

Abstract
Streptococcus pneumoniae commonly inhabits the nasopharynx as a member of the commensal biofilm. Infection with respiratory viruses, such as influenza A virus, induces commensal S. pneumoniae to disseminate beyond the nasopharynx and to elicit severe infections of the middle ears, lungs, and blood that are associated with high rates of morbidity and mortality. Current preventive strategies, including the polysaccharide conjugate vaccines, aim to eliminate asymptomatic carriage with vaccine-type pneumococci. However, this has resulted in serotype replacement with, so far, less fit pneumococcal strains, which has changed the nasopharyngeal flora, opening the niche for entry of other virulent pathogens (e.g., Streptococcus pyogenes, Staphylococcus aureus, and potentially Haemophilus influenzae). The long-term effects of these changes are unknown. Here, we present an attractive, alternative preventive approach where we subvert virus-induced pneumococcal disease without interfering with commensal colonization, thus specifically targeting disease-causing organisms. In that regard, pneumococcal surface protein A (PspA), a major surface protein of pneumococci, is a promising vaccine target. Intradermal (i.d.) immunization of mice with recombinant PspA in combination with LT-IIb(T13I), a novel i.d. adjuvant of the type II heat-labile enterotoxin family, elicited strong systemic PspA-specific IgG responses without inducing mucosal anti-PspA IgA responses. This response protected mice from otitis media, pneumonia, and septicemia and averted the cytokine storm associated with septic infection but had no effect on asymptomatic colonization. Our results firmly demonstrated that this immunization strategy against virally induced pneumococcal disease can be conferred without disturbing the desirable preexisting commensal colonization of the nasopharynx.
INTRODUCTION
Upper respiratory tract infections are often polymicrobial in nature, where infections with respiratory viruses are highly associated with concomitant bacterial infection with organisms residing in the nasopharynx (1). One of the most important of these biological pairings is the interaction between influenza A virus (IAV) and Streptococcus pneumoniae (the pneumococcus) (2). In children, the most frequent complication following IAV infections is acute otitis media, of which S. pneumoniae is one of the most common etiologic agents (3–5). S. pneumoniae is also the most frequently identified pathogen in hospitalized children and adults who present with primary or secondary community-acquired pneumonia (6). Thirty-three percent of all episodes of pneumonia and 54% of all viral coinfections are associated with infections by S. pneumoniae (7). In these cases, the principal viral contributor is IAV. During typical influenza seasons, superinfections with S. pneumoniae are the primary causes of mortality (8).
A variety of models have been proposed to explain the mechanisms by which IAV infections enhance pneumococcal colonization, promote invasion of the respiratory tract, and alter host responses to bacterial infection (8). Until recently, the synergistic effect between pneumococci and IAV was assumed to be unidirectional. New studies, however, indicate that host signals in response to viral infection stimulate dispersal of pneumococci from harmless nasopharyngeal biofilms that disseminate to otherwise sterile tissues where the bacterium is more pathogenic (9). This virus-dependent dispersal of pneumococci from the biofilm is the initial step in promulgating invasive disease and produces a phenotypic shift in the bacterium that promotes survival and pathogenicity in other host niches, a phenotypic shift that is dramatically different from that of broth-grown pneumococci commonly used for infection experiments (10).
To control for pneumococcal infections and to reduce the prevalence of virally induced pneumococcal invasive disease, the pneumococcal conjugate vaccine (PCV) was developed and licensed in 2000. The initial PCV protected against seven (PCV-7) of the most common pneumococcal serotypes. In 2010, two new PCV formulations were released that conferred protection against an additional three (PCV-10) or six (PCV-13) serotypes emerging in the population as a result of serotype replacement or more commonly caused disease in low- and middle-income countries. Yet the large number of pneumococcal serotypes (currently 96) and the poor immunological responses against most nonvaccine serotypes inherently limit the utility of these vaccines in broadly protecting against disease (11). There is a critical need, therefore, to identify novel strategies and immunogens to expand coverage and to improve vaccine efficacy, especially in older and younger populations who are at highest risk. Potential strategies include the use of conserved protein antigens as vaccine targets and employing alternative routes of vaccine delivery (12).
Among the leading vaccine candidates currently under consideration is PspA, the pneumococcal surface protein A (13). PspA is a choline-binding surface protein that inhibits complement-mediated phagocytosis (14). Furthermore, PspA binds to and prevents killing of the pneumococci by binding lactoferrin (15). PspA is an important virulence factor that is expressed by all clinical pneumococcal isolates and is essential for full virulence during local and invasive disease although its role during colonization is less clear (16–19). Other studies have revealed that PspA, when employed as an immunogen in mouse models, protects against primary pneumococcal infection (20–23) and against pneumococcal challenge subsequent to a viral infection (24, 25). The vast majority of these investigations, however, examined the protection offered by PspA immunization against invasive disease caused by challenge with planktonic, broth-grown pneumococci, which do not represent either the biofilm community of S. pneumoniae normally found residing in the nasopharynx prior to contact with IAV or the bacteria released from biofilms in response to virus infection (4). Thus, the capacity of PspA, when used as an immunogen, to protect against invasive disease produced by pneumococci that are residing in a commensal nasopharyngeal biofilm subsequent to infection with IAV and its protective role against pneumonia and otitis media have not been adequately evaluated.
There is a growing literature that supports the model that, in the nasopharynx, S. pneumoniae is a harmless commensal bacterium which by “competitive exclusion” inhibits colonization by other harmful potential pathogens, such as Streptococcus pyogenes and Staphylococcus aureus (26, 27), that are more prevalent in children immunized with the conjugate vaccines. Although cocolonization of S. pneumoniae and Haemophilus influenzae is common, it is unclear whether the observed pneumococcal serotype replacement in the nasopharynx results in an increased bacterial burden of H. influenzae (27, 28) or not (26, 29). Niche exclusion has been described in other systems. Intestinal disease caused by Clostridium spp. has been attributed to invasion of the pathogen into vacated niches produced by broad-spectrum antibiotics (1, 11); there is an inverse relationship between the presence of commensal Lactobacillus spp. and pathogenic Escherichia coli in the likelihood of inducing recurrent urogenital tract infections (30); and Neisseria lactamica effectively excludes colonization of the oropharynx by Neisseria meningitidis (31).
In this study, we investigated the capacity of PspA to protect against the development of pneumococcal disease from virulent pneumococci dispersed from colonizing biofilms following IAV infection. Intradermal (i.d.) immunization with PspA and LT-IIb(T13I), a novel detoxified i.d. adjuvant (32), elicited a strong, systemic antigen (Ag)-specific immune response and protected mice against development of secondary otitis media, pneumonia, and associated bacteremia. These effects were obtained without disrupting stable and asymptomatic biofilm colonization by the pneumococci. Additionally, this immunization strategy protected against sepsis challenge and significantly attenuated the cytokine storm that is commonly observed with disseminated pneumococcal infection. Thus, inducing protection against virally induced invasive pneumococcal disease without disrupting desirable asymptomatic nasopharyngeal colonization is an intriguing possibility with important clinical implications.
MATERIALS AND METHODS
Ethics statement.
Experiments were performed in strict accordance with the recommendations in the Guide for the Care and Use of Laboratory Animals of the National Institutes of Health (33). The protocols were approved by the Institutional Animal Care and Use Committee at the University at Buffalo, Buffalo, NY, USA. All bacterial inoculations and treatments were performed under conditions to minimize any potential suffering of the animals.
Chemicals and reagents.
Recombinant PspA fragment UAB055 consisting of the N-terminal 302 amino acids of PspA from the unencapsulated strain Rx1, a derivative of the type 2 encapsulated strain D39, and LT-IIb(T13I) were purified using previously described methods (34, 35). All preparations were determined to be essentially free of lipopolysaccharide (<0.03 ng/μg of protein) (Limulus amebocyte assay kit, Endosafe system; Charles River Laboratories, Charleston, SC). Cell culture reagents were obtained from Invitrogen, Carlsbad, CA. Bacterial and cell culture media and reagents were purchased from VWR, Inc., Radnor, PA. Chemically defined bacterial growth medium (CDM) was obtained from JRH Biosciences, Lexera, KS. Sheep blood was purchased from BioLink, Inc., Liverpool, NY. Remaining reagents were obtained from Sigma-Aldrich, St. Louis, MO.
Bacterial strains.
Pneumococcal strains were grown in chemically defined medium (CDM) or in Todd-Hewitt medium containing 0.5% yeast extract (THY) as described previously (9). This study used the serotype 19F strain EF3030 (36), the serotype 2 strain D39 (37), and strain JY53, an isogenic mutant of D39 lacking PspA (D39-PspA null) (38). Pneumococci were verified by their sensitivity to optochin, using an optochin diffusion assay on blood agar, resulting in a clearing zone around the optochin disc (Fluka Analytical/Sigma-Aldrich) of >15 mm (39).
Static biofilm model.
