doi: 10.1016/j.biocel.2007.03.018. [DOI] [PubMed] [Google Scholar]
- 24.Clarke AJ. 1993. Compositional analysis of peptidoglycan by high performance anion-exchange chromatography. Anal Biochem 212:344–350. doi: 10.1006/abio.1993.1339. [DOI] [PubMed] [Google Scholar]
- 25.Osorio-Valeriano M, de la Mora J, Camarena L, Dreyfus G. 2015. Biochemical characterization of the flagellar rod components of Rhodobacter sphaeroides: properties and interactions. J Bacteriol 198:544–552. doi: 10.1128/JB.00836-15. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Hall RA. 2004. Studying protein-protein interactions via blot overlay or far Western blot. Methods Mol Biol 261:167–174. [DOI] [PubMed] [Google Scholar]
- 27.Hash JH. 1967. Measurement of bacteriolytic enzymes. J Bacteriol 93:1201–1202. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Gasteiger E, Hoogland C, Gattiker A, Duvaud S, Wilkins MR, Appel RD, Bairoch A. 2005. Protein identification and analysis tools on the ExPASy server, p 571–607. In Walker JM. (ed), The proteomics protocols handbook. Humana Press, Totowa, NJ. [Google Scholar]
- 29.Markowitz VM, Chen IM, Palaniappan K, Chu K, Szeto E, Pillay M, Ratner A, Huang J, Woyke T, Huntemann M, Anderson I, Billis K, Varghese N, Mavromatis K, Pati A, Ivanova NN, Kyrpides NC. 2014. IMG 4 version of the Integrated Microbial Genomes comparative analysis system. Nucleic Acids Res 42(Database issue):D560–D567. doi: 10.1093/nar/gkt963. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Edgar RC. 2004. MUSCLE: multiple sequence alignment with high accuracy and high throughput. Nucleic Acids Res 32:1792–1797. doi: 10.1093/nar/gkh340. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Bennett-Lovsey RM, Herbert AD, Sternberg MJ, Kelley LA. 2008. Exploring the extremes of sequence/structure space with ensemble fold recognition in the program Phyre. Proteins 70:611–625. [DOI] [PubMed] [Google Scholar]
- 32.Laemmli UK. 1970. Cleavage of structural proteins during the assembly of the head of bacteriophage T4. Nature 227:680–685. doi: 10.1038/227680a0. [DOI] [PubMed] [Google Scholar]
- 33.Louis-Jeune C, Andrade-Navarro MA, Perez-Iratxeta C. 2012. Prediction of protein secondary structure from circular dichroism using theoretically derived spectra. Proteins 80:374–381. doi: 10.1002/prot.23188. [DOI] [PubMed] [Google Scholar]
- 34.van Asselt EJ, Thunnissen AM, Dijkstra BW. 1999. High resolution crystal structures of the Escherichia coli lytic transglycosylase Slt70 and its complex with a peptidoglycan fragment. J Mol Biol 291:877–898. doi: 10.1006/jmbi.1999.3013. [DOI] [PubMed] [Google Scholar]
- 35.van Asselt EJ, Kalk KH, Dijkstra BW. 2000. Crystallographic studies of the interactions of Escherichia coli lytic transglycosylase Slt35 with peptidoglycan. Biochemistry 39:1924–1934. doi: 10.1021/bi992161p. [DOI] [PubMed] [Google Scholar]
- 36.Lee M, Hesek D, Llarrull LI, Lastochkin E, Boggess PHB, Mobashery S. 2013. Reactions of all E. coli lytic transglycosylases with bacterial cell wall. J Am Chem Soc 135:3311–3314. doi: 10.1021/ja309036q. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Sudiarta IP, Fukushima T, Sekiguchi J. 2010. Bacillus subtilis CwlQ (previously YjbJ) is a bifunctional enzyme exhibiting muramidase and soluble-lytic transglycosylase activities. Biochem Biophys Res Commun 398:606–612. doi: 10.1016/j.bbrc.2010.07.001. [DOI] [PubMed] [Google Scholar]
