

ABSTRACT
Certain wood decay basidiomycetes, collectively referred to as brown rot fungi, rapidly depolymerize cellulose while leaving behind the bulk of cell wall lignin as a modified residue. The mechanism(s) employed is unclear, but considerable evidence implicates the involvement of diffusible oxidants generated via Fenton-like chemistry. Toward a better understanding of this process, we have examined the transcriptome and secretome of Wolfiporia cocos when cultivated on media containing glucose, purified crystalline cellulose, aspen (Populus grandidentata), or lodgepole pine (Pinus contorta) as the sole carbon source. Compared to the results obtained with glucose, 30, 183, and 207 genes exhibited 4-fold increases in transcript levels in cellulose, aspen, and lodgepole pine, respectively. Mass spectrometry identified peptides corresponding to 64 glycoside hydrolase (GH) proteins, and of these, 17 corresponded to transcripts upregulated on one or both woody substrates. Most of these genes were broadly categorized as hemicellulases or chitinases. Consistent with an important role for hydroxyl radical in cellulose depolymerization, high transcript levels and upregulation were observed for genes involved in iron homeostasis, iron reduction, and extracellular peroxide generation. These patterns of regulation differ markedly from those of the closely related brown rot fungus Postia placenta and expand the number of enzymes potentially involved in the oxidative depolymerization of cellulose.
IMPORTANCE The decomposition of wood is an essential component of nutrient cycling in forest ecosystems. Few microbes have the capacity to efficiently degrade woody substrates, and the mechanism(s) is poorly understood. Toward a better understanding of these processes, we show that when grown on wood as a sole carbon source the brown rot fungus W. cocos expresses a unique repertoire of genes involved in oxidative and hydrolytic conversions of cell walls.
INTRODUCTION
A central process in nutrient recycling in forest ecosystems, wood decomposition involves the combined activities of abiotic and biotic processes. The composition and porosity of wood represent formidable barriers to microbial decay, but it has long been known that certain Agaricomycotina efficiently depolymerize the major constituents, cellulose, hemicellulose, and lignin (reviewed in reference 1). Mechanistic aspects of wood decay remain uncertain, but genome annotations of a growing number of these fungi have revealed genetic repertoires seemingly consistent with two basic forms of decay: white rot and brown rot (2, 3).
Efficient ligninolysis by white rot fungi is typically associated with the maintenance of genes encoding class II peroxidases, and cellulose hydrolysis is attributed to the concerted actions of exo- and endoglucanases. These glycoside hydrolases (GHs) are often linked to cellulose binding modules, particularly those categorized as family 1 carbohydrate binding modules (CBM1s) (http://www.cazy.org). Exceptions to these generalities exist, especially among less efficient lignin degraders, such as Schizophyllum commune, Botryobasidium botryosum, and Jaapia argillacea (4).
In sharp contrast, brown rot fungi, such as Wolfiporia cocos, lack class II peroxidases and conventional cellobiohydrolases with CBM1s. Consistent with this, these fungi modify only lignin, and cellulose is rapidly depolymerized. The latter is commonly ascribed to low-molecular-weight oxidants, chiefly hydroxyl radical generated by the Fenton reaction (H2O2 + Fe2+ + H+ → H2O + Fe3+ + ˙OH) (5–8). Plausible redox systems capable of sustaining this reaction have been proposed (reviewed in references 9 and 10), including hydroquinone redox cycling (11, 12). Genes potentially involved in generating the key Fenton reactants include those encoding benzoquinone reductases (BQRs), laccases (Lac), various H2O2-producing oxidoreductases (e.g., copper radical oxidases [CROs]), and iron reductases (GLPs). Ferroxidase (FET3), ferric reductase transmembrane component (FRE), iron permease (FTE), and oxalate decarboxylase (ODC) would be expected to play an important indirect role through the modulation of iron homeostasis.
Toward a mechanistic understanding of the process, the transcriptomes and secretomes of W. cocos were examined when cultured for 5 days on microcrystalline cellulose (MCC), lodgepole pine (LP), and aspen (Asp).
MATERIALS AND METHODS
Culture conditions.
Basal medium contained, per liter, 2 g of NH4NO3, 2 g of KH2PO4, 0.5 g of MgSO4·7H2O, 0.1 g of CaCl2·2H2O, 1 mg of thiamine hydrochloride, and 10 ml of mineral solution. Mineral solution contained, per liter, 1.5 g of nitrilotriacetic acid, 3 g of MgSO4·7H2O, 0.5 g of MnSO4·H2O, 1 g of NaCl, 0.1 g of FeSO4·H2O, 0.1 g of CoSO4, 0.1 g of CaCl2, 0.1 g of ZnSO4·7H2O, 0.01 g of CuSO4, 0.01 g of AlK(SO4)2·12H2O, 0.01 g of H3BO3, and 0.01 g of NaMoO4·2H2O (13). Two-liter Erlenmeyer flasks containing 250 ml of this medium were supplemented with 0.5% (wt/vol) debarked, Wiley-milled (1-mm mesh) Populus grandidentata (aspen) or Pinus contorta (lodgepole pine) as the sole carbon source. For defined cultures, 0.5% (wt/vol) microcrystalline cellulose (MCC; Avicel PH-101; Fluka Chemika) or glucose (Glc) served as the carbon source. Triplicate cultures for each medium were inoculated with mycelia scraped from agar plates and incubated on a rotary shaker (150 rpm) at room temperature. Ferrozine measurements (14, 15) showed no significant increase in iron reduction after 5 days incubation when cultures were harvested by filtration through Miracloth (Calbiochem, La Jolla, CA) as described previously (16, 17).
Whole-transcriptome shotgun sequencing.
Harvested mycelium was immediately snap-frozen in liquid nitrogen and the total RNA was purified as described previously (18). Samples were submitted to the University of Wisconsin Madison Biotechnology Center for verification of purity and integrity on an Agilent 2100 BioAnalyzer, followed by library preparation (Illumina TruSeq RNA [Rev.A]) and sample multiplexing for sequencing on the HiSeq2000 as described previously (19). DNAStar Inc. (Madison, WI) modules SeqNGen and ArrayStar (v4) were used to map reads, visualize results, and apply statistical analyses.
LC-MS/MS secretome analysis.
Nano-liquid chromatography-tandem mass spectrometry (nano-LC-MS/MS) was used to identify proteins in culture filtrates as described previously (17, 19, 20). In short, ∼300 μg of trichloroacetic acid (TCA)-acetone-precipitated protein was trypsin digested and∼7.5 μg loaded for nano-LC-MS/MS analysis using an Agilent 1100 nanoflow system (Agilent Technologies) connected to a hybrid linear ion trap-Orbitrap mass spectrometer (LTQ-Orbitrap XL; Thermo Fisher Scientific) equipped with a nanoelectrospray ion source. High-performance liquid chromatography (HPLC) was performed using an in-house-fabricated 15-cm C18 column packed with MAGIC C18AQ 3-μm particles (MICHROM Bioresources Inc., Auburn, CA) and laser pulled tip (P-2000; Sutter Instrument) using 360-μm by 75-μm fused silica tubing. The LTQ-Orbitrap was set to acquire MS/MS spectra in data-dependent mode, and raw MS/MS data were searched against the W. cocos amino acid sequence database available at http://genome.jgi.doe.gov/pages/dynamicOrganismDownload.jsf?organism=Wolco1 (12,746 protein entries) using in-house Mascot search engine 2.2.07 (Matrix Science) with variable methionine oxidation and asparagine and glutamine deamidation. Peptide mass tolerance was set at 15 ppm and fragment mass at 0.6 Da. Significance-of-identification analysis and spectrum-based quantification were performed with Scaffold software (version 4.0.4; Proteome Software Inc., Portland, OR). Acceptable identification was set to more than 95.0% probability within a 0.9% false-discovery rate and containing at least two identified peptides. The Protein Prophet algorithm (21) was used to assign protein probabilities. Proteins containing peptides that could not be differentiated based on MS/MS analyses were grouped to satisfy the principles of parsimony. Approximate protein abundance in each of the cultures was expressed as the exponentially modified protein abundance index (emPAI) value (22). It should be noted that under the conditions employed, mass spectrometry detection of extracellular proteins is subject to false-negative results due to the frequency of trypsin cleavage, low molecular weight, substrate binding, and/or proteolytic degradation. False-positive results, in contrast, are unlikely.
Microarray data accession number.
Raw transcriptome data and normalized RPKM values (reads per kilobase per million mapped reads) have been deposited to the NCBI Gene Expression Omnibus (GEO) database under accession number GSE78007.
RESULTS
Identification of differentially regulated genes.
