

Abstract
Borrelia burgdorferi possesses a sophisticated chemotaxis signaling system; however, the roles of the majority of the chemotaxis proteins in the infectious life cycle have not yet been demonstrated. Specifically, the role of CheD during host colonization has not been demonstrated in any bacterium. Here, we systematically characterized the B. burgdorferi CheD homolog using genetics and biochemical and mouse-tick-mouse infection cycle studies. Bacillus subtilis CheD plays an important role in chemotaxis by deamidation of methyl-accepting chemotaxis protein receptors (MCPs) and by increasing the receptor kinase activity or enhancing CheC phosphatase activity, thereby regulating the levels of the CheY response regulator. Our biochemical analysis indicates that B. burgdorferi CheD significantly enhances CheX phosphatase activity by specifically interacting with the phosphatase. Moreover, CheD specifically binds two of the six MCPs, indicating that CheD may also modulate the receptor proteins. Although the motility of the cheD mutant cells was indistinguishable from that of the wild-type cells, the mutant did exhibit reduced chemotaxis. Importantly, the mutant showed significantly reduced infectivity in C3H/HeN mice via needle inoculation. Mouse-tick-mouse infection assays indicated that CheD is dispensable for acquisition or transmission of spirochetes; however, the viability of cheD mutants in ticks is marginally reduced compared to that of the wild-type or complemented cheD spirochetes. These data suggest that CheD plays an important role in the chemotaxis and pathogenesis of B. burgdorferi. We propose potential connections between CheD, CheX, and MCPs and discuss how these interactions play critical roles during the infectious life cycle of the spirochete.
INTRODUCTION
Borrelia burgdorferi, the causative agent of Lyme disease, cycles between Ixodes ticks and a mammalian host. Motility and chemotaxis are known to be required for host tissue colonization or disease production by many bacteria, including the spirochetes; however, these processes have not been rigorously characterized in any spirochete (1–13). B. burgdorferi is a spirochetal motile bacterium that contains a sophisticated and complicated chemotaxis signal transduction system. This spirochete is a long (10- to 20-μm) and thin (0.33-μm) organism with flat-wave morphology, and its motility is provided by flagella that are located within the periplasmic space. There are 7 to 11 periplasmic flagella inserted at each pole of the cell, forming a ribbon-like structure that wraps around the cell cylinder as they extend toward the other pole of the cell (5, 6, 9). When the periplasmic flagellar motors rotate asymmetrically (i.e., flagella at one pole rotate clockwise [CW] and the flagella at other pole rotate counterclockwise [CCW]), the spirochete runs. When the flagellar motors at both poles of the cell rotate in the same direction (both CW or CCW), B. burgdorferi cells flex with no net translocation (14). This manner of swimming also produces an unusual “corkscrew-like” locomotion. This distinctive motility pattern is believed to be important for efficient migration within the dense tissues through which these spirochetes preferentially disseminate in both vertebrate and arthropod hosts (5, 15). Interestingly, where most externally flagellated bacteria either slow down or stop within viscous material (16, 17), the velocities of spirochetes actually increase within viscous media (18–20), thus empowering the spirochetes to penetrate into tissues that other bacteria fail to invade.
Chemotaxis is also believed to be very important for the complex vector-host life cycle utilized by B. burgdorferi. After transmission from tick to mammalian host, B. burgdorferi must quickly perceive its new local environment and make the appropriate adaptations, rapidly disseminating through skin tissues to evade the cellular immune response and migrating to appropriate target tissues, which allows evasion of the expanding adaptive immune response and survival. After residing in the mammalian host for weeks to years, B. burgdorferi must then be able to detect that a new tick vector has attached to the host's skin and then rapidly migrate to the bite site and enter the vector (21).
Chemotaxis allows bacteria to follow gradients of nutrients and other environmental stimuli by governing their motility. The components of the chemotaxis signal transduction systems that mediate these responses are highly conserved among prokaryotes. The best-studied system is found in Escherichia coli (or Salmonella enterica serovar Typhimurium) (22–24). In this model organism, membrane-spanning methyl-accepting chemotaxis protein receptors (MCPs) detect environmental signals. These MCP proteins are coupled to a histidine kinase (CheA) and a linker protein (CheW). The catalytic activity of CheA is regulated by occupancy of these MCPs with a ligand (e.g., an attractant or repellent). CheA uses ATP to autophosphorylate, and the phosphate group is then transferred to the response regulator, CheY. The phosphorylated CheY (CheY-P) then binds to the flagellar motor switch proteins FliM and FliN (25). This binding results in the change of direction of the flagella from the default CCW (running motility) to CW rotation, resulting in tumbling motility. Binding of attractant to the MCPs decreases the kinase activity of CheA, resulting in reduced CheY-P, as well as tumbling frequency. The concentration of CheY-P determines whether a cell runs (which requires low CheY-P) or tumbles (high CheY-P). CheY-P is able to autodephosphorylate; however, the phosphatase CheZ efficiently dephosphorylates the levels of CheY-P in E. coli (26). This phosphorylation-dephosphorylation is crucial for the bacterial cells to respond appropriately to environmental stimuli. The chemotaxis system adapts to persistent modification via receptor methylation by the methyltransferase CheR (positive stimuli) and demethylation by the methylesterase CheB (negative stimuli). CheR and CheB are the only known chemoreceptor modification enzymes in this E. coli model (23).
Even though the components of the chemotaxis signal transduction system are conserved among prokaryotes, the chemotaxis pathway in B. burgdorferi differs from the other well-studied bacterial models and is much more complicated due to the presence of multiple copies of chemotaxis genes, including six mcp, two cheA, three cheY, two cheB, two cheR, and three cheW genes, which may contribute to the asymmetric motility of the spirochete. Moreover, B. burgdorferi has a putative gene encoding CheD, which is not found in E. coli (5). CheD is relatively well characterized in Bacillus subtilis and Thermotoga maritima (27–30). In B. subtilis, CheD plays an important role in motility and chemotaxis. A cheD mutant displays a distinctive motility pattern: wild-type (WT) cells exhibit running and tumbling motility, whereas the ΔcheD mutant constantly tumbles. At the molecular level, CheD interacts with the receptor MCPs. MCP proteins contain the well-conserved substrate-binding site of CheD, A/S-X-X-Q/E-Q/E-X-X-A/S (A, alanine; S, serine; Q, glutamine; E, glutamate; X, any amino acid). CheD modifies the MCPs by deamidating glutamine to glutamate residues. The deamidation by CheD is required for the B. subtilis chemoreceptors to effectively transduce signals to the CheA kinase (29, 31). Additionally, CheD interacts with the phosphatase CheC, enhancing its enzymatic activity and fine-tuning the intracellular concentration of CheY-P (27, 29). B. burgdorferi possesses a phosphatase, CheX, that has been previously reported to possess CheY-P-dephosphorylating activity, and the cheX mutant cells constantly flex (32–34). It is noteworthy that the “flex” of B. burgdorferi is considered to be equivalent to the tumbling phenotype seen in E. coli. Moreover, the MCPs of B. burgdorferi contain the well-conserved potential substrate-binding sites of CheD (29, 31, 35). These observations led us to hypothesize that CheD in B. burgdorferi plays a role similar to that of its counterpart in B. subtilis. Most importantly, even though the role of CheD in motility and chemotaxis has been demonstrated in other bacteria, its role in pathogenesis has not been reported in any organism. To date, only one chemotaxis (cheA2) gene has been investigated in the B. burgdorferi infectious life cycle (i.e., tick-to-mouse transmission) (7). A rigorous study is therefore warranted in order to understand the functions of other putative chemotaxis genes.
In this study, we determined the requirement for cheD in motility and chemotaxis in B. burgdorferi by inactivating the cheD gene, as well as by demonstrating its enzymatic role in the chemotaxis signaling system. Moreover, we defined the role of cheD in the disease process (mouse-tick-mouse infection cycles) for the first time. We showed that CheD not only is important in chemotaxis, as it enhances the CheX phosphatase activity, but also is essential for the infectious life cycle of B. burgdorferi.
MATERIALS AND METHODS
Ethics statement.
East Carolina University is accredited by the International Association for Assessment and Accreditation of Laboratory Animal Care. Protocols for tick and animal experimentation were approved by the East Carolina University animal care and use committee.
Bacterial strains and growth conditions.
The low-passage, virulent B. burgdorferi strain B31-A3 was used as a wild-type clone throughout the study (36, 37). The genome of the virulent B31 strain was found to contain a total of 21 plasmids, with 12 linear and 9 circular plasmids, in addition to its 960-kbp linear chromosome (38, 39). Clone B31-A3 lacks circular plasmid 9 (cp9) but remains infectious in tick-mouse cycle studies (37, 40). The construction of cheD mutants and complemented strains is described below. B. burgdorferi cells were cultured in liquid Barbour-Stoenner-Kelly (BSK-II) medium, and plating BSK was prepared using 0.5% agarose (8, 41, 42). Cells were grown at 35°C in a 2.5% CO2 humidified incubator as described previously (8, 37, 41). Antibiotics, when required, were included in the B. burgdorferi culture medium at the following concentrations: 200 μg/ml kanamycin, 40 μg/ml gentamicin, and 100 μg/ml streptomycin. E. coli cells were grown at 37°C in Luria-Bertani (LB) broth or LB agar (43). Antibiotics, when required, were included in the E. coli culture medium at the following concentrations: 100 μg/ml ampicillin, 35 μg/ml chloramphenicol, and 100 μg/ml spectinomycin.
