

Abstract
Trypanosoma cruzi species is categorized into six discrete typing units (TcI to TcVI) of which TcI is most abundantly noted in the sylvatic transmission cycle and considered the major cause of human disease. In our study, the TcI strains Colombiana (COL), SylvioX10/4 (SYL), and a cultured clone (TCC) exhibited different biological behavior in a murine model, ranging from high parasitemia and symptomatic cardiomyopathy (SYL), mild parasitemia and high tissue tropism (COL), to no pathogenicity (TCC). Proteomic profiling of the insect (epimastigote) and infective (trypomastigote) forms by two-dimensional gel electrophoresis/matrix-assisted laser desorption ionization–time of flight mass spectrometry, followed by functional annotation of the differential proteome data sets (≥2-fold change, P < 0.05), showed that several proteins involved in (i) cytoskeletal assembly and remodeling, essential for flagellar wave frequency and amplitude and forward motility of the parasite, and (ii) the parasite-specific antioxidant network were enhanced in COL and SYL (versus TCC) trypomastigotes. Western blotting confirmed the enhanced protein levels of cytosolic and mitochondrial tryparedoxin peroxidases and their substrate (tryparedoxin) and iron superoxide dismutase in COL and SYL (versus TCC) trypomastigotes. Further, COL and SYL (but not TCC) were resistant to exogenous treatment with stable oxidants (H2O2 and peroxynitrite [ONOO−]) and dampened the intracellular superoxide and nitric oxide response in macrophages, and thus these isolates escaped from macrophages. Our findings suggest that protein expression conducive to increase in motility and control of macrophage-derived free radicals provides survival and persistence benefits to TcI isolates of T. cruzi.
INTRODUCTION
Chagas disease, caused by the unicellular protozoan Trypanosoma cruzi, is ranked as the third most important parasitic disease in terms of disability-adjusted life years (1, 2). T. cruzi is naturally transmitted by Triatomine insects. In recent years, a significant proportion of the infected population has emigrated from rural areas, leading to the urbanization of Chagas disease in countries where the disease is endemic, as well as internationally (1). Chagas disease is therefore an emergent global public health problem associated with congenital transmission (3, 4), blood transfusions (5), and organ transplantations (6). Current estimates suggest that ∼70 million people are at risk of infection (7, 8) and that ∼6 million individuals are infected with T. cruzi in Latin America and Mexico. However, these estimates may not be accurate, since they are not derived from detailed epidemiological studies. For example, in Argentina, the Chagas Disease National Control Program reported 13 provinces located in the Central-Northeast area as endemic for Chagas disease in 2010 (9). Vectorial transmission was reported to be absent in Jujuy, Entre Ríos, La Pampa, Neuquén, and Río Negro but active in provinces such as Formosa, Chaco, Córdoba, Santiago del Estero, and others, with a Triatoma infestans infestation index of 18.1% (national average, 10.2%) (9). In the state of Salta that shares borders with Bolivia and Paraguay, national data indicated a 7.5% seroprevalence (5.7% for blood donors and 1.82% among young children); however, several research groups reported up to 27% seropositivity from field surveys in restricted geographical areas (10, 11). These observations may mean the existence of heterogeneity in the epidemiologic situation related to Chagas disease in regions where it is endemic.
Acute Chagas disease carries a mortality rate of <5% (12), and an asymptomatic or indeterminate stage is most commonly presented in those surviving the acute infection. However, several decades later, infected individuals develop clinically symptomatic cardiac (∼30%) or digestive (8 to 10%) forms of Chagas disease (13). The chagasic megacolon is primarily presented in cases in Central Brazil and Chile but is largely nonexistent in those in Venezuela and parts of Central America (14). The situation in Argentina is not very clear; however, epidemiological data suggest that genetic variations of both host and the parasite play an important role in determining the development of clinical disease in infected individuals.
Trypanosoma (Schizotrypanum) cruzi belongs to the order Kinetoplastida, which comprises flagellar organisms with a kinetoplast organelle located in the cell mitochondrion. T. cruzi is included in the stercorarian section, together with the group of trypanosomes whose infective stage develops in the vectors' digestive tract and contaminates the mammalian hosts through feces (15). The subgenus Schizotrypanum has been adopted to identify the intracellular replicative stage. At present, based upon molecular and multilocus enzyme electrophoresis analyses, T. cruzi isolates are classified into six discrete typing units (DTU) referred to as TcI to TcVI (16). Of these, TcI has the widest distribution and is the most common cause of disease in Colombia and Venezuela (17), and in the northern region of South America and the United States (4, 18). Although T. cruzi isolates belonging to other DTU (e.g., TcII, TcV, and TcVI) have been identified in peripheral blood of seropositive patients in Southern Cone countries of South America (19), TcI isolates were mainly reported in heart biopsy specimens from chronic chagasic patients in the Southern Cone countries (20, 21). These findings suggested to us that patients could be carrying a mixed infection, with TcI associated with the development of clinical cardiac disease. Other studies have, however, failed to find a reliable association between cardiomyopathy and TcI DTU (22).
In this study, we aimed to identify whether diversity at the virulence level exists among the TcI isolates of T. cruzi, and we investigated the potential molecular mechanism(s) responsible for diversity in virulence and pathogenicity of the TcI isolates. We examined the infectivity and virulence of three TcI strains chosen based upon their diverse origin, which included the Colombiana strain (COL) recovered from a chronically infected human patient from Colombia (23); the Sylvio-X10 strain (SYL) isolated from an acutely infected human patient in Brazil (24); and the TCC laboratory strain (TCC) cloned from the continuous axenic culture of T. cruzi isolated from an infected human patient in Chile (25). We performed global protein expression profiling of the insect (epimastigote) and infective (trypomastigote) stages of the three strains to identify the proteins that might be differentially expressed in a stage- or strain-specific manner and contribute to differences in the disease outcome. We validated the proteome results by Western blotting and performed in vitro and in vivo studies to look at the protein networks that might be responsible for variations in parasites' survival in macrophages and virulence in mice. Our results suggested that a gene/protein expression profile conducive to enhanced flagellar motility and peroxiredoxin-dependent decomposition of macrophage-derived free radicals provided survival and persistence benefits to TcI DTU isolates. We discuss the significance of our observations in relation to T. cruzi modulation of host responses for ensuring its intracellular survival, dissemination, and persistence resulting in chronic disease.
MATERIALS AND METHODS
Parasites, cell culture, and mice.
All chemicals were of molecular grade and purchased from Sigma-Aldrich (St. Louis, MO) unless stated otherwise. Epimastigotes of TcI strains of T. cruzi (COL, SYL, and TCC) were cultured in liver infusion tryptose–10% fetal bovine serum (FBS) medium supplemented with 25 μg of hemin/ml, 100 U of penicillin/ml, and 100 mg of streptomycin/ml. T. cruzi trypomastigotes were maintained and propagated by continuous in vitro passage in C2C12 cells. RAW 264.7 macrophages were cultured in Dulbecco modified Eagle medium high glucose–10% FBS medium (Life Technologies, Grand Island, NY). Macrophages were seeded in six-well (5 × 104/well), 12-well (2 × 104/well), or 96-well (0.5 × 104/well) plates or Nunc Lab-Tek II chamber slides (1 × 104/well), infected with T. cruzi trypomastigotes (cell/parasite ratio, 1:3) for 3 h, washed to remove free parasites, and then incubated according to experimental requirements.
All animal experiments were performed according to the National Institutes of Health Guide for the Care and Use of Experimental Animals and approved by the institutional ethics committee at the Universidad Nacional de Salta. Swiss mice were bred at the animal facility at the School of Health Science, National University of Salta, Salta, Argentina. T. cruzi epimastigotes were incubated in the presence of 1% triatomine gut homogenate to facilitate differentiation to metacyclic trypomastigotes (26). Complement-resistant metacyclic trypomastigotes were purified by incubation of stationary culture in the presence of normal human serum. Swiss mice (6 to 8 weeks old) were infected by intraperitoneal injection with T. cruzi (104 metacyclic trypomastigotes per mouse). Survival from infection was monitored daily, and blood parasitemia was monitored at 2-day intervals for up to 56 days postinfection (p.i.). Mice were sacrificed in the chronic disease phase (>120 days p.i.), and blood and tissue samples (heart, skeletal muscle) were harvested and stored at −80°C.
Histology.
Tissue sections were fixed in 10% buffered formalin for 24 h, dehydrated in absolute ethanol, cleared in xylene, and embedded in paraffin. Then, 5-μm sections of the heart and skeletal muscle tissue (≥3 tissue sections/organ/mouse) were stained with hematoxylin and eosin (H&E) and evaluated by two observers in a blinded fashion by light microscopy using an Olympus BX-15 microscope (Center Valley, PA) equipped with a digital camera and Simple PCI software (v.6.0; Compix, Sewickley, PA). Inflammatory lesions (myocarditis) were scored as follows: 0, absent; 1, focal or mild with ≤1 foci; 2, moderate with ≥2 inflammatory foci; 3, severe with extensive generalized coalescing of inflammatory foci or disseminated inflammation; or 4, diffused inflammation with severe tissue necrosis, interstitial edema, and loss of integrity (27). Inflammatory infiltrate was characterized as diffused or focal depending upon how closely the inflammatory cells were associated. The parasitic or inflammatory foci in skeletal muscle and heart tissue sections were scored as follows: 0, absent; 1, 0 to 1 foci; 2, 1 to 5 foci; and 3, >5 foci.
T. cruzi homogenates, two-dimensional electrophoresis (2DE), and image analysis.
T. cruzi epimastigotes or trypomastigotes were washed with cold phosphate-buffered saline and lysed in 7 M urea, 2 M thiourea, 4% CHAPS {3-[(3-cholamidopropyl)-dimethylammonio]-1-propanesulfonate}, 12 mM EDTA, and 40 mM Tris (pH 7.6) containing protease inhibitor cocktail (1 mM phenylmethylsulfonyl fluoride, 0.001 mM pepstatin, and 0.1 mM leupeptin). Parasite pellets were added to the lysis buffer (1 g [wet weight] per 3 ml of lysis buffer), vortexed for 3 min, incubated on ice for 10 min, and centrifuged at 1,000 × g for 5 min to remove the cell debris. Protein concentrations were determined by using a Bio-Rad protein assay (Bio-Rad, Hercules, CA).
T. cruzi lysates (100 μg of protein) were separated by 2DE, employing an IPGphor multiple sample isoelectric focusing (IEF) device (GE Healthcare, Little Chalfont, United Kingdom) in the first dimension and a Criterion Dodeca cell (Bio-Rad) in the second dimension, as described previously (28). Briefly, samples were loaded onto 11-cm dehydrated, precast, immobilized pH gradient (IPG) strips (pH 3 to 10 and pH 5 to 8; Bio-Rad), and the strips were rehydrated overnight in 1 M thiourea, 8 M urea, 2% CHAPS, 1% dithiothreitol, and 0.2% ampholyte rehydration buffer containing 0.002% bromophenol blue. The IEF was performed at 20°C using the following parameters: 50 V, 11 h; 250 V, 1 h; 500 V, 1 h; 1000 V, 1 h; 8,000 V, 2 h; and then at 8,000 V (for a total of 50,000 V). The IPG strips were transferred to equilibration buffer (50 mM Tris-HCl [pH 6.8], 6 M urea, 20% glycerol) and sequentially incubated for 15 min each in the presence of 2% dithiothreitol–2% sodium dodecyl sulfate (SDS; reducing conditions) and 2.5% iodoacetamide–2% SDS (alkylating conditions). Equilibrated IPG strips were subjected to 2DE by using 8 to 10% linear gradient precast Tris-HCl gels (Bio-Rad) in Tris-glycine-SDS buffer (25 mM Tris-HCl, 192 mM glycine, 0.1% SDS [pH 8.3]). Electrophoresis was performed on a Protean plus Dodeca cell system at 75 V for 1 h and then at 120 V until the dye front reached the bottom of the gel.
Gels were fixed in 10% methanol–7% acetic acid, stained with SYPRO Ruby (Bio-Rad), and destained in 10% ethanol–7% acetic acid to remove the background. Gel images were acquired at an excitation wavelength of 488 nm and an emission wavelength of 560 nm by using a high-resolution ProXPRESS proteomic imaging system (Perkin-Elmer, Waltham, MA). The exposure time was adjusted to achieve a value of ∼55,000 to 63,000 pixel intensity (16-bit saturation) from the most intense protein spots on the gel (28).
All 2D gels, representing at least four samples per stage (epimastigote and trypomastigote) from the COL, SYL, and TCC isolates, were scanned and analyzed with the Progenesis SameSpots software 2.0 (NonLinear Dynamics, Durham, NC). After manual and automated pixel-to-pixel alignment, the program performed automatic spot detection on all images. Normalized spot volumes, i.e., the volume of each spot over the volume of all spots in the gel, were used to compare the different groups, and candidates were identified as protein spots that changed in expression level by ≥2-fold versus their specific control. Statistical significance was assessed by a two-tailed Student t test and analysis of variance test (analysis of variance [ANOVA]), and P values of <0.05 were considered significant for comparisons between control and experimental data.
Mass spectrometry (MS) and protein identification.