Transcriptome sequencing (RNA-seq) identified a total of 38 genes whose transcripts accumulated >4-fold (RPKM values > 10; P < 0.01) in pairwise comparisons between glucose (Glc)-grown and MCC-grown organisms. Comparisons of aspen (Asp) or lodgepole pine (LP) against Glc at the same threshold identified regulated transcripts corresponding to 190 and 214 genes, respectively. Few transcripts were more abundant in Glc-grown cultures than in cultures grown in MCC (8 genes), Asp (7 genes), and LP (7 genes), and none corresponded to genes implicated in wood decay (see Data Set S1 in the supplemental material). The distribution of genes with increased transcript levels in MCC, Asp, and LP relative to Glc (Fig. 1A) showed considerable overlap. For example, 146 genes were upregulated >4-fold in both Asp and LP. Moreover, the number of genes seemingly influenced by a single substrate (e.g., 30 genes in Asp [Fig. 1A]) fell sharply at lower thresholds, such as 2-fold. Of course, highly expressed but unregulated genes may also play a key role in cellulose depolymerization. For perspective, RPKM scores greater than 782, 829, 812, and 799 represent the upper 1% of genes expressed in Glc, MCC, Asp, and LP, respectively (see full listing in Data Set S1).
FIG 1.
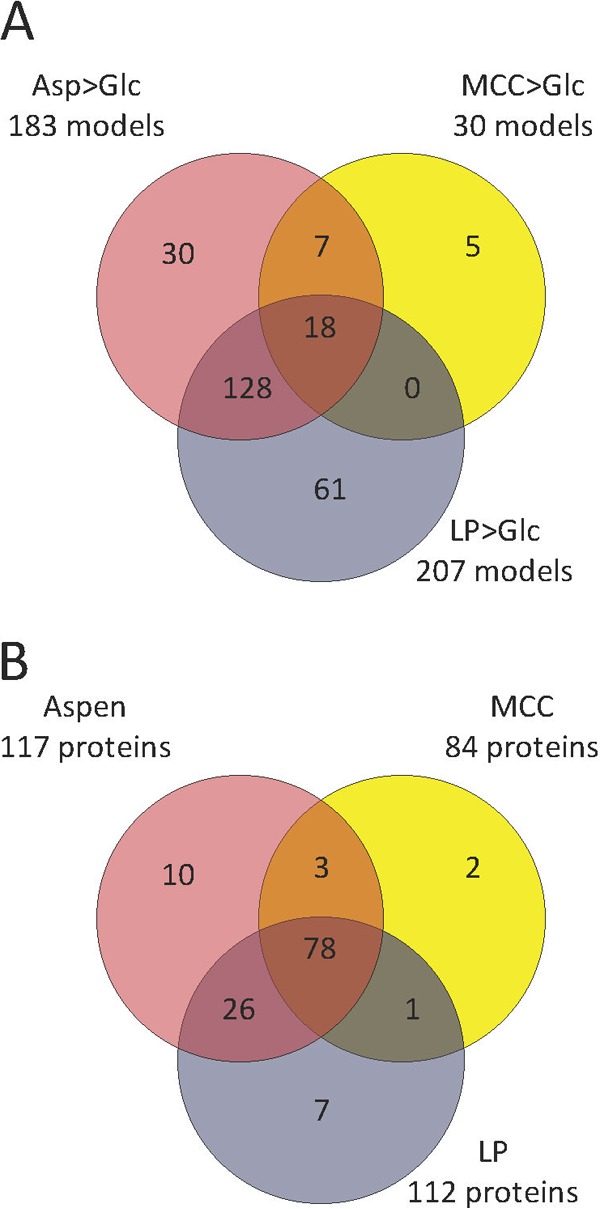
Regulated transcripts (A) and LC-MS/MS-identified protein (B) in media containing aspen (Asp), microcrystalline cellulose (MCC), and lodgepole pine (LP). RNA-seq results (A) exclude genes with transcripts accumulating <4-fold relative to Glc or with RPKM values of <10. Protein identifications include only those with ≥2 unique peptides. Substantial overlap between substrates was observed. For examples, 146 (128 plus 18) genes were upregulated and 104 (26 plus 78) proteins were identified in both Asp and LP.
Shotgun LC-MS/MS analysis of culture filtrates identified a total of 704 W. cocos proteins. Cell lysis and high sensitivity of mass spectrometry led to the identification of many enzymes and structural proteins likely involved in intracellular processes, e.g., ribosomal proteins and glycolytic enzymes (see Data Set S1 in the supplemental material for a complete listing). Focusing attention on the 959 W. cocos proteins with predicted secretion signals (Neural Network scores ≥ 4), we unambiguously detected 132 proteins, often in more than one medium. The distribution of those detected in MCC, Asp, and LP is shown in Fig. 1B. In medium containing Glc as the sole carbon source, 51 proteins were identified. Of these, an epoxide hydrolase (Wolco_137580), a protease family A1 protein (Wolco_116315), and a protease family S8 protein (Wolco_140059) were identified exclusively in Glc medium (see Data Set S1). In connection with this, it should be mentioned that inaccurate 5′ termini and nonclassical signals may increase the number of false negatives. A relevant example include laccase model Wolco_139080, which, due to a truncated N terminus, features no secretion signal. (Model 90916 properly extends the region.) Also pertinent, the methanol oxidase (Wolco_24953) sequence predicts a nonclassical signal (SecP score = 0.82; http://www.cbs.dtu.dk/services/SecretomeP/) (23).
Carbohydrate-active enzymes.
W. cocos is predicted (http://www.cazy.org) (24) to encode 148 glycoside hydrolases, 14 carbohydrate esterase, and two polysaccharide lyases (2). Twenty-seven of these genes were significantly (P < 0.01) upregulated greater than 4-fold on cellulose-based media (MCC, Asp, and LP) relative to Glc (Table 1). These genes were broadly characterized as hemicellulase- or chitinase-encoding genes, and genetic multiplicity and substrate-induced differential regulation were observed. For example, transcripts of the endoxylanase Wolco_164484 were the most highly upregulated in Asp, and the corresponding protein was detected in all filtrates. By comparison, another GH10, Wolco_103638, showed more modest upregulation and the protein was not identified in MCC-containing medium. Similarly, paralogous gene pairs encoding exopolygalacturonases Wolco_128405 and Wolco_79505, α-rhamnosidases Wolco_102857 and Wolco_74146, α-glucuronidases Wolco_26703 and Wolco_138209, and β-glucosidases Wolco_137577 and Wolco_23559 exhibited different expression patterns. Three putative chitinase-encoding genes exhibited higher transcript levels on cellulose-based media than with Glc, but only Wolco_146767 and Wolco_76203 peptides were identified in culture filtrates. Interestingly, upregulated transcript levels were also observed for two putative chitin deacetylases. No peptides or elevated transcripts were observed for polysaccharide lyases Wolco_104113 and Wolco_161668. Lytic polysaccharide monooxygenases (LPMOs) Wolco_134838 and Wolco_57339, formerly designated members of glycoside hydrolase family 61 (25–30), were not highly expressed or regulated under the conditions employed (see Data Set S1).
TABLE 1.