Construction and complementation of the cheD mutant.
Construction of the cheD inactivation plasmids, electroporation, and plating conditions were described previously (8, 44). Briefly, the cheD gene (locus number bb0606) and flanking DNA were first amplified by PCR from the chromosomal DNA of B. burgdorferi strain B31-A3 using primers CheD-KO-F (GGAATCAGGCTTAAATCTTG) and CheD-KO-R (AAGCATGGAAAGTTGAAACC). The PCR product was cloned into plasmid pGEM-T Easy (Promega Inc.). The cheD gene was inactivated using a kanamycin resistance cassette (PflgB-aph1) (36), which was inserted at the HindIII sites located within cheD. Competent B31-A3 cells were electroporated with cheD-PflgB-aph1 DNA that was linearized by NotI restriction digestion to remove the ampicillin restriction marker of the vector, preventing it from being introduced into B. burgdorferi (8, 44). The transformants were selected with kanamycin, and the kanamycin-resistant transformants were confirmed by PCR to have PflgB-aph1 integrated within cheD (8, 44, 45). Confirmation of cheD gene inactivation was achieved by the lack of cheD transcripts in the cheD mutant (ΔcheD) cells by quantitative reverse transcriptase (qRT)-PCR (see below).
The cheD mutant was complemented in cis by genomic integration using a pXLF14301 suicide vector (35, 46, 47). To complement the mutant, the cheD gene and its native promoter (PcheD) DNA sequences were PCR amplified from genomic DNA of B. burgdorferi strain B31-A3 using primers CheDcom.-F (AATTAAAATGATTTAACATATTTCCCAATAACATAGATAC) and CheDcom.-R (GCGGCCGCTTAAAAAACCTTTGTTCC) and PcheD-F (ACTAGTATTGGCCATATCCCCATTAAGGC) and PcheD-R (GTATCTATGTTATTGGGAAATATGTTAAATCATTTTAATT), respectively. The amplified DNA fragments were ligated by overlapping PCR, yielding a PcheD-cheD DNA fragment, and then cloned into plasmid pGEM-T Easy (Promega Inc.). Finally, PcheD-cheD was excised from pGEM-T Easy using SpeI and NotI restriction digestions and then cloned into pXLF14301, yielding plasmid pXLFCheD. The plasmid was then electroporated into the ΔcheD mutant cells, followed by selection with gentamicin and kanamycin. The resistant clones were analyzed by PCR for integration of PcheD-cheD within the intergenic region of bb0445 and bb0446. Restoration of cheD expression in the complemented cheD (cheDcom) clones was also verified by qRT-PCR (see below). Retention of B. burgdorferi endogenous plasmids in the wild-type, mutant, and complemented strains was verified by PCR as described previously (8, 35, 37, 45).
RT-PCR.
Exponentially growing B. burgdorferi wild-type cells (2 × 107 cells/ml) were treated with RNAprotect, followed by total RNA isolation using the RNeasy minikit (Qiagen Inc.). Contaminating DNA in the RNA samples was removed by RNase-free Turbo DNase I (Ambion Inc.) digestion for 3 h at 37°C, followed by RNeasy minipurification. For reverse transcription (RT)-PCR, cDNA was prepared from 1 μg RNA using the AffinityScript QPCR cDNA synthesis kit (Agilent Technologies Inc.) according to the manufacturer's protocol. The iCycler detection system (Bio-Rad Inc.) was used to measure cheD transcript levels according to the manufacturer's instructions. The B. burgdorferi enolase gene was used as a reference gene (35, 45, 48, 49). The gene-specific primers (5′-3′) were RT-enolase-F (TGGAGCGTACAAAGCCAACATT), RT-enolase-R (TGAAAAACCTCTGCTGCCATTC), CheD-qRT-F (CCTGGTGAAGCTTTTGTTTC), and CheD-qRT-R (TTGATCAGGAGATATGTCAAGATC). The relative level of expression was calculated using the 2−ΔΔCT method (35, 45, 49–51).
Recombinant protein expression in E. coli.
In order to express B. burgdorferi cheD in E. coli, a DNA fragment harboring the entire cheD open reading frame was amplified from chromosomal DNA of B31-A3 using primers R.CheD-F (TTAAATCATTTTAATTTTAA) and R. CheD-R (TTAAAAAACCTTTGTTCCGT) and cloned into the pASK-IBA7+ expression vector (IBA Inc.) to generate Strep-Tactin-tagged CheD (Strep-CheD). E. coli BL21(DE3) harboring pASK-IBA7+::cheD was induced with 200 ng/ml of anhydrotetracycline. Expression and purification of E. coli CheA and B. burgdorferi His-CheY3 and His-CheX proteins were described previously (33, 52, 53). B. burgdorferi mcp3, mcp4, and mcp5 open reading frames were cloned separately in the expression vector pMAL c2x (NEB Inc.) to generate MBP-MCP3, MBP-MCP4, and MBP-MCP5 recombinant proteins, respectively. E. coli codon-plus cells harboring pMAL c2x::mcp3, mcp4, or mcp5 was expressed and purified according to the manufacturer's protocol (NEB Inc.). The purity of the recombinant proteins was verified by sodium dodecyl sulfate-polyacrylamide gel electrophoresis (SDS-PAGE) and quantified by comparison with standard bovine serum albumin (BSA).
SDS-PAGE and immunoblot analyses.
SDS-PAGE and immunoblotting with an enhanced-chemiluminescence detection method (GE Health Inc.) were carried out as reported previously (8, 44). The concentrations of protein in cell lysates were determined with a Bio-Rad protein assay kit using BSA as a standard. Unless otherwise noted, 5 μg of lysate protein was subjected to SDS-PAGE and immunoblotting using B. burgdorferi-specific antibodies.
Affinity blotting.
Protein-protein interactions were determined using affinity blotting (54). Briefly, 2 μg of purified recombinant Strep-CheD protein was subjected to SDS-PAGE and transferred to polyvinylidene difluoride (PDVF) membranes. The membranes were blocked overnight with shaking at 4°C in blocking solution (5% skim milk, 0.9% NaCl, 10 mM Tris, pH 7.4). After incubation, 10 μg of purified His-CheX, His-CheY3, MBP-MCP3, MBP-MCP4, or MBP-MCP5 was added to the blocking solution for 3 h (54). The membranes were washed 4 times with immunoblotting wash buffer and then probed with the proper antibodies. Detection was performed with the ECL immunoblotting detection kit (GE Healthcare Inc.).
Phosphorylation assays.
Phosphorylation of CheA, phosphotransfer from CheA-P to CheY3, and dephosphorylation of CheY3-P by CheX phosphatase have been described previously (32, 33, 53). B. burgdorferi has two CheA proteins, CheA1 and CheA2, and both are capable of autophosphorylation by ATP (53). However, the expression level of B. burgdorferi CheA1 or CheA2 in E. coli is poor, and they are also difficult to purify under native conditions (32). Alternatively, we performed the autophosphorylation assays using E. coli CheA. Purified E. coli CheA was kindly provided by Ruth Silversmith (University of North Carolina, Chapel Hill, NC) (55). To generate radiolabeled phosphorylated CheA (CheA-32P), 20 pmol of purified CheA was incubated with 10 μCi of [γ-32P]ATP and 30 mM nonradiolabeled ATP for 30 min at room temperature. Autophosphorylated CheA was applied to Bio-Spin6 columns according to the manufacturer's instructions to remove unincorporated [γ-32P]ATP (33, 53). CheA-32P was then incubated with 160 pmol of purified CheY3 for 5 min, as described previously (32, 53). The CheA and CheY3 reaction mixture was then incubated with 0.15 pmol of CheX or 0.15 pmol of CheX plus 15 pmol of CheD for various times. In vitro phosphorylation reactions were conducted in TKM buffer (50 mM Tris-HCl, 50 mM KCl, 5 mM MgCl2, pH 8.5), and the reactions were stopped using stop buffer (50 mM Tris-HCl, 100 mM dithiothreitol, 2% [wt/vol] SDS, 0.1% [wt/vol] bromophenol blue, 10% [vol/vol] glycerol, 5% [vol/vol] 2-mercaptoethanol, pH 6.8). The reaction mixtures were subjected to SDS-PAGE. The gels were dried, and the CheY3-32P dephosphorylation levels were analyzed by phosphorimage analysis using a Typhoon 9410 phosphorimager/fluorescence imager (GE Healthcare Inc.) (32, 53). The intensities of phosphorylated proteins (PI) were calculated using Quantity One 1-D analysis software ver. 4.6.9 (Bio-Rad Inc.) and are expressed as the relative “PI volume.”
Dark-field microscopy, swarm plate chemotaxis assays, and colony swarm plate assays.