Static biofilms were produced as described previously (9). In short, pneumococci grown in CDM were seeded onto round, glass coverslips in the bottom of polystyrene 24-well plates with or without a substratum of confluent human respiratory epithelial cells (HREC). Biofilms were cultured at 34°C in 5% CO2 as indicated in the legend for Fig. 1, with a change of culture medium every 12 h. Biofilm integrity was tested by exposure to gentamicin (500 μg/ml for 3 h at 34°C in 5% CO2) or to phosphate-buffered saline (PBS) alone as a control to determine total initial biomass, followed by viable plate counts.
FIG 1.

Validation of PspA as a target for vaccination in biofilm dispersed bacteria. Biomass (A) and susceptibility (B) to gentamicin exposure (500 μg/ml for 3 h) of in vitro biofilms produced by wild-type and pspA-negative D39 bacteria following 48 h of growth over prefixed HREC. The bars show the geometric mean and 95% confidence interval of four biofilms. (C) Scanning electron microscopy images of WT and pspA-negative D39 biofilms formed on primary ciliated respiratory cells. (D) Colonization of D39 wild-type and pspA null pneumococci in mice at 48 h postinoculation. The dots represent individual mice, and the line represents the geometric mean of the data set. NP, nasopharyngeal. (E) Gene expression levels of pspA in biofilm and heat-dispersed EF3030 pneumococci. The graph presents the means and standard deviations. (F) Levels of PspA in whole-cell protein extracts from biofilm (BF) and released pneumococci from heat-dispersed (HD) and influenza (IAV)-treated biofilms grown over fixed or live HREC. (G) Quantification of pspA protein levels in biofilm (BF), heat-dispersed (HD), and influenza (IAV)-dispersed biofilm bacteria. Statistical significance of data in panels A, B, D, and E was determined using a Mann-Whitney test (**, P < 0.01; ns, not significant). Graphs in panels A, B, and E include data from at least two independent experiments. The graph in panel G represents the results from measurement of the bands shown in panel F.
Biofilm structure was assessed using scanning electron microscopy (SEM) studies. Biofilms were fixed using 2.5% glutaraldehyde, 0.075% ruthenium red, and 0.075 M lysine acetate in 0.1 M sodium cacodylate buffer (pH 7.2) for 1 h at room temperature. This procedure has been shown to retain carbohydrate structures and improve preservation of biofilm morphology. Samples were washed three times without shaking for 15 min at room temperature in 0.075% ruthenium red in 0.2 M sodium cacodylate buffer and were then dehydrated with a graded series of ethanol solutions (10, 30, 50, 75, 95, and 100%) at room temperature, with 15 min used for each step. Samples were exchanged into 100% hexamethyldisilazane and allowed to air dry before being analyzed using an SU70 scanning electron microscope at an acceleration voltage of 5.0 kV (available through the South Campus Instrumentation Center, University at Buffalo, NY).
To measure bacterial viability in these biofilms, Live/Dead stain (Invitrogen) was added to 48-h biofilms according to the manufacturer's instructions. Biofilm layers were inspected by confocal microscopy using a Zeiss LSM5 laser scanning confocal microscope (Zeiss, Inc., Thornwood, NY).
Transcriptional expression analysis (quantitative reverse transcription-PCR [qRT-PCR]).
Bacterial pellets from each bacterial population were resuspended in 0.5 ml of 0.9% NaCl and 1 ml of RNAprotect (Qiagen, Valencia, CA), and the mixture was incubated at room temperature for 5 min. Cells were pelleted at 9,000 × g for 2 min at room temperature. RNA was purified from the cells using QIAshredder columns and an RNeasy minikit (40).
To analyze the relative gene expression, qRT-PCR was performed using primers homologous to the gyrase A (gyrA) gene (forward primer, 5′-ATGGTCTCAAAGCGCTGAAT; reverse primer, 5′-TGGCGATACGACTCATACCA) and pspA (forward primer, 5′-CGCTAATGGTGCTATGGCTA; reverse primer, 5′-CGTTGACTTTAGCCCAACCT). The qRT-PCR data were analyzed by using the 2−ΔΔCT (where CT is threshold cycle) method (41). Gyrase A was chosen as the reference gene for data normalization as this gene did not exhibit significant changes in expression in our previous transcriptome sequencing (RNA-Seq) analyses (10).
PspA immunoblotting.
Static EF3030 pneumococcal biofilms were formed on an epithelial substratum of HREC that had been fixed by incubation in 4% paraformaldehyde in a 24-well plate, as described previously (9). For heat dispersal, 48-h static biofilms were shifted from 34°C to the febrile-range temperature of 38.5°C for 4 h. Dispersed bacteria were then removed from each individual well and centrifuged. For IAV dispersal, 48-h static biofilms were transferred onto confluent live epithelial substratum grown in 24-well plates and allowed to reestablish for 24 h. The cells were then infected with 1 × 105 PFU/ml of IAV. Following 1 h of incubation, the free virus was removed and replaced with fresh RPMI medium. After 24 h of incubation, planktonic bacteria were removed from each individual well and centrifuged, and protein preparations were made.
To create protein preparations, the pelleted bacteria were resuspended in lysis buffer (10 mM Tris, 1 mM EDTA, 1% SDS) and incubated at 55°C for 30 min. Samples were vortexed for 15 s to ensure a homogenous mixture. Proteins (50 μg of protein per sample) were resolved in a 10% SDS-PAGE gel and transferred to a nitrocellulose membrane. The membrane was blocked at room temperature for 1 h using a solution of 2.5% bovine serum albumin (BSA; Amresco, Solon, OH) in Tris-buffered saline (TBS)-Tween (0.1%) (TBS-T) buffer. After the membrane was washed in TBS-T, it was probed overnight at room temperature with mouse PspA antiserum (1:5,000) diluted in TBS-T–2.5% BSA. The next morning, the membrane was washed with TBS-T–2.5% BSA and incubated for 1 h at room temperature with goat anti-mouse IgG DyLight 680-conjugated antibody (Ab; 1:10,000) (ThermoFisher Scientific). After the membrane was washed, it was scanned, and the signals were quantified using an Odyssey CLx infrared imaging system (Licor).
Intradermal and intranasal (i.n.) immunizations and sample collection.
For intradermal (i.d.) immunization, 8-week-old female BALB/cByJ mice (Jackson Laboratories) were immunized by the i.d. route, as previously described (32). Briefly, groups of mice were anesthetized with 75 mg/kg of ketamine and 10 mg/kg of xylazine, fur was removed on the dorsum, and the underlying skin was swabbed with alcohol. With the use of insulin syringes, mice were intradermally immunized with PBS (vehicle control) or PspA (5.0 μg) and LT-IIb(T13I) (1.0 μg) in 30-μl volumes. The i.d. immunization regimen consisted of a primary immunization followed by booster immunizations administered at day 10 and day 20. The mice were injected all three times in the same dorsal area but on alternating sides each time.
For i.n. immunization, 8-week-old female unanesthetized BALB/cByJ mice were immunized intranasally with PBS (vehicle control) or PspA in combination with 1.0 μg of LT-IIb(T13I). Immunizations were administered into both external nostrils (5 μl/naris), with mice allowed to rest for 5 min between each nasal administration. Similar to the i.d. immunization regimen, the i.n. immunization regimen consisted of a primary immunization followed by booster immunizations administered at day 10 and day 20.
Blood collected on day 27 from the tail vein of immunized mice was centrifuged at 4°C for 20 min at 16,000 relative centrifugal force (RCF) to pellet cells, after which serum fractions were collected and stored at −80°C. Saliva samples were collected on day 27 with a micropipette after stimulation of salivary flow by injecting each mouse intraperitoneally (i.p.) with 5.0 μg of carbachol (Sigma-Aldrich Co., St. Louis, MO).
PspA antibody analysis.
Levels of isotype and subclass anti-PspA Ab in serum and saliva were measured by enzyme-linked immunosorbent assay (ELISA) (35). Nunc Immulon 2HB polystyrene 96-well microtiter plates (ThermoFisher Scientific) coated with 100 μl of PspA (5 μg/ml) per well were incubated overnight at room temperature. To determine total IgA concentrations, plates were coated with 100 μl of unlabeled goat anti-mouse IgA-specific Ab (1 mg/ml) (Southern Biotechnology Associates, Birmingham, AL). After plates were blocked with PBS containing 0.15% Tween 20 and 1% BSA, serial 2-fold dilutions of serum or secretion samples were added in duplicate, and plates were incubated overnight at room temperature. Plates were washed with PBS-Tween and incubated at room temperature for 4 h with the appropriate alkaline phosphatase-conjugated goat anti-mouse Ig isotype or subclass-specific Ab (Southern Biotechnology). Plates were washed and developed with nitrophenyl phosphate substrate (1 mg/ml) (Amresco), and the reaction was terminated by the addition of 100 μl/well of 2 N NaOH. ELISA plates were read on a VersaMax microplate reader at a 405-nm wavelength and analyzed with SoftMax Pro, version 5.4 (Molecular Devices, Sunnydale, CA). Concentrations of Ag-specific and total IgA Ab were calculated by interpolation of calibration curves generated using a mouse Ig reference serum (ICN Biomedicals, Aurora, IL). Salivary IgA responses are reported as the percentage of PspA-specific IgA of the total IgA to compensate for variation in salivary flow rate (35, 42). As we have described previously, the concentration of salivary IgA is indicative of the concentration of mucosal IgA in other compartments, such as in nasal secretions, yet easier to determine (35, 42).