Glycoside hydrolases and carbohydrate esterases with transcripts accumulating ≥4 fold (P < 0.01) on media containing microcrystalline cellulose, aspen, or lodgepole pine relative to glucose (Glc)a
| IDb | Putative functionc | emPAId |
RPKMe |
MCC/Glc |
Asp/Glc |
LP/Glc |
LP/Asp |
||||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
| Glc | MCC | Asp | LP | Glc | MCC | Asp | LP | R | P | R | P | R | P | R | P | ||
| 164484 | GH10 endoxylanase | 0.51 | 0.98 | 3.45 | 2.08 | 32 | 612 | 857 | 853 | 19.1 | 0.02 | 26.8 | 0.00 | 26.7 | 0.00 | 1.0 | 1.00 |
| 139561 | GH5_22 β-glycanase | 0.00 | 0.00 | 0.77 | 0.60 | 5 | 40 | 125 | 266 | 8.4 | 0.01 | 26.4 | 0.00 | 56.0 | 0.00 | 2.1 | 0.14 |
| 128405* | GH28 exopolygalacturonase | 0.00 | 0.00 | 0.30 | 0.45 | 3 | 9 | 64 | 130 | 3.5 | 0.00 | 25.3 | 0.00 | 51.1 | 0.00 | 2.0 | 0.40 |
| 146767 | GH18/CBM5 chitinase | 0.00 | 0.64 | 7.06 | 4.80 | 119 | 1206 | 2667 | 2044 | 10.2 | 0.00 | 22.5 | 0.00 | 17.2 | 0.00 | 0.8 | 0.22 |
| 26703 | GH115 α-glucuronidase | 0.00 | 0.00 | 1.03 | 1.29 | 29 | 88 | 275 | 397 | 3.1 | 0.00 | 9.6 | 0.00 | 13.8 | 0.00 | 1.4 | 0.45 |
| 106510 | GH5_22 β-glycanase | 1 | 5 | 9 | 17 | 4.4 | 0.17 | 7.3 | 0.01 | 13.9 | 0.00 | 1.9 | 0.42 | ||||
| 21871 | GH16 1,3(4)-β-glucanase | 0.33 | 0.93 | 0.35 | 0.40 | 84 | 468 | 613 | 721 | 5.6 | 0.00 | 7.3 | 0.00 | 8.6 | 0.00 | 1.2 | 0.66 |
| 137577 | GH3 β-glucosidase | 0.00 | 0.00 | 0.22 | 0.38 | 4 | 15 | 31 | 68 | 3.3 | 0.07 | 6.9 | 0.00 | 15.1 | 0.00 | 2.2 | 0.10 |
| 24959 | GH79 glycoside hydrolase | 0.42 | 0.77 | 0.38 | 0.67 | 44 | 132 | 238 | 333 | 3.0 | 0.00 | 5.4 | 0.00 | 7.6 | 0.00 | 1.4 | 0.19 |
| 76203 | GH18 possible chitinase | 4.78 | 8.09 | 5.40 | 3.68 | 1103 | 2784 | 5855 | 4934 | 2.5 | 0.01 | 5.3 | 0.00 | 4.5 | 0.00 | 0.8 | 0.62 |
| 89691 | GH18/CBM5 chitinase | 118 | 424 | 575 | 488 | 3.6 | 0.01 | 4.9 | 0.00 | 4.1 | 0.00 | 0.8 | 0.48 | ||||
| 26848 | GH5_15 β-1,6-glucanase | 0.38 | 0.45 | 1.61 | 2.19 | 76 | 135 | 368 | 373 | 1.8 | 0.05 | 4.8 | 0.00 | 4.9 | 0.01 | 1.0 | 1.00 |
| 103638 | GH10 endoxylanase | 0.00 | 0.00 | 1.11 | 1.64 | 25 | 56 | 119 | 129 | 2.3 | 0.42 | 4.8 | 0.00 | 5.2 | 0.01 | 1.1 | 0.96 |
| 138209 | GH115 α-glucuronidase | 0.00 | 0.00 | 0.12 | 0.11 | 2 | 3 | 8 | 17 | 1.7 | 0.13 | 4.3 | 0.00 | 9.8 | 0.00 | 2.3 | 0.13 |
| 78411 | GH1 β-glucosidase | 3 | 10 | 15 | 16 | 3.0 | 0.09 | 4.2 | 0.00 | 4.7 | 0.00 | 1.1 | 0.81 | ||||
| 19345 | GH51 α-arabinofuranosidase | 0.35 | 0.53 | 0.20 | 0.23 | 22 | 100 | 88 | 85 | 4.6 | 0.00 | 4.0 | 0.01 | 3.9 | 0.05 | 1.0 | 0.99 |
| 24785 | GH2 β mannosidase | 0.00 | 0.00 | 0.00 | 0.12 | 30 | 62 | 117 | 163 | 2.1 | 0.03 | 3.9 | 0.00 | 5.5 | 0.00 | 1.4 | 0.05 |
| 137392 | GH125 α-mannosidase | 0.60 | 1.21 | 0.45 | 0.59 | 79 | 152 | 248 | 317 | 1.9 | 0.05 | 3.1 | 0.00 | 4.0 | 0.00 | 1.3 | 0.34 |
| 102857 | GH78 possible α-rhamnosidase | 6 | 7 | 17 | 31 | 1.3 | 0.68 | 2.8 | 0.00 | 5.3 | 0.00 | 1.9 | 0.05 | ||||
| 125762 | GH28 exopolygalacturonase | 3 | 3 | 7 | 29 | 0.9 | 0.82 | 2.7 | 0.00 | 10.4 | 0.01 | 3.8 | 0.12 | ||||
| 149294 | GH31 α-xylosidase | 19 | 34 | 49 | 122 | 1.8 | 0.27 | 2.5 | 0.00 | 6.3 | 0.00 | 2.5 | 0.00 | ||||
| 147868 | GH5_7 endo-β-1,4-mannanase | 0.14 | 0.21 | 0.16 | 0.18 | 24 | 42 | 59 | 105 | 1.7 | 0.33 | 2.5 | 0.00 | 4.3 | 0.00 | 1.8 | 0.02 |
| 23559 | GH3 β-glucosidase | 0.00 | 0.00 | 0.06 | 0.00 | 23 | 33 | 56 | 99 | 1.4 | 0.53 | 2.4 | 0.00 | 4.3 | 0.00 | 1.8 | 0.02 |
| 79505* | GH28 exopolygalaturonase | 0.00 | 0.00 | 0.00 | 0.16 | 4 | 6 | 7 | 17 | 1.6 | 0.02 | 1.8 | 0.02 | 4.1 | 0.01 | 2.3 | 0.17 |
| 31035* | CE4 chitin deacetylase | 3 | 18 | 146 | 161 | 5.1 | 0.00 | 42.5 | 0.00 | 46.8 | 0.00 | 1.1 | 0.75 | ||||
| 165643 | CE8 possible pectinesterase | 0.00 | 0.00 | 0.20 | 0.31 | 1 | 3 | 15 | 40 | 3.7 | 0.00 | 16.9 | 0.00 | 46.1 | 0.00 | 2.7 | 0.08 |
| 86713 | CE4 possible chitin deacetylase | 38 | 98 | 173 | 158 | 2.6 | 0.01 | 4.6 | 0.00 | 4.2 | 0.00 | 0.9 | 0.75 | ||||
Genes exhibiting relatively low expression levels in all four substrates (RPKM values < 10) are excluded. Expression ratios (R) and moderated t test-derived probability (P) are shown. Proteins are ranked by transcript ratios on Asp relative to Glc.
Protein model identifiers (ID) derived from the Joint Genome Institute's databases (44). Models with asterisks need editing.
Putative function based on CAZy classification (http://www.cazy.org) and, in the case of GH5 family members, the analysis of Aspeborg et al. (45). Excluding “possible” designations, SwissProt BLASTP score swere >500. CE, carbohydrate esterase.
emPAI, exponentially modified protein abundance index (22).
RNA-seq-normalized reads per kilobase per million (RPKM). For perspective, 11,190 and 11,205 genes had at least one transcript detected in Asp and LP samples, respectively. Moreover, genes with Asp/Glc and LP/Glc ratios of >7.1 and >9.2, respectively, were within the top 1%.
BLASTP queries of the Postia placenta database (http://genome.jgi.doe.gov/pages/blast-query.jsf?db=Pospl1) (31) identified at least one homolog (bit scores >1000) for each of the carbohydrate-active enzyme-encoding genes listed in Table 1. Most of these genes are broadly categorized as hemicellulases, and patterns of regulation were similar in both species (see Data Set S2 in the supplemental material for detailed comparisons). Notable exceptions include highly expressed and regulated W. cocos chitinases with family 5 carbohydrate binding modules (Wolco_146767 and Wolco_89691). The P. placenta homologs exhibit no significant transcript accumulation when cultured on ball-milled aspen (P. grandidentata) or white pine (Pinus strobus) (see Data Set S2) (32).
Oxidative systems.
Various models have implicated Fenton-generated hydroxyl radicals in cellulose depolymerization by brown rot fungi. Iron homeostasis and reduction play a key role, and consistent with this process, genes encoding ferroxidase and iron permease were substantially upregulated in Asp and LP relative to Glc (Table 2). Transcript levels of the P. placenta Fet3 and Frt1 orthologs also increased in woody substrates (see Data Set S2) (32), but the upregulation of W. cocos ferric reductase transmembrane component (Wolco_109665), a putative siderophore-iron transporter (Wolco_166774), and oxalate decarboxylase (Wolco_163340) was not observed in the P. placenta cultures (Table 2; see also Data Set S2) (32).
TABLE 2.