Growing B. burgdorferi cells were imaged using a Zeiss Imager M1 dark-field microscope connected to a digital camera to determine morphology and motility. Swarm plate assays were performed to determine the spirochete's chemotactic ability as described previously (33, 41). Briefly, approximately 1 × 106 cells in a 5-μl volume were spotted into 0.35% agarose plates containing BSK medium diluted 1:10 in Dulbecco's phosphate-buffered saline. Since B. burgdorferi is a slow-growing organism with a 5- to 12-h generation time (56), the swarm plates were incubated for 5 days. Swarming images were documented using a digital camera (8, 45, 49, 57). To determine an individual colony's swarming ability, B. burgdorferi clones were plated on separate semisolid BSK plates (20 to 50 colonies per plate). Approximately 1 month after inoculation, the swarming diameter of each individual colony was measured. Each isolate was assayed in at least two independent experiments.
Mouse infection studies.
Six- to 7-week-old Mus musculus C3H/HeN mice were used for infection studies, as described previously (8, 35, 37, 49, 58). For infection via needle, 5 × 102 to 5 × 106 in vitro-grown spirochetes were injected intraperitoneally (8, 37, 59). The number of spirochetes was determined using a Petroff-Hausser chamber, and each clone was verified for its retention of endogenous plasmids before injection. The mice were bled 2 weeks postinoculation for immunoblot analysis with mouse sera against B. burgdorferi antigens to determine infectivity, as described previously (35, 45, 59, 60). Reisolation of B. burgdorferi from mouse skin, bladder, and joint tissues was performed 3 weeks postinjection to assess the ability of spirochetes to infect mice (35, 37, 45, 58, 61). Tissues of euthanized mice were placed in BSK-II growth medium and incubated for up to 35 days, and the presence of spirochetes was determined by dark-field microscopy. The 50% infectious dose (ID50) was calculated as described below.
Determination of ID50 and statistical analysis.
The dose required to infect 50% of the mice was experimentally determined for the wild-type, ΔcheD mutant, and cheDcom strains, as described previously (35, 49). The data from the ID50 infection experiment and the single-dose infection experiment for each strain were combined for the estimations of the 50% infectious dose. Comparison between strain ID50 values was done using a generalized linear model with a probit link function and the log dose. This method is also known as probit regression, and in it, we assume identical slopes in the response-log dose relationship but different intercepts for each strain. Graphically, the assumptions manifest themselves as dose-response curves with lateral shifts corresponding to the changes in the intercept. Additionally, an overdispersion parameter was fitted in order to accommodate greater homogeneity in infection rates than would otherwise be permitted by the model. All calculations were carried out using JMP V12 software (SAS Institute, Inc., Cary, NC). A P value of ≤0.05 between samples was considered significant.
Assessment of spirochete acquisition and transmissions using mouse-tick-mouse infection assays.
To assess if naive Ixodes scapularis ticks are able to acquire B. burgdorferi from infected C3H/HeN mice and if the spirochetes can survive in the ticks and then be transmitted from the ticks to naive mice, we performed mouse-tick-mouse cycle experiments (35, 45, 49). Briefly, naive mice were injected with in vitro-grown spirochetes to infect 100% of the mice (100 × ID50). Before performing any infection studies, the spirochetes were verified by PCR to ensure that each clone retained all the endogenous plasmids required for persistent infection in mouse and tick hosts (8, 35, 37, 45, 62). To verify the infection status, the mice were bled 2 weeks postinoculation and the sera were tested by immunoblotting using B. burgdorferi cell lysates. Three weeks postinjection, naive larval ticks were fed to repletion on separate spirochete-infected mice (3 mice per bacterial strain; ∼200 larvae/mouse) for 5 to 7 days and collected once they dropped off the mice to determine acquisition of spirochetes from the mice by the ticks. To determine the percentage of spirochete-positive ticks for each B. burgdorferi clone, fed ticks were squashed individually to isolate genomic DNA, followed by B. burgdorferi flaB gene-specific PCR, as described previously (35, 46, 49, 63). Seven days after drop-off, the fed larvae were surface sterilized using 3% H2O2 followed by 70% ethanol, crushed individually in BSK-II medium, and plated in semisolid BSK to determine the number of viable spirochetes per tick (i.e., acquisition of spirochetes from infected mice by larval ticks) (8, 37, 49, 58, 61). In determining the total number of spirochetes per fed tick, 10 ticks were analyzed for each strain, and the results were expressed as a scatter plot with the mean. Statistical analysis was performed as described below. A second subset of fed larvae was allowed to molt. Two to 3 weeks after the molt, the nymphal ticks were allowed to feed on naive mice to determine transmission of spirochetes from infected nymphs to mice (8, 9). Naive C3H/HeN mice were anesthetized, and groups of nymphs were confined to capsules affixed to the shaved backs of the mice (3 mice per strain; 15 nymphs/mouse) (8, 64, 65). The ticks were allowed to feed until repletion (3 to 5 days) and then collected from the capsules. At 7 days postrepletion, the ticks were squashed individually, and spirochete burdens were determined as described above. Statistical analysis was performed as described below. The mice were euthanized 3 weeks postrepletion, and the transmission of the spirochetes was evaluated by reisolation.
Statistical analysis.
The significance of the difference between the mean values of the groups was analyzed as follows. Unless otherwise stated, for all data, normality was checked using a Shapiro-Wilk normality test. If the data passed normality, a multiple-comparison analysis was performed by using one-way analysis of variance (ANOVA), followed by Tukey's post hoc test. If the data did not pass normality, a multiple-comparison analysis was performed by using a Kruskal-Wallis test, followed by a Dunn test. A P value of ≤0.05 between samples was considered significant.
RESULTS
Phosphatase activity of CheX is enhanced by CheD.
Amino acid sequence analysis of B. burgdorferi CheD indicated that it shares approximately 30% identity with B. subtilis and T. maritima CheD proteins. Most importantly, the essential residues of B. subtilis and T. maritima CheD are also conserved in B. burgdorferi (data not shown) (29). In B. subtilis, CheD plays an important role in chemotaxis by modification of MCPs by deamidation of glutamine to glutamate residues or by enhancing CheC phosphatase chemotaxis activity to efficiently dephosphorylate the response regulator CheY, thereby regulating the levels of CheY-P (27–30). Because of its possession of the conserved residues, we predicted that B. burgdorferi CheD may exhibit functions similar to those seen in other bacteria (27, 29, 31). The B. burgdorferi genome lacks a homolog of the CheC gene but encodes a functional phosphatase, CheX, that efficiently dephosphorylates CheY3-P (33, 34). In order to test if B. burgdorferi CheD is able to stimulate the phosphatase activity, we first performed protein-protein interaction assays using affinity blotting, as we supposed that CheD should bind to CheX in order to enhance the phosphatase activity. The affinity-blotting data indicated that CheD interacts with CheX but not with the chemotaxis response regulator CheY3 (Fig. 1). These observations suggest that CheD may be able to stimulate the phosphatase activity.
FIG 1.
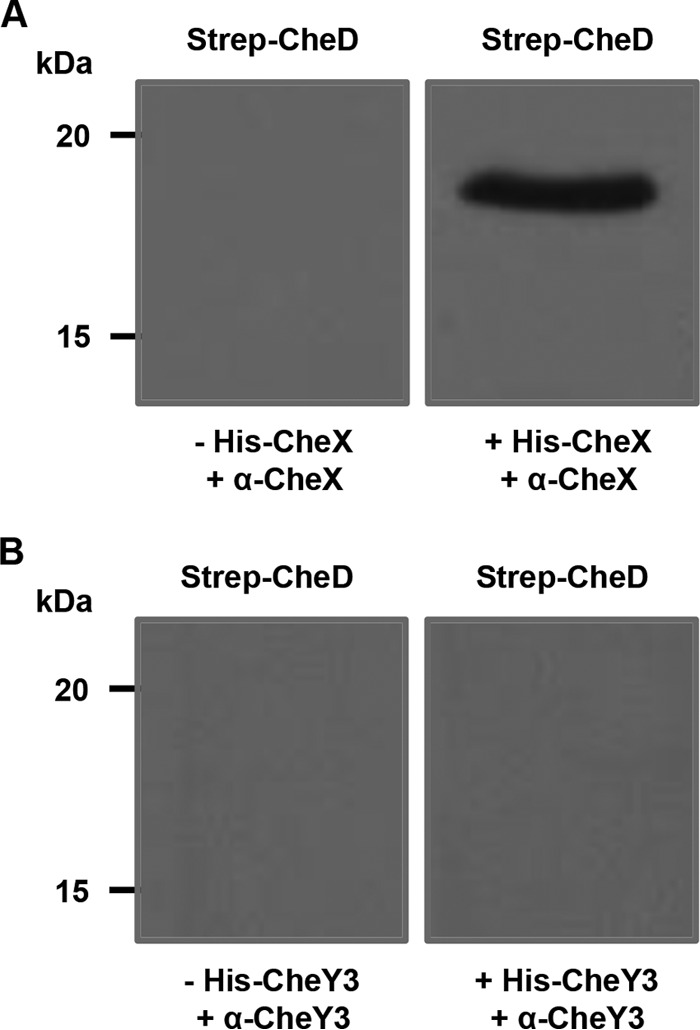
Affinity blot analysis indicates that CheD specifically interacts with CheX. (A) Purified Strep-CheD was incubated with (right) or without (left) His-CheX, followed by immunoblotting with polyclonal anti-CheX. The band at an apparent molecular mass of 19 kDa (Strep-CheD) was observed in blots probed with His-CheX. (B) Purified Strep-CheD was probed with (right) or without (left) His-CheY3, followed by Western blotting with polyclonal anti-CheY3. The bands at an apparent molecular mass of 19 kDa (Strep-CheD) were not detected with the His-CheY3 probe.