Selected spots (<1 mm) on the 2D gels containing the protein of interest were excised and trypsin digested as described previously by us (29, 30). Briefly, gel spots were incubated at 37°C for 30 min in 50 mM NH4HCO3 and dehydrated twice for 5 min each time in 100 μl of acetonitrile, and the proteins were digested in gel at 37°C overnight with 10 μl of trypsin solution (1% trypsin in 25 mM ammonium bicarbonate). Peptide mixtures (1 μl) were directly spotted onto a matrix-assisted laser desorption ionization–time of flight (MALDI-TOF) tandem MS (MS/MS) target plate with 1 μl of alpha-cyano-4-hydroxycinnamic acid matrix solution (5 mg/ml in 50% acetonitrile) and analyzed by using a MALDI-TOF/TOF ABI 4800 proteomics analyzer (Applied Biosystems, Foster City, CA). The Applied Biosystems software package included 4000 Series Explorer (v3.6 RC1) with Oracle Database Schema Version (v. 3.19.0) and Data Version (3.80.0) to acquire and analyze MS and MS/MS spectral data. The instrument was operated in positive ion reflectron mode with the focus mass set at 1,700 Da (the mass range was 850 to 3,000 Da). For MS data, 2,000 to 4,000 laser shots were acquired and averaged from each protein spot. A peptide mixture with reference masses of 904.468, 1,296.685, 1,570.677, and 2,465.199 kDa was used for automatic external calibration. After MALDI MS analysis, MALDI MS/MS was performed on several (5 to 10) abundant ions from each sample spot. A 1-kV positive-ion MS/MS method was used to acquire data under post-source-decay conditions. The instrument precursor selection window was ±3 Da. For MS/MS data, 2,000 laser shots were acquired and averaged from each sample spot. Automatic external calibration was performed using reference fragment masses 175.120, 480.257, 684.347, 1,056.475, and 1,441.635 (28).
For protein identification, the MS and MS/MS spectral data were searched against the National Center for Biotechnology Information, UniProtKB/Swiss-Prot, and TryTryp databases (accessed on 21 July 2013) using AB Sciex GPS Explorer (v.3.6) software in conjunction with MASCOT (v2.2.07) as described previously (28). Briefly, protein match probabilities were determined by using expectation values and/or MASCOT protein scores. The MS peak filtering included the indicated parameters—mass range, 800 to 4,000 Da; minimum signal-to-noise ratio (S/N) filter, 10; mass exclusion list tolerance, 0.5 Da—and the mass exclusion list for some trypsin and keratin-containing compounds included masses of 842.51, 870.45, 1,045.56, 1,179.60, 1,277.71, 1,475.79, and 2,211.1 Da. The MS/MS peak filtering included the following parameters: minimum S/N filter, 10; maximum missed cleavages, 1; fixed modification of carbamidomethyl (C); variable modifications due to oxidation (M); precursor tolerance, 0.2 Da; MS/MS fragment tolerance, 0.3 Da; mass, monoisotopic; and peptide charges, +1. The significance of a protein match, based on both the peptide mass fingerprint (PMF) in the MS and the MS/MS data from several precursor ions, is presented as the expectation value. The expectation value is the number of matches with equal or better scores that are expected to occur by chance alone. The default significance threshold is P < 0.05; however, we used a stringent threshold of 10−3 for protein identification: the lower the expectation value, the more significant the score.
Western blotting.
Polyclonal antibodies against T. cruzi cytosolic tryparedoxin peroxidase (cTXNPx) and mitochondrial tryparedoxin peroxidase (mTXNPx) were obtained as described previously (31, 32). Polyclonal antibodies against the iron superoxide dismutase (FeSOD) isoforms, FeSODa and FeSODb, and the tryparedoxin isoforms, TXN I and TXN II, were produced as described previously (31, 32).
T. cruzi epimastigotes (5 × 108 cells/ml) and trypomastigotes (1 × 109 cells/ml) were lysed in 10 mM Tris-HCl (pH 7.5), 5 mM EDTA, and 1% Nonidet P-40 containing protease inhibitor cocktail (Sigma-Aldrich). Parasite lysates (30 μg/10 μl) were mixed with equal volumes of 2× loading buffer (100 mM Tris-Cl [pH 6.8], 4% SDS, 0.2% bromophenol blue, 20% glycerol, and 200 mM β-mercaptoethanol). Samples were resolved by 10% SDS-PAGE, and the proteins were transferred onto polyvinylidene difluoride (PVDF) membranes by using a wet vertical Criterion blotter (Bio-Rad). Membranes were blocked for 1 h with 5% nonfat dry milk (Bio-Rad) in 50 mM Tris-HCl (pH 7.4), 150 mM NaCl, and 0.05% Tween 20. Antibodies were diluted in Tris-buffered saline (TBS; 25 mM Tris-HCl [pH 7.6], 140 mM NaCl, and 3 mM KCl) containing 1% bovine serum albumin and 0.1% Tween 20. Membranes were incubated overnight at 4°C with antibodies against mTXNPx (1:2,000 dilution), cTXNPx (1:5,000 dilution), FeSODa and FeSODb (1:2,000 dilution), and TXN1 and TXN2 (1:1,000 dilution). Membranes were then washed three times in TBS and incubated for 1 h with horseradish peroxidase (HRP)-conjugated anti-rabbit IgG (1:10,000 dilution; Sigma-Aldrich). Signal was developed by using an enhanced chemiluminescence detection system (GE Healthcare). Membranes were stained with Ponceau S to confirm equal loading of samples. Images were visualized and digitized, and densitometry analysis was performed on a Fluorchem HD2 system (Alpha Innotech, Santa Clara, CA).
Parasite susceptibility to oxidants.
Activated macrophages exert cytotoxic effects against microorganisms via NADPH oxidase-dependent production of superoxide (O2˙−) that then produces stable and diffusible H2O2 oxidant. The reaction of superoxide with nitric oxide produces peroxynitrite (ONOO−) that is a strong cytotoxic oxidant. To assess the susceptibility of T. cruzi isolates to these oxidants, trypomastigotes were distributed to 96-well plates (105/100 μl/well), followed by incubation in the presence of 250 μM hydrogen peroxide (H2O2) or peroxynitrite (ONOO−). The selected concentrations of oxidants were chosen based upon the dose response curves using 0 to 500 μg of H2O2 and ONOO− that showed <10% of SylvioX10 trypomastigotes died when treated with the highest concentration for 2 h. alamarBlue (10% final concentration; Invitrogen) was added, and its reduction by mitochondrial dehydrogenases of the live parasites, resulting in the formation of resazurin (red, highly fluorescent; excitation, 560 nm/emission, 590 nm), was monitored by using a SpectraMax M5 microplate reader (Molecular Devices, Sunnyvale, CA). The results are expressed as the percentage of viability, which was calculated as follows: (the mean fluorescence of parasites treated with oxidants/the mean fluorescence of parasites untreated) × 100.
Activation of macrophages by T. cruzi: oxidative/nitrosative burst and cytokine release.
RAW 264.7 macrophages were infected with T. cruzi for 3 h (cell/parasite ratio, 1:3), washed with RPMI to remove free parasites, and then incubated for 24, 36, or 48 h. The cells were incubated with 10 μM CM-H2DCF-DA (Molecular Probes, Eugene, OR), and its oxidation by intracellular reactive oxygen species (ROS), resulting in the formation of fluorescent dichlorodihydrofluorescein (DCF; excitation, 498 nm/emission, 598 nm), was monitored on a SpectraMax M5 microplate reader. Fluorescence microscopy for intracellular DCF was performed on an Olympus BX-15 microscope, and images were captured with a mounted digital camera (magnification, ×40).
ROS release in culture supernatants from infected macrophages was measured by an Amplex red assay. Briefly, supernatants (50 μl) were added in triplicate to 96-well, black flat-bottom plates and mixed with 100 μM Amplex red (Molecular Probes) and HRP at 0.3 U/ml. The HRP-catalyzed Amplex red oxidation by H2O2, resulting in fluorescent resorufin formation, was monitored at an excitation wavelength of 563 nm and an emission wavelength of 587 nm (standard curve, 50 nM to 5 μM H2O2) (33).
The inducible nitric oxide synthase (iNOS) activity was monitored by the Griess reagent assay by using a nitrate/nitrite colorimetric assay kit (Cayman, Ann Arbor, MI). Briefly, culture supernatants (80 μl) were reduced by the addition of 0.01 U of nitrite reductase, and 100 μl of 1% sulfanilamide made in 5% phosphoric acid–0.1% N-(1-naphthyl) ethylenediamine dihydrochloride (1:1 ratio [vol/vol]) was added. After incubation for 10 min, the formation of diazonium salt was monitored at 545 nm (standard curve, 2 to 100 μM sodium nitrite). Culture supernatants from infected macrophages were also utilized to measure secreted gamma interferon (IFN-γ), tumor necrosis factor alpha (TNF-α), and interleukin-6 (IL-6) cytokines by using optEIA enzyme-linked immunosorbent assay (ELISA) kits (Pharmingen, San Diego, CA), according to the manufacturer's specifications.
Parasite replication and release in macrophages.
RAW 264.7 macrophages were cultured in 12-well plates, infected with T. cruzi isolates for 3 h, washed to remove free parasites, and then incubated for 6, 12, or 18 h. Total DNA was isolated from infected and uninfected macrophages by using a QIAamp DNA minikit (Qiagen, Chatsworth, CA). The total DNA (50 ng) was used as a template, and real-time PCR performed on an iCycler thermal cycler with SYBR green Supermix (Bio-Rad) and Tc18S-specific (forward, 5′-TAGTCATATGCTTGTTTC-3′; reverse, 5′-GCAACAGCATTAATATACGC-3′) oligonucleotides. Data were normalized to murine GAPDH (glyceraldehyde-3-phosphate dehydrogenase; forward, 5′-TGTGATGGGTGTGAACCACGAGA-3′; reverse, 5′-GAGCCCTTCCACAATGCCAAAGT-3′), and the fold change in T. cruzi burden calculated as 2−ΔΔCT, where ΔCT represents the CT (sample) − the CT (control) (27). To assess parasite release, metacyclic trypomastigotes derived from stationary-phase epimastigote cultures of the different T. cruzi isolates were added to RAW 264.7 macrophages, and the trypomastigote release in the supernatant was monitored for 2 weeks by light microscopy.
Data analysis.
Data (means ± the standard deviations) were derived from at least three independent experiments (triplicate observations per experiment) performed on different days. All data were analyzed by using Prism 5 (GraphPad Software, La Jolla, CA). Data were tested for normal distribution by histogram and Q-Q test and analyzed by using a Student t test (comparison of two groups), and one-way analysis of variance (ANOVA) with Tukey's test (comparison of multiple groups). The level of significance was accepted at P < 0.05.
RESULTS
We used a well-established experimental model to assess the virulence of TcI isolates of T. cruzi. Swiss mice infected with SYL exhibited patent blood parasitemia that peaked at 21 to 28 days p.i. and was controlled by day 49 p.i. (Fig. 1A). Mice infected with COL exhibited a delayed and up to 2-fold lower level of peak parasitemia compared to that noted in SYL-infected mice (P < 0.01 [t test]). In comparison, mice infected with TCC exhibited no detectable parasitemia by light microscopy, although blood PCR for Tc18SrDNA (Fig. 1A) and hemoculture (data not shown) confirmed that mice were infected. Normal Swiss mice were used as controls in all experiments.
FIG 1.
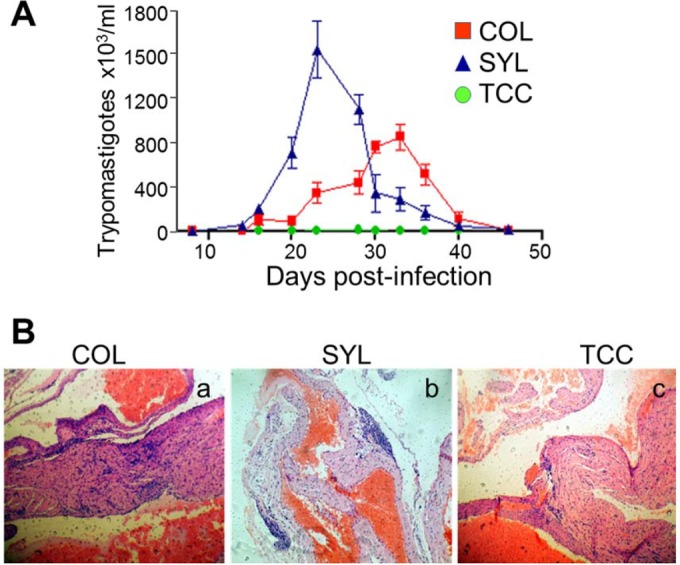
Biological behavior of selected TcI isolates of T. cruzi in mice. Swiss mice were infected with Colombiana (COL), Sylvio X10 (SYL), and TCC strains of T. cruzi (n = 6 per group), as described in Materials and Methods. (A) Blood parasitemia was monitored by light microscopy (10 microscopic fields per sample). (B) Representative H&E staining (blue, nuclear; pink, muscle, cytoplasm, or keratin) of heart tissue sections from COL-, SYL-, and TCC-infected mice, harvested during the chronic disease phase (>120 days postinfection [p.i.]; magnification, ×10). In all figures, data are presented as means ± the SD, and significance is indicated as follows: *, P < 0.05; **, P < 0.01; and ***, P < 0.001.