Peptides and transcripts corresponding to genes implicated in the generation of extracellular oxidants when W. cocos or P. placenta was grown for 5 days in media containing glucose, microcrystalline cellulose, aspen, or lodgepole pine as the sole carbon source
| IDa | Putative functionb | emPAIc |
RPKMd |
MCC/Glc |
Asp/Glc |
LP/Glc |
|||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
| Glc | MCC | Asp | LP | Glc | MCC | Asp | LP | R | P | R | P | R | P | ||
| 27193 | Benzoquinone reductase | 1.44 | 3.27 | 1.93 | 0.99 | 167 | 197 | 160 | 169 | 1.2 | 0.62 | 1.0 | 0.93 | 1.0 | 0.96 |
| 24953 | Methanol oxidase | 0.10 | 0.00 | 0.07 | 0.00 | 150 | 128 | 64 | 281 | 0.9 | 0.69 | 0.4 | 0.04 | 1.9 | 0.21 |
| 80969 | Copper radical oxidase 1 | 2 | 2 | 1 | 2 | 0.8 | 0.54 | 0.5 | 0.08 | 0.7 | 0.64 | ||||
| 106250 | Copper radical oxidase 5 | 0.00 | 0.00 | 0.52 | 0.69 | 5 | 32 | 372 | 343 | 7.1 | 0.00 | 81.0 | 0.00 | 74.7 | 0.00 |
| 83651 | Copper radical oxidase 2 | 1.41 | 2.96 | 0.99 | 1.29 | 111 | 111 | 214 | 211 | 1.0 | 0.99 | 1.9 | 0.01 | 1.9 | 0.01 |
| 87683 | Copper radical oxidase 6 | 1 | 1 | 1 | 1 | 0.5 | 0.02 | 1.0 | 0.70 | 0.7 | 0.03 | ||||
| 109665 | Ferric reductase transmembrane | 9 | 122 | 446 | 175 | 13.2 | 0.03 | 48.5 | 0.00 | 19.0 | 0.01 | ||||
| 123970 | Ferroxidase | 18 | 19 | 68 | 65 | 1.1 | 0.93 | 3.7 | 0.00 | 3.5 | 0.01 | ||||
| 76446 | Glycopeptide iron reductase | 1 | 2 | 13 | 138 | 2.0 | 0.30 | 11.8 | 0.02 | 130.1 | 0.00 | ||||
| 26263 | Glycopeptide iron reductase | 0.00 | 0.00 | 0.27 | 0.00 | 109 | 141 | 339 | 358 | 1.3 | 0.18 | 3.1 | 0.00 | 3.3 | 0.00 |
| 156960 | Glycopeptide iron reductase | 0 | 0 | 1 | 1 | 1.5 | 0.38 | 3.0 | 0.02 | 6.9 | 0.15 | ||||
| 40810 | Glycopeptide iron reductase | 611 | 617 | 923 | 790 | 1.0 | 0.96 | 1.5 | 0.03 | 1.3 | 0.17 | ||||
| 156965 | Glycopeptide iron reductase | 2 | 1 | 3 | 4 | 0.6 | 0.23 | 1.1 | 0.66 | 1.5 | 0.18 | ||||
| 105249 | Glycopeptide iron reductase | 7 | 7 | 6 | 4 | 0.9 | 0.67 | 0.8 | 0.41 | 0.6 | 0.04 | ||||
| 105247 | Glycopeptide iron reductase | 1 | 1 | 1 | 1 | 0.9 | 0.72 | 0.6 | 0.42 | 0.9 | 0.72 | ||||
| 76567 | Glycopeptide iron reductase | 1 | 0 | 0 | 0 | 0.4 | 0.33 | 0.5 | 0.18 | 0.6 | 0.40 | ||||
| 164149 | Glycopeptide iron reductase | 6 | 2 | 2 | 1 | 0.4 | 0.06 | 0.3 | 0.00 | 0.2 | 0.04 | ||||
| 139155 | Iron permease (FTR1) | 13 | 13 | 72 | 60 | 1.0 | 1.00 | 5.4 | 0.00 | 4.5 | 0.00 | ||||
| 157228 | Laccase | 2 | 2 | 41 | 79 | 0.9 | 0.56 | 18.7 | 0.00 | 36.2 | 0.00 | ||||
| 165246 | Laccase | 0.00 | 0.00 | 0.44 | 1.05 | 47 | 20 | 240 | 200 | 0.4 | 0.29 | 5.2 | 0.01 | 4.3 | 0.02 |
| 139080 | Laccase | 0.00 | 0.00 | 0.00 | 0.47 | 43 | 38 | 44 | 78 | 0.9 | 0.48 | 1.0 | 0.83 | 1.8 | 0.16 |
| 163340 | Oxalate decarboxylase | 0.00 | 0.00 | 0.00 | 0.35 | 7 | 7 | 52 | 118 | 1.0 | 0.86 | 7.5 | 0.02 | 17.0 | 0.00 |
| 106770 | Oxalate decarboxylase | 0.58 | 0.39 | 0.00 | 0.00 | 3 | 3 | 2 | 2 | 0.8 | 0.22 | 0.6 | 0.02 | 0.7 | 0.06 |
| 139266 | Tyrosinase | 9 | 20 | 74 | 270 | 2.2 | 0.09 | 8.0 | 0.03 | 29.4 | 0.01 | ||||
| 140963 | Tyrosinase | 3 | 46 | 38 | 114 | 14.6 | 0.00 | 12.0 | 0.04 | 36.1 | 0.03 | ||||
| 166774 | Siderophore iron transporter | 6 | 5 | 71 | 56 | 0.8 | 0.29 | 14.9 | 0.00 | 11.8 | 0.00 | ||||
Protein model identifiers (ID) derived from the Joint Genome Institute's databases (44). Expression ratios (R) and moderated t test-derived probability (P) were computed against the Glc reference.
Putative function based on best hit scores and percent amino acid similarity over aligned regions of known proteins.
emPAI, exponentially modified protein abundance index (22).
RNA-seq-normalized reads per kilobase per million (RPKM).
Nine genes with similarity to a purported Phanerochaete chrysosporium hydroxyl radical-producing glycopeptide (GLP1; GenBank accession number AB236889) (33, 34) were identified. Three W. cocos GLP genes were highly expressed and peptides were detected for Wolco_26263 in Asp. Four GLP-encoding genes were identified in the P. placenta genome, although only Pospl_128971 showed modest upregulation on microcrystalline cellulose (1.9-fold; P < 0.01 [31]), Asp (1.6-fold; P < 0.01; see Data Set S2), and white pine (1.8-fold; P < 0.01; see Data Set S2).
Another proposed pathway for sustained hydroxyl radical formation involves hydroquinone redox cycling and, in this connection, high levels of W. cocos benzoquinone reductase (BQR) transcripts and the corresponding peptides were detected in all media. Potentially, a complete Fenton system could be generated via the oxidation of quinones by the observed upregulated laccases and tyrosinases (Table 2) (35). A similar repertoire of genes was identified in P. placenta, but BQR transcripts increased substantially on woody substrates relative to Glc and no Pospl_124517 peptides were detected (see Data Set 2).
Several potential sources of H2O2 were evident. The highly expressed Wolco_24953 protein is 88% identical to Gloeophyllum trabeum methanol oxidase (GenBank accession number DQ835989). Possibly utilizing methanol from lignin demethylation during incipient brown rot decay, the enzyme has been implicated in hydroxyl radical generation via H2O2 production (36). In addition, four H2O2-generating copper radical oxidases (37) were also expressed. Of these, CRO5 was highly expressed and, impressively, the fourth most highly upregulated gene among 11,191 detected in Asp (Table 2; see also Data Set S1). This observation stands in distinct contrast to the findings for P. placenta, for which CRO5 regulation was less than 2-fold in Asp or white pine relative to Glc (see Data Set S2) (32). Conversely, the methanol oxidase of P. placenta was substantially upregulated on the woody substrates but transcript levels of the W. cocos ortholog (Wolco_24953) were unaffected.
Regulated extracellular proteins with uncertain roles.
Six genes were significantly (P < 0.01) upregulated (>4-fold) in Asp and LP relative to Glc, and their peptides were detected in the same woody substrates (Table 3). RPKM values for the gene encoding protein Wolco_97131 were within the top 1% on Asp and LP. The protein features cupredoxin domains and a predicted COOH transmembrane domain but otherwise is of unknown or “hypothetical” function. FAD binding domains were predicted in Wolco_139157 and Wolco_139466, and among Swiss-Prot entries, the latter is most closely related (34% identity with 94% coverage) to the 3-hydroxybenzoate 6-hydroxylase of Klebsiella oxytoca. Wolco_167014 features functionally diverse metallo-dependent phosphatase domains, and while the protein sequence is highly conserved, it is also uncharacterized. Wolco_22757 encodes lysophospholipase with significant homology to numerous fungal enzymes, although here, too, a clear relationship to lignocellulose conversion is lacking. This W. cocos protein, and all those listed in Table 3, had unambiguous P. placenta homologs (bit scores >1350). However, elevated transcript levels and detectable peptides were limited to the aromatic ring hydroxylase (Pospl_56710) on white pine and the metallophosphoesterase (Pospl_128158) on Asp, respectively (see Data Set S2).
TABLE 3.