To determine if CheD is able to enhance phosphatase activity, we performed phosphorylation-dephosphorylation assays using recombinant purified CheA, CheY3, and CheX. When CheY3 was incubated with CheA-32P for 5 min, CheY3 was phosphorylated, as we have reported previously (Fig. 2A, lane 2) (32, 33, 53). Moreover, incubation of CheY3-32P with CheX for 2.5 min resulted in dephosphorylation of CheY3-32P by CheX (Fig. 2A, lane 3) (33). Importantly, when CheD was added in the CheA-CheY3-CheX reaction mixture for 2.5 min, CheY3-32P dephosphorylation was enhanced (Fig. 2A, lane 4) and the level of CheY3-32P was completely diminished within 5 min (Fig. 2A, lane 8). CheD itself did not affect CheY3-32P dephosphorylation (Fig. 2A, lanes 5 and 9). Moreover, the enhancement of CheX phosphatase activity was dependent on the CheD concentration (Fig. 2B, lanes 4 to 8), indicating that CheD efficiently enhances CheX phosphatase activity.
FIG 2.

CheD enhances phosphatase activity of CheX. Shown are phosphorylation-dephosphorylation assays using purified recombinant CheA (A), CheY3 (Y), CheX (X), and CheD (D) proteins. (A) Twenty picomoles of CheA was autophosphorylated using 10 μCi of [γ-32P]ATP (lane 1). CheA-32P was then incubated separately with 160 pmol of CheY3 for 5 min (lanes 2 and 6). Radiolabeled CheY3-32P was incubated with 0.15 pmol of CheX for 2.5 min or 5 min (lanes 3 and 7, respectively). The CheY3-32P and CheX reaction mixture was incubated with (lanes 4 land 8) or without (lanes 5 and 9) 15 pmol of CheD. The arrowheads indicate the positions of CheA-32P and CheY3-32P. (B) CheX phosphatase activity was efficiently enhanced by CheD. CheA was autophosphorylated as described above (lane 1). CheA-32P was then incubated with 160 pmol of CheY3 for 10 min (lane 2). The radiolabeled CheY3-32P was mixed with 0.15 pmol of CheX for 5 min without CheD (lane 3) or with 0.2 pmol (lane 4), 2 pmol (lane 5), 5 pmol (lane 6), 10 pmol (lane 7), or 20 pmol (lane 8) of CheD. CheY3-32P was also incubated with 20 pmol of CheD to confirm that CheD did not interfere with CheY3-P dephosphorylation (lane 9). The arrowheads indicate the positions of CheA-32P, CheY3-32P, and CheD. The relative intensities (“PI volumes”) of CheY3-32P are shown on the right with the corresponding phosphorimage lane numbers above.
CheD interacts with MCP3 and MCP4.
To determine if CheD is able to modify a B. burgdorferi chemoreceptor MCP protein, we performed deamidation assays using recombinant purified proteins, as described previously (29, 31). Even though we were able to detect deamidation with the positive-control (B. subtilis chemoreceptor) MCP-A, our multiple attempts under various assay conditions failed to deamidate any B. burgdorferi MCP. Since we were unable to detect deamidation of an MCP by CheD, we performed CheD-MCP protein-protein interaction assays alternatively, reasoning that CheD must first bind to an MCP in order to deamidate it. Using recombinant purified MCP and CheD proteins in affinity blotting, we were able to determine CheD-MCP interactions. Specifically, we found that CheD interacts with MCP3 and MCP4, but not MCP5 (Fig. 3).
FIG 3.
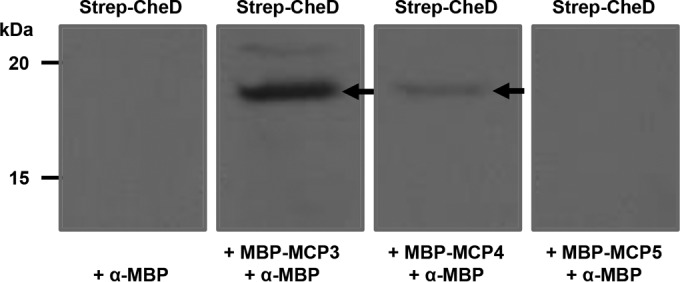
CheD specifically interacts with MCP3 and MCP4. Affinity blotting was performed as described in the text. Purified Strep-CheD was probed with MBP-MCP3, MBP-MCP4, or MBP-MCP5, followed by immunoblotting with monoclonal MBP antibodies (Thermo Scientific). The bands at an apparent molecular mass of 19 kDa (Strep-CheD; arrows) were always observed in blots probed with MCP3 and MCP4, but not MCP5.
Construction of cheD mutant and complemented strains.
In order to determine the role of CheD in motility, chemotaxis, or the infectious cycle of B. burgdorferi, we inactivated the gene using a kanamycin resistance cassette (PflgB-aph1) in the low-passage-number virulent wild-type clone B31-A3 (see Fig. S1 in the supplemental material) (35, 45, 49, 57). PCR analysis of the kanamycin-resistant clones indicated that the PflgB-aph1 cassette was inserted in the cheD gene (not shown), resulting in successful construction of the mutant. Furthermore, we confirmed by PCR that the cheD mutant retained all the linear and circular endogenous plasmids seen in the parental wild-type cells (data not shown).
The cheD mutant (ΔcheD) was complemented in cis by genomic integration to ensure that the phenotype of the mutant was solely attributed to the mutation and not due to a secondary mutation elsewhere in the genome (see Fig. S1 in the supplemental material) (35, 57). Accordingly, the cheD gene and its native promoter (PcheD) were fused together and inserted within bb0445 and bb0446 in the suicide vector pXLFCheD (46, 47). The insertion of PcheD-cheD was downstream of both bb0445 and bb0446 and less likely to affect their expression, as reported previously (35, 46, 47). To detect the expression of cheD in the wild-type, ΔcheD mutant, and complemented cheD (cheDcom) cells, we performed qRT-PCR using gene-specific primers. Real-time PCR detected the cheD transcripts in wild-type and cheDcom cells, but not in the cheD mutant cells (see Fig. S2 in the supplemental material). Moreover, the level of cheD transcripts synthesized in the cheDcom cells was approximately 84% of that in wild-type cells, indicating the restoration of cheD synthesis in the cheDcom cells (see Fig. S2 in the supplemental material).
In vitro motility and chemotaxis phenotypes of the ΔcheD mutant cells.
We analyzed the ΔcheD mutant to determine any alteration of motility or chemotaxis behavior and compared the results with the wild-type or cheDcom cells. Since we showed that CheD enhanced the phosphatase activity of CheX, we postulated that the cheD mutant cells might display an altered motility or chemotaxis phenotype. Dark-field microscopic analysis indicated that the motility patterns of the cheD mutant (run-flex/pause-reversal) and its flexing frequency during swimming were not noticeably different from those of the wild-type cells (9 to 12 flexes per minute [data not shown]). However, the chemotaxis was found to be significantly reduced compared to the wild-type (P = 0.002) or the cheDcom (P = 0.007) cells (Fig. 4A). In swarm plate assays, the average swarm diameters of the ΔcheD mutant were approximately 18% smaller than those of the wild-type or cheDcom cells (Fig. 4A). The swarm plate assay is a group event where millions of bacteria attempt to migrate out from the initial site of inoculation (in a semisolid plate) as they chemotax and metabolize neighboring nutrients, resulting in a swarm ring. However, the results obtained from such an assay may not accurately determine the chemotactic ability of an individual spirochete. Accordingly, we plated 20 to 50 B. burgdorferi cells in a semisolid plate (the same plates used for the swarm plate assay) to determine the chemotactic ability of individual cells by measuring their colony (swarm) size. Prolonged incubation of the plates produced colony sizes that were significantly smaller than those of the wild-type (P = 0.0001) or cheDcom (P = 0.014) cells. On average, the colony size of ΔcheD mutants was approximately 13% smaller than that of wild-type or cheDcom cells (Fig. 4B). Together, our results indicate that cheD plays an important role in B. burgdorferi chemotaxis.
FIG 4.