Staining of surface PspA protein on S. pneumoniae.
Planktonic D39 wild-type (WT) and PspA-negative (D39-PspA null) pneumococci were cultured to mid-log phase and washed twice with PBS. Cells were stained for 30 min at 4°C in PBS and 2% fetal calf serum (FCS) by incubation with pooled, heat-treated serum from PspA-immunized mice (1:50). Cells were washed twice in PBS and 2% FCS and stained for 30 min at 4°C with anti-mouse IgG AF488 secondary antibody (Life Technologies) at 1:400 in PBS and 2% FCS. Cells were washed twice with PBS and resuspended in 4% paraformaldehyde for fixation at room temperature for 15 min. Fixed cells were washed and resuspended in PBS and 2% FCS and analyzed by flow cytometry using an LSR Fortessa (BD).
Murine colonization and IAV infection.
Six- to 8-week-old female BALB/cByJ mice (Jackson Laboratories) were maintained in filter-top cages on standard laboratory chow and water ad libitum until use. Mice were colonized as described previously (43). In brief, 20 μl of a bacterial suspension containing 5 × 106 CFU of pneumococci in phosphate-buffered saline (PBS) was pipetted into the nares of nonanesthetized mice. Uninfected control mice received PBS alone. The absence of symptoms associated with pneumonia and invasive disease was confirmed by lack of visual signs, such as ruffled fur, inactivity, and labored breathing, as well as lack of fever measured through the abdominal surface temperature. At 48 h postinoculation, animals were either infected with IAV or euthanized by CO2 asphyxiation, and tissues were dissected as described below. For IAV infection following colonization, 40 PFU of influenza A/PR8/34 (H3N2) virus was pipetted into the nares of nonanesthetized mice; 1 or 5 days later mice were euthanized, and tissues were collected as described below.
Tissue collection following colonization and IAV infection.
Various tissues and samples were collected from immunized and unimmunized mice following euthanization. Harvesting of mouse nasal septum and lungs was done as previously described (9, 43). To harvest mouse nasal septum, the skin was first dissected and completely removed from the skull and nose, and the skull was sectioned in the coronal plane. The remainder of the anterior and posterior skull was then removed, and the suture line was incised bilaterally to reveal the nasal septum. Scissors were inserted into the posterior nasal cavity and used to separate the nasal septum from the maxillae (laterally) and the ethmoid bone (medially). Tissue present in the nasal conchae was then harvested with forceps. Lungs were harvested with a midline chest incision, and skin was peeled away to expose the chest. Scissors were used to cut around the ribcage and expose the lungs where the trachea was cut, and the lungs were removed from the chest cavity. To obtain middle ear tissue, the outer ears were removed with scissors, and the middle ear on each side was extracted from the cranium using forceps. Blood was obtained by puncturing the submandibular vein, collecting the blood with a lancet, and collecting the flow in microcentrifuge tubes. Harvested tissue was homogenized and serially diluted on tryptic soy agar supplemented with 5% sheep blood (blood agar) to measure the number of CFU per mouse nasopharynx, ear, lung, or blood tissue as a measure of colonization load. In some experiments, intranasal wash samples (nasopharyngeal lavage fluid) were obtained by injecting PBS retrograde through the trachea prior to the harvesting of tissue. Bacterial load was measured by determining viable colony counts after plating.
Septicemia challenge following i.d. immunization.
Sham-immunized mice and mice immunized with PspA plus LT-IIb(T13I) were challenged by i.p. injection of 1 × 105 CFU of biofilm-dispersed, virulent S. pneumoniae. Mice were monitored for 24 h for signs of septicemia. Mice observed to be overtly moribund were euthanized by CO2 asphyxiation and cervical dislocation after collection of blood from the submandibular vein. Blood samples were assayed for bacterial burden. Serum fractions were used for inflammatory cytokine analysis.
Inflammatory cytokine analysis.
Serum from immunized mice collected after direct bacterial challenge was analyzed for levels of inflammatory cytokines using a mouse inflammatory cytokine cytometric bead array kit (BD Biosciences). For sham-immunized mice, sera were collected at the time of euthanasia once the mice had become moribund. For PspA-immunized mice, sera were collected 24 h after i.p. injection of bacteria. Concentrations of cytokines in the sera were determined using a BD FACSArray bioanalyzer, and FCAP (Flow Cytometric Analysis Program) Array software (BD Biosciences, San Jose, CA).
Statistical analysis.
Column comparisons were analyzed for statistical significance using a two-tailed Mann-Whitney test for unpaired data, based on the non-Gaussian distribution of the data sets. Multivariate analysis was done using one-way analysis of variance (ANOVA) that was corrected for variance using a Bartlett variance test and using a Bonferroni multiple-comparison test for multiple comparisons. Survival curves were analyzed using a log rank test. For all tests a P value of <0.05 was considered significant. Statistical analysis was performed using GraphPad Prism software (version 6.0 h; GraphPad Software, Inc., La Jolla, CA).
RESULTS
Validating PspA as a vaccine target in pneumococci that are dispersed from biofilms.
To examine the usefulness of PspA as an immunogen that specifically targets disease-causing bacteria without targeting biofilm colonization, the role of PspA in biofilm formation and nasopharyngeal colonization and the expression of PspA in biofilm and biofilm-dispersed bacteria were determined. First, D39 wild-type (WT) pneumococci and an isogenic PspA-deficient (pspA null) mutant were used to evaluate the role of PspA during biofilm formation. After 48 h of growth on prefixed epithelia, D39 lacking pspA was not impaired in biofilm biomass or in development of biofilm-specific resistance to the antibiotic gentamicin (500 μg/ml for 3 h) compared to results with WT D39 bacteria (Fig. 1A and B). Examination by scanning electron microscopy of biofilms cultured over primary ciliated respiratory cells revealed no identifiable impairment in formation of biofilm structures. Both D39 WT and D39-PspA null bacteria formed complex communities with visible towers and water channels throughout the structure (Fig. 1C). Additionally, D39 and D39-PspA null bacteria colonized the mouse nasopharynx with similar bacterial burdens 48 h after inoculation, suggesting that PspA played no significant role in colonization (Fig. 1D).
Next, PspA expression was evaluated using the clinical isolate EF3030, a strain that was employed in subsequent in vivo mouse infection experiments. Our recent studies of the total transcriptome of EF3030 pneumococci showed that pspA had an increase in expression in bacteria dispersed from biofilms in response to virus and virus-induced host conditions, such as increased temperature and ATP, compared to expression in EF3030 biofilm bacteria (10). Here, we show that although EF3030 biofilm bacteria expressed pspA, the expression level was 3.03-fold higher in heat-dispersed bacteria (P < 0.01) (Fig. 1E). To investigate the expression state of PspA protein in biofilm-residing and biofilm-dispersed bacteria, immunoblotting was performed on whole-cell extracts from EF3030 obtained from biofilms grown on epithelial cell monolayers and from EF3030 that had been dispersed from equivalent biofilms by febrile temperature or by infection with IAV. Dispersed bacteria collected from biofilms that were exposed to febrile temperature or to IAV exhibited 2.43-fold and 2.73-fold increases, respectively, in PspA protein expression in comparison to the levels of PspA expressed by biofilm-residing bacteria (Fig. 1F and G).
Collectively, these data suggest that PspA does not have a significant role in biofilm formation or biofilm colonization of the mouse nasopharynx and is expressed at lower levels in pneumococci that reside in biofilms. Yet PspA is upregulated in pneumococci that are dispersed from biofilms produced on epithelial monolayers by febrile temperatures or by IAV infection. These results validated the consideration of PspA as a vaccine target for evoking immune protection specifically against disseminated pneumococci without altering the desirable commensal colonization of the nasopharynx.
Intradermal immunization with PspA and LT-IIb(T13I) produces a robust systemic anti-PspA immune response that does not affect colonization.