Relative protein abundances and transcript accumulations of genes not previously implicated in plant cell wall degradationa
| IDb | Functional domainsc | IPR ID | emPAId |
RPKMe |
MCC/Glc |
Asp/Glc |
LP/Glc |
|||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
| Glc | MCC | Asp | LP | Glc | MCC | Asp | LP | R | P | R | P | R | P | |||
| 139466 | Aromatic-ring hydroxylase | IPR003042 | 0.00 | 0.00 | 0.36 | 0.58 | 23 | 25 | 126 | 176 | 1.1 | 0.67 | 5.5 | 0.00 | 7.7 | 0.00 |
| 167014 | Metallophosphoesterase | IPR004843 | 0.00 | 0.00 | 0.58 | 0.34 | 39 | 125 | 210 | 177 | 3.2 | 0.00 | 5.4 | 0.00 | 4.6 | 0.00 |
| 22757 | Lysophospholipase | IPR002642 | 0.00 | 0.00 | 0.45 | 0.31 | 22 | 38 | 110 | 110 | 1.8 | 0.11 | 5.1 | 0.00 | 5.1 | 0.00 |
| 139157* | FAD-linked oxidase | IPR006094 | 0.00 | 0.00 | 0.18 | 0.20 | 10 | 9 | 39 | 51 | 0.9 | 0.53 | 4.0 | 0.00 | 5.2 | 0.00 |
| 97131* | Hypothetical (cupredoxins) | IPR008972 | 0.00 | 0.00 | 0.13 | 0.15 | 66 | 291 | 1733 | 1259 | 4.4 | 0.00 | 26.3 | 0.00 | 19.1 | 0.00 |
| 138259 | Hypothetical | 0.25 | 0.49 | 0.64 | 0.91 | 22 | 49 | 106 | 112 | 2.2 | 0.04 | 4.8 | 0.00 | 5.1 | 0.00 | |
Limited to genes with regulated transcript levels (>4-fold; P < 0.01), with predicted secretion signals and with at least two unique peptides detected by mass spectrometry. Genes exhibiting relatively low transcript levels in all four substrates (RPKM values < 10) are excluded. Expression ratios (R) and moderated t test-derived probability (P) are shown. IPR, InterPro domain.
Protein model identifiers (ID) derived from the Joint Genome Institute's databases (44). Models with asterisks each have a possible transmembrane helix.
Possible functional domains and InterPro database identifier.
emPAI, exponentially modified protein abundance index (22).
RNA-seq-normalized reads per kilobase per million (RPKM).
Complete metabolism of wood extractives and carbohydrate-derived molecules involves intracellular, often membrane-bound, cytochrome P450 monooxygenases (Table 4) and transporters (Table 5). Of the estimated 206 P450 genes, transcripts of 10 were significantly (P < 0.01) upregulated in Asp and LP relative to Glc (Table 4). None of these genes were closely linked. Wolco_24176 and Wolco_136784 were differentially regulated in Asp and LP, a phenomenon previously observed by comparing Asp and white pine cultures of P. placenta (32). Four Wolco_24176 homologs with bit scores >1000 (Pospl_99219, Pospl_97728, Pospl_56490, and Pospl_89660) were more highly expressed in white pine than in Asp (see Data Set S2).
TABLE 4.
Cytochrome P450s with transcripts accumulating >4-fold in one or more pairwise comparisonsa
| IDb | Classc | Locationd | Commente | RPKMf |
MCC/Glc |
Asp/Glc |
LP/Glc |
LP/Asp |
|||||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
| Glc | MCC | Asp | LP | R | P | R | P | R | P | R | P | ||||
| 144633 | E class, group 1 | 7:1232140–1234411 | Wolco_75851 + _90431 | 2 | 5 | 57 | 149 | 2.4 | 0.01 | 28.6 | 0.00 | 74.4 | 0.00 | 2.6 | 0.22 |
| 24176 | E class, group 1 | 3:3470288–3472772 | 2 | 12 | 42 | 160 | 7.0 | 0.02 | 24.7 | 0.00 | 93.4 | 0.00 | 3.8 | 0.04 | |
| 138085 | P450, CYP52 | 6:714438–717341 | TMH; emPAI 0.2 in Asp | 33 | 172 | 744 | 345 | 5.2 | 0.00 | 22.5 | 0.00 | 10.4 | 0.00 | 0.5 | 0.25 |
| 97631 | E class, group 1 | 2:3721700–3724067 | TMHs | 4 | 16 | 66 | 38 | 4.2 | 0.08 | 17.2 | 0.01 | 10.0 | 0.02 | 0.6 | 0.82 |
| 78067 | E class, group 1 | 9:1999436–2001658 | TMHs | 4 | 7 | 34 | 70 | 1.8 | 0.03 | 9.0 | 0.01 | 18.3 | 0.00 | 2.0 | 0.66 |
| 136784 | E class, group 1 | 3:2527152–2530169 | 19 | 41 | 156 | 54 | 2.2 | 0.01 | 8.3 | 0.00 | 2.9 | 0.01 | 0.3 | 0.01 | |
| 22844 | P450, CYP52 | 2:470855–474456 | TMHs | 134 | 215 | 865 | 1235 | 1.6 | 0.20 | 6.5 | 0.00 | 9.2 | 0.00 | 1.4 | 0.07 |
| 104840 | E class, group 1 | 8:1170047–1173084 | TMH, IVS | 11 | 9 | 66 | 127 | 0.8 | 0.59 | 6.1 | 0.00 | 11.6 | 0.00 | 1.9 | 0.45 |
| 136925 | E class, group 1 | 3:3544211–3545493 | 5′ truncated | 5 | 6 | 28 | 14 | 1.1 | 0.84 | 5.3 | 0.00 | 2.6 | 0.01 | 0.5 | 0.13 |
| 136367 | E class, group 1 | 2:3126858–3128993 | 13 | 15 | 55 | 66 | 1.2 | 0.42 | 4.2 | 0.00 | 5.0 | 0.00 | 1.2 | 0.63 | |
Genes exhibiting relatively low expression levels in Asp and LP (RPKM values < 10) are excluded. Expression ratios (R) and moderated t test-derived probability (P) are shown.
Protein model identifiers (ID) derived from the Joint Genome Institute's databases (44).
Classification based on the Fungal Cytochrome P450 Database (FCPD; http://P450.riceblast.snu.ac.kr/) (46).
Assembly location presented as Scaffold:coordinates.
TMH, transmembrane helices. Gene modes encoding protein models Wolco_75851 and Wolco_90431 are truncated, but covered by alternative model Wolco_144633; mPAI, exponentially modified protein abundance index (22).
RNA-seq-normalized reads per kilobase per million (RPKM).
TABLE 5.
Putative transporters with transcripts accumulating >4-fold in one or more pairwise comparisonsa
| IDb | Putative transporter(s)c | Commentsd | RPKMe |
MCC/Glc |
Asp/Glc |
LP/Glc |
LP/Asp |
|||||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
| Glc | MCC | Asp | LP | R | P | R | P | R | P | R | P | |||
| 131978 | Cytosine-purine permease | TMHs | 10 | 18 | 42 | 40 | 1.79 | 0.02 | 4.11 | 0.00 | 3.93 | 0.00 | 0.96 | 0.93 |
| 102298 | Quinate permease | TMHs | 5 | 51 | 815 | 1278 | 10.21 | 0.00 | 164.87 | 0.00 | 258.47 | 0.00 | 1.57 | 0.26 |
| 104972 | Monosaccharides | TMH, peptides | 186 | 451 | 1034 | 875 | 2.43 | 0.01 | 5.56 | 0.00 | 4.71 | 0.00 | 0.85 | 0.72 |
| 125778 | Oligopeptides | TMHs | 48 | 206 | 727 | 810 | 4.31 | 0.00 | 15.24 | 0.00 | 16.98 | 0.00 | 1.11 | 0.78 |
| 109849 | ABC | 28 | 30 | 146 | 280 | 1.08 | 0.79 | 5.24 | 0.00 | 10.06 | 0.00 | 1.92 | 0.07 | |
| 106642 | Amino acid | TMHs | 13 | 21 | 60 | 55 | 1.68 | 0.18 | 4.80 | 0.00 | 4.38 | 0.00 | 0.91 | 0.82 |
| 105323 | Amino acid permease | 4 | 10 | 56 | 77 | 2.74 | 0.03 | 15.24 | 0.00 | 21.16 | 0.00 | 1.39 | 0.69 | |
| 139405 | Ammonium | TMHs, IVS | 29 | 47 | 136 | 130 | 1.62 | 0.10 | 4.63 | 0.00 | 4.42 | 0.00 | 0.96 | 0.92 |
| 25027 | High-affinity glucose | 7 | 25 | 154 | 277 | 3.71 | 0.01 | 23.25 | 0.00 | 41.73 | 0.00 | 1.79 | 0.05 | |
| 27321 | High-affinity glucose | TMHs | 48 | 102 | 370 | 341 | 2.13 | 0.06 | 7.74 | 0.00 | 7.15 | 0.00 | 0.92 | 0.77 |
| 137336 | Lactose permease | TMHs | 68 | 136 | 470 | 888 | 2.01 | 0.15 | 6.93 | 0.00 | 13.09 | 0.00 | 1.89 | 0.02 |
| 17021 | MFS | TMHs | 13 | 13 | 61 | 67 | 1.01 | 0.99 | 4.86 | 0.00 | 5.31 | 0.00 | 1.09 | 0.85 |
| 21787 | MFS | TMHs | 281 | 471 | 1144 | 1039 | 1.68 | 0.04 | 4.08 | 0.00 | 3.70 | 0.00 | 0.91 | 0.89 |
| 26276 | MFS | 118 | 252 | 741 | 898 | 2.14 | 0.04 | 6.29 | 0.00 | 7.63 | 0.00 | 1.21 | 0.65 | |
| 82841 | MFS | TMHs | 93 | 104 | 451 | 820 | 1.11 | 0.83 | 4.86 | 0.01 | 8.82 | 0.01 | 1.82 | 0.43 |
| 89075 | MFS | TMHs | 6 | 15 | 83 | 125 | 2.35 | 0.01 | 13.08 | 0.00 | 19.66 | 0.00 | 1.50 | 0.42 |
| 94573 | MFS | TMHs | 75 | 248 | 319 | 447 | 3.32 | 0.00 | 4.29 | 0.00 | 5.99 | 0.00 | 1.40 | 0.58 |