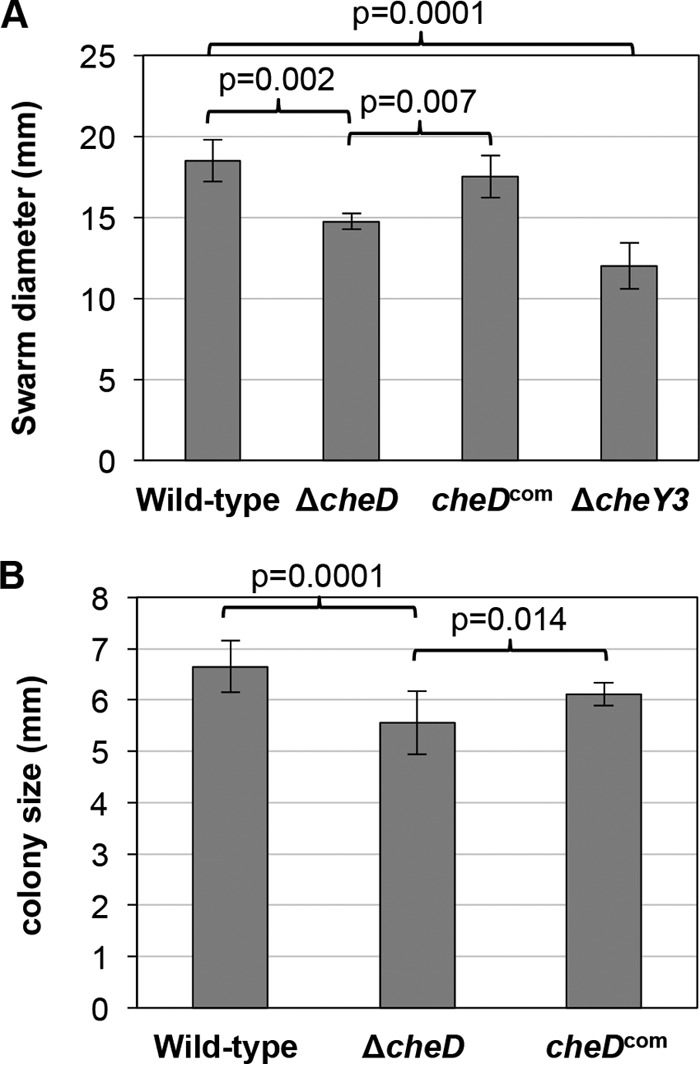
ΔcheD mutant cells are deficient in chemotaxis. (A) Swarm plate assays indicated a defect in chemotaxis exhibited by ΔcheD cells. Approximately 1 × 106 B. burgdorferi cells from the indicated strains were spotted into 0.35% soft agarose plates, which were then incubated for 5 days. The swarm diameter of each clone was measured in millimeters. A nonchemotactic cheY3 mutant was used as a control (53). The bars represent the means ± standard deviations of the mean for each clone (with data from at least 3 plates per clone). Statistical analysis was performed by using ANOVA, as described in Materials and Methods. P values between samples are shown at the top. (B) ΔcheD mutants display reduced colony size compared to the wild-type or complemented strain. B. burgdorferi cells from the indicated strains were plated on 0.4% soft agarose plates. An individual colony's swarming ability was determined 4 weeks after inoculation. The colony size of each clone was measured in millimeters. P values were determined using ANOVA. The values are the means ± standard deviations of the averages of at least 3 plates (20 individual colonies) per strain. P values between samples are shown at the top; a P value of ≤0.05 was considered significant.
B. burgdorferi ΔcheD mutant cells can establish infection in C3H/HeN mice by needle inoculation but display reduced infectivity.
While the role of CheD in motility and chemotaxis has been demonstrated in other bacteria, its role in pathogenesis has not been determined in any bacterium. In order to evaluate the infection potential of the mutant, groups of C3H/HeN mice were challenged with 10-fold-increasing doses of wild-type, ΔcheD, or cheDcom cells to determine the ID50. Immediately before the injections, we confirmed by PCR that these strains retained their endogenous plasmids (data not shown) (8, 35, 37, 45). Two weeks postinoculation, the mice were bled and their sera were assessed for reactivity with B. burgdorferi antigen membrane protein A, also known as P39 (59, 60). Furthermore, to confirm the serology results, mice were euthanized 3 weeks postinoculation and spirochetes were reisolated from ear, bladder, and joint tissues. The serology results indicated that cheD mutant cells were attenuated in establishing an infection in mice (not shown) and correlated well with reisolation of spirochetes from the tissues examined (Table 1). Whereas the cheDcom cells did not show a significant increase in the ID50 compared to the parental wild-type cells (P = 0.3), the ID50 for the cheD mutant was approximately 1 logarithm higher than that of wild-type or cheDcom cells, indicating significant attenuation in virulence (P = 0.012 and P = 0.0005, respectively) (Table 1).
TABLE 1.
ΔcheD mutant cells are able to establish infection in C3H/HeN mice, but infectivity is considerably reduceda
| Strain | Dose (CFU) | No. of mice infected/no. tested | ID50 |
|---|---|---|---|
| WT | 7.5 × 102 | 0/7 | 1.8080 × 104 |
| 7.5 × 103 | 1/7 | ||
| 7.5 × 104 | 5/7 | ||
| 9.3 × 105 | 7/7 | ||
| ΔcheD | 6.2 × 102 | 0/7 | 1.5694 × 105 |
| 6.2 × 103 | 1/7 | ||
| 6.2 × 104 | 1/7 | ||
| 1.00 × 106 | 6/7 | ||
| 1.00 × 107 | 7/7 | ||
| cheDcom | 5.6 × 102 | 0/7 | 1.9483 × 104 |
| 5.6 × 103 | 2/7 | ||
| 5.6 × 104 | 5/7 | ||
| 8.2 × 105 | 7/7 |
Mice were injected with the indicated B. burgdorferi clones and doses. Infectivity was determined by reisolation of B. burgdorferi from the tissues of euthanized mice (ear skin, ankle joint, and urinary bladder). ID50 and statistical analyses were performed using probit regression, as described in Materials and Methods. The ΔcheD mutant spirochetes were significantly attenuated compared to the ID50 of the wild type (P = 0.0125) or the complemented B. burgdorferi (P = 0.0005) (significance was defined as a P value of ≤0.05).
I. scapularis ticks are able to acquire and transmit mutant B. burgdorferi.
Because infection of and survival within the mammalian host represents only one aspect of the B. burgdorferi enzootic life cycle, a more comprehensive evaluation of the activities of these mutants in the tick vector was justified. Naive I. scapularis larvae were allowed to feed on infected mice in order to determine acquisition, as well viability, of B. burgdorferi in larval ticks. Seven days after feeding on infected mice, a subset of larvae were squashed individually, and genomic DNA was purified in order to determine spirochete-positive ticks by PCR using B. burgdorferi flaB gene-specific primers. Another set of fed larvae were squashed individually for plating in semisolid medium in order to determine the number of viable spirochetes per tick. The PCR data shown in Fig. 5A indicate that 100% of the larvae that fed on wild-type-, ΔcheD-, or cheDcom-infected mice were able to acquire the spirochetes (i.e., were spirochete positive). The burden of the mutant spirochetes per tick was not significantly lower than that of the wild type (P = 0.129) (Fig. 5B). However, the burden of mutant B. burgdorferi spirochetes was statistically less than the burden seen in larval ticks infected with the complemented cheDcom (P = 0.007).
FIG 5.
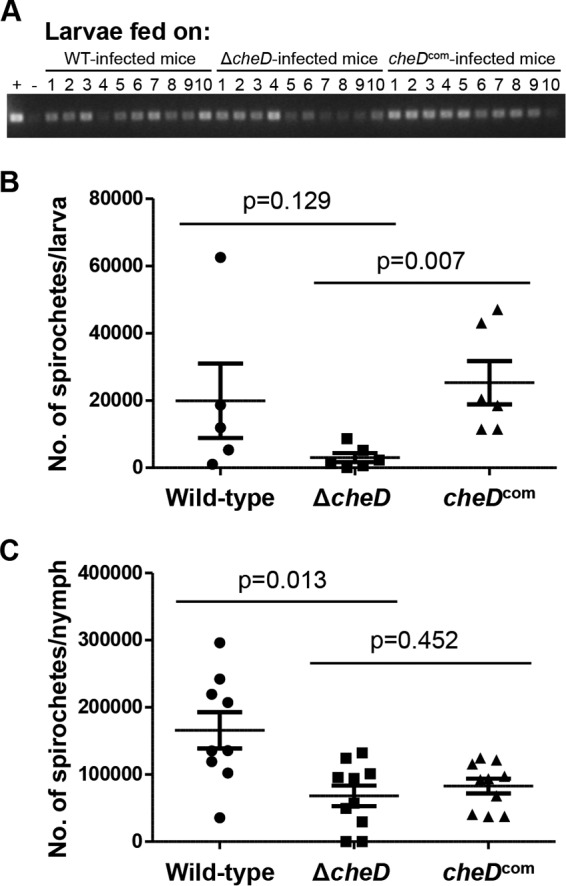
Naive ticks are able to acquire and transmit ΔcheD mutant spirochetes during mouse-tick-mouse infection studies. (A) Naive ticks were allowed to feed on infected mice to determine acquisition of spirochetes. Ten larvae fed upon the wild-type, ΔcheD, or complemented-spirochete-infected mice were squashed individually to isolate DNA. The DNA samples were then used to detect B. burgdorferi flaB by PCR. The PCR data indicated that 100% of the larval ticks were able to acquire the spirochetes from B. burgdorferi-infected mice (all groups). Tick DNA that we had previously confirmed to be spirochete positive was used as a positive control (+), and double-distilled water (ddH2O) was used as a negative control (−). (B) ΔcheD mutant spirochetes are able to survive in fed larvae. Viable ΔcheD spirochetes in fed larvae are reduced compared to the wild-type or complemented cells. The P values shown were determined by using ANOVA, as described in Materials and Methods. A P value of ≤0.05 between samples was considered significant. (C) Mutant spirochetes are able to survive in fed nymphs, but the number of viable ΔcheD cells in fed nymphs is considerably less than that of the wild type (P = 0.013) but not statistically different from that of the cheDcom cells (P = 0.452). The P values were determined as for panel B, and significance was defined as a P value of ≤0.05. A representative result from two independent studies with different batches of ticks is shown. The bars represent the numbers of mean viable spirochetes per tick ± standard errors of the mean (SEM).