Given the positive findings regarding the increased expression of PspA in biofilm-dispersed pneumococci, we formulated an immunization strategy with the goal of generating a strong systemic anti-PspA antibody (Ab) response with little or no mucosal Ab response in the hope of providing protection against disseminated pneumococcal disease without affecting biofilm colonization in the nasopharynx. To maximize the anti-PspA IgG response, the adjuvant LT-IIb(T13I) was utilized. This detoxified adjuvant has been shown to be superior to aluminum salt adjuvants with respect to antibody responses to various Ags, including an Ag derived from ricin toxin (32, 63). The well-described N-terminal portion of PspA from strain Rx1 (UAB055) (see Materials and Methods for details) was used as an immunogen (34). Immunization with PspA in the absence of the adjuvant produced relatively low levels of Ag-specific serum IgG. In contrast, the addition of LT-IIb(T13I) increased by 30-fold the amount of Ag-specific IgG Ab (P < 0.01) (Fig. 2A). Examining IgG subclass distribution in mice that were immunized with PspA plus LT-IIb(T13I) revealed that the vast majority of PspA-specific IgG was of the IgG1 subclass and at a level ∼2 logs greater than that of the IgG2a or IgG2b subclass (P < 0.01) (Fig. 2B). To demonstrate that the Abs generated by immunization with PspA were PspA-specific, WT D39 pneumococci and pspA-negative pneumococci (D39-PspA null) were analyzed by flow cytometry using polyclonal Ab obtained from sera of mice that had been immunized with PspA plus LT-IIb(T13I). WT D39 pneumococci exhibited a high level of positive staining with PspA antiserum, whereas no significant staining above background was observed for the D39-PspA null strain. Thus, Abs generated by immunization of mice with PspA plus LT-IIb(T13I) were PspA specific (Fig. 2C) and did not recognize other proteins exposed on the surface of the bacteria.
FIG 2.

Intradermal immunization with PspA does not affect colonization. (A) Mice were immunized by the i.d. route on days 0, 10, and 20 with 5.0 μg of PspA alone or with 1.0 μg of LT-IIb(T13I). One week after the final booster, serum samples were taken, and PspA-specific IgG was determined by ELISA. Each dot represents serum from an individual mouse (n = 6). ***, P < 0.001 for results of immunization with PspA in the presence or absence of adjuvant. (B) Pooled serum from mice immunized with PspA plus LT-IIb(T13I) was evaluated for Ag-specific IgG subclasses. Each dot represents serum pooled samples of 2 mice (n = 8). ***, P < 0.001 for a comparison of the concentration of IgG1 with either IgG2a or IgG2b. (C) Ab specificity to PspA was confirmed by labeling D39 WT and D39 pspA null bacteria with serum Ab collected after i.d. immunization followed by a goat anti-mouse secondary Ab conjugated to Alexa Fluor 488, and samples were analyzed by flow cytometry. (D) Two weeks after the final booster immunization (day 34), mice were inoculated intranasally with biofilm pneumococci. Forty-eight hours after inoculation, tissues, blood, and nasopharyngeal lavage samples were collected and measured for bacterial burden. There was no difference in colonization burdens between sham-immunized and PspA-immunized animals and no dissemination to the lungs or middle ear. Statistical analysis was performed using a Mann-Whitney test (**, P < 0.01). Panels A and B include data from one experiment, whereas panel D includes data from two separate experiments.
While high levels of anti-PspA IgG were induced in the sera of mice that had received i.d. immunization with PspA plus LT-IIb(T13I), no PspA-specific IgA Ab was detected in the saliva of those mice (Table 1). As a positive control for PspA-specific salivary IgA, mice were immunized mucosally through the i.n. route with PspA plus LT-IIb(T13I) or vehicle alone (sham). This strategy resulted in a strong Ag-specific IgA Ab response in the saliva of mice immunized with PspA, whereas no Ab responses were detected in the sham-immunized mice (Table 1). Intranasal immunization with PspA alone induced a very poor mucosal immune response, with less than 3% for the Ag-specific/total Ab concentration (data not shown) compared to 51% for the Ag-specific/total Ab concentration in mice immunized with PspA plus LT-IIb(T13I) (Table 1). Mice immunized mucosally through the i.n. route with PspA plus LT-IIb(T13I) also produced a robust IgG response in serum (Table 1). PspA-specific immune responses were not detected in any of the sham-immunized groups of mice.
TABLE 1.
Levels of PspA-specific Ab on day 27 following intradermal or intranasal immunization
| Immunization regimea | Serum IgG (μg/ml)b | Salivary IgA (%)c |
|---|---|---|
| i.d. route | ||
| Sham | NDd | ND |
| PspA + LT-IIb(T13I) | 907.6 ± 74.91 | ND |
| i.n. route | ||
| Sham | ND | ND |
| PspA + LT-IIb(T13I) | 1,128.0 ± 114.3 | 51.7 ± 7.1 |
Mice were immunized with PBS (sham) or 5.0 μg PspA plus 1.0 μg LT-IIb(T13I) on days 0, 10, and 20 for both i.d. and i.n. immunizations.
Data are representative of four separate groups with 5 mice per group (n = 20). Values are means ± standard errors of the means.
Data represent the ratio of specific to total antibody. Values (means ± standard errors of the means) are representative of one group of 5 mice (n = 5).
ND, not detected.
Mice immunized i.d. with vehicle (sham) or PspA plus LT-IIb(T13I) were subsequently employed in pneumococcal challenge experiments. Two weeks following a second booster immunization of PspA plus LT-IIb(T13I), mice were inoculated intranasally with biofilm-derived EF3030 pneumococci. Nasopharyngeal tissues were collected 48 h after challenge and assessed for pneumococcal burden. In comparison to sham-immunized mice, mice that had received i.d. immunizations with PspA plus LT-IIb(T13I) (vaccinated) had similar colonization burdens of biofilm bacteria on nasal tissues (Fig. 2D). These results demonstrated that i.d. immunization with PspA plus LT-IIb(T13I) generated a robust systemic humoral immune response yet did not disrupt nasopharyngeal colonization of biofilm pneumococci.
Intradermal immunization with PspA prevents secondary pneumococcal disease following IAV infection.
To evaluate the role of PspA in secondary bacterial disease following IAV infection, pneumococcal challenge was performed on sham-immunized mice and mice immunized with PspA plus LT-IIb(T13I). We first attempted to use PspA null pneumococci to verify the specificity of the protective immune response. Although PspA null pneumococci dispersed to a similar degree from in vitro biofilms as wild-type pneumococci in response to virus-induced stimuli (Fig. 3A) and disseminated to the lungs and middle ears of mice after infection with IAV, they were quickly eradicated in these environments (Fig. 3B and C). This confirmed the important role for PspA in bacterial survival outside the nasopharyngeal environment even in association with virus infection and made it impossible to verify that PspA immunization would fail to protect against bacteria lacking PspA.
FIG 3.
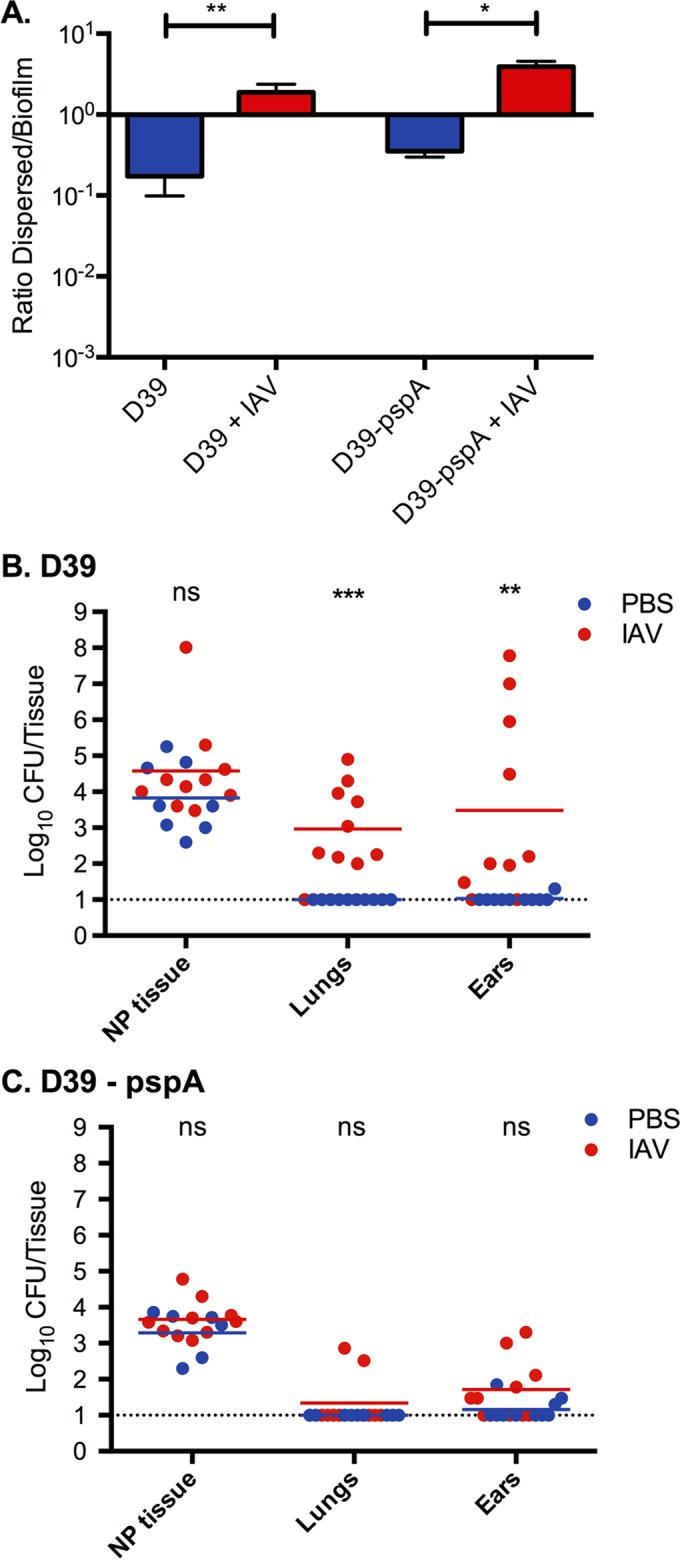
Role of PspA in biofilm release and survival in tissue after IAV infection. (A) D39 and D39 pspA null bacteria biofilms were made on top of respiratory epithelial cells for 48 h, and the cells were infected with IAV for 24 h. The ratio of dispersed (released) biofilm bacteria in the medium to the remaining bacteria in the biofilm was measured in the presence or absence of IAV. The bars show ratios with standard deviations. (B and C) Mice were colonized for 48 h with D39 wild-type bacteria and D39 lacking PspA and infected with 40 PFU of IAV. After 24 h the bacterial burden was measured in nasopharyngeal (NP) tissue and lung tissue and in the middle ears. D39 bacteria caused pneumonia and otitis media after IAV infection, whereas D39 pspA null pneumococci, even though they were released equally well in vitro (panel A), survived poorly in the lungs and middle ear tissues. Each experiment was performed twice with 5 mice in each group. Each dot in the graphs represents a single mouse. The lines represent the geometric mean. Statistics for all data sets were done using a Mann Whitney test (ns, not significant; *, P < 0.05; **, P < 0.01; ***, P < 0.001).