| 106117 | MFS | TMHs | 5 | 5 | 22 | 24 | 0.98 | 0.93 | 4.50 | 0.00 | 5.03 | 0.00 | 1.12 | 0.89 |
| 106119 | MFS | TMHs | 15 | 34 | 284 | 465 | 2.30 | 0.18 | 19.13 | 0.00 | 31.27 | 0.00 | 1.64 | 0.10 |
| 127030 | MFS | 5 | 22 | 259 | 303 | 4.17 | 0.00 | 49.64 | 0.00 | 58.08 | 0.00 | 1.17 | 0.59 | |
| 133564 | MFS | TMHs | 4 | 6 | 27 | 20 | 1.38 | 0.75 | 6.64 | 0.00 | 4.78 | 0.00 | 0.72 | 0.48 |
| 139794 | MFS | TMHs | 8 | 12 | 33 | 48 | 1.51 | 0.39 | 4.21 | 0.00 | 6.13 | 0.00 | 1.46 | 0.22 |
| 139522 | MFS | TMHs | 5 | 8 | 56 | 110 | 1.52 | 0.07 | 10.26 | 0.00 | 20.11 | 0.00 | 1.96 | 0.18 |
| 123017 | Oligopeptide | TMHs | 3 | 11 | 61 | 85 | 3.37 | 0.02 | 19.32 | 0.00 | 27.06 | 0.00 | 1.40 | 0.58 |
| 166774 | Siderophore iron transporter | 5 | 4 | 71 | 56 | 0.76 | 0.29 | 14.90 | 0.00 | 11.75 | 0.00 | 0.79 | 0.66 | |
| 107257 | Sugar MFS | 6 | 21 | 293 | 590 | 3.23 | 0.00 | 46.06 | 0.00 | 92.67 | 0.00 | 2.01 | 0.17 | |
| 135536 | Sugar MFS | TMHs | 27 | 38 | 166 | 377 | 1.44 | 0.12 | 6.21 | 0.00 | 14.14 | 0.00 | 2.28 | 0.01 |
| 96986 | Sulfate permease | TMHs | 8 | 6 | 50 | 48 | 0.82 | 0.60 | 6.59 | 0.00 | 6.33 | 0.00 | 0.96 | 0.92 |
| 133004 | Sulfite efflux pump | 10 | 41 | 102 | 59 | 3.97 | 0.00 | 9.79 | 0.00 | 5.64 | 0.00 | 0.58 | 0.37 | |
| 29361 | Sugar MFS | TMHs | 42 | 42 | 107 | 225 | 1.00 | 0.99 | 2.55 | 0.00 | 5.55 | 0.00 | 2.10 | 0.02 |
| 163632 | Sugar MFS | TMHs | 12 | 21 | 43 | 115 | 1.82 | 0.09 | 3.71 | 0.00 | 9.95 | 0.00 | 2.68 | 0.00 |
| 139839 | MFS | TMHs | 24 | 40 | 86 | 101 | 1.60 | 0.40 | 3.67 | 0.00 | 4.29 | 0.00 | 1.17 | 0.80 |
| 138483 | MFS | TMHs | 25 | 27 | 80 | 123 | 1.10 | 0.68 | 3.21 | 0.00 | 4.96 | 0.00 | 1.54 | 0.31 |
| 100664 | MFS | TMHs | 4 | 4 | 13 | 17 | 1.08 | 0.80 | 3.57 | 0.00 | 4.86 | 0.00 | 1.36 | 0.65 |
| 100215 | MFS | TMHs | 4 | 6 | 13 | 18 | 1.04 | 0.84 | 3.00 | 0.00 | 4.18 | 0.00 | 1.40 | 0.37 |
Limited to genes with Asp and LP RPKM values of >10. Expression ratios (R) and moderated t test-derived probability (P) are shown.
Protein model identifiers (ID) derived from the Joint Genome Institute's databases (44).
Classification based on SwissProt best hits. Abbreviations: MFS, major facilitator superfamily; ABC, ATP-binding cassette superfamily.
Predicted transmembrane helices (THHs) and gene models with extended intervening sequences (IVS) indicated. Peptides were detected in Asp and LP filtrates with emPAI values of 0.14 and 0.16, respectively.
RNA-seq-normalized reads per kilobase per million (RPKM).
The influence of wood species was less pronounced among transporters, although transcripts of the major facilitator superfamily (MFS) sugar transporter Wolco_135536 were 2.3-fold more abundant in LP versus Asp. Of the 35 upregulated genes (Table 5), all had homologs (bit scores >500) in P. placenta and, in 16 cases, at least one homolog was upregulated in white pine and/or Asp (see Data Set S2). The most highly upregulated transporter in Asp and LP was Wolco_102298. The likely P. placenta ortholog of this gene, Pospl_108735 (bit score of 2207, with 81.5% amino acid identity), was also upregulated in white pine or Asp relative to Glc. The precise function of this and other transporters is difficult to assign based on secondary structure, although the best hit (33% identity at 80% coverage, with a bit score of 623) within the Swiss-Prot database is the Neurospora crassa quinate transporter (UniProt accession number P11636). The above-mentioned putative siderophore iron transporter (Table 2) Wolco_166774 showed only distant homology (score = 507; 39% identity) to Pospl_105993. Transcript levels corresponding to the P. placenta gene were not significantly altered by substrate.
DISCUSSION
Relative to Glc, the substrate substantially influenced transcript levels. Only 30 genes were upregulated greater than 4-fold in MCC, whereas 183 and 207 genes were upregulated in Asp and LP, respectively (Fig. 1A). However, mass spectrometry unambiguously identified 84 proteins in the MCC culture filtrates, while Asp and LP yielded a modest increase of 117 and 112, respectively (Fig. 1B). Given the defined composition of MCC, the medium may be preferred for assessing the transcriptional impact of supplements and/or for protein purification.
As representatives of hardwoods and conifers, Asp and LP differ substantially in their structures and compositions (32, 38, 39). Nevertheless, W. cocos gene expression patterns were remarkably similar on these substrates. For example, transcripts of 146 genes were upregulated greater than 4-fold in both Asp and LP, while only 30 were “uniquely” upregulated in Asp and only 61 in LP (Fig. 1A). If the threshold is reduced from 4-fold (P < 0.01) to 2-fold (P < 0.05), the overlap is even more obvious, with the number of upregulated genes reduced from 30 to 3 in Asp and from 61 to 5 in LP.
Thus, only 5 genes were differentially expressed on the two woody substrates and all accumulated >4-fold (P < 0.01) on LP relative to Asp (see Data Set 1 in the supplemental material). Possibly reflecting metabolism of conifer resins, transcripts corresponding to cytochrome P450s Wolco_104855, Wolco_24177, and Wolco_99035 accumulated 4.4-fold, 8.1-fold, and 9.8-fold, respectively, on LP relative to Asp. Similarly, the observed 4.3-fold upregulation of Wolco_74146, a GH78 α-l-rhamnosidase, is likely related to softwood hemicellulose composition. Defying such simple explanations, elevated transcript levels were also observed for a dihydrodipicolinate synthase (Wolco_104457), an enzyme involved in amino acid biosynthesis (IPR 002220).
Genome annotations of a growing number of wood decay fungi have revealed genetic repertoires seemingly consistent with decay characteristics (2, 3). Brown rot fungi such as W. cocos are widely thought to employ nonenzymatic Fenton chemistry to generate hydroxyl radicals (H2O2 + Fe2+ + H+ → H2O + Fe3+ + ˙OH) (5–8). Various redox systems, including hydroquinone redox cycling (11, 12), have been proposed (reviewed in references 9 and 10).
Our analyses examined the transcriptome and secretome of W. cocos, with particular focus on the production of key reactants (H2O2 and Fe2+). These included genes encoding benzoquinone reductases (BQR), laccases (Lac), various H2O2-producing oxidoreductases (e.g., CROs), and iron reductases (GLPs). We also report ferroxidase (FET3), ferric reductase transmembrane component (FRE), iron permease (FTE), and oxalate decarboxylase (ODC) as possibly modulating iron homeostasis.
Based on ferrozine measurements of iron reduction (14, 15), cultures were harvested after 5 days on Glc, MCC, LP, or Asp. The last substrate was selected because it was previously used for microarray-based transcriptome studies of P. placenta (32) and for low-resolution LC-MS/MS secretome studies of Fomitopsis pinicola (2). These three brown rot fungi are closely related members of the Antrodia clade (40) and would reasonably be expected to employ similar strategies for the conversion of lignocellulose.