In order to determine if the infected ticks were able to survive the molting and to transmit the mutant B. burgdorferi in naive mice, a subset of fed larvae were allowed to molt. Two to 3 weeks after molting, the infected nymphs were allowed to feed on naive C3H/HeN mice (3 mice per strain; 15 nymphs per mouse). Seven days postrepletion, the nymphs were analyzed individually by plating on semisolid plates as described above. The plating results indicated that the mutant spirochete burden in fed nymphs was considerably reduced compared to that of the wild type (P = 0.013); however, the ΔcheD mutant load was not statistically different from that of the cheDcom-infected nymphs (P = 0.452) (Fig. 5C) despite the fact that 100% of the nymphs were spirochete positive by all clones (data not shown).
To determine the transmission of B. burgdorferi from infected nymphs to mice, the tick-fed animals were bled at 2 weeks postfeeding to perform serology, followed by euthanization of the mice to reisolate B. burgdorferi from ear, ankle joint, and urinary bladder tissues at 3 weeks postrepletion. Serology, as well as bacterial outgrowth analyses, indicated that the ΔcheD mutants were able to transmit and establish infection in all the mice with no detectable deficiency (Table 2; see Fig. S3 in the supplemental material) even though we performed these in vivo studies twice using separate batches of ticks and mice.
TABLE 2.
ΔcheD spirochete-infected nymphal ticks are able to transmit the organisms into naive C3H/HeN micea
| Strain | No. of tissues positive/no. examined | No. of mice infected/no. tested |
|---|---|---|
| Wild type | 8/9 | 3/3 |
| ΔcheD | 9/9 | 3/3 |
| cheDcom | 8/9 | 3/3 |
Fifteen infected nymphs molted from larvae (from an acquisition assay) were allowed to feed on each C3H/HeN mouse. The mice were euthanized 3 weeks postrepletion. Infectivity was determined by reisolation of B. burgdorferi from the tissues of the euthanized mice. A representative result from two independent studies is shown.
DISCUSSION
In this study, we systematically demonstrated that CheD specifically interacts with CheX and enhances phosphatase activity (Fig. 1 and 2). It is notable that CheC-type phosphatases are monomers that possess two CheY-P dephosphorylation sites whereas the CheX-type phosphatases are dimers containing only one dephosphorylation site (29, 33, 34, 66). Considering this distinction, this is the first report demonstrating the stimulation of CheX-type phosphatases by CheD. CheD was reported to possess various functions in B. subtilis (27, 31, 67). While we were able to determine the CheD-CheX relationship, our multiple attempts to deamidate B. burgdorferi MCPs using CheD were unsuccessful despite the fact that our positive-control B. subtilis MCP deamidation was successful (27, 29), and we performed the assays under various conditions (data not shown). However, we were able to show that CheD specifically interacts with MCP3 and MCP4, but not MCP5 (Fig. 3). It is possible that CheD binds to other MCPs, but we chose to use these three MCPs because MCP3 and MCP5 are homologs of the major chemoreceptors Tar and Tsp of E. coli, respectively, and MCP4 possesses three CheD binding sites (see Table S1 in the supplemental material). CheD binding to MCP3 and MCP4 suggests that either our deamidation assay conditions were not optimized or the CheD/MCP proteins purified from E. coli cells were nonfunctional/inactive.
Because we found that CheD enhances CheX phosphatase activity and the cheX mutant constantly flexes (33), we supposed that the cheD mutant would also exhibit a motility phenotype. However, we failed to detect any noticeable difference in motility behavior exhibited by the ΔcheD cells. Even though the motility phenotype of the cheD mutant was indistinguishable from that of the wild-type cells, we demonstrated that CheD is important for chemotaxis (Fig. 4). The chemotaxis effect is unrelated to the mutant's growth rate in vitro, since the cells grow normally in the BSK-II growth medium (not shown). Based on these findings, we propose that the enhancer function associated with CheD is not likely to have a noticeable effect on CheX function in motility in vitro.
Our mouse infection studies demonstrated that the mutant cells are able to establish infection in C3H/HeN mice; however, the mutant was significantly attenuated (Table 1). These results were not completely surprising given the fact that the nonchemotactic cheX mutant cells are noninfectious in mice (M. A. Motaleb, unpublished data). In ΔcheD mutant cells, CheX, the only phosphatase in B. burgdorferi, is intact. Deletion of CheD likely had a minor effect on the CheX protein's ability to dephosphorylate CheY-P. Thus, the attenuation in mice that we observed with the cheD mutant spirochetes is reasonable, as the mutant did exhibit a reduced chemotaxis phenotype. It is also plausible that the attenuation we found with the mutant spirochetes could be a result of the chemoreceptors not being modified/deamidated normally due to the deletion of cheD. Moreover, we found that CheD is dispensable for acquisition or transmission of B. burgdorferi (Table 2; see Fig. S3 in the supplemental material) despite the fact that the viability of mutant spirochetes was reduced in ticks (both larvae and nymphs), and this reduced burden was detected consistently even though the P values of the wild type versus ΔcheD or cheDcom versus ΔcheD were not always statistically significant (Fig. 5).
Chemotaxis has been found to be important for the disease processes of many bacteria, including the spirochetes (1, 2, 4, 5, 7, 68); however, our findings with the ΔcheD mutant suggest that either the in vitro assay conditions do not reflect the in vivo host conditions (in vivo, the cheD mutant may be normal with respect to its chemotactic ability) or the chemotaxis defect we observed with the ΔcheD mutant in vitro was not reduced enough to affect the acquisition or transmission of spirochetes. However, our coherent observation of marginally reduced burdens of cheD mutant spirochetes in ticks could be related to its activity in CheX, as the cheX mutant spirochete burdens in ticks are also marginally reduced compared to those of the wild-type (Motaleb, unpublished). Moreover, we postulate that this phenotype is linked to fewer mutant spirochetes having been acquired by the ticks while feeding or that the viability defect is connected to the receptor MCP modification function. Further evaluation of the roles of MCP proteins in B. burgdorferi or how CheD affects the receptors and their subsequent effect on virulence needs to be done.
The Lyme disease spirochete, B. burgdorferi, exists primarily in a zoonotic cycle involving Ixodes ticks and mammalian hosts. Effective chemotaxis in these disparate habitats potentially explains the need for multiple chemotaxis systems in the organism. We propose that different adaptation systems (CheD or CheR1/CheR2 mediated) are likely employed to sense gradients of different scales and contours in these diverse milieus. However, exactly how these chemotaxis systems contribute to the fitness of the Lyme disease spirochete is an open question.
Supplementary Material
ACKNOWLEDGMENTS
Research in our laboratory is supported by National Institutes of Health grants 1R01AR060834 and 1R21AI113014.
We thank Elizabeth Novak for critical reading of the manuscript. We also thank Ruth Silversmith and George Ordal for sharing reagents.
Footnotes
Supplemental material for this article may be found at http://dx.doi.org/10.1128/IAI.01347-15.