Elicitation of inflammatory response is important for control of acute T. cruzi infection, but its chronic persistence can contribute to tissue injury (27). Histological studies were performed in chronic stage (>120 days p.i.) to assess the tissue inflammation and myocarditis. The inflammatory foci were observed in all tissue sections (heart and skeletal muscle) of COL- and SYL-infected mice. Representative images of H&E-stained heart tissue sections are presented in Fig. 1B. Mice infected with COL isolate (versus normal/uninfected mice) exhibited extensive and diffused inflammation consisting mainly of interstitial inflammatory infiltrate throughout the heart (histological score, 2.75 ± 0.4 versus 0.3 ± 0.03; P < 0.05 [t test]; Fig. 1Ba) and skeletal muscle (histological score, 3 ± 0.43 versus 0.32 ± 0.03; P < 0.05 [t test]) tissue sections. The extent of inflammatory infiltrate in the heart tissue (score, 2.5 ± 0.45; Fig. 1Bb) and skeletal muscle (score, 3 ± 0.33) of SYL-infected mice was not statistically different than that observed in COL-infected mice. The degeneration of muscle fibers in the heart (Fig. 1B) and skeletal muscle tissue (data not shown) of COL- and SYL-infected mice was also evident. The TCC-infected mice exhibited no overt tissue pathology and minimal inflammatory infiltrate (histological score, 0.25 ± 0.03 and 0.5 ± 0.053 in heart and skeletal muscle, respectively) that was significantly lower than that noted in COL- and SYL-infected mice (P < 0.05 [ANOVA-Tukey]; Fig. 1Bc). These data suggested that COL and SYL (but not TCC) strains elicited blood parasitemia during the acute infection period and persistent inflammatory infiltrate and injury in the myocardium and skeletal tissue during the chronic phase. The extent of blood parasitemia was not directly correlated with chronic persistence of the parasite in the tissues.
To begin to understand why TcI isolates may exhibit a differing virulence in mice, we performed proteomic analysis of the infective trypomastigotes of COL, SYL, and TCC isolates. The proteome profile of the epimastigote (insect, replicative) stage was included to evaluate whether changes in the proteome profile are stage or strain specific. In initial analysis, when using pH 3 to 10 strips, most of the proteins were resolved in the middle or on the acidic side of the second-dimension 8 to 16% gradient gel (data not shown). The narrow-range IPG strips (pH 5 to 8) yielded enhanced resolution of T. cruzi proteins and detection of 2-fold-more spots than were detected by using other IPG strips. We therefore used pH 5 to 8 IPG strips for developing the proteome profile of COL, SYL, and TCC strains (n ≥ 4 per sample). Representative gel images for the epimastigote (Fig. 2Aa to c) and trypomastigote (Fig. 2Ba to c) forms of each T. cruzi strain are shown in Fig. 2. All protein spots were within the relative molecular size of 10 to 250 kDa.
FIG 2.

Proteomic profiling of TcI strains of T. cruzi by 2DE. Epimastigote (A) and trypomastigote (B) stages of T. cruzi COL, SYL, and TCC strains were cultured as described in Materials and Methods. Parasite lysates were separated in the first dimension by IEF on 11-cm linear, pH 5 to 8 immobilized pH gradient strips, and in the second dimension by SDS-PAGE on an 8 to 10% gradient gel. Gel images were obtained by using a high-resolution ProXPRESS proteomic imaging system. Representative gel images of protein lysates from COL (Aa and Ba), SYL (Ab and Bb), and TCC (Ac and Bc) are shown.
The 2D gel images were assessed for quality control by SameSpots software, and gel images were aligned both manually and automatically against the reference epimastigote stage and trypomastigote stage gels (Fig. 3). Spot boundaries were then detected to identify the protein spots reproducibly found in all samples within a group, as well as those that were differentially abundant between any two groups. On average, 1,153, 1,347, and 1,106 protein spots were observed in epimastigotes, and 1,463, 1,172, and 1,475 protein spots were observed in trypomastigotes of COL, SYL, and TCC isolates, respectively.
FIG 3.

Identification of differentially abundant protein spots in T. cruzi TcI strains. Protein spots that exhibited significant changes in abundance in epimastigote (A) and trypomastigote (B) stages of COL and SYL (versus TCC, P < 0.05) are marked on representative gel images of SYL lysate. Protein spots were subjected to MALDI-TOF MS analysis for protein identification (see Tables 1 and 2).
For the purpose of selecting protein spots for identification by MS, the data sets were analyzed in a pairwise manner by a t test with Welch's correction that accounts for unequal variances. This analysis yielded 55, 47, and 30 differentially abundant protein spots (fold change, ≥2.0; P < 0.05 [t test]) in COL-versus-SYL, SYL-versus-TCC, and COL-versus-TCC epimastigotes, respectively. Likewise, we identified 50, 33, and 40 differentially abundant protein spots (fold change, ≥2.0; P < 0.05 [t test]) in COL-versus-SYL, SYL-versus-TCC, and COL-versus-TCC trypomastigotes, respectively. These protein spots were subjected to MALDI-TOF MS/MS analysis, and homology searches were conducted against the TryTryp database for protein identification. The protein spots that were differentially abundant in epimastigotes and trypomastigotes of COL, SYL, or TCC (fold change, ≥2.0, P < 0.05) and identified with high confidence (E value < 0.001) are listed in Tables 1 and 2, respectively.
TABLE 1.
Protein spots that were differentially expressed in epimastigote stage of TcI isolates of T. cruzia
| Protein spot | Protein name (gene) | NCBI accession no. | Mol mass (Da) | Peptide count | Protein score | Protein score (CI%) | pI | E value |
|---|---|---|---|---|---|---|---|---|
| Spots conserved in the three isolates | ||||||||
| 1 | Chain A, tyrosine aminotransferase (TcTAT) | gi|6137418 | 46,120.3 | 12 | 188 | 100 | 5.72 | 3.155E–13 |
| 3 | Tyrosine aminotransferase (TcTAT) | gi|71407311 | 46,238.5 | 13 | 442 | 100 | 5.82 | 1.256E–38 |
| 4 | Tyrosine aminotransferase N-term (TcTAT) | gi|71655339 | 27,698.9 | 12 | 400 | 100 | 5.71 | 1.991E–34 |
| 5 | l-Tyrosine:2-oxoglutarate aminotransferase (TcTAT) | gi|1168606 | 46,137.4 | 20 | 737 | 100 | 5.82 | 3.972E–68 |
| 6 | Tyrosine aminotransferase (TcTAT) | gi|71413938 | 46,100.4 | 16 | 537 | 100 | 5.66 | 3.972E–48 |
| 7 | NADPH flavin oxidoreductase (old yellow enzyme, TcOYE) | gi|61741942 | 42,191.6 | 19 | 902 | 100 | 6.03 | 1.256E–84 |
| 8 | NADPH flavin oxidoreductase (old yellow enzyme, TcOYE) | gi|61741950 | 42,233.6 | 18 | 889 | 100 | 5.97 | 2.506E–83 |
| 9 | l-Threonine 3-dehydrogenase (TcTDH) | gi|71406160 | 36,880.7 | 8 | 181 | 100 | 6.71 | 1.581E–12 |
| 10 | NADPH flavin oxidoreductase (old yellow enzyme, TcOYE) | gi|61741952 | 42,080.5 | 20 | 749 | 100 | 6.03 | 2.506E–69 |
| 11 | Aldo-keto reductase/quinone oxido-reductase (AKR) | gi|189396135 | 32,485.5 | 13 | 499 | 100 | 7.18 | 1.256E–43 |
| 12 | Aldo-keto reductase/quinone oxido-reductase (AKR) | gi|14279174 | 31,764.2 | 13 | 376 | 100 | 8.58 | 2.506E–31 |
| Protein spots differentially expressed in COL (compared to TCC) | ||||||||
| 13 | Proline racemase B (TcPRACB) | gi|74812220 | 38,440.6 | 12 | 442 | 100 | 5.44 | 1.256E–38 |
| 14 | Proline racemase N-term (TcPRACB) | gi|71400794 | 32,411.5 | 11 | 435 | 100 | 5.63 | 6.295E–38 |
| 15 | Major paraflagellar rod protein 2 (TcPAR2) | gi|162179 | 69,504.9 | 19 | 355 | 100 | 5.85 | 6.295E–30 |
| 16 | Paraflagellar rod protein N-term (TcPAR) | gi|71411387 | 42,471.9 | 16 | 355 | 100 | 6.12 | 6.295E–30 |
| 17 | Paraxonemal (paraflagellar) rod protein (TcPAR2) | gi|71398801 | 31,336.1 | 10 | 164 | 100 | 5.95 | 7.924E–11 |
| 18 | 2-Hydroxyacid dehydrogenase N-term, d-isomer (2-Hacid-dh) | gi|71406187 | 28,516.7 | 16 | 567 | 100 | 6.17 | 3.972E–51 |
| 20 | Short interspersed repetitive element (TcSIRE) | gi|1911172 | 11,565 | 10 | 530 | 100 | 5.08 | 1.991E–47 |
| 21 | 2-Hydroxyacid dehydrogenase, d-isomer (2-Hacid-dh) | gi|71406189 | 22,228.2 | 11 | 296 | 100 | 5.87 | 5E–24 |
| 23 | HpcH/HpaI aldolase (TcP28) | gi|4558456 | 29,886.2 | 7 | 156 | 100 | 6.06 | 5E–10 |
| 24 | Pyridoxal kinase (TcPDXK) | gi|71418579 | 33,273.6 | 4 | 125 | 100 | 5.93 | 6.295E–07 |
| 25 | NADP-dependent malic enzyme N term (TcME1) | gi|71662506 | 62,705.5 | 12 | 307 | 100 | 6.55 | 3.972E–25 |
| 26 | NADP-dependent malic enzyme N term (TcME1) | gi|71651465 | 62,634.4 | 14 | 300 | 100 | 6.37 | 1.991E–24 |
| 27 | Cytochrome c oxidase subunit IV (TcCOXIV) | gi|71660419 | 38,924.4 | 16 | 351 | 100 | 5.72 | 1.581E–29 |
| 28 | Tryparedoxin peroxidase (TcTXNPx) | gi|17224953 | 22,246.1 | 11 | 413 | 100 | 5.96 | 9.976E–36 |
| 29 | Tryparedoxin peroxidase (TcTXNPx) | gi|71399514 | 22,375.1 | 11 | 411 | 100 | 6.32 | 1.581E–35 |
| 30 | Tryparedoxin peroxidase (TcTXNPx) | gi|4388655 | 25,545.3 | 22 | 167 | 100 | 7.56 | 2.506E–28 |
| 31 | Calpain-like cysteine peptidase (TcCAPN1) | gi|71411006 | 12,798.3 | 6 | 179 | 100 | 5.07 | 1.256E–11 |
| 32 | Beta tubulin of T. cyclops (TcTUBB) | gi|91983197 | 44,496.8 | 13 | 410 | 100 | 5.68 | 1.991E–35 |
| 33 | Beta tubulin of T. danilewskyi (TdTUBB) | gi|74229926 | 49,695.8 | 12 | 391 | 100 | 4.74 | 1.581E–33 |
| 34 | Alpha tubulin of T. cyclops (TcTUBA) | gi|91983196 | 49,777.5 | 19 | 677 | 100 | 4.93 | 1.991E–61 |