- 38.Dworkin J. 2009. Cellular polarity in prokaryotic organisms. Cold Spring Harb Perspect Biol 1:a003368. doi: 10.1101/cshperspect.a003368. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Chaput C, Labigne A, Boneca IG. 2007. Characterization of Helicobacter pylori lytic transglycosylases Slt and MltD. J Bacteriol 189:422–429. doi: 10.1128/JB.01270-06. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Pfeffer JM, Moynihan PJ, Clarke CA, Clarke AJ. 2012. Control of lytic transglycosylase activity within bacterial cell walls, p 55–68. In Reid CW, Twine SM, Read AN (ed) Bacterial glycomics. Caister Academic Press, Norfolk, United Kingdom. [Google Scholar]
- 41.Cohen EJ, Hughes KT. 2014. Rod-to-hook transition for extracellular flagellum assembly is catalyzed by the L-ring dependent rod scaffold removal. J Bacteriol 196:2387–2395. doi: 10.1128/JB.01580-14. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Minamino T, Yamaguchi S, Macnab RM. 2000. Interaction between FliE and FlgB, a proximal rod component of the flagellar basal body of Salmonella. J Bacteriol 182:3029–3036. doi: 10.1128/JB.182.11.3029-3036.2000. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.García-Gómez E, Espinosa N, de la Mora J, Dreyfus G, González-Pedrajo B. 2011. The muramidase EtgA from enteropathogenic Escherichia coli is required for efficient type III secretion. Microbiology 157:1145–1160. doi: 10.1099/mic.0.045617-0. [DOI] [PubMed] [Google Scholar]
- 44.Erhardt M, Namba K, Hughes KT. 2010. Bacterial nanomachines: the flagellum and type III injectisome. Cold Spring Harb Perspect Biol 2:a000299. doi: 10.1101/cshperspect.a000299. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Kawamoto A, Morimoto YV, Miyata T, Minamino T, Hughes KT, Kato T, Namba K. 2013. Common and distinct structural features of Salmonella injectisome and flagellar basal body. Sci Rep 3:3369. doi: 10.1038/srep03369. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Creasey EA, Delahay RM, Daniell SJ, Frankel G. 2003. Yeast two-hybrid system survey of interactions between LEE-encoded proteins of enteropathogenic Escherichia coli. Microbiology 149:2093–2106. doi: 10.1099/mic.0.26355-0. [DOI] [PubMed] [Google Scholar]
- 47.Burkinshaw BJ, Deng W, Lameignère E, Wasney GA, Zhu H, Worrall LJ, Finlay BB, Strynadka NC. 2015. Structural analysis of a specialized type III secretion system peptidoglycan-cleaving enzyme. J Biol Chem 290:10406–10417. doi: 10.1074/jbc.M115.639013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Poggio S, Osorio A, Dreyfus G, Camarena L. 2005. The flagellar hierarchy of Rhodobacter sphaeroides is controlled by the concerted action of two enhancer-binding proteins. Mol Microbiol 58:969–983. doi: 10.1111/j.1365-2958.2005.04900.x. [DOI] [PubMed] [Google Scholar]
- 49.Viollier PH, Shapiro L. 2003. A lytic transglycosylase homologue, PleA, is required for the assembly of pili and the flagellum at the Caulobacter crescentus cell pole. Mol Microbiol 49:331–345. doi: 10.1046/j.1365-2958.2003.03576.x. [DOI] [PubMed] [Google Scholar]
- 50.Nambu T, Inagaki Y, Kutsukake K. 2006. Plasticity of the domain structure in FlgJ, a bacterial protein involved in flagellar rod formation. Genes Genet Syst 81:381–389. doi: 10.1266/ggs.81.381. [DOI] [PubMed] [Google Scholar]
- 51.Scheurwater EM, Clarke AJ. 2008. The C-terminal domain of Escherichia coli YfhD functions as a lytic transglycosylase. J Biol Chem 283:8363–8373. doi: 10.1074/jbc.M710135200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Yunck R, Cho C, Bernhardt TG. 2016. Identification of MltG as a potential terminase for peptidoglycan polymerization in bacteria. Mol Microbiol 99:700–718. doi: 10.1111/mmi.13258. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Höppner C, Carle A, Sivanesan D, Hoeppner S, Baron C. 2005. The putative lytic transglycosylase VirB from Brucella suis interacts with the type IV secretion system core components VirB8, VirB9 and VirB11. Microbiology 151:3469–3482. doi: 10.1099/mic.0.28326-0. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.