Immunized mice were instead colonized with biofilm-derived EF3030 bacteria and challenged intranasally 48 h later with IAV. On day 1 post-viral infection, immunization with PspA plus LT-IIb(T13I) significantly lowered the bacterial burden of disseminated bacteria in the lungs and middle ears (P < 0.05 and P < 0.001, respectively) in response to viral infection in comparison with the responses of sham-immunized controls (Fig. 4A). There was no detectable dissemination to the bloodstream in either group at this time point (Fig. 4A). Immunized animals appeared completely healthy, whereas sham-immunized animals showed slightly ruffled fur and lower activity in the cages, suggesting initial stages of infection. At day 5 post-viral infection, immunization with PspA plus LT-IIb(T13I) continued to significantly control dissemination of pneumococci to the lungs and middle ears (P < 0.001 and P < 0.05, respectively) (Fig. 4B), thus preventing development of pneumonia and otitis media. Importantly, at day 5 post-viral infection, bacteria were not detected in the circulation of mice immunized with PspA plus LT-IIb(T13I) (P < 0.001 compared to results with sham-immunized mice). Visual monitoring of these mice indicated no signs of infection; mice displayed normal activity, respiration, and abdominal temperature. In stark contrast, sham-immunized mice developed both otitis media and pneumonia, with significant levels of disseminating bacteremia (Fig. 4B), which resulted in lethargy, increased abdominal temperature, and ruffled fur, suggesting a febrile response. Respiration was shallower and labored. Clearly, i.d. immunization against PspA prevented development of disseminated pneumococcal disease induced by IAV infection.
FIG 4.

Protection from secondary pneumococcal disease following influenza virus infection by i.d. immunization with PspA and LT-IIb(T13I). Mice were immunized with 5.0 μg of PspA plus 1.0 μg of LT-IIb(T13I) (vaccinated) or with PBS (sham). Two weeks after the final booster immunization (day 34), mice were inoculated intranasally with biofilm-derived EF3030 pneumococci. Forty-eight hours after inoculation, mice received 40 PFU of IAV by the intranasal route. Tissues and nasal lavage samples were collected at 1 day (A) and 5 days (B) post-viral infection and measured for bacterial burden. Each dot represents an individual mouse (n = 10). Statistical analysis was performed using a Mann-Whitney test (ns, not significant; *, P < 0.05; ***, P < 0.001). Graphs include data from two independent experiments.
Intradermal immunization against PspA prevents mortality due to septicemia.
To investigate if i.d. immunization with PspA plus LT-IIb(T13I) would protect against an overwhelming septic infection, biofilm-dispersed EF3030 pneumococci were employed again based on their significantly increased virulence in invasive disease compared with that of broth-grown EF3030 that are completely cleared from the circulation (10). This approach was used, first, to mimic the phenotype of the bacteria more closely associated with infection in vivo and, second, to verify our novel immunization strategy in a classical experimental model used by other investigators (23, 27). Whereas 90% of sham-immunized mice died within 20 h of infection, immunization with PspA plus LT-IIb(T13I) conferred complete protection against septicemia-induced death by dispersed EF3030 bacteria (P < 0.001) (Fig. 5A). Low levels of bacteremia were observed after intraperitoneal challenge with dispersed bacteria following immunization with PspA plus LT-IIb(T13I). The level of bacteremia was significantly lower than the level of bacteremia observed in the sham-immunized group (P < 0.001) (Fig. 5B). The small bacterial burden remaining in the group immunized with PspA plus LT-IIb(T13I) was attributed to the inability of the mice to completely clear the large amount of injected EF3030 bacteria within 24 h.
FIG 5.
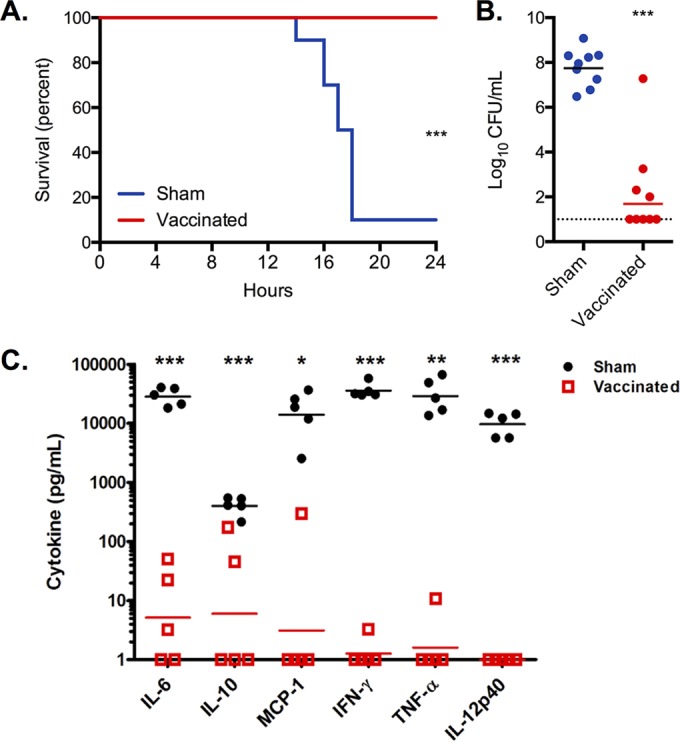
Intradermal immunization with PspA provides protection against septicemia. Mice were immunized with 5.0 μg of PspA plus 1.0 μg of LT-IIb(T13I) (vaccinated) or with PBS (sham). Survival (A) and bacterial burden in the bloodstream (B) were measured for mice injected i.p. with EF3030 bacteria dispersed from biofilms in response to heat. (C) Levels of inflammatory cytokines in the blood at the time of euthanasia up to 24 h following bacterial challenge. Each dot represents an individual mouse (n = 5). Statistical analysis was performed using a log rank test for survival data in panel A or a Mann-Whitney U test for bacterial burden and cytokine data (panels B and C). *, P < 0.05; **, P < 0.01; ***, P < 0.001. Panels A and B include data from two independent experiments, whereas panel C includes data from one of the experiments included in the other panels.
Sham-immunized mice infected with EF3030 had significantly elevated levels of the inflammatory cytokines interleukin-6 (IL-6), monocyte chemotactic protein 1 (MCP-1), gamma interferon (IFN-γ), tumor necrosis factor alpha (TNF-α), and IL-12p40, a response that indicated a cytokine storm environment that is typical in a lethal systemic infection (Fig. 4C). No similar cytokine storm was evident in the EF3030-infected mice immunized with PspA plus LT-IIb(T13I). Thus, the immunity established by i.d. immunization with PspA plus LT-IIb(T13I) reduced the inflammation induced in the bloodstream, which was at least partly based on the rapid elimination of bacteria and the subsequent low bacterial burden compared to that in infected sham-immunized mice. These data strongly indicated that i.d. immunization against PspA dramatically reduced mortality due to septicemia induced by biofilm-dispersed, invasive S. pneumoniae.