| 35 | Alpha tubulin of T. brucei (TbTUBA) | gi|115504283 | 49,755.5 | 21 | 646 | 100 | 4.93 | 2.506E–58 |
| 36 | Elongation factor 1α (TcEF1A) | gi|71408910 | 49,083.4 | 19 | 416 | 100 | 8.93 | 5E–36 |
| 37 | Ribokinase (TcRBKS) | gi|71659743 | 35,262.6 | 7 | 158 | 100 | 6.95 | 3.155E–10 |
| 38 | Heat shock protein 70 partial (TcHSP70) | gi|10626 | 74,233.7 | 31 | 626 | 100 | 5.64 | 2.506E–56 |
| 39 | Heat shock protein HSP70 (TcHSP70) | gi|162117 | 71,210.1 | 30 | 623 | 100 | 5.42 | 5E–56 |
| 40 | Heat shock 70-kDa protein, mitochondrial (TcHSP70-10) | gi|71407515 | 70,946 | 24 | 516 | 100 | 5.75 | 2.506E–45 |
| Spots differentially expressed in SYL (compared to TCC) | ||||||||
| 53 | Cytochrome c oxidase subunit IV (TcCOXIV) | gi|71667854 | 38,884.4 | 15 | 339 | 100 | 5.72 | 2.506E–28 |
| 54 | Tryparedoxin peroxidase (TcTXNPx) | gi|71413207 | 22,376.1 | 10 | 357 | 100 | 5.69 | 3.972E–30 |
| 55 | Tryparedoxin peroxidase (TcTXNPx) | gi|4388655 | 25,506.1 | 6 | 134 | 100 | 7.62 | 7.924E–08 |
| 56 | Elongation factor alpha G5 (TcEFA-G5) | gi|52424046 | 49,067.4 | 18 | 405 | 100 | 8.93 | 6.295E–35 |
| 57 | Heat shock protein 70 (TcHSP70) | gi|50659756 | 71,329.9 | 30 | 524 | 100 | 5.06 | 3.972E–46 |
| 58 | Glucose-regulated protein 78 (TcHSPA5 or TcGRP78) | gi|71415505 | 71,271.9 | 29 | 510 | 100 | 5.09 | 9.976E–45 |
| 59 | Glucose-regulated protein 78 (TcHSPA5 or TcGRP78) | gi|349838 | 71,488 | 25 | 365 | 100 | 5.07 | 3.155E–30 |
| 75- to 77-kDa antigen (TcHSP70) | gi|1097750 | 41,716.5 | 17 | 273 | 100 | 4.99 | 5E–21 | |
| 60 | Mitochondrial heat shock protein (TcHSP70mt) | gi|205278870 | 70,969.1 | 25 | 527 | 100 | 5.87 | 1.99E–46 |
| 61 | Heat shock protein 70 (TcHSP70) | gi|205278868 | 71,193 | 26 | 517 | 100 | 5.32 | 2.51E–73 |
| 62 | Alpha tubulin (TcTUBA) | gi|71397525 | 49,767.5 | 22 | 806 | 100 | 4.94 | 2.51E–74 |
| 62 | Alpha tubulin (TcTUBA) | gi|1314208 | 46,927.4 | 20 | 787 | 100 | 5.49 | 1.99E–72 |
| 63 | Beta tubulin 1.9 (TcTUBB) | gi|18568139 | 49,667.8 | 16 | 643 | 100 | 4.74 | 5.00E–58 |
| 64 | Beta tubulin of T. cyclops (TcTUBB) | gi|91983197 | 44,496.8 | 14 | 626 | 100 | 5.68 | 2.51E–56 |
| 65 | Calpain-like cysteine peptidase (TcCAPN1) | gi|71409962 | 12,839.3 | 5 | 170 | 100 | 5.08 | 1.26E–11 |
| 66 | Tryparedoxin peroxidase family (TcGPX1) | gi|19171158 | 16,057.1 | 5 | 102 | 99.9 | 5.27 | 9.98E–51 |
| 67 | Histone H2B of T. brucei (TbHIST1H2B) | gi|71748436 | 12,588.8 | 4 | 84 | 96.1 | 11.77 | 1.26E–54 |
| 68 | Beta tubulin (TcTUBB) | gi|71656281 | 49,667.7 | 15 | 570 | 100 | 4.7 | 9.51E–56 |
| 69 | Alpha tubulin of T. danilewskyi (TdTUBA) | gi|74229924 | 49,765.6 | 21 | 722 | 100 | 4.94 | 6.29E–66 |
| 70 | Cofilin-actin depolymerizing factor (TcCFL1) | gi|71417693 | 15,735.8 | 9 | 300 | 100 | 5.69 | 9.98E–24 |
| 71 | Prostaglandin F synthase (TcPGFS) | gi|71651365 | 32,489.5 | 16 | 532 | 100 | 7.71 | 6.29E–47 |
| 72 | Hypothetical protein | gi|71657351 | 129,881.1 | 18 | 77 | 79.8 | 6.84 | 7.92E–41 |
| 73 | Lipophosphoglycan biosynthetic protein (TcLPG3) | gi|71663313 | 86,968.5 | 17 | 248 | 100 | 5.26 | 1.58E–18 |
| 74 | 3-Hydroxy-3-methylglutaryl coenzyme A synthase-like (TcHMGCS2) | gi|71661816 | 54,719.2 | 14 | 445 | 100 | 6.17 | 3.15E–38 |
| 75 | Carboxypeptidase (TcCPD) | gi|71667929 | 57,626.9 | 19 | 136 | 100 | 5.92 | 2.51E–07 |
| Metallocarboxypeptidase (TcCPD) | gi|47076074 | 57,686.9 | 18 | 127 | 100 | 5.85 | 1.99E–06 | |
T. cruzi epimastigotes (COL, SYL, and TCC isolates) were cultured in LIT media. Protein homogenates were resolved by two-dimensional gel electrophoresis. Gel images were analyzed on Progenesis SameSpots software, and normalized spot volumes were used to compare different groups. Proteins spots with a >2-fold change were subjected to MALDI-TOF MS/MS analysis. The number of peptides for each protein spot for which peptide mass fingerprint was obtained by MALDI-TOF MS is listed. Protein spots that were identified with high confidence by Mascot search engine (a protein score of >62) and statistically significant at the 95% confidence interval (CI%) are listed. The probability of match of the peptides in database search was accepted as significant at an E value of <0.001. pI, isoelectric pH. Underlined text indicates proteins involved in host cell invasion and cytoskeletal remodeling and motility. Boldface text indicates proteins of the antioxidant family involved in redox homeostasis.
Bioinformatics analysis using SMART and TARGET software showed that several of the proteins that participate in host cell invasion (e.g., cysteine peptidases, surface glycoprotein gp82, and cruzipain) and cytoskeletal remodeling and motility (e.g., α- and β-isoforms of actin and tubulin, cofilin-1, KMP11, and paraflagellar rod proteins) were significantly increased in abundance in both the epimastigote and trypomastigote stages of the SYL and COL compared to that noted in the TCC strain (Tables 1 and 2). The antioxidant proteins involved in redox homeostasis (e.g., tryparedoxin peroxidases, peroxiredoxins, thioredoxin-dependent peroxide reductase [PRDX3], and iron superoxide dismutases [FeSODa/b]) were primarily increased in abundance in the trypomastigote stage of the SYL and COL compared to that noted in TCC trypomastigotes (Table 2).
TABLE 2.
Protein spots that were differentially expressed in trypomastigote stage of TcI isolates of T. cruzia
| Protein spot | Protein name (gene) | NCBI accession no. | Mol mass (Da) | Peptide count | Protein score | Protein score (CI%) | pI | E value |
|---|---|---|---|---|---|---|---|---|
| Spots differentially expressed in COL isolate compared to TCC | ||||||||
| 1 | Tryparedoxin peroxidase (TcTXNPx) | gi|17224953 | 22,246.1 | 10 | 340 | 100 | 5.96 | 1.99054E–28 |
| 2 | Tryparedoxin peroxidase (TcTXNPx) | gi|71399514 | 22,375.1 | 10 | 304 | 100 | 6.32 | 3.97164E–24 |
| 5 | Calpain-like cysteine peptidase (Tc CL) | gi|71411006 | 12,798.3 | 5 | 164 | 100 | 5.07 | 7.92447E–11 |
| 6 | Tryparedoxin peroxidase family (TcGPX1) | gi|19171158 | 16,057.1 | 4 | 82 | 98.73 | 5.27 | 1.25674E–05 |
| 7 | Paraflagellar rod protein 3 (TcPFR3) | gi|2226084 | 68,705.3 | 24 | 288 | 100 | 5.86 | 3.15479E–23 |
| 8 | Paraflagellar rod protein, 69 kDa (TcPFR) | gi|71650963 | 69,518.9 | 26 | 215 | 100 | 5.85 | 6.29463E–16 |
| 9 | Paraflagellar rod protein 2 (TcPFR2) | gi|162179 | 69,504.9 | 24 | 198 | 100 | 5.85 | 3.15479E–14 |
| 10 | Paraflagellar rod protein of T. brucei (TbPFR-C/TbPFR-D) | gi|72387193 | 68,640.3 | 19 | 175 | 100 | 5.78 | 6.29463E–12 |
| 11 | Heat shock protein 70 (TcHSP70/TcHSPA5) | gi|50659756 | 71,329.9 | 33 | 877 | 100 | 5.06 | 3.97164E–82 |
| 12 | Glucose-regulated protein, 78 kDa (TcGRP78) | gi|349838 | 71,488 | 29 | 603 | 100 | 5.07 | 9.97631E–55 |
| 13 | Alpha tubulin (TcTUBA) | gi|71397525 | 49,767.5 | 19 | 405 | 100 | 4.94 | 6.29463E–35 |
| 14 | Alpha tubulin (TcTUBA) | gi|1314208 | 46,927.4 | 18 | 398 | 100 | 5.49 | 3.15479E–34 |
| 15 | Beta tubulin of T. grayi (TgTUBB) | gi|91983201 | 42,806.9 | 13 | 288 | 100 | 5.29 | 3.15479E–23 |
| 16 | Beta tubulin of T. danilewskyi (TdTUBB) | gi|74229926 | 49,695.8 | 13 | 281 | 100 | 4.74 | 1.58114E–22 |
| 17 | Cytochrome c oxidase subunit IV (TcCOXIV) | gi|71667854 | 38,884.4 | 15 | 339 | 100 | 5.72 | 2.50594E–28 |
| 18 | Kinetoplastid membrane protein 11 (TcKMP11) | gi|71413455 | 10,876.3 | 8 | 349 | 100 | 5.73 | 1.25594E–28 |
| 19 | Kinetoplastid membrane protein 11 of T. rangeli (TrKMP11) | gi|32892042 | 11,003.4 | 8 | 347 | 100 | 6.52 | 1.99054E–28 |
| 20 | Phosphoglycerate kinase 1 (TcPGK1) | gi|71411859 | 44,441.1 | 16 | 240 | 100 | 6.19 | 9.97631E–18 |
| 21 | 3-Phosphoglycerate kinase, glycosomal (TcPGKC) | gi|71654900 | 54,867.6 | 15 | 122 | 100 | 6.94 | 6.29463E–06 |
| 22 | Chain A, tryparedoxin peroxidase (TcTXNPx) | gi|58176947 | 22,743.3 | 8 | 305 | 100 | 5.71 | 3.15479E–24 |
| 23 | Peroxiredoxin, antioxidant oxidoreductase (TcPRX) | gi|4388655 | 20,235.2 | 9 | 440 | 100 | 5.07 | 9.97631E–38 |
| 24 | Ubiquitin precursor (TcUBQ) | gi|162337 | 40,105.6 | 5 | 409 | 100 | 9.39 | 1.25594E–34 |
| 25 | Fe-superoxide dismutase, chain A (TcFeSODa) | gi|158428656 | 21,800.8 | 7 | 441 | 100 | 6.65 | 7.92447E–38 |
| 26 | Fe-superoxide dismutase (TcFeSOD) | gi|1066332 | 21,818.9 | 7 | 440 | 100 | 6.64 | 9.97631E–38 |
| 27 | Calpain-like cysteine peptidase (TcCAPN1) | gi|71409962 | 12,839.3 | 4 | 159 | 100 | 5.08 | 1.25594E–09 |
| 29 | Ascorbate-dependent peroxidase (TcAPX) | gi|24370982 | 31,855.2 | 7 | 269 | 100 | 6.69 | 1.25594E–20 |
| Spots differentially expressed in SYL isolate compared to TCC | ||||||||
| 30 | Beta tubulin of T. cyclops (TcTUBB) | gi|91983197 | 44,496.8 | 14 | 295 | 100 | 5.68 | 6.29463E–24 |
| 31 | Beta tubulin 1.9 (TcTUBB) | gi|18568139 | 49,667.8 | 14 | 291 | 100 | 4.74 | 1.58114E–23 |
| 32 | Prostaglandin F2 alpha synthase (TcPGFS) | gi|71661165 | 14,648.3 | 4 | 119 | 100 | 6.05 | 2.50594E–06 |
| 33 | Amidinotransferase-like (TcGATM) | gi|71661567 | 43,051.9 | 9 | 184 | 100 | 6 | 7.92447E–13 |
| 34 | Amidinotransferase-like (TcGATM) | gi|71415202 | 28,667.4 | 5 | 128 | 100 | 5.21 | 3.15479E–07 |
| 35 | 10-kDa heat shock protein (TcHSPE1) | gi|71425263 | 10,692.8 | 2 | 66 | 49.41 | 9.05 | 2.50594E–18 |
| 36 | Tryparedoxin peroxidase, chain A (TcTXNPX) | gi|58176947 | 22,743.3 | 9 | 328 | 100 | 5.71 | 3.15479E–27 |