Our transcriptome profiles of W. cocos and P. placenta provided only limited support for this view. Enhanced iron acquisition, discerned as upregulation of FET3 and FTR1, was observed in these brown rot species (Table 2; see also Data Set S1) in distinct contrast to the white rot fungus P. chrysosporium (16). This strategy is entirely consistent with a Fenton-based system. On the other hand, W. cocos was clearly differentiated from P. placenta by dramatic upregulation of CRO5 and by increased numbers and regulation of genes encoding GLPs and Lacs. While BQR and MOX orthologs are present in both genomes, significant upregulation in Asp and conifer wood is observed only in P. placenta. These observations argue against the relative importance of hydroxyquinone redox cycling in W. cocos.
Although roles for GLPs, oxalate decarboxylase, and CRO5 might be envisioned for a Fenton-based system (e.g., in Fe3+ reduction, metal ion speciation, and peroxide generation, respectively), the mechanistic details are very much uncertain. In the case of GLP, Amadori-modified amino acids in GLP are proposed to serve as redox centers mediating ferric ion and oxygen reduction (34), while NADH is suggested as the physiological source of reductant based on detection of the dinucleotide in cultures (41). Production of other low-molecular-weight Fe3+-reducing compounds has been characterized in W. cocos cultures, which produce both hydroxamate-type and catechol-type chelators (14), as do other brown-rot fungi (see reference 10 for a review). Reduction of Fe3+ in a biomimetic catechol-type system is highly dependent on oxalate concentration since Fe3+ is reduced only as the Fe3+-mono-oxalate (42). This suggests that oxalate decarboxylase could have a role in modulating iron redox reactions, in addition to iron homeostasis. In regard to peroxide production in W. cocos cultures, CRO5 is a likely candidate based on its homology with GLX, a phylogenetically related but distinct copper radical oxidase (2, 37). However, in contrast to the case with GLX (43), nothing is known of the substrate specificity of CRO5. Therefore, any possible contribution of CRO5 substrates and products in iron chelation and redox chemistry, in addition to peroxide production, remains unclear.
Of course, this uncertainty is widespread among the “hypothetical” proteins, which constitute a major portion of fungal genomes. For perspective on the dimensions of this issue, of the 12,746 predicted W. cocos genes, only 48% have assigned Pfam domains (2). Toward narrowing the number of candidates for further consideration, genes corresponding to transcripts upregulated on woody substrates and/or to detectable extracellular peptides are most likely involved in plant cell wall degradation. Further investigation is warranted for those with intriguing redox domains, such as the two cupredoxins predicted in Wolco_97131 (Table 3). This widely distributed domain (IPR008972) is common to functionally divergent enzymes, including multicopper oxidases, nitrite reductases, and laccases. In this context, 33 hypothetical proteins with predicted secretion signals were upregulated (>4-fold; P < 0.01) on MCC, Asp, or LP relative to Glc. Further supporting their potential role, 6 of these genes featured RPKM values well within the upper centile (see Data Set S1). Such expression patterns strongly support an important role in cell wall depolymerization.
Further progress will be made through the integration of transcript patterns over time along with compositional analysis as recently demonstrated with P. placenta colonization of biochemically distinct poplar lines (47). Such profiles could be complemented by simultaneous examination of the fungal secretomes and microscopic assessments of decay. Purification and characterization of the native or recombinant enzymes would clarify questions of substrate preference, but as of this writing, powerful genetic tools for gene disruption or suppression are not available for any brown rot fungus.
Supplementary Material
ACKNOWLEDGMENT
We thank Michael Mozuch for technical assistance.
Funding Statement
This work was supported in part by the National Research Initiative of the U.S. Department of Agriculture Cooperative State, Research Education and Extension Service grant 2007-35504-18257, to D.C. and R.A.B.
Footnotes
Supplemental material for this article may be found at http://dx.doi.org/10.1128/AEM.00639-16.
REFERENCES
- 1.Cullen D. 2014. Wood decay, p 41–62. In Martin F. (ed), Ecological genomics of fungi. Wiley-Blackwell, New York, NY. [Google Scholar]
- 2.Floudas D, Binder M, Riley R, Barry K, Blanchette RA, Henrissat B, Martinez AT, Otillar R, Spatafora JW, Yadav JS, Aerts A, Benoit I, Boyd A, Carlson A, Copeland A, Coutinho PM, de Vries RP, Ferreira P, Findley K, Foster B, Gaskell J, Glotzer D, Gorecki P, Heitman J, Hesse C, Hori C, Igarashi K, Jurgens JA, Kallen N, Kersten P, Kohler A, Kues U, Kumar TK, Kuo A, LaButti K, Larrondo LF, Lindquist E, Ling A, Lombard V, Lucas S, Lundell T, Martin R, McLaughlin DJ, Morgenstern I, Morin E, Murat C, Nagy LG, Nolan M, Ohm RA, et al. 2012. The Paleozoic origin of enzymatic lignin decomposition reconstructed from 31 fungal genomes. Science 336:1715–1719. doi: 10.1126/science.1221748. [DOI] [PubMed] [Google Scholar]
- 3.Kohler A, Kuo A, Nagy LG, Morin E, Barry KW, Buscot F, Canback B, Choi C, Cichocki N, Clum A, Colpaert J, Copeland A, Costa MD, Dore J, Floudas D, Gay G, Girlanda M, Henrissat B, Herrmann S, Hess J, Hogberg N, Johansson T, Khouja HR, LaButti K, Lahrmann U, Levasseur A, Lindquist EA, Lipzen A, Marmeisse R, Martino E, Murat C, Ngan CY, Nehls U, Plett JM, Pringle A, Ohm RA, Perotto S, Peter M, Riley R, Rineau F, Ruytinx J, Salamov A, Shah F, Sun H, Tarkka M, Tritt A, Veneault-Fourrey C, Zuccaro A, et al. 2015. Convergent losses of decay mechanisms and rapid turnover of symbiosis genes in mycorrhizal mutualists. Nat Genet 47:410–415. doi: 10.1038/ng.3223. [DOI] [PubMed] [Google Scholar]
- 4.Riley R, Salamov AA, Brown DW, Nagy LG, Floudas D, Held BW, Levasseur A, Lombard V, Morin E, Otillar R, Lindquist EA, Sun H, LaButti KM, Schmutz J, Jabbour D, Luo H, Baker SE, Pisabarro AG, Walton JD, Blanchette RA, Henrissat B, Martin F, Cullen D, Hibbett DS, Grigoriev IV. 2014. Extensive sampling of basidiomycete genomes demonstrates inadequacy of the white-rot/brown-rot paradigm for wood decay fungi. Proc Natl Acad Sci U S A 111:9923–9928. doi: 10.1073/pnas.1400592111. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Xu G, Goodell B. 2001. Mechanisms of wood degradation by brown-rot fungi: chelator-mediated cellulose degradation and binding of iron by cellulose. J Biotechnol 87:43–57. doi: 10.1016/S0168-1656(00)00430-2. [DOI] [PubMed] [Google Scholar]
- 6.Cohen R, Suzuki MR, Hammel KE. 2004. Differential stress-induced regulation of two quinone reductases in the brown rot basidiomycete Gloeophyllum trabeum. Appl Environ Microbiol 70:324–331. doi: 10.1128/AEM.70.1.324-331.2004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Cohen R, Jensen KA, Houtman CJ, Hammel KE. 2002. Significant levels of extracellular reactive oxygen species produced by brown rot basidiomycetes on cellulose. FEBS Lett 531:483–488. doi: 10.1016/S0014-5793(02)03589-5. [DOI] [PubMed] [Google Scholar]
- 8.Arantes V, Milagres AM, Filley TR, Goodell B. 2011. Lignocellulosic polysaccharides and lignin degradation by wood decay fungi: the relevance of nonenzymatic Fenton-based reactions. J Ind Microbiol Biotechnol 38:541–555. doi: 10.1007/s10295-010-0798-2. [DOI] [PubMed] [Google Scholar]
- 9.Goodell B. 2003. Brown rot fungal degradation of wood: our evolving view, p 97–118. In Goodell B, Nicholas D, Schultz T (ed), Wood deterioration and preservation. American Chemical Society, Washington, DC. [Google Scholar]