DISCUSSION
PspA is a crucial virulence factor of planktonic cell populations that are extant during invasive disease. Infection experiments using planktonic pneumococci have clearly demonstrated that intranasal immunization with PspA can protect against colonization; however, the contribution of PspA to colonization is unclear (34, 44). Similarly, little is known about the role of PspA in the nasopharyngeal pneumococcal life cycle, including biofilm formation and dispersion. Here, we demonstrate that PspA has little, if any, role in the formation of a pneumococcal biofilm or in establishment of asymptomatic colonization. Yet expression of PspA is dramatically increased in biofilm-dispersed bacteria, which represent the major physiological pneumococcal phenotype that disseminates and elicits disease at otherwise sterile sites.
These features made PspA a good candidate to test the hypothesis that it is possible to immunize specifically against disease-causing organisms leaving the nasopharyngeal environment without affecting asymptomatic colonization. To evaluate the role of PspA in the development of secondary infections, we infected mice with IAV intranasally 48 h after the mice had been colonized with biofilm-derived pneumococci in the nasopharynx to mimic the natural course of virus-induced disease progression in children and other populations with high pneumococcal carriage rates (45). This procedure has been shown to produce populations of bacteria with a major change in transcriptional profiles that differ significantly from both biofilm bacteria and broth-grown bacteria and produce pneumococci with significantly increased virulence in otherwise sterile sites (9, 10). Intradermal immunization with PspA in the presence of LT-IIb(T13I), a detoxified heat-labile enterotoxin, elicited a strong PspA-specific systemic immune response but did not disturb nasopharyngeal colonization. Importantly, immunization with PspA significantly inhibited development of secondary pneumococcal superinfections and protected against mortality due to septicemia and its associated cytokine storm, supporting our hypothesis. As this was the main purpose of the studies, cross-protection studies were outside the scope of these studies as PspA has been well characterized in this regard.
Clearance of microbial pathogens typically occurs by Fcγ receptor or by complement receptor (CR)-mediated phagocytosis. PspA inhibits activation of the alternative pathway of the complement system, a system that is critical for mediating clearance of S. pneumoniae by the innate immune system (46). In the mouse, clearance of PspA null pneumococci by this pathway is mediated by CRs located on the surfaces of macrophages and neutrophils (17). In this study, we showed that i.d. immunization against PspA induced high levels of PspA-specific serum IgG, which protected against pneumococcal disease. While the precise mechanism(s) responsible for the protection is unknown, the response is likely mediated by PspA-specific IgG Abs and Fcγ receptor phagocytosis. The possibility cannot be ignored, however, that binding of PspA with high-affinity Abs neutralized the complement-inhibiting properties of S. pneumoniae to promote effective CR-mediated clearance. The latter hypothesis is supported by a previous study that reported that addition of anti-PspA antibodies enhanced binding of complement components to PspA-positive pneumococci (47).
Two recent studies on the role of PspA in virus-induced secondary pneumococcal infection demonstrated that i.n. immunization with PspA administered in the presence of cholera toxin B subunit as an adjuvant or with polyinosinic-poly(C), a Toll-like receptor 3 agonist, reduced the bacterial load of pneumococci in the lungs of influenza A virus-infected mice. Yet neither strategy eradicated the pneumococci in the lungs (24, 25). While these studies did not examine nasopharyngeal colonization, previous research suggested that mucosal immunization with PspA was protective against colonization of the nasopharynx (34, 44). These findings, however, suffered from the fact that these colonization studies were performed using planktonic broth-grown bacteria in which PspA is highly expressed. In contrast, it is believed that colonization is biofilm associated, under which condition expression of PspA is low (10).
Three families of PspA have been identified to date, with less than 50% sequence divergence within each family (48, 49). Anti-PspA Abs are usually cross-reactive and cross-protective against invasive disease, at least within families (31, 50). The potential cross-reactivity and cross-protection of antibodies induced by i.d. immunization with PspA in combination with LT-IIb(T131I) will be pursued in future studies. Since many individuals will undergo successive episodes of pneumococcal carriage throughout their lifetimes (51–54), it seems unlikely that mucosal Abs to PspA that are generated during nasopharyngeal carriage can prevent successive acquisition of other strains. Our results indicate that biofilm pneumococci represent the dominant physiological state of the bacteria during initial colonization, whether or not biofilm bacteria or broth-grown bacteria are used for intranasal inoculation, and suggest that nasopharyngeal carriage of pneumococci in biofilm communities has a pathogenically quiescent phenotype that does not elicit cross-protective immunity against PspA.
Overall, these observations indicated that the effects of a vaccine developed against acquisition of carriage and for prevention of invasive disease are two distinct endpoints that should be considered separately. Determining if nasopharyngeal eradication of S. pneumoniae is either a beneficial or a harmful intervention is not an easy question to address (55). The advantage of colonization eradication of invasive serotypes is herd immunity: the indirect protection from infection based on the lower level of disease-associated strains in circulation that can spread between individuals. Herd immunity has been demonstrated after the introduction of the conjugate vaccines in both young children and the elderly (56, 57). But there is a caveat. Many other respiratory pathogens compete for the same ecological niche as the pneumococci. Therefore, pneumococcal mucosal colonization per se may, in fact, protect against colonization by other virulent organisms and keep the ecological niche balanced. Several observational studies have shown that S. aureus prevalence is negatively associated with the presence of S. pneumoniae (3). Successful clearance of the S. pneumoniae from the nasopharyngeal niche by vaccination using the pneumococcal conjugate vaccine-7 (PCV-7) has been correlated with increased colonization of S. aureus (29, 58) and, depending on the level of serotype replacement, in some studies also increased colonization with H. influenzae (26, 27). The odds of acquiring otitis media and staphylococcal pneumonia are increased 3-fold in individuals who received PCV-7, resulting in diminished pneumococcal colonization with the most fit serotypes (59–62). A vaccine targeting only disease-causing organisms would have the benefit of not interfering with the nasopharyngeal microbiome but would not induce herd immunity and would therefore require administering the vaccine to a larger population of at-risk individuals.
In conclusion, our results suggest that bacterial factors, such as PspA, that are unique to bacteria that leave colonizing biofilms as a result of host factors induced by virus infection or other immunological assaults but that are not required for biofilm formation and colonization are excellent vaccine candidates: they can induce immune responses that will inhibit the onset and development of pneumococcal disease without interfering with potentially desirable nasopharyngeal colonization by the bacteria. This strategy represents a major paradigm shift in the prevention of pneumococcal disease. It will be critical to obtain additional information in regard to microbial interactions in the nasopharynx and the conditions that are conducive for progression to infection to better understand the benefits of maintaining harmless nasopharyngeal colonization by potentially pathogenic bacteria.
ACKNOWLEDGMENT
We thank David Briles for providing the plasmid pUAB055 that expressed the N-terminal portion of PspA from strain Rx1.
Funding Statement
Funds from the NIDCD, NIDCR, VR, and Oishei Foundation were used to pay for the in vitro and in vivo experiments of the study. The grant from NIAID was an institutional training grant that supported Ryan Reddinger during the studies.