| 37 | Alpha tubulin of T. danilewskyi (TdTUBAi) | gi|74229924 | 49,765.6 | 18 | 394 | 100 | 4.94 | 7.92447E–34 |
| 38 | Tubulin alpha (TcTUBA) | gi|3915082 | 49,696.5 | 17 | 358 | 100 | 4.9 | 3.15479E–30 |
| 39 | NADPH flavin oxidoreductase (old yellow enzyme, TcOYE) | gi|61741942 | 42,191.6 | 12 | 250 | 100 | 6.03 | 1.99054E–19 |
| 40 | Glutamate dehydogenase (TcGLUD1) | gi|71414987 | 41,680.5 | 11 | 239 | 100 | 7.1 | 2.50594E–18 |
| 41 | Calmodulin (TcCALM1) | gi|71407658 | 26,740.2 | 5 | 89 | 100 | 5.1 | 3.15479E–17 |
| 42 | Cysteine peptidase inhibitor (TcSERPIN) | gi|71664690 | 12,114 | 2 | 89 | 98.63 | 6.58 | 0.001256238 |
| 43 | Ubiquitin homolog (TcUBQ) | gi|10673 | 14,648.8 | 5 | 404 | 100 | 9.82 | 3.97164E–34 |
| 44 | Vacuolar ATP synthase subunit B-like (TcATP5B) | gi|70870795 | 104,474.5 | 30 | 656 | 100 | 5.75 | 2.50594E–59 |
| 47 | Heat shock protein 85 (TcHSP85) | gi|70876495 | 82,887.1 | 18 | 680 | 100 | 5.79 | 9.97631E–62 |
| 48 | Ubiquitin-like (TcUBQ) | gi|136677 | 8,503.6 | 5 | 427 | 100 | 6.56 | 1.99054E–36 |
| 49 | Ubiquitin-like (TcUBQ) | gi|10674 | 8,762.7 | 5 | 425 | 100 | 6.56 | 3.15479E–36 |
| 50 | Kinetoplastid membrane protein 11 (TcKMP11) | gi|6166388 | 9,923.9 | 8 | 351 | 100 | 6.58 | 7.92447E–29 |
| 51 | Kinetoplastid membrane protein 11 of T. rangeli (TrKMP11) | gi|24286543 | 11,048.4 | 8 | 349 | 100 | 5.96 | 1.25594E–28 |
| 52 | Cysteine-protease inhibitor/Chagasin (TcSERPIN) | gi|118137894 | 14,448 | 3 | 185 | 100 | 6.66 | 3.15479E–12 |
| 53 | Cysteine peptidase inhibitor (TcSERPIN) | gi|71664690 | 12,114 | 2 | 127 | 100 | 6.58 | 1.99054E–06 |
| 54 | Hypothetical protein (Tc CL) | gi|71410630 | 12,677.5 | 2 | 71 | 23.03 | 6.58 | 1.99054E–12 |
| 55 | Tryparedoxin peroxidase (TcTXNPX) | gi|71413207 | 22,376.1 | 8 | 419 | 100 | 5.69 | 1.25594E–35 |
| 56 | Thioredoxin-dependent peroxide reductase (PRDX3) | gi|241785831 | 25,789.7 | 4 | 214 | 100 | 9.08 | 3.97164E–15 |
| 57 | Iron superoxide dismutase (TcFeSOD) | gi|71414374 | 23,339.6 | 7 | 439 | 100 | 6.7 | 1.25594E–37 |
| 58 | Iron superoxide dismutase B (TcFeSODB) | gi|2149612 | 21,745.8 | 6 | 285 | 100 | 6.37 | 3.15479E–22 |
| 59 | Heat shock 70-kDa protein (TcHSP70) | gi|71406304 | 40,831 | 18 | 720 | 100 | 6.19 | 9.97631E–66 |
| 60 | Heat shock protein HSP70 (TcHSP70) | gi|162117 | 71,210.1 | 19 | 685 | 100 | 5.42 | 3.15479E–62 |
| 61 | Heat shock protein 70 (TcHSP70) | gi|10626 | 74,233.7 | 17 | 609 | 100 | 5.64 | 1.25594E–54 |
| 62 | Ubiquitin-conjugating enzyme-like (Tc CL) | gi|71414042 | 15,754.8 | 9 | 157 | 100 | 5.81 | 1.99054E–09 |
| 63 | Beta tubulin 1.9 (TcTUBB) | gi|18568139 | 49,667.8 | 12 | 227 | 100 | 4.74 | 1.99054E–16 |
| 64 | Beta tubulin (TcTUBB) | gi|3915883 | 49,540.7 | 11 | 262 | 100 | 4.69 | 1.25594E–20 |
| 65 | Alpha tubulin (TcTUBA) | gi|1314208 | 46,927.4 | 18 | 400 | 100 | 5.49 | 9.97631E–34 |
| 66 | Alpha tubulin (TcTUBA) | gi|71397525 | 49,767.5 | 18 | 396 | 100 | 4.94 | 2.50594E–33 |
| 67 | Calpain-like cysteine peptidase (TcCAPN1) | gi|71411006 | 12,798.3 | 5 | 177 | 100 | 5.07 | 1.99054E–11 |
| 68 | 3-Phosphoglycerate kinase (TcPGK) | gi|16945527 | 54,767.4 | 15 | 121 | 100 | 6.94 | 7.92447E–06 |
| 69 | Phosphoglycerate kinase (TcPGKCL) | gi|71411862 | 64,528.8 | 14 | 114 | 100 | 8.04 | 3.97164E–05 |
| 70 | Tryparedoxin peroxidase (TcTXNPX) | gi|17224953 | 22,246.1 | 9 | 492 | 100 | 5.96 | 6.29463E–43 |
| 71 | Cruzipain | gi|1136308 | 47,852.1 | 3 | 348 | 100 | 5.35 | 1.58114E–28 |
| 72 | 69-kDa paraflagellar rod protein (TcPFR) | gi|71411387 | 42,471.9 | 18 | 153 | 100 | 6.12 | 9.97631E–10 |
| 73 | Surface glycoprotein, 82 kDa (TcGP82) | gi|6049148 | 59,591.9 | 20 | 170 | 100 | 6.05 | 9.97631E–11 |
T. cruzi trypomastigotes (COL, SYL, and TCC isolates) were cultured in C2C12 cells. Parasite protein homogenates were resolved by two-dimensional gel electrophoresis. Gel images were analyzed on Progenesis SameSpots software, and normalized spot volumes were used for comparison of different groups. Proteins spots with >2-fold change were subjected to MALDI-TOF MS/MS analysis. The significance of a protein match, based on the peptide mass fingerprint in the MS and the MS/MS data, is presented as expectation values (P < 0.01). pI, isoelectric pH. Underlined text indicates proteins involved in host cell invasion and cytoskeletal remodeling and motility. Boldface text indicates proteins of the antioxidant family that are involved in redox homeostasis.
We chose to confirm the expression profile of the antioxidant proteins because these exhibited a differential abundance in a stage- (epimastigote versus trypomastigote) as well as strain-specific manner. Parasite lysates were probed by using antibodies against cytosolic (cTXNPx) and mitochondrial (mTXNPx) isoforms of tryparedoxin peroxidase, tryparedoxin isoforms TXN I and TXN II, and iron superoxide dismutase isoforms FeSODa and FeSODb (31, 32). Due to the finding of a differential abundance of α-tubulin and β-actin in T. cruzi isolates (Table 1 and Table 2), we could not use these proteins for normalizing the antioxidant protein content. Instead, equal protein loading was confirmed by Ponceau S staining of the PVDF membranes, as before (34). Representative Western blot images and densitometry evaluation of the protein abundance in the epimastigote stage of the Tc1 isolates are shown in Fig. 4. The cTXNPx expression was increased by 46% in COL epimastigotes (versus TCC, P < 0.05 [ANOVA-Tukey]), whereas the abundance of all other members of the tryparedoxin family and FeSOD isoforms was not changed significantly in the epimastigote stage of the three TcI isolates (Fig. 4A and B). The Ponceau S-stained gel image (Fig. 4C) confirmed equal loading of all samples.
FIG 4.
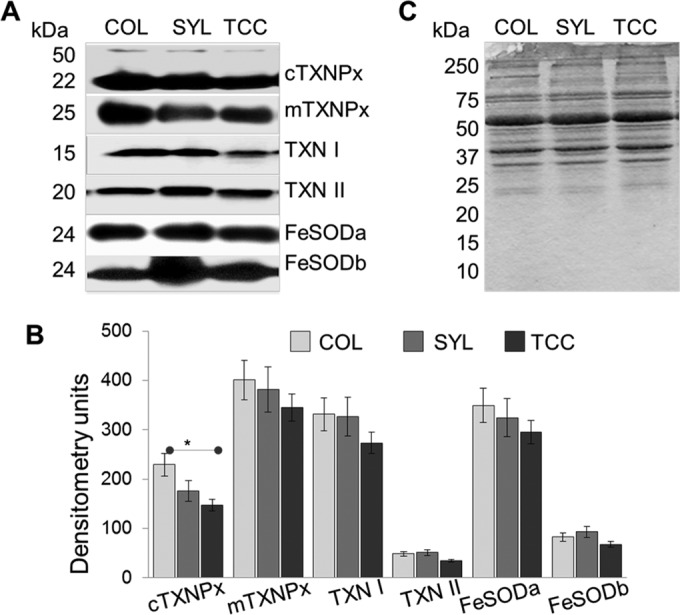
Western blotting for selected proteins of the antioxidant network in TcI epimastigotes. (A) Homogenates of the COL, SYL, and TCC epimastigotes were resolved by SDS-PAGE. Membranes were probed by using specific antibodies against mitochondrial and cytosolic tryparedoxin peroxidases (mTXNPx and cTXNPx, respectively), tryparedoxin substrates (TXN I and TXN II), and Fe2+ SODs (FeSODa and FeSODb). (B) Bar graphs show densitometric analysis of the band intensities from Western blotting, normalized to total loaded protein. (C) Ponceau S staining shows equal loading of all samples.
The expanded views of the corresponding spot for FeSOD and TXNPx from representative 2D gel images of SYL and TCC trypomastigotes are shown in Fig. 5A. As determined by Western blotting, COL and SYL trypomastigotes exhibited increases in the protein levels of cTXNPx (2.5- and 2-fold, respectively; P < 0.05 [ANOVA]), mTXNPx (32 and 28%, respectively; P < 0.05 to 0.01 [ANOVA]); TXN I (2.0- and 2.14-fold, respectively; P < 0.01), TXN II (39.7 and 31.5%, respectively; P < 0.05 [ANOVA]); and FeSODa (33.1 and 49.5%, respectively; P < 0.05 [ANOVA]) compared to that noted in TCC trypomastigotes (Fig. 5B and C). The protein level of FeSODb was not statistically different in trypomastigotes of the three isolates. The Ponceau S-stained gel image (Fig. 5D) confirmed equal loading of all samples. Together, the results presented in Fig. 4 and 5 validate the 2DE/MS findings and suggest that the antioxidant network was upregulated in the trypomastigote stage of the virulent TcI isolates (COL and SYL) of T. cruzi.
FIG 5.
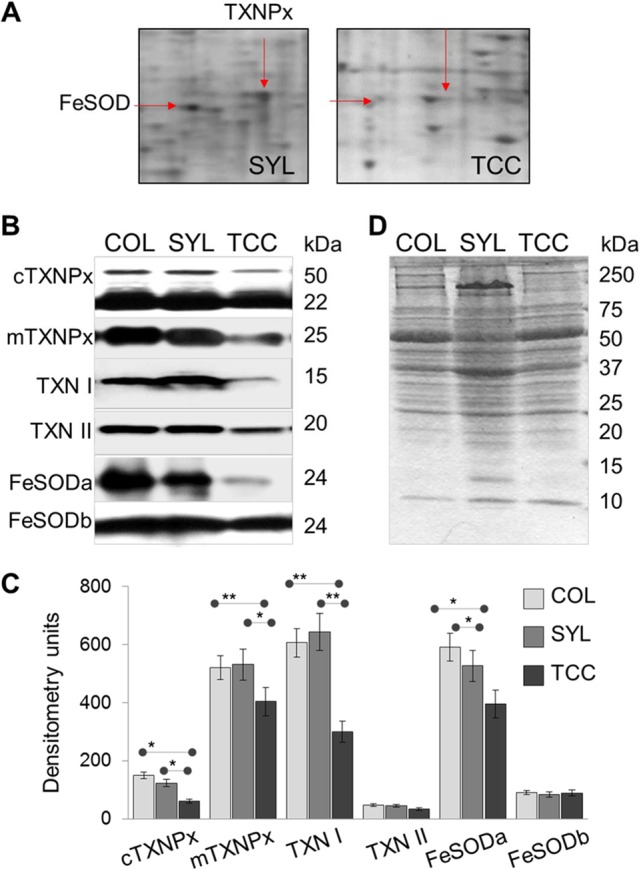
Validation of expression profile of selected proteins of the antioxidant network in the trypomastigote stage of TcI strains. (A) Expanded views of the corresponding spots for TXNPx (23 kDa) and FeSOD (21 kDa) from representative 2D gel images of SYL and TCC trypomastigotes are shown. (B) Homogenates of COL, SYL, and TCC trypomastigotes were resolved by SDS-PAGE. Membranes were probed by using specific antibodies against mitochondrial and cytosolic tryparedoxin peroxidases (mTXNPx and cTXNPx, respectively), tryparedoxin substrates (TXN I and TXN II), and Fe2+ SODs (FeSODa and FeSODb). (C) The band intensities from Western blot images were digitized and normalized to total protein loaded. (D) Ponceau S staining shows equal loading of all samples.
To assess the biological significance of the antioxidant status in TcI isolates, we took a three-pronged approach. First, we sought to determine whether the selected TcI strains differed in their ability to resist exogenous oxidative/nitrosative stress (Fig. 6). Trypomastigotes were challenged for 2 and 4 h with biologically relevant doses of H2O2 and ONOO−, the main oxidants present in the phagosomal environment (35, 36), and parasite viability was determined by an alamarBlue assay. Up to 84.9, 95.7, and 66.8% of COL, SYL, and TCC trypomastigotes, respectively, were viable after H2O2 treatment for 2 h, and their viabilities were not further decreased after 4 h (Fig. 6A). Likewise, 80.6, 93.2, and 49.7%, of COL, SYL, and TCC trypomastigotes, respectively, were viable after ONOO− treatment for 2 h, and parasite viability was not further reduced at 4 h (Fig. 6B). In comparison to TCC, the SYL and COL isolates exhibited significantly higher levels of resistance to exogenous H2O2 (27 and 46%, respectively; P < 0.01 [ANOVA-Tukey]) and ONOO− (64.5% and 2.1-fold, respectively; P < 0.001 [ANOVA-Tukey]) stress (Fig. 6).
FIG 6.