REFERENCES
- 1.Butler SM, Camilli A. 2005. Going against the grain: chemotaxis and infection in Vibrio cholerae. Nat Rev Microbiol 3:611–620. doi: 10.1038/nrmicro1207. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Lertsethtakarn P, Ottemann KM, Hendrixson DR. 2011. Motility and chemotaxis in Campylobacter and Helicobacter. Annu Rev Microbiol 65:389–410. doi: 10.1146/annurev-micro-090110-102908. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Novak EA, Sultan SZ, Motaleb MA. 2014. The cyclic-di-GMP signaling pathway in the Lyme disease spirochete, Borrelia burgdorferi. Front Cell Infect Microbiol 4:56. doi: 10.3389/fcimb.2014.00056. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Sampedro I, Parales RE, Krell T, Hill JE. 2015. Pseudomonas chemotaxis. FEMS Microbiol Rev 39:17–46. doi: 10.1111/1574-6976.12081. [DOI] [PubMed] [Google Scholar]
- 5.Charon NW, Cockburn A, Li C, Liu J, Miller KA, Miller MR, Motaleb MA, Wolgemuth CW. 2012. The unique paradigm of spirochete motility and chemotaxis. Annu Rev Microbiol 66:349–370. doi: 10.1146/annurev-micro-092611-150145. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Motaleb M, Liu J, Wooten RM. 2015. Spirochetal motility and chemotaxis in the natural enzootic cycle and development of Lyme disease. Curr Opin Microbiol 28:106–113. doi: 10.1016/j.mib.2015.09.006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Sze CW, Zhang K, Kariu T, Pal U, Li C. 2012. Borrelia burgdorferi needs chemotaxis to establish infection in mammals and to accomplish its enzootic cycle. Infect Immun 80:2485–2492. doi: 10.1128/IAI.00145-12. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Sultan SZ, Manne A, Stewart PE, Bestor A, Rosa PA, Charon NW, Motaleb MA. 2013. Motility is crucial for the infectious life cycle of Borrelia burgdorferi. Infect Immun 81:2012–2021. doi: 10.1128/IAI.01228-12. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Sultan SZ, Sekar P, Zhao X, Manne A, Liu J, Wooten RM, Motaleb MA. 2015. Motor rotation is essential for the formation of the periplasmic flagellar ribbon, cellular morphology, and Borrelia burgdorferi persistence within Ixodes scapularis tick and murine hosts. Infect Immun 83:1765–1777. doi: 10.1128/IAI.03097-14. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Li C, Xu H, Zhang K, Liang FT. 2010. Inactivation of a putative flagellar motor switch protein FliG1 prevents Borrelia burgdorferi from swimming in highly viscous media and blocks its infectivity. Mol Microbiol 75:1563–1576. doi: 10.1111/j.1365-2958.2010.07078.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Lambert A, Picardeau M, Haake DA, Sermswan RW, Srikram A, Adler B, Murray GA. 2012. FlaA proteins in Leptospira interrogans are essential for motility and virulence but are not required for formation of the flagellum sheath. Infect Immun 80:2019–2025. doi: 10.1128/IAI.00131-12. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Guyard C, Raffel SJ, Schrumpf ME, Dahlstrom E, Sturdevant D, Ricklefs SM, Martens C, Hayes SF, Fischer ER, Hansen BT. 2013. Periplasmic flagellar export apparatus protein, FliH, is involved in post-transcriptional regulation of FlaB, motility and virulence of the relapsing fever spirochete Borrelia hermsii. PLoS One 8:e72550. doi: 10.1371/journal.pone.0072550. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Lin T, Gao L, Zhao X, Liu J, Norris SJ. 2015. Mutations in the Borrelia burgdorferi flagellar type III secretion system genes fliH and fliI profoundly affect spirochete flagellar assembly, morphology, motility, structure, and cell division. mBio 6:e00579-15. doi: 10.1128/mBio.00579-15. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Li C, Bakker RG, Motaleb MA, Sartakova ML, Cabello FC, Charon NW. 2002. Asymmetrical flagellar rotation in Borrelia burgdorferi nonchemotactic mutants. Proc Natl Acad Sci U S A 99:6169–6174. doi: 10.1073/pnas.092010499. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Li C, Motaleb A, Sal M, Goldstein SF, Charon NW. 2000. Spirochete periplasmic flagella and motility. J Mol Microbiol Biotechnol 2:345–354. [PubMed] [Google Scholar]
- 16.Schneider WR, Doetsch RN. 1974. Effect of viscosity on bacterial motility. J Bacteriol 117:696–701. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Shoesmith J. 1960. The measurement of bacterial motility. J Gen Microbiol 22:528–535. doi: 10.1099/00221287-22-2-528. [DOI] [Google Scholar]
- 18.Goldstein SF, Charon NW. 1988. Motility of the spirochete Leptospira. Cell Motil Cytoskeleton 9:101–110. doi: 10.1002/cm.970090202. [DOI] [PubMed] [Google Scholar]
- 19.Klitorinos A, Noble P, Siboo R, Chan E. 1993. Viscosity-dependent locomotion of oral spirochetes. Oral Microbiol Immunol 8:242–244. doi: 10.1111/j.1399-302X.1993.tb00567.x. [DOI] [PubMed] [Google Scholar]
- 20.Nakamura S, Adachi Y, Goto T, Magariyama Y. 2006. Improvement in motion efficiency of the spirochete Brachyspira pilosicoli in viscous environments. Biophys J 90:3019–3026. doi: 10.1529/biophysj.105.074336. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Bockenstedt LK, Wormser GP. 2014. Review: unraveling Lyme disease. Arthritis Rheumatol 66:2313–2323. doi: 10.1002/art.38756. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Micali G, Endres RG. 2016. Bacterial chemotaxis: information processing, thermodynamics, and behavior. Curr Opin Microbiol 30:8–15. doi: 10.1016/j.mib.2015.12.001. [DOI] [PubMed] [Google Scholar]
- 23.Sourjik V, Wingreen NS. 2012. Responding to chemical gradients: bacterial chemotaxis. Curr Opin Cell Biol 24:262–268. doi: 10.1016/j.ceb.2011.11.008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Bi S, Lai L. 2015. Bacterial chemoreceptors and chemoeffectors. Cell Mol Life Sci 72:691–708. doi: 10.1007/s00018-014-1770-5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Sarkar MK, Paul K, Blair D. 2010. Chemotaxis signaling protein CheY binds to the rotor protein FliN to control the direction of flagellar rotation in Escherichia coli. Proc Natl Acad Sci U S A 107:9370–9375. doi: 10.1073/pnas.1000935107. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Silversmith RE, Bourret RB. 1999. Throwing the switch in bacterial chemotaxis. Trends Microbiol 7:16–22. doi: 10.1016/S0966-842X(98)01409-7. [DOI] [PubMed] [Google Scholar]
- 27.Kristich CJ, Ordal GW. 2002. Bacillus subtilis CheD is a chemoreceptor modification enzyme required for chemotaxis. J Biol Chem 277:25356–25362. doi: 10.1074/jbc.M201334200. [DOI] [PubMed] [Google Scholar]
- 28.Yuan W, Glekas GD, Allen GM, Walukiewicz HE, Rao CV, Ordal GW. 2012. The importance of the interaction of CheD with CheC and the chemoreceptors compared to its enzymatic activity during chemotaxis in Bacillus subtilis. PLoS One 7:e50689. doi: 10.1371/journal.pone.0050689. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Chao X, Muff TJ, Park S, Zhang S, Pollard AM, Ordal GW, Bilwes AM, Crane BR. 2006. A receptor-modifying deamidase in complex with a signaling phosphatase reveals reciprocal regulation. Cell 124:561–571. doi: 10.1016/j.cell.2005.11.046. [DOI] [PubMed] [Google Scholar]
- 30.Rao CV, Glekas GD, Ordal GW. 2008. The three adaptation systems of Bacillus subtilis chemotaxis. Trends Microbiol 16:480–487. doi: 10.1016/j.tim.2008.07.003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Glekas GD, Plutz MJ, Walukiewicz HE, Allen GM, Rao CV, Ordal GW. 2012. Elucidation of the multiple roles of CheD in Bacillus subtilis chemotaxis. Mol Microbiol 86:743–756. doi: 10.1111/mmi.12015. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Motaleb MA, Miller MR, Li C, Charon NW. 2007. Phosphorylation assays of chemotaxis two-component system proteins in Borrelia burgdorferi. Methods Enzymol 422:438–447. doi: 10.1016/S0076-6879(06)22022-6. [DOI] [PubMed] [Google Scholar]
- 33.Motaleb MA, Miller MR, Li C, Bakker RG, Goldstein SF, Silversmith RE, Bourret RB, Charon NW. 2005. CheX is a phosphorylated CheY phosphatase essential for Borrelia burgdorferi chemotaxis. J Bacteriol 187:7963–7969. doi: 10.1128/JB.187.23.7963-7969.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Pazy Y, Motaleb MA, Guarnieri MT, Charon NW, Zhao R, Silversmith RE. 2010. Identical phosphatase mechanisms achieved through distinct modes of binding phosphoprotein substrate. Proc Natl Acad Sci U S A 107:1924–1929. doi: 10.1073/pnas.0911185107. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Pitzer JE, Sultan SZ, Hayakawa Y, Hobbs G, Miller MR, Motaleb MA. 2011. Analysis of the Borrelia burgdorferi cyclic-di-GMP-binding protein PlzA reveals a role in motility and virulence. Infect Immun 79:1815–1825. doi: 10.1128/IAI.00075-11. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Bono JL, Elias AF, Kupko JJ III, Stevenson B, Tilly K, Rosa P. 2000. Efficient targeted mutagenesis in Borrelia burgdorferi. J Bacteriol 182:2445–2452. doi: 10.1128/JB.182.9.2445-2452.2000. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Elias AF, Stewart PE, Grimm D, Caimano MJ, Eggers CH, Tilly K, Bono JL, Akins DR, Radolf JD, Schwan TG, Rosa P. 2002. Clonal polymorphism of Borrelia burgdorferi strain B31 MI: implications for mutagenesis in an infectious strain background. Infect Immun 70:2139–2150. doi: 10.1128/IAI.70.4.2139-2150.2002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Fraser CM, Casjens S, Huang WM, Sutton GG, Clayton R, Lathigra R, White O, Ketchum KA, Dodson R, Hickey EK. 1997. Genomic sequence of a Lyme disease spirochaete, Borrelia burgdorferi. Nature 390:580–586. doi: 10.1038/37551. [DOI] [PubMed] [Google Scholar]