REFERENCES
- 1.Bosch AA, Biesbroek G, Trzcinski K, Sanders EA, Bogaert D. 2013. Viral and bacterial interactions in the upper respiratory tract. PLoS Pathog 9:e1003057. doi: 10.1371/journal.ppat.1003057. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Short KR, Habets MN, Hermans PW, Diavatopoulos DA. 2012. Interactions between Streptococcus pneumoniae and influenza virus: a mutually beneficial relationship? Future Microbiol 7:609–624. doi: 10.2217/fmb.12.29. [DOI] [PubMed] [Google Scholar]
- 3.van den Bergh MR, Biesbroek G, Rossen JW, de Steenhuijsen Piters WA, Bosch AA, van Gils EJ, Wang X, Boonacker CW, Veenhoven RH, Bruin JP, Bogaert D, Sanders EA. 2012. Associations between pathogens in the upper respiratory tract of young children: interplay between viruses and bacteria. PLoS One 7:e47711. doi: 10.1371/journal.pone.0047711. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Pettigrew MM, Gent JF, Pyles RB, Miller AL, Nokso-Koivisto J, Chonmaitree T. 2011. Viral-bacterial interactions and risk of acute otitis media complicating upper respiratory tract infection. J Clin Microbiol 49:3750–3755. doi: 10.1128/JCM.01186-11. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Heikkinen T, Chonmaitree T. 2003. Importance of respiratory viruses in acute otitis media. Clin Microbiol Rev 16:230–241. doi: 10.1128/CMR.16.2.230-241.2003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Madhi SA, Klugman KP, Vaccine TG. 2004. A role for Streptococcus pneumoniae in virus-associated pneumonia. Nat Med 10:811–813. doi: 10.1038/nm1077. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Michelow IC, Olsen K, Lozano J, Rollins NK, Duffy LB, Ziegler T, Kauppila J, Leinonen M, McCracken GHJ. 2004. Epidemiology and clinical characteristics of community-acquired pneumonia in hospitalized children. Pediatrics 113:701–707. doi: 10.1542/peds.113.4.701. [DOI] [PubMed] [Google Scholar]
- 8.McCullers JA. 2006. Insights into the interaction between influenza virus and pneumococcus. Clin Microbiol Rev 19:571–582. doi: 10.1128/CMR.00058-05. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Marks LR, Davidson BA, Knight PR, Hakansson AP. 2013. Interkingdom signaling induces Streptococcus pneumoniae biofilm dispersion and transition from asymptomatic colonization to disease. mBio 4:e00438–13. doi: 10.1128/mBio.00438-13. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Pettigrew MM, Marks LR, Kong Y, Gent JF, Roche-Hakansson H, Hakansson AP. 2014. Dynamic changes in the Streptococcus pneumoniae transcriptome during transition from biofilm formation to invasive disease upon influenza A virus infection. Infect Immun 82:4607–4619. doi: 10.1128/IAI.02225-14. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Klugman KP. 2011. Contribution of vaccines to our understanding of pneumococcal disease. Philos Trans R Soc Lond B Biol Sci 366:2790–2798. doi: 10.1098/rstb.2011.0032. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Barocchi MA, Censini S, Rappuoli R. 2007. Vaccines in the era of genomics: the pneumococcal challenge. Vaccine 25:2963–2973. doi: 10.1016/j.vaccine.2007.01.065. [DOI] [PubMed] [Google Scholar]
- 13.Tai SS. 2006. Streptococcus pneumoniae protein vaccine candidates: properties, activities and animal studies. Crit Rev Microbiol 32:139–153. doi: 10.1080/10408410600822942. [DOI] [PubMed] [Google Scholar]
- 14.Tu AH, Fulgham RL, McCrory MA, Briles DE, Szalai AJ. 1999. Pneumococcal surface protein A inhibits complement activation by Streptococcus pneumoniae. Infect Immun 67:4720–4724. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Hakansson A, Roche H, Mirza S, McDaniel LS, Brooks-Walter A, Briles DE. 2001. Characterization of binding of human lactoferrin to pneumococcal surface protein A. Infect Immun 69:3372–3381. doi: 10.1128/IAI.69.5.3372-3381.2001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Berry AM, Paton JC. 2000. Additive attenuation of virulence of Streptococcus pneumoniae by mutation of the genes encoding pneumolysin and other putative pneumococcal virulence proteins. Infect Immun 68:133–140. doi: 10.1128/IAI.68.1.133-140.2000. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Li J, Glover DT, Szalai AJ, Hollingshead SK, Briles DE. 2007. PspA and PspC minimize immune adherence and transfer of pneumococci from erythrocytes to macrophages through their effects on complement activation. Infect Immun 75:5877–5885. doi: 10.1128/IAI.00839-07. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Ogunniyi AD, LeMessurier KS, Graham RM, Watt JM, Briles DE, Stroeher UH, Paton JC. 2007. Contributions of pneumolysin, pneumococcal surface protein A (PspA), and PspC to pathogenicity of Streptococcus pneumoniae D39 in a mouse model. Infect Immun 75:1843–1851. doi: 10.1128/IAI.01384-06. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Ren B, Szalai AJ, Thomas O, Hollingshead SK, Briles DE. 2003. Both family 1 and family 2 PspA proteins can inhibit complement deposition and confer virulence to a capsular serotype 3 strain of Streptococcus pneumoniae. Infect Immun 71:75–85. doi: 10.1128/IAI.71.1.75-85.2003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Moore QC, Bosarge JR, Quin LR, McDaniel LS. 2006. Enhanced protective immunity against pneumococcal infection with PspA DNA and protein. Vaccine 24:5755–5761. doi: 10.1016/j.vaccine.2006.04.046. [DOI] [PubMed] [Google Scholar]
- 21.Ogunniyi AD, Grabowicz M, Briles DE, Cook J, Paton JC. 2007. Development of a vaccine against invasive pneumococcal disease based on combinations of virulence proteins of Streptococcus pneumoniae. Infect Immun 75:350–357. doi: 10.1128/IAI.01103-06. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Coats MT, Benjamin WH, Hollingshead SK, Briles DE. 2005. Antibodies to the pneumococcal surface protein A, PspA, can be produced in splenectomized and can protect splenectomized mice from infection with Streptococcus pneumoniae. Vaccine 23:4257–4262. doi: 10.1016/j.vaccine.2005.03.039. [DOI] [PubMed] [Google Scholar]
- 23.Briles DE, Hollingshead SK, Paton JC, Ades EW, Novak L, van Ginkel FW, Benjamin WHJ. 2003. Immunizations with pneumococcal surface protein A and pneumolysin are protective against pneumonia in a murine model of pulmonary infection with Streptococcus pneumoniae. J Infect Dis 188:339–348. doi: 10.1086/376571. [DOI] [PubMed] [Google Scholar]
- 24.King QO, Lei B, Harmsen AG. 2009. Pneumococcal surface protein A contributes to secondary Streptococcus pneumoniae infection after influenza virus infection. J Infect Dis 200:537–545. doi: 10.1086/600871. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Ezoe H, Akeda Y, Piao Z, Aoshi T, Koyama S, Tanimoto T, Ishii KJ, Oishi K. 2011. Intranasal vaccination with pneumococcal surface protein A plus poly(I:C) protects against secondary pneumococcal pneumonia in mice. Vaccine 29:1754–1761. doi: 10.1016/j.vaccine.2010.12.117. [DOI] [PubMed] [Google Scholar]
- 26.Lewnard JA, Givon-Lavi N, Huppert A, Pettigrew MM, Regev-Yochay G, Dagan R, Weinberger DM. 23 December 2015. Epidemiological markers for interactions among Streptococcus pneumoniae, Haemophilus influenzae, and Staphylococcus aureus in upper respiratory tract carriage. J Infect Dis Epub ahead of print. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Biesbroek G, Wang X, Keijser BJ, Eijkemans RM, Trzcinski K, Rots NY, Veenhoven RH, Sanders EA, Bogaert D. 2014. Seven-valent pneumococcal conjugate vaccine and nasopharyngeal microbiota in healthy children. Emerg Infect Dis 20:201–210. doi: 10.3201/eid2002.131220. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Xu Q, Almudervar A, Casey JR, Pichichero ME. 2012. Nasopharyngeal bacterial interactions in children. Emerg Infect Dis 18:1738–1745. doi: 10.3201/eid1811.111904. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Madhi SA, Adrian P, Kuwanda L, Cutland C, Albrich WC, Klugman KP. 2007. Long-term effect of pneumococcal conjugate vaccine on nasopharyngeal colonization by Streptococcus pneumoniae—and associated interactions with Staphylococcus aureus and Haemophilus influenzae colonization—in HIV-infected and HIV-uninfected children. J Infect Dis 196:1662–1666. doi: 10.1086/522164. [DOI] [PubMed] [Google Scholar]
- 30.Klugman KP, Madhi SA, Adegbola RA, Cutts F, Greenwood B, Hausdorff WP. 2011. Timing of serotype 1 pneumococcal disease suggests the need for evaluation of a booster dose. Vaccine 29:3372–3373. doi: 10.1016/j.vaccine.2011.02.089. [DOI] [PubMed] [Google Scholar]
- 31.Briles DE, Hollingshead SK, Nabors GS, Paton JC, Brooks-Walter A. 2000. The potential for using protein vaccines to protect against otitis media caused by Streptococcus pneumoniae. Vaccine 19(Suppl 1):S87–S95. doi: 10.1016/S0264-410X(00)00285-1. [DOI] [PubMed] [Google Scholar]