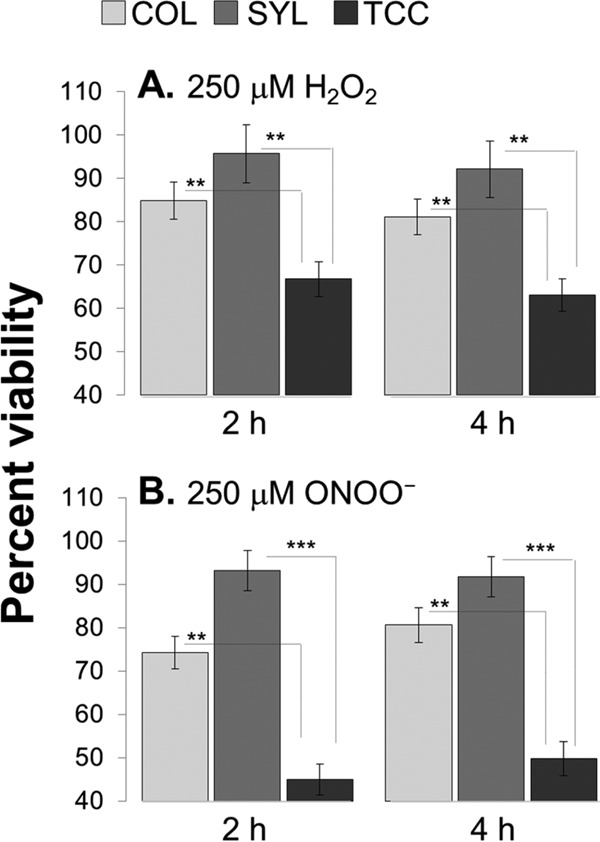
Variable resistance of TcI strains to oxidants. The COL, SYL, and TCC trypomastigotes were in vitro incubated with 250 μM H2O2 (A) or 250 μM ONOO− (B). Parasite viability was determined at 2 and 4 h postincubation by an alamarBlue assay. Data are presented as the percentage of viability with respect to untreated parasites.
Next, we sought to determine whether TcI isolates exhibited differing capacities to modulate intracellular oxidative/nitrosative stress. For this, macrophages were incubated with T. cruzi trypomastigotes for 24, 36, and 48 h, and the intracellular ROS was measured by using a CM-H2DCF-DA fluorescence probe. Macrophages infected with COL and SYL for 24 h exhibited minimal intracellular DCF fluorescence, while TCC-infected macrophages demonstrated a significant increase in DCF fluorescence compared to that noted in normal controls (Fig. 7A, compare panels a and b to panel c). A similar pattern of increased intracellular DCF fluorescence in TCC-infected macrophages, compared to COL- or SYL-infected macrophages, was observed at 36 h and 48 h p.i. (data not shown).
FIG 7.

Macrophage activation of oxidative/nitrosative response, but not inflammatory cytokine response, is deregulated by the antioxidant status of the TcI strains of T. cruzi. RAW 264.7 macrophages were infected with COL, SYL, and TCC trypomastigotes for 24, 36, or 48 h. Normal (uninfected) cells, incubated for similar time periods, were used as controls. (A) Cells were incubated for 24 h and then loaded with H2DCFDA. The oxidation of H2DCFDA by intracellular ROS, resulting the in formation of fluorescent DCF, was monitored by fluorescence microscopy. (B and C) Supernatants were used to measure ROS (B) and nitrite (C) levels by an Amplex red and a Griess reagent assay, respectively. (D and E) TNF-α (D) and IL-6 (E) cytokines in supernatant samples were monitored by an ELISA.
Subsequently, we measured the release of H2O2 (Amplex red assay) and nitrate/nitrite (Griess assay) from the infected macrophages. The H2O2 levels in supernatants of macrophages infected with COL and SYL isolates for 24 or 48 h were not statistically different from that noted in normal controls (Fig. 7B). TCC-infected (versus SYL- or COL-infected) macrophages exhibited up to 1.5- and 2.3-fold increases in H2O2 levels at 24 and 48 h, respectively (Fig. 7B; all, P < 0.01 [ANOVA-Tukey]). The nitrate/nitrite release, an indicator of iNOS activity, was increased by 1.6- to 2.7-fold, 1.7- to 2.9-fold, and 3.7- to 21-fold in COL-, SYL-, and TCC-infected macrophages, respectively (Fig. 7C). The maximal difference in nitrate/nitrite level between COL- and SYL-infected versus TCC-infected macrophages was noted at 24 h p.i. (Fig. 7C, P < 0.001 [ANOVA-Tukey]).
To verify that macrophages infected with TCC may simply not be activated, we monitored cytokine (IFN-γ, TNF-α, and IL-6) release in the supernatants of infected cells by an ELISA. These data showed that, irrespective of the parasite strain used, patent increases in TNF-α (33.5- to 35.5-fold) and IL-6 (>100-fold) release were mounted by macrophages within 24 h p.i., and the cytokine release was maintained at 36 and 48 h p.i. (Fig. 7D and E). No increase in IFN-γ release was observed in any of the infected macrophages. Together, the data presented in Fig. 7 suggested that COL and SYL (versus TCC) trypomastigotes faced compromised oxidative and nitrosative stress in macrophages. The differences in antioxidant capacity of the parasite isolates had no effect on cytokine signaling in macrophages.
Finally, we sought to determine whether the differential abundance of antioxidants conferred a survival advantage to COL and SYL trypomastigotes in macrophages. For this, RAW 264.7 macrophages were infected with T. cruzi isolates for 6, 12, and 18 h. Infected cells were submitted to Giemsa staining, and >200 cells/slide (triplicate experiments) were evaluated by light microscopy to determine the number of infected macrophages. We observed no statistically significant difference in the parasite uptake by macrophages incubated with COL, SYL, or TCC isolates (data not shown). A quantitative measure of intracellular parasites by real-time quantitative PCR showed a significant increase in intracellular SYL, followed by COL and TCC, from 0 to 18 h p.i. (Fig. 8A). The rapid replication of SYL in macrophages was evidenced by detection of Tc18SrDNA signal that was 12.6- to 13.5-fold and >27-fold higher than that noted in COL- and TCC-infected macrophages, respectively, from 6 to 12 h p.i. (P < 0.01 to 0.001 [ANOVA-Tukey]). By 18 h p.i., intracellular COL level was 7.6-fold higher than that noted for TCC (P < 0.01 [t test]) but lagged behind that of SYL by 3.2-fold (P < 0.01 [t test]).
FIG 8.
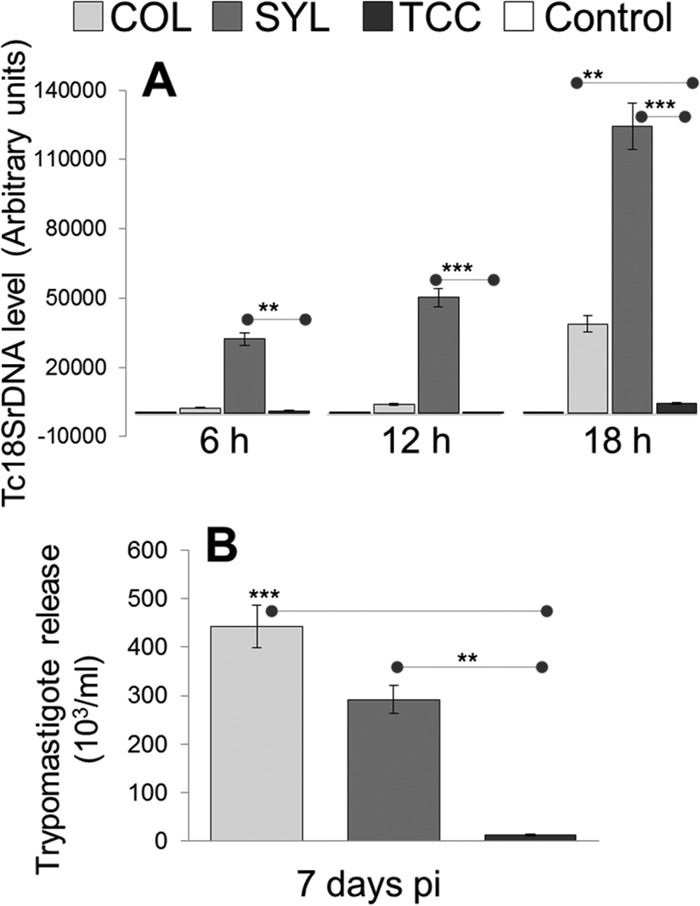
Intracellular replication of TcI isolates and release from infected macrophages. (A) RAW 264.7 macrophages were infected with COL, SYL, and TCC for 6, 12, and 18 h. Real-time quantitative PCR analysis of parasite burden in infected macrophages was performed by using Tc18SrDNA-specific oligonucleotides. The data were normalized to GAPDH expression. Normal/uninfected cells were used as controls. (B) RAW 264.7 macrophages were infected with metacyclic form of COL, SYL, and TCC, and the release of trypomastigotes in supernatants was determined 7 days p.i. by light microscopy.
We used metacyclic trypomastigotes derived from stationary-phase epimastigote cultures of COL, SYL, and TCC isolates to monitor their ability to escape from macrophages. For this, parasites were added to monolayers of RAW 264.7 macrophages and, after infection for 3 h, the cells were washed and incubated for 2 weeks. The hemacytometer counting of parasites in supernatants of infected cells at 7 days p.i. showed 36.8- and 24.3-fold-higher levels of parasite release from macrophages infected with COL and SYL isolates, respectively, compared to that observed in supernatants of TCC-infected macrophages (Fig. 8B, P < 0.01 to 0.001 [ANOVA-Tukey]). By the end of the second week postinfection, all cells infected with SYL and COL had ruptured, releasing parasites, whereas TCC-infected macrophages continued to exhibit intracellular parasites (data not shown). These data suggested that (i) macrophages were equally competent in their ability to phagocytose different isolates of T. cruzi, (ii) COL and SYL (versus TCC) were better equipped to survive and replicate in macrophages, and (iii) COL and SYL trypomastigotes succeeded in efficient release from macrophages.
DISCUSSION
Reported cases of Chagas disease from Latin America have been associated with TcI isolates (10, 11, 37, 38). It has been postulated that TcI isolates exhibit more tissue tropism than do other lineages of T. cruzi (39–41) and are therefore responsible for severe pathology of the heart. In this context, we performed biological characterization of three T. cruzi isolates of the TcI DTU and showed that COL, SYL, and TCC isolates exhibited differing growth and virulence behavior in mice (Fig. 1). The striking difference in the infectivity and disease severity pattern of COL and SYL versus that of TCC made them an ideal model for global proteomic profiling for the identification of candidates involved with the mechanism of parasite invasion, replication, and survival in the host cells.
The comparative proteome profiling of the three TcI strains that exhibited different virulence levels in mice yielded novel findings. One, the expression profiles of numerous proteins in COL and SYL epimastigotes and trypomastigotes were consistent with enhanced motility and contractility. For example, upregulation of α-actin, α-tubulin, β-tubulin, cofilin-1, and KMP11 that are central to remodeling of the cytoskeleton and modulation of cell shape for migration (42, 43) was noted in COL and SYL (versus TCC) strains (Tables 1 and 2). Likewise, members of the paraflagellar rod (PFR) and paraxonemal protein families, known to be essential for flagellar wave frequency and amplitude and forward motility of the parasite (reviewed in reference 44), were enhanced in abundance in COL and SYL (versus TCC) strains. Further, SYL and COL trypomastigotes also exhibited an increased abundance of calpain-like cysteine peptidases, surface glycoprotein gp82, and cruzipain that are involved in host cell attachment and invasion (45, 46). We surmise that virulence of the COL, SYL, and possibly other isolates of the TcI DTU is determined by the motility capacity that ensures parasite transmission from triatomines and dissemination to tissues in the mammalian host. Indeed, several studies using RNA interference against PFR2 in T. brucei have shown that ablation of PFR2 resulted in sluggish parasites that lacked cytokinesis capability and were monstrous in size, also showing multiple nuclei, kinetoplasts, and flagella; these PFR2-deficient cells were unable to survive in vitro or in vivo (47, 48). Further studies will be required to validate the importance of cytoskeletal remodeling and flagellar function as new targets for possible intervention against T. cruzi infection.