- 39.Casjens S, Palmer N, Van Vugt R, Mun Huang W, Stevenson B, Rosa P, Lathigra R, Sutton G, Peterson J, Dodson RJ. 2000. A bacterial genome in flux: the twelve linear and nine circular extrachromosomal DNAs in an infectious isolate of the Lyme disease spirochete Borrelia burgdorferi. Mol Microbiol 35:490–516. [DOI] [PubMed] [Google Scholar]
- 40.Jewett MW, Lawrence KA, Bestor A, Byram R, Gherardini F, Rosa PA. 2009. GuaA and GuaB are essential for Borrelia burgdorferi survival in the tick-mouse infection cycle. J Bacteriol 191:6231–6241. doi: 10.1128/JB.00450-09. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Motaleb MA, Miller MR, Bakker RG, Li C, Charon NW. 2007. Isolation and characterization of chemotaxis mutants of the Lyme disease spirochete Borrelia burgdorferi using allelic exchange mutagenesis, flow cytometry, and cell tracking. Methods Enzymol 422:421–437. doi: 10.1016/S0076-6879(06)22021-4. [DOI] [PubMed] [Google Scholar]
- 42.Stewart PE, Rosa PA. 2008. Transposon mutagenesis of the Lyme disease agent Borrelia burgdorferi. Methods Mol Biol 431:85–95. [DOI] [PubMed] [Google Scholar]
- 43.Bertani G. 1951. Studies on lysogenesis. I. The mode of phage liberation by lysogenic Escherichia coli. J Bacteriol 62:293–300. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Motaleb MA, Corum L, Bono JL, Elias AF, Rosa P, Samuels DS, Charon NW. 2000. Borrelia burgdorferi periplasmic flagella have both skeletal and motility functions. Proc Natl Acad Sci U S A 97:10899–10904. doi: 10.1073/pnas.200221797. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Sultan SZ, Pitzer JE, Miller MR, Motaleb MA. 2010. Analysis of a Borrelia burgdorferi phosphodiesterase demonstrates a role for cyclic-di-guanosine monophosphate in motility and virulence. Mol Microbiol 77:128–142. doi: 10.1111/j.1365-2958.2010.07191.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Zhang X, Yang X, Kumar M, Pal U. 2009. BB0323 function is essential for Borrelia burgdorferi virulence and persistence through tick-rodent transmission cycle. J Infect Dis 200:1318–1330. doi: 10.1086/605846. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Li X, Pal U, Ramamoorthi N, Liu X, Desrosiers DC, Eggers C, Anderson JF, Radolf JD, Fikrig E. 2007. The Lyme disease agent Borrelia burgdorferi requires BB0690, a Dps homologue, to persist within ticks. Mol Microbiol 63:694–710. [DOI] [PubMed] [Google Scholar]
- 48.Motaleb MA, Sal MS, Charon NW. 2004. The decrease in FlaA observed in a flaB mutant of Borrelia burgdorferi occurs posttranscriptionally. J Bacteriol 186:3703–3711. doi: 10.1128/JB.186.12.3703-3711.2004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Sultan SZ, Pitzer JE, Boquoi T, Hobbs G, Miller MR, Motaleb MA. 2011. Analysis of the HD-GYP domain cyclic dimeric GMP phosphodiesterase reveals a role in motility and the enzootic life cycle of Borrelia burgdorferi. Infect Immun 79:3273–3283. doi: 10.1128/IAI.05153-11. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Simm R, Remminghorst U, Ahmad I, Zakikhany K, Romling U. 2009. A role for the EAL-like protein STM1344 in regulation of CsgD expression and motility in Salmonella enterica serovar Typhimurium. J Bacteriol 191:3928–3937. doi: 10.1128/JB.00290-09. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Livak KJ, Schmittgen TD. 2001. Analysis of relative gene expression data using real-time quantitative PCR and the 2−ΔΔCT method. Methods 25:402–408. doi: 10.1006/meth.2001.1262. [DOI] [PubMed] [Google Scholar]
- 52.Smith JG, Latiolais JA, Guanga GP, Citineni S, Silversmith RE, Bourret RB. 2003. Investigation of the role of electrostatic charge in activation of the Escherichia coli response regulator CheY. J Bacteriol 185:6385–6391. doi: 10.1128/JB.185.21.6385-6391.2003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Motaleb MA, Sultan SZ, Miller MR, Li C, Charon NW. 2011. CheY3 of Borrelia burgdorferi is the key response regulator essential for chemotaxis and forms a long-lived phosphorylated intermediate. J Bacteriol 193:3332–3341. doi: 10.1128/JB.00362-11. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Toker AS, Macnab RM. 1997. Distinct regions of bacterial flagellar switch protein FliM interact with FliG, FliN and CheY. J Mol Biol 273:623–634. doi: 10.1006/jmbi.1997.1335. [DOI] [PubMed] [Google Scholar]
- 55.Silversmith RE, Appleby JL, Bourret RB. 1997. Catalytic mechanism of phosphorylation and dephosphorylation of CheY: kinetic characterization of imidazole phosphates as phosphodonors and the role of acid catalysis. Biochemistry 36:14965–14974. doi: 10.1021/bi9715573. [DOI] [PubMed] [Google Scholar]
- 56.Rosa PA, Tilly K, Stewart PE. 2005. The burgeoning molecular genetics of the Lyme disease spirochaete. Nat Rev Microbiol 3:129–143. doi: 10.1038/nrmicro1086. [DOI] [PubMed] [Google Scholar]
- 57.Motaleb MA, Pitzer JE, Sultan SZ, Liu J. 2011. A novel gene inactivation system reveals altered periplasmic flagellar orientation in a Borrelia burgdorferi fliL mutant. J Bacteriol 193:3324–3331. doi: 10.1128/JB.00202-11. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Stewart PE, Bestor A, Cullen JN, Rosa PA. 2008. A tightly regulated surface protein of Borrelia burgdorferi is not essential to the mouse-tick infectious cycle. Infect Immun 76:1970–1978. doi: 10.1128/IAI.00714-07. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.Jewett MW, Lawrence K, Bestor AC, Tilly K, Grimm D, Shaw P, VanRaden M, Gherardini F, Rosa PA. 2007. The critical role of the linear plasmid lp36 in the infectious cycle of Borrelia burgdorferi. Mol Microbiol 64:1358–1374. doi: 10.1111/j.1365-2958.2007.05746.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60.Simpson WJ, Burgdorfer W, Schrumpf ME, Karstens RH, Schwan TG. 1991. Antibody to a 39-kilodalton Borrelia burgdorferi antigen (P39) as a marker for infection in experimentally and naturally inoculated animals. J Clin Microbiol 29:236–243. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61.Grimm D, Elias AF, Tilly K, Rosa PA. 2003. Plasmid stability during in vitro propagation of Borrelia burgdorferi assessed at a clonal level. Infect Immun 71:3138–3145. doi: 10.1128/IAI.71.6.3138-3145.2003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62.Purser JE, Norris SJ. 2000. Correlation between plasmid content and infectivity in Borrelia burgdorferi. Proc Natl Acad Sci U S A 97:13865–13870. doi: 10.1073/pnas.97.25.13865. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63.Yang XF, Pal U, Alani SM, Fikrig E, Norgard MV. 2004. Essential role for OspA/B in the life cycle of the Lyme disease spirochete. J Exp Med 199:641–648. doi: 10.1084/jem.20031960. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64.Mulay VB, Caimano MJ, Iyer R, Dunham-Ems S, Liveris D, Petzke MM, Schwartz I, Radolf JD. 2009. Borrelia burgdorferi bba74 is expressed exclusively during tick feeding and is regulated by both arthropod- and mammalian host-specific signals. J Bacteriol 191:2783–2794. doi: 10.1128/JB.01802-08. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65.Patton TG, Dietrich G, Dolan MC, Piesman J, Carroll JA, Gilmore RD Jr. 2011. Functional analysis of the Borrelia burgdorferi bba64 gene product in murine infection via tick infestation. PLoS One 6:e19536. doi: 10.1371/journal.pone.0019536. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66.Park S, Chao X, Gonzalez-Bonet G, Beel BD, Bilwes AM, Crane BR. 2004. Structure and function of an unusual family of protein phosphatases: the bacterial chemotaxis proteins CheC and CheX. Mol Cell 16:563–574. [DOI] [PubMed] [Google Scholar]
- 67.Walukiewicz HE, Tohidifar P, Ordal GW, Rao CV. 2014. Interactions among the three adaptation systems of Bacillus subtilis chemotaxis as revealed by an in vitro receptor-kinase assay. Mol Microbiol 93:1104–1118. doi: 10.1111/mmi.12721. [DOI] [PubMed] [Google Scholar]
- 68.Lux R, Miller JN, Park NH, Shi W. 2001. Motility and chemotaxis in tissue penetration of oral epithelial cell layers by Treponema denticola. Infect Immun 69:6276–6283. doi: 10.1128/IAI.69.10.6276-6283.2001. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.