- 32.Greene CJ, Chadwick CM, Mandell LM, Hu JC, O'Hara JM, Brey RN, Mantis NJ, Connell TD. 2013. LT-IIb(T13I), a nontoxic type II heat-labile enterotoxin, augments the capacity of a ricin toxin subunit vaccine to evoke neutralizing antibodies and protective immunity. PLoS One 8:e69678. doi: 10.1371/journal.pone.0069678. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.National Research Council. 1996. Guide for the care and use of laboratory animals. National Academy Press, Washington, DC. [Google Scholar]
- 34.Briles DE, Ades E, Paton JC, Sampson JS, Carlone GM, Huebner RC, Virolainen A, Swiatlo E, Hollingshead SK. 2000. Intranasal immunization of mice with a mixture of the pneumococcal proteins PsaA and PspA is highly protective against nasopharyngeal carriage of Streptococcus pneumoniae. Infect Immun 68:796–800. doi: 10.1128/IAI.68.2.796-800.2000. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Nawar HF, Arce S, Russell MW, Connell TD. 2005. Mucosal adjuvant properties of mutant LT-IIa and LT-IIb enterotoxins that exhibit altered ganglioside-binding activities. Infect Immun 73:1330–1342. doi: 10.1128/IAI.73.3.1330-1342.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Andersson B, Dahmen J, Frejd T, Leffler H, Magnusson G, Noori G, Eden CS. 1983. Identification of an active disaccharide unit of a glycoconjugate receptor for pneumococci attaching to human pharyngeal epithelial cells. J Exp Med 158:559–570. doi: 10.1084/jem.158.2.559. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Avery OT, Macleod CM, McCarty M. 1944. Studies on the chemical nature of the substance inducing transformation of pneumococcal types: induction of transformation by a deoxyribonucleic acid fraction isolated from pneumococcus type III. J Exp Med 79:137–158. doi: 10.1084/jem.79.2.137. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.McDaniel LS, Yother J, Vijayakumar M, McGarry L, Guild WR, Briles DE. 1987. Use of insertional inactivation to facilitate studies of biological properties of pneumococcal surface protein A (PspA). J Exp Med 165:381–394. doi: 10.1084/jem.165.2.381. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Bowers EF, Jeffries LR. 1955. Optochin in the identification of str. pneumoniae. J Clin Pathol 8:58–60. doi: 10.1136/jcp.8.1.58. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Tyx RE, Roche-Hakansson H, Hakansson AP. 2011. Role of dihydrolipoamide dehydrogenase in regulation of raffinose transport in Streptococcus pneumoniae. J Bacteriol 193:3512–3524. doi: 10.1128/JB.01410-10. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Schmittgen TD, Livak KJ. 2008. Analyzing real-time PCR data by the comparative CT method. Nat Protoc 3:1101–1108. doi: 10.1038/nprot.2008.73. [DOI] [PubMed] [Google Scholar]
- 42.Martin M, Metzger DJ, Michalek SM, Connell TD, Russell MW. 2000. Comparative analysis of the mucosal adjuvanticity of the type II heat-labile enterotoxins LT-IIa and LT-IIb. Infect Immun 68:281–287. doi: 10.1128/IAI.68.1.281-287.2000. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Marks LR, Reddinger RM, Hakansson AP. 2012. High levels of genetic recombination during nasopharyngeal carriage and biofilm formation in Streptococcus pneumoniae. mBio 3:e00200–12. doi: 10.1128/mBio.00200-12. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Wu HY, Nahm MH, Guo Y, Russell MW, Briles DE. 1997. Intranasal immunization of mice with PspA (pneumococcal surface protein A) can prevent intranasal carriage, pulmonary infection, and sepsis with Streptococcus pneumoniae. J Infect Dis 175:839–846. doi: 10.1086/513980. [DOI] [PubMed] [Google Scholar]
- 45.Short KR, Diavatopoulos DA, Thornton R, Pedersen J, Strugnell RA, Wise AK, Reading PC, Wijburg OL. 2011. Influenza virus induces bacterial and nonbacterial otitis media. J Infect Dis 204:1857–1865. doi: 10.1093/infdis/jir618. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Quin LR, Moore QC, McDaniel LS. 2007. Pneumolysin, PspA, and PspC contribute to pneumococcal evasion of early innate immune responses during bacteremia in mice. Infect Immun 75:2067–2070. doi: 10.1128/IAI.01727-06. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Ren B, Szalai AJ, Hollingshead SK, Briles DE. 2004. Effects of PspA and antibodies to PspA on activation and deposition of complement on the pneumococcal surface. Infect Immun 72:114–122. doi: 10.1128/IAI.72.1.114-122.2004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Hollingshead SK, Baril L, Ferro S, King J, Coan P, Briles DE. 2006. Pneumococcal surface protein A (PspA) family distribution among clinical isolates from adults over 50 years of age collected in seven countries. J Med Microbiol 55:215–221. doi: 10.1099/jmm.0.46268-0. [DOI] [PubMed] [Google Scholar]
- 49.Briles DE, Tart RC, Swiatlo E, Dillard JP, Smith P, Benton KA, Ralph BA, Brooks-Walter A, Crain MJ, Hollingshead SK, McDaniel LS. 1998. Pneumococcal diversity: considerations for new vaccine strategies with emphasis on pneumococcal surface protein A (PspA). Clin Microbiol Rev 11:645–657. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Briles DE, Hollingshead SK, King J, Swift A, Braun PA, Park MK, Ferguson LM, Nahm MH, Nabors GS. 2000. Immunization of humans with recombinant pneumococcal surface protein A (rPspA) elicits antibodies that passively protect mice from fatal infection with Streptococcus pneumoniae bearing heterologous PspA. J Infect Dis 182:1694–1701. doi: 10.1086/317602. [DOI] [PubMed] [Google Scholar]
- 51.Gwaltney JM, Sande MA, Austrian R, Hendley JO. 1975. Spread of Streptococcus pneumoniae in families. II. Relation of transfer of S. pneumoniae to incidence of colds and serum antibody. J Infect Dis 132:62–68. [DOI] [PubMed] [Google Scholar]
- 52.Lipsitch M, Abdullahi O, D'Amour A, Xie W, Weinberger DM, Tchetgen Tchetgen E, Scott JA. 2012. Estimating rates of carriage acquisition and clearance and competitive ability for pneumococcal serotypes in Kenya with a Markov transition model. Epidemiology 23:510–519. doi: 10.1097/EDE.0b013e31824f2f32. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Hill PC, Townend J, Antonio M, Akisanya B, Ebruke C, Lahai G, Greenwood BM, Adegbola RA. 2010. Transmission of Streptococcus pneumoniae in rural Gambian villages: a longitudinal study. Clin Infect Dis 50:1468–1476. doi: 10.1086/652443. [DOI] [PubMed] [Google Scholar]
- 54.Levine OS, O'Brien KL, Knoll M, Adegbola RA, Black S, Cherian T, Dagan R, Goldblatt D, Grange A, Greenwood B, Hennessy T, Klugman KP, Madhi SA, Mulholland K, Nohynek H, Santosham M, Saha SK, Scott JA, Sow S, Whitney CG, Cutts F. 2006. Pneumococcal vaccination in developing countries. Lancet 367:1880–1882. doi: 10.1016/S0140-6736(06)68703-5. [DOI] [PubMed] [Google Scholar]
- 55.Gonzalez BE, Jacobs MR. 2013. The potential of human nasal colonization with Streptococcus pneumoniae as a universal pneumococcal vaccine. Am J Respir Crit Care Med 187:794–795. doi: 10.1164/rccm.201302-0361ED. [DOI] [PubMed] [Google Scholar]
- 56.Black S, Shinefield H, Baxter R, Austrian R, Elvin L, Hansen J, Lewis E, Fireman B. 2006. Impact of the use of heptavalent pneumococcal conjugate vaccine on disease epidemiology in children and adults. Vaccine 24(Suppl S2:S79–S80. doi: 10.1016/j.vaccine.2005.01.132. [DOI] [PubMed] [Google Scholar]
- 57.Jackson LA, Janoff EN. 2008. Pneumococcal vaccination of elderly adults: new paradigms for protection. Clin Infect Dis 47:1328–1338. doi: 10.1086/592691. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Bogaert D, van Belkum A, Sluijter M, Luijendijk A, de Groot R, Rumke HC, Verbrugh HA, Hermans PW. 2004. Colonisation by Streptococcus pneumoniae and Staphylococcus aureus in healthy children. Lancet 363:1871–1872. doi: 10.1016/S0140-6736(04)16357-5. [DOI] [PubMed] [Google Scholar]
- 59.Veenhoven R, Bogaert D, Uiterwaal C, Brouwer C, Kiezebrink H, Bruin J, IJ E, Hermans P, de Groot R, Zegers B, Kuis W, Rijkers G, Schilder A, Sanders E. 2003. Effect of conjugate pneumococcal vaccine followed by polysaccharide pneumococcal vaccine on recurrent acute otitis media: a randomised study. Lancet 361:2189–2195. doi: 10.1016/S0140-6736(03)13772-5. [DOI] [PubMed] [Google Scholar]
- 60.CDC. 2009. Surveillance for pediatric deaths associated with 2009 pandemic influenza A (H1N1) virus infection—United States, April-August 2009. MMWR Morb Mortal Wkly Rep 58:941–947. [PubMed] [Google Scholar]
- 61.Tasher D, Stein M, Simões EA, Shohat T, Bromberg M, Somekh E. 2011. Invasive bacterial infections in relation to influenza outbreaks, 2006-2010. Clin Infect Dis 53:1199–1207. doi: 10.1093/cid/cir726. [DOI] [PubMed] [Google Scholar]
- 62.Cunha BA, Syed U, Strollo S. 2011. Swine influenza (H1N1) pneumonia in hospitalized adults: chest film findings. Heart Lung 40:253–256. doi: 10.1016/j.hrtlng.2010.07.013. [DOI] [PubMed] [Google Scholar]
- 63.Vance DJ, Greene CJ, Rong Y, Mandell LM, Connell TD, Mantis NJ. 2015. Comparative adjuvant effects of type II heat-labile enterotoxins in combination with two different candidate ricin toxin vaccine antigens. Clin Vaccine Immunol 22:1285–1293. doi: 10.1128/CVI.00402-15. [DOI] [PMC free article] [PubMed] [Google Scholar]