Comparative proteome analysis of the two parasite forms showed that the differentiation of epimastigotes to trypomastigotes was accompanied by a shift from histidine catabolism to glutamate, a decline in ribosomal proteins, and an increase in the expression level of a battery of antioxidant enzymes (compare Tables 1 and 2). Three major families of peroxidases identified in trypanosomatids included the 2-cysteine peroxiredoxins, as well as the non-selenium glutathione and ascorbate peroxidases (49). Notably, we showed a significant correlation between the abundance of the antioxidant enzymes and the virulent behavior of the COL and SYL trypomastigotes. The increase in tryparedoxin perdoxidases occurred in infective trypomastigote and replicative amastigote stages irrespective of the cell types (e.g., muscle cells in this study, and LLC-MK2 and macrophages in other studies [34, 50]) used for culturing the parasites. Regeneration of the reduced TXNPx depends on cytosolic (TXN I)- and membrane (TXN II)-localized thioredoxin substrates (31, 51). Consistent with the increased abundance of TXNPx, the TXN I level was also increased in COL and SYL trypomastigotes (Fig. 5). COL and SYL trypomastigotes also showed an increased abundance of the FeSODa (mitochondrial) enzyme (Fig. 5). Others have also documented the differential expression of peroxiredoxin and FeSOD proteins of the antioxidant network in parasites in correlation with in vivo infectivity and pathological behavior (32, 34, 52). The question arises as to how the TXNPx/TXN antioxidant network is associated with the virulence of the TcI isolates of T. cruzi. One explanation could be that, upon infection, T. cruzi faces the hostile environment of phagocytes, and an increase in antioxidant response would allow adaptation of the infective forms to the oxidative environment of phagocytic cells in the mammalian host (45, 46). Activated macrophages provide a key defense response to the invading pathogens and exert cytotoxic effects via NADPH oxidase (NOX)-mediated production of superoxide and ROS that result in enhanced oxidative modification of DNA, proteins, and lipids (53, 54). Alternatively, superoxide may combine with nitric oxide to produce peroxynitrite and peroxynitrous acid that can directly kill T. cruzi (55). Indeed, COL and SYL (versus TCC) exhibited an enhanced capacity to survive in and escape from macrophages (Fig. 8), and these responses were associated with detection of low levels of oxidative and nitrosative stress in COL-infected and SYL-infected (versus TCC-infected) macrophages (Fig. 7) . The in vitro (Fig. 7) and in vivo (Fig. 1) data presented in this study do not clarify whether the antioxidant network enhanced the capacity of the SYL and COL isolates to efficiently scavenge ROS and nitric oxide (NO) or inhibit the macrophage activation of oxidative and nitrosative response. Yet, our finding of increased resistance of COL and SYL trypomastigotes to exogenous H2O2 and peroxynitrite-mediated killing (Fig. 6) implies that the parasite's ability to scavenge ROS/NO, at least partially, provides it a survival advantage in immune cells. Others have provided direct biochemical evidence showing that TXNPx acts as a hub to neutralize free radicals through peroxynitrite reductase activity (56). An enhanced association between the protein level and activity of tryparedoxin peroxidases (TXNPx) and virulence of other isolates of DTU TcI-TcIV has also been noted (35, 56). Thus, we surmise that intraphagosomal survival from ROS and ˙NO is the key to the parasite's existence in the host and that, irrespective of the phylogenetic lineage, T. cruzi isolates capable of utilizing an antioxidant network to avoid oxidative damage would likely ensure their survival, dissemination, and persistence in the host. The increase in the expression of FeSODa in SYL and COL is in line with our interpretation, since the dismutation of superoxide will lead to a decrease in the concentration of oxidative species.
It is important to note that after escaping from macrophages, T. cruzi faces oxidative stress in other tissues as well. For example, we have shown enhanced mitochondrial production of ROS in cardiomyocytes during T. cruzi infection (33). It is plausible that T. cruzi utilizes antioxidant enzyme network to prevent mitochondrial ROS-mediated direct killing or ROS activation of the apoptosis pathway for removal of the infected (or damaged) cells (57), thereby ensuring its persistence in the host. A consequence of prolonged survival of infected (damaged) cells is the signaling of cell repair and proliferative responses, culminating in cardiac fibrosis (58). Further studies are required to demonstrate the novel role of TXNPx/TXN in establishing a cardiac remodeling environment in Chagas disease; however, if such is the case, inhibiting the parasite antioxidant activity will provide a novel strategy to manage the homeostatic clearance of damaged cells (and parasite) and control the fibrotic processes that culminate in cardiomyopathy in chronic Chagas disease.
In summary, we conclude that T. cruzi virulent isolates ensure survival and persistence in the host via peroxiredoxin-dependent decomposition of macrophage-derived free radicals. This is evidenced by our findings in the infective stage of the virulent parasite isolates (COL and SYL), i.e., an upregulation of the protein levels for cTXNPx, mTXNPx, and TXN I that readily detoxified cytotoxic and diffusible reactive oxygen and nitrogen species generated in vitro or released by the macrophages. The absence of differences in inflammatory cytokine levels (i.e., TNF-α and IL-6) in macrophages infected with virulent or nonvirulent isolates of T. cruzi suggests that the peroxiredoxin-based antioxidant system only helps the parasite to cope with the oxidative challenge without interfering with the cytokine signaling. Further characterization of the parasite's antioxidant network in tissues from infected experimental animals at different stages of infection and disease development would support its role in the pathogenesis and parasite persistence and provide a strong basis for the development of new chemotherapeutic options against T. cruzi.
T. cruzi isolates have been characterized as belonging to the TcI to TcVI lineages. Efforts establishing an association between parasite lineage and pathogenicity have not been successful. We demonstrate that parasite isolates of the same lineage exhibit a variable virulence in mice. An unbiased proteomic approach, followed by targeted studies, showed that the differential abundance of the peroxiredoxin antioxidants and the flagellar proteins allowed intraphagosomal survival from oxidative/nitrosative stress and parasite dissemination and persistence. Thus, gene expression conducive to enhanced antioxidant capacity and motility, irrespective of phylogenetic lineage, provided virulence to T. cruzi and should be evaluated for identifying virulent isolates circulating in the Americas.
ACKNOWLEDGMENTS
We thank the Biomedical Resource Facility and Mass Spectrometry Laboratory at the University of Texas Medical Branch at Galveston (UTMB) for mass spectrometric analysis, G. Suarez for the helpful critiques on the two-dimensional gel electrophoresis experiments, and S. Andrade, Laboratory of Experimental Chagas Disease Autoimmunity and Cellular Immunology at the Centro de Pesquisas Gonçalo Moniz/Fiocruz, Salvador Bahia, Brazil, for kindly providing the Colombian strain of T. cruzi.
This study was supported in part by grants from the National Institutes of Health/National Institute of Allergy and Infectious Diseases (2R01AI0545780) and the Institute for Human Infections and Immunity (IHII), UTMB, to N.J.G.; CONICET (PIP 2010-2012 11420090100297) to M.P.Z.; Flight Attendant Medical Research Institute Young Clinical Scientist Award to Y.M.H. (123385); and Agencia Nacional de Investigación e Innovación (Uruguay) grant DCI-ALA/2011/023-502 to C.R. M.P.Z. also received a CONICET young researcher fellowship that helped initiate this study. S.-J.K. received a predoctoral fellowship from the UTMB McLaughlin Endowment, IHII. The funders had no role in study design, data collection and analysis, decision to publish, or preparation of the manuscript.
Funding Statement
This work was supported in part by grants from the National Institutes of Health/National Institute of Allergy and Infectious Diseases (2R01AI0545780) and the Institute for Human Infections and Immunity (IHII), UTMB, Galveston to NJG; CONICET (PIP 2010-2012 11420090100297) to MPZ, Flight Attendant Medical Research Institute Young Clinical Scientist Award to YMH (123385), and Agencia Nacional de Investigación e Innovación (Uruguay) grant DCI-ALA/2011/023-502 to CR. MPZ was also a recipient of a CONICET young researcher fellowship that helped initiate this study. SJK is the recipient of a predoctoral fellowship from the UTMB McLaughlin Endowment, IHII. The funders had no role in study design, data collection and analysis, decision to publish, or preparation of the manuscript.
REFERENCES
- 1.Tanowitz HB, Weiss LM, Montgomery SP. 2011. Chagas disease has now gone global. PLoS Negl Trop Dis 5:e1136. doi: 10.1371/journal.pntd.0001136. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Lee BY, Bacon KM, Bottazzi ME, Hotez PJ. 2013. Global economic burden of Chagas disease: a computational simulation model. Lancet Infect Dis 13:342–348. doi: 10.1016/S1473-3099(13)70002-1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Bern C, Martin DL, Gilman RH. 2011. Acute and congenital Chagas disease. Adv Parasitol 75:19–47. doi: 10.1016/B978-0-12-385863-4.00002-2. [DOI] [PubMed] [Google Scholar]
- 4.Messenger LA, Miles MA, Bern C. 2015. Between a bug and a hard place: Trypanosoma cruzi genetic diversity and the clinical outcomes of Chagas disease. Expert Rev Anti Infect Ther 13:995–1029. doi: 10.1586/14787210.2015.1056158. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Centers for Disease Control and Prevention. 2007. Blood donor screening for Chagas disease–United States, 2006-2007. MMWR Morb Mortal Wkly Rep 56:141–143. [PubMed] [Google Scholar]
- 6.Centers for Disease Control and Prevention. 2006. Chagas disease after organ transplantation–Los Angeles, California, 2006. MMWR Morb Mortal Wkly Rep 55:798–800. [PubMed] [Google Scholar]
- 7.Bhatia V, Wen J-J, Zacks MA, Garg NJ. 2009. American trypanosomiasis and perspectives on vaccine development, p 1407–1434. In Stanberry LR, Barrett AD (ed), Vaccines for biodefense and emerging and neglected diseases. Academic Press, New York, NY. [Google Scholar]
- 8.Hotez PJ, Dumonteil E, Woc-Colburn L, Serpa JA, Bezek S, Edwards MS, Hallmark CJ, Musselwhite LW, Flink BJ, Bottazzi ME. 2012. Chagas disease: “the new HIV/AIDS of the Americas.” PLoS Negl Trop Dis 6:e1498. doi: 10.1371/journal.pntd.0001498. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Lent H, Wygodzisnky P. 1979. Revision of the Triatominae (Hemiptera, Reduviidae) and their significance as vectors of Chagas' disease. Bull Am Museum Natural History 163:123–520. [Google Scholar]
- 10.Diosque P, Padilla AM, Cimino RO, Cardozo RM, Negrette OS, Marco JD, Zacca R, Meza C, Juarez A, Rojo H, Rey R, Corrales RM, Nasser JR, Basombrio MA. 2004. Chagas disease in rural areas of Chaco Province, Argentina: epidemiologic survey in humans, reservoirs, and vectors. Am J Trop Med Hyg 71:590–593. [PubMed] [Google Scholar]
- 11.Cura CI, Lucero RH, Bisio M, Oshiro E, Formichelli LB, Burgos JM, Lejona S, Bruses BL, Hernandez DO, Severini GV, Velazquez E, Duffy T, Anchart E, Lattes R, Altcheh J, Freilij H, Diez M, Nagel C, Vigliano C, Favaloro L, Favaloro RR, Merino DE, Sosa-Estani S Schijman AG. 2012. Trypanosoma cruzi discrete typing units in Chagas disease patients from endemic and non-endemic regions of Argentina. Parasitology 139:516–521. doi: 10.1017/S0031182011002186. [DOI] [PubMed] [Google Scholar]
- 12.Rassi A Jr, Rassi A, Marin-Neto JA. 2010. Chagas disease. Lancet 375:1388–1402. doi: 10.1016/S0140-6736(10)60061-X. [DOI] [PubMed] [Google Scholar]
- 13.Tanowitz HB, Machado FS, Spray DC, Friedman JM, Weiss OS, Lora J, Nascimento D, Nunes MC, Garg NJ, Ribeiro AL. 2015. Developments in the management of Chagas cardiomyopathy. Expert Rev Cardiovasc Ther 13:1393–1340. doi: 10.1586/14779072.2015.1103648. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Macedo AM, Machado CR, Oliveira RP, Pena SD. 2004. Trypanosoma cruzi: genetic structure of populations and relevance of genetic variability to the pathogenesis of Chagas disease. Mem Inst Oswaldo Cruz 99:1–12. [DOI] [PubMed] [Google Scholar]
- 15.De Souza W. 2002. Basic cell biology of Trypanosoma cruzi. Curr Pharm Des 8:269–285. doi: 10.2174/1381612023396276. [DOI] [PubMed] [Google Scholar]
- 16.Zingales B, Miles MA, Campbell DA, Tibayrenc M, Macedo AM, Teixeira MM, Schijman AG, Llewellyn MS, Lages-Silva E, Machado CR, Andrade SG, Sturm NR. 2012. The revised Trypanosoma cruzi subspecific nomenclature: rationale, epidemiological relevance, and research applications. Infect Genet Evol 12:240–253. doi: 10.1016/j.meegid.2011.12.009. [DOI] [PubMed] [Google Scholar]
- 17.Carrasco HJ, Segovia M, Llewellyn MS, Morocoima A, Urdaneta-Morales S, Martinez C, Martinez CE, Garcia C, Rodriguez M, Espinosa R, de Noya BA, Diaz-Bello Z, Herrera L, Fitzpatrick S, Yeo M, Miles MA, Feliciangeli MD. 2012. Geographical distribution of Trypanosoma cruzi genotypes in Venezuela. PLoS Negl Trop Dis 6:e1707. doi: 10.1371/journal.pntd.0001707. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Guhl F, Ramirez JD. 2011. Trypanosoma cruzi I diversity: towards the need of genetic subdivision? Acta Trop 119:1–4. doi: 10.1016/j.actatropica.2011.04.002. [DOI] [PubMed] [Google Scholar]
- 19.Barnabe C, Neubauer K, Solari A, Tibayrenc M. 2001. Trypanosoma cruzi: presence of the two major phylogenetic lineages and of several lesser discrete typing units (DTUs) in Chile and Paraguay. Acta Trop 78:127–137. doi: 10.1016/S0001-706X(00)00183-2. [DOI] [PubMed] [Google Scholar]
- 20.Montamat EE, De Luca D'Oro GM, Gallerano RH, Sosa R, Blanco A. 1996. Characterization of Trypanosoma cruzi populations by zymodemes: correlation with clinical picture. Am J Trop Med Hyg 55:625–628. [DOI] [PubMed] [Google Scholar]
- 21.Abolis NG, Araujo SM, Toledo MJ, Fernandez MA, Gomes ML. 2011. Trypanosoma cruzi I-III in southern Brazil causing individual and mixed infections in humans, sylvatic reservoirs, and triatomines. Acta Trop 120:167–172. doi: 10.1016/j.actatropica.2011.08.001. [DOI] [PubMed] [Google Scholar]
- 22.del Puerto R, Nishizawa JE, Kikuchi M, Iihoshi N, Roca Y, Avilas C, Gianella A, Lora J, Velarde FU, Renjel LA, Miura S, Higo H, Komiya N, Maemura K, Hirayama K. 2010. Lineage analysis of circulating Trypanosoma cruzi parasites and their association with clinical forms of Chagas disease in Bolivia. PLoS Negl Trop Dis 4:e687. doi: 10.1371/journal.pntd.0000687. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Federici EE, Abelmann WH, Neva FA. 1964. Chronic and progressive myocarditis and myositis in C3H mice infected with Trypanosoma cruzi. Am J Trop Med Hyg 13:272–280. [DOI] [PubMed] [Google Scholar]
- 24.Postan M, Dvorak JA, McDaniel JP. 1983. Studies of Trypanosoma cruzi clones in inbred mice. I. A comparison of the course of infection of C3H/HEN mice with two clones isolated from a common source. Am J Trop Med Hyg 32:497–506. [DOI] [PubMed] [Google Scholar]
- 25.Cura CI, Duffy T, Lucero RH, Bisio M, Peneau J, Jimenez-Coello M, Calabuig E, Gimenez MJ, Valencia Ayala E, Kjos SA, Santalla J, Mahaney SM, Cayo NM, Nagel C, Barcan L, Malaga Machaca ES, Acosta Viana KY, Brutus L, Ocampo SB, Aznar C, Cuba Cuba CA, Gurtler RE, Ramsey JM, Ribeiro I, VandeBerg JL, Yadon ZE, Osuna A, Schijman AG. 2015. Multiplex real-time PCR assay using TaqMan probes for the identification of Trypanosoma cruzi DTUs in biological and clinical samples. PLoS Negl Trop Dis 9:e0003765. doi: 10.1371/journal.pntd.0003765. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Isola EL, Lammel EM, Gonzalez Cappa SM. 1986. Trypanosoma cruzi: differentiation after interaction of epimastigotes and Triatoma infestans intestinal homogenate. Exp Parasitol 62:329–335. doi: 10.1016/0014-4894(86)90039-1. [DOI] [PubMed] [Google Scholar]
- 27.Gupta S, Garg NJ. 2013. TcVac3 induced control of Trypanosoma cruzi infection and chronic myocarditis in mice. PLoS One 8:e59434. doi: 10.1371/journal.pone.0059434. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Garg NJ, Soman KV, Zago MP, Koo SJ, Spratt H, Stafford S, Blell ZN, Gupta S, Nunez Burgos J, Barrientos N, Brasier AR, Wiktorowicz JE. 2016. Changes in proteome profile of peripheral blood mononuclear cells in chronic Chagas disease. PLoS Negl Trop Dis 10:e0004490. doi: 10.1371/journal.pntd.0004490. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Dhiman M, Zago MP, Nunez S, Nunez-Burgio F, Garg NJ. 2012. Cardiac oxidized antigens are targets of immune recognition by antibodies and potential molecular determinants in Chagas disease pathogenesis. PLoS One 7:e28449. doi: 10.1371/journal.pone.0028449. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Wen JJ, Zago MP, Nunez S, Gupta S, Nunez Burgos F, Garg NJ. 2012. Serum proteomic signature of human chagasic patients for the identification of novel protein biomarkers of disease. Mol Cell Proteomics 11:435–452. doi: 10.1074/mcp.M112.017640. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Pineyro MD, Parodi-Talice A, Portela M, Arias DG, Guerrero SA, Robello C. 2011. Molecular characterization and interactome analysis of Trypanosoma cruzi tryparedoxin 1. J Proteomics 74:1683–1692. doi: 10.1016/j.jprot.2011.04.006. [DOI] [PubMed] [Google Scholar]
- 32.Pineyro MD, Parodi-Talice A, Arcari T, Robello C. 2008. Peroxiredoxins from Trypanosoma cruzi: virulence factors and drug targets for treatment of Chagas disease? Gene 408:45–50. doi: 10.1016/j.gene.2007.10.014. [DOI] [PubMed] [Google Scholar]
- 33.Gupta S, Bhatia V, Wen JJ, Wu Y, Huang MH, Garg NJ. 2009. Trypanosoma cruzi infection disturbs mitochondrial membrane potential and ROS production rate in cardiomyocytes. Free Radic Biol Med 47:1414–1421. doi: 10.1016/j.freeradbiomed.2009.08.008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Piacenza L, Zago MP, Peluffo G, Alvarez MN, Basombrio MA, Radi R. 2009. Enzymes of the antioxidant network as novel determiners of Trypanosoma cruzi virulence. Int J Parasitol 39:1455–1464. doi: 10.1016/j.ijpara.2009.05.010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Alvarez MN, Peluffo G, Piacenza L, Radi R. 2011. Intraphagosomal peroxynitrite as a macrophage-derived cytotoxin against internalized Trypanosoma cruzi: consequences for oxidative killing and role of microbial peroxiredoxins in infectivity. J Biol Chem 286:6627–6640. doi: 10.1074/jbc.M110.167247. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Martinez A, Peluffo G, Petruk AA, Hugo M, Pineyro D, Demicheli V, Moreno DM, Lima A, Batthyany C, Duran R, Robello C, Marti MA, Larrieux N, Buschiazzo A, Trujillo M, Radi R, Piacenza L. 2014. Structural and molecular basis of the peroxynitrite-mediated nitration and inactivation of Trypanosoma cruzi iron-superoxide dismutases (Fe-SODs) A and B: disparate susceptibilities due to the repair of Tyr35 radical by Cys83 in Fe-SODB through intramolecular electron transfer. J Biol Chem 289:12760–12778. doi: 10.1074/jbc.M113.545590. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Burgos JM, Altcheh J, Petrucelli N, Bisio M, Levin MJ, Freilij H, Schijman AG. 2009. Molecular diagnosis and treatment monitoring of congenital transmission of Trypanosoma cruzi to twins of a triplet delivery. Diagn Microbiol Infect Dis 65:58–61. doi: 10.1016/j.diagmicrobio.2009.04.010. [DOI] [PubMed] [Google Scholar]
- 38.Schijman AG, Vigliano CA, Viotti RJ, Burgos JM, Brandariz S, Lococo BE, Leze MI, Armenti HA, Levin MJ. 2004. Trypanosoma cruzi DNA in cardiac lesions of Argentinean patients with end-stage chronic Chagas heart disease. Am J Trop Med Hyg 70:210–220. [PubMed] [Google Scholar]
- 39.Burgos JM, Begher SB, Freitas JM, Bisio M, Duffy T, Altcheh J, Teijeiro R, Lopez Alcoba H, Deccarlini F, Freilij H, Levin MJ, Levalle J, Macedo AM, Schijman AG. 2005. Molecular diagnosis and typing of Trypanosoma cruzi populations and lineages in cerebral Chagas disease in a patient with AIDS. Am J Trop Med Hyg 73:1016–1018. [PubMed] [Google Scholar]
- 40.Burgos JM, Diez M, Vigliano C, Bisio M, Risso M, Duffy T, Cura C, Brusses B, Favaloro L, Leguizamon MS, Lucero RH, Laguens R, Levin MJ, Favaloro R, Schijman AG. 2010. Molecular identification of Trypanosoma cruzi discrete typing units in end-stage chronic Chagas heart disease and reactivation after heart transplantation. Clin Infect Dis 51:485–495. doi: 10.1086/655680. [DOI] [PubMed] [Google Scholar]
- 41.Burgos JM, Begher S, Silva HM, Bisio M, Duffy T, Levin MJ, Macedo AM, Schijman AG. 2008. Molecular identification of Trypanosoma cruzi I tropism for central nervous system in Chagas reactivation due to AIDS. Am J Trop Med Hyg 78:294–297. [PubMed] [Google Scholar]
- 42.Chang L, Goldman RD. 2004. Intermediate filaments mediate cytoskeletal crosstalk. Nat Rev Mol Cell Biol 5:601–613. doi: 10.1038/nrm1438. [DOI] [PubMed] [Google Scholar]
- 43.Thomas MC, Garcia-Perez JL, Alonso C, Lopez MC. 2000. Molecular characterization of KMP11 from Trypanosoma cruzi: a cytoskeleton-associated protein regulated at the translational level. DNA Cell Biol 19:47–57. doi: 10.1089/104454900314708. [DOI] [PubMed] [Google Scholar]
- 44.Portman N, Gull K. 2010. The paraflagellar rod of kinetoplastid parasites: from structure to components and function. Int J Parasitol 40:135–148. doi: 10.1016/j.ijpara.2009.10.005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Parodi-Talice A, Monteiro-Goes V, Arrambide N, Avila AR, Duran R, Correa A, Dallagiovanna B, Cayota A, Krieger M, Goldenberg S, Robello C. 2007. Proteomic analysis of metacyclic trypomastigotes undergoing Trypanosoma cruzi metacyclogenesis. J Mass Spectrom 42:1422–1432. doi: 10.1002/jms.1267. [DOI] [PubMed] [Google Scholar]
- 46.Atwood JA III, Weatherly DB, Minning TA, Bundy B, Cavola C, Opperdoes FR, Orlando R, Tarleton RL. 2005. The Trypanosoma cruzi proteome. Science 309:473–476. doi: 10.1126/science.1110289. [DOI] [PubMed] [Google Scholar]
- 47.Griffiths S, Portman N, Taylor PR, Gordon S, Ginger ML, Gull K. 2007. RNA interference mutant induction in vivo demonstrates the essential nature of trypanosome flagellar function during mammalian infection. Eukaryot Cell 6:1248–1250. doi: 10.1128/EC.00110-07. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Broadhead R, Dawe HR, Farr H, Griffiths S, Hart SR, Portman N, Shaw MK, Ginger ML, Gaskell SJ, McKean PG, Gull K. 2006. Flagellar motility is required for the viability of the bloodstream trypanosome. Nature 440:224–227. doi: 10.1038/nature04541. [DOI] [PubMed] [Google Scholar]
- 49.Castro H, Tomas AM. 2008. Peroxidases of trypanosomatids. Antioxid Redox Signal 10:1593–1606. doi: 10.1089/ars.2008.2050. [DOI] [PubMed] [Google Scholar]
- 50.Gadelha FR, Goncalves CC, Mattos EC, Alves MJ, Pineyro MD, Robello C, Peloso EF. 2013. Release of the cytosolic tryparedoxin peroxidase into the incubation medium and a different profile of cytosolic and mitochondrial peroxiredoxin expression in H2O2-treated Trypanosoma cruzi tissue culture-derived trypomastigotes. Exp Parasitol 133:287–293. doi: 10.1016/j.exppara.2012.12.007. [DOI] [PubMed] [Google Scholar]
- 51.Arias DG, Marquez VE, Chiribao ML, Gadelha FR, Robello C, Iglesias AA, Guerrero SA. 2013. Redox metabolism in Trypanosoma cruzi: functional characterization of tryparedoxins revisited. Free Radic Biol Med 63:65–77. doi: 10.1016/j.freeradbiomed.2013.04.036. [DOI] [PubMed] [Google Scholar]
- 52.Andrade HM, Murta SM, Chapeaurouge A, Perales J, Nirde P, Romanha AJ. 2008. Proteomic analysis of Trypanosoma cruzi resistance to benznidazole. J Proteome Res 7:2357–2367. doi: 10.1021/pr700659m. [DOI] [PubMed] [Google Scholar]
- 53.Cardoni RL, Antunez MI, Morales C, Nantes IR. 1997. Release of reactive oxygen species by phagocytic cells in response to live parasites in mice infected with Trypanosoma cruzi. Am J Trop Med Hyg 56:329–334. [DOI] [PubMed] [Google Scholar]
- 54.Piacenza L, Alvarez MN, Peluffo G, Radi R. 2009. Fighting the oxidative assault: the Trypanosoma cruzi journey to infection. Curr Opin Microbiol 12:415–421. doi: 10.1016/j.mib.2009.06.011. [DOI] [PubMed] [Google Scholar]
- 55.Alvarez MN, Piacenza L, Irigoin F, Peluffo G, Radi R. 2004. Macrophage-derived peroxynitrite diffusion and toxicity to Trypanosoma cruzi. Arch Biochem Biophys 432:222–232. doi: 10.1016/j.abb.2004.09.015. [DOI] [PubMed] [Google Scholar]
- 56.Pineyro MD, Arcari T, Robello C, Radi R, Trujillo M. 2011. Tryparedoxin peroxidases from Trypanosoma cruzi: high efficiency in the catalytic elimination of hydrogen peroxide and peroxynitrite. Arch Biochem Biophys 507:287–295. doi: 10.1016/j.abb.2010.12.014. [DOI] [PubMed] [Google Scholar]
- 57.Budzko DB, Kierszenbaum F. 1974. Isolation of Trypanosoma cruzi from blood. J Parasitol 60:1037–1038. doi: 10.2307/3278544. [DOI] [PubMed] [Google Scholar]
- 58.Gupta S, Dhiman M, Wen JJ, Garg NJ. 2011. ROS signalling of inflammatory cytokines during Trypanosoma cruzi infection. Adv Parasitol 76:153–170. doi: 10.1016/B978-0-12-385895-5.00007-4. [DOI] [PubMed] [Google Scholar]