- 10.Arantes V, Jellison J, Goodell B. 2012. Peculiarities of brown-rot fungi and biochemical Fenton reaction with regard to their potential as a model for bioprocessing biomass. Appl Microbiol Biotechnol 94:323–338. doi: 10.1007/s00253-012-3954-y. [DOI] [PubMed] [Google Scholar]
- 11.Paszczynski A, Crawford R, Funk D, Goodell B. 1999. De novo synthesis of 4,5-dimethoxycatechol and 2, 5-dimethoxyhydroquinone by the brown rot fungus Gloeophyllum trabeum. Appl Environ Microbiol 65:674–679. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Suzuki MR, Hunt CG, Houtman CJ, Dalebroux ZD, Hammel KE. 2006. Fungal hydroquinones contribute to brown rot of wood. Environ Microbiol 8:2214–2223. doi: 10.1111/j.1462-2920.2006.01160.x. [DOI] [PubMed] [Google Scholar]
- 13.Highley TL. 1973. Influence of carbon source on cellulase activity of white rot and brown rot fungi. Wood Fiber 5:50–58. [Google Scholar]
- 14.Arantes V, Milagres AM. 2008. Response of Wolfiporia cocos to iron availability: alterations in growth, expression of cellular proteins, Fe3+-reducing activity and Fe3+-chelators production. J Appl Microbiol 104:185–193. [DOI] [PubMed] [Google Scholar]
- 15.Stookey LL. 1970. Ferrozine: a new spectrophotometric reagent for iron. Anal Chem 42:779–781. doi: 10.1021/ac60289a016. [DOI] [Google Scholar]
- 16.Vanden Wymelenberg A, Gaskell J, Mozuch MD, Sabat G, Ralph J, Skyba O, Mansfield S, Blanchette RA, Martinez D, Grigoriev I, Kersten P, Cullen D. 2010. Comparative transcriptome and secretome analysis of wood decay fungi Postia placenta and Phanerochaete chrysosporium. Appl Environ Microbiol 76:3599–3610. doi: 10.1128/AEM.00058-10. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Fernandez-Fueyo E, Ruiz-Duenas FJ, Ferreira P, Floudas D, Hibbett DS, Canessa P, Larrondo LF, James TY, Seelenfreund D, Lobos S, Polanco R, Tello M, Honda Y, Watanabe T, Ryu JS, Kubicek CP, Schmoll M, Gaskell J, Hammel KE, St John FJ, Vanden Wymelenberg A, Sabat G, Splinter BonDurant S, Syed K, Yadav JS, Doddapaneni H, Subramanian V, Lavin JL, Oguiza JA, Perez G, Pisabarro AG, Ramirez L, Santoyo F, Master E, Coutinho PM, Henrissat B, Lombard V, Magnuson JK, Kues U, Hori C, Igarashi K, Samejima M, Held BW, Barry KW, LaButti KM, Lapidus A, Lindquist EA, Lucas SM, Riley R, Salamov AA, Hoffmeister D, Schwenk D, Hadar Y, Yarden O, et al. 2012. Comparative genomics of Ceriporiopsis subvermispora and Phanerochaete chrysosporium provide insight into selective ligninolysis. Proc Natl Acad Sci U S A 109:5458–5463. doi: 10.1073/pnas.1119912109. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Vanden Wymelenberg A, Gaskell J, Mozuch MD, Kersten P, Sabat G, Martinez D, Cullen D. 2009. Transcriptome and secretome analysis of Phanerochaete chrysosporium reveal complex patterns of gene expression. Appl Environ Microbiol 75:4058–4068. doi: 10.1128/AEM.00314-09. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Hori C, Ishida T, Igarashi K, Samejima M, Suzuki H, Master E, Ferreira P, Ruiz-Duenas FJ, Held B, Canessa P, Larrondo LF, Schmoll M, Druzhinina IS, Kubicek CP, Gaskell JA, Kersten P, St John F, Glasner J, Sabat G, Splinter BonDurant S, Syed K, Yadav J, Mgbeahuruike AC, Kovalchuk A, Asiegbu FO, Lackner G, Hoffmeister D, Rencoret J, Gutierrez A, Sun H, Lindquist E, Barry K, Riley R, Grigoriev IV, Henrissat B, Kues U, Berka RM, Martinez AT, Covert SF, Blanchette RA, Cullen D. 2014. Analysis of the Phlebiopsis gigantea genome, transcriptome and secretome provides insight into its pioneer colonization strategies of wood. PLoS Genet 10:e1004759. doi: 10.1371/journal.pgen.1004759. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Ryu JS, Shary S, Houtman CJ, Panisko EA, Korripally P, St John FJ, Crooks C, Siika-Aho M, Magnuson JK, Hammel KE. 2011. Proteomic and functional analysis of the cellulase system expressed by Postia placenta during brown rot of solid wood. Appl Environ Microbiol 77:7933–7941. doi: 10.1128/AEM.05496-11. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Nesvizhskii AI, Keller A, Kolker E, Aebersold R. 2003. A statistical model for identifying proteins by tandem mass spectrometry. Anal Chem 75:4646–4658. doi: 10.1021/ac0341261. [DOI] [PubMed] [Google Scholar]
- 22.Ishihama Y, Oda Y, Tabata T, Sato T, Nagasu T, Rappsilber J, Mann M. 2005. Exponentially modified protein abundance index (emPAI) for estimation of absolute protein amount in proteomics by the number of sequenced peptides per protein. Mol Cell Proteomics 4:1265–1272. doi: 10.1074/mcp.M500061-MCP200. [DOI] [PubMed] [Google Scholar]
- 23.Bendtsen JD, Kiemer L, Fausboll A, Brunak S. 2005. Non-classical protein secretion in bacteria. BMC Microbiol 5:58. doi: 10.1186/1471-2180-5-58. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Lombard V, Golaconda Ramulu H, Drula E, Coutinho PM, Henrissat B. 2014. The carbohydrate-active enzymes database (CAZy) in 2013. Nucleic Acids Res 42:D490–D495. doi: 10.1093/nar/gkt1178. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Agger JW, Isaksen T, Varnai A, Vidal-Melgosa S, Willats WG, Ludwig R, Horn SJ, Eijsink VG, Westereng B. 2014. Discovery of LPMO activity on hemicelluloses shows the importance of oxidative processes in plant cell wall degradation. Proc Natl Acad Sci U S A 111:6287–6292. doi: 10.1073/pnas.1323629111. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Bey M, Zhou S, Poidevin L, Henrissat B, Coutinho PM, Berrin JG, Sigoillot JC. 2013. Cello-oligosaccharide oxidation reveals differences between two lytic polysaccharide monooxygenases (family GH61) from Podospora anserina. Appl Environ Microbiol 79:488–496. doi: 10.1128/AEM.02942-12. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Levasseur A, Drula E, Lombard V, Coutinho PM, Henrissat B. 2013. Expansion of the enzymatic repertoire of the CAZy database to integrate auxiliary redox enzymes. Biotechnol Biofuels 6:41. doi: 10.1186/1754-6834-6-41. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Quinlan RJ, Sweeney MD, Lo Leggio L, Otten H, Poulsen JC, Johansen KS, Krogh KB, Jorgensen CI, Tovborg M, Anthonsen A, Tryfona T, Walter CP, Dupree P, Xu F, Davies GJ, Walton PH. 2011. Insights into the oxidative degradation of cellulose by a copper metalloenzyme that exploits biomass components. Proc Natl Acad Sci U S A 108:15079–15084. doi: 10.1073/pnas.1105776108. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Westereng B, Ishida T, Vaaje-Kolstad G, Wu M, Eijsink VG, Igarashi K, Samejima M, Stahlberg J, Horn SJ, Sandgren M. 2011. The putative endoglucanase PcGH61D from Phanerochaete chrysosporium is a metal-dependent oxidative enzyme that cleaves cellulose. PLoS One 6:e27807. doi: 10.1371/journal.pone.0027807. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Yakovlev I, Vaaje-Kolstad G, Hietala AM, Stefanczyk E, Solheim H, Fossdal CG. 2012. Substrate-specific transcription of the enigmatic GH61 family of the pathogenic white-rot fungus Heterobasidion irregulare during growth on lignocellulose. Appl Microbiol Biotechnol 95:979–990. doi: 10.1007/s00253-012-4206-x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Martinez D, Challacombe J, Morgenstern I, Hibbett D, Schmoll M, Kubicek CP, Ferreira P, Ruiz-Duenas FJ, Martinez AT, Kersten P, Hammel KE, Vanden Wymelenberg A, Gaskell J, Lindquist E, Sabat G, Bondurant SS, Larrondo LF, Canessa P, Vicuna R, Yadav J, Doddapaneni H, Subramanian V, Pisabarro AG, Lavin JL, Oguiza JA, Master E, Henrissat B, Coutinho PM, Harris P, Magnuson JK, Baker SE, Bruno K, Kenealy W, Hoegger PJ, Kues U, Ramaiya P, Lucas S, Salamov A, Shapiro H, Tu H, Chee CL, Misra M, Xie G, Teter S, Yaver D, James T, Mokrejs M, Pospisek M, Grigoriev IV, Brettin T, Rokhsar D, Berka R, Cullen D. 2009. Genome, transcriptome, and secretome analysis of wood decay fungus Postia placenta supports unique mechanisms of lignocellulose conversion. Proc Natl Acad Sci U S A 106:1954–1959. doi: 10.1073/pnas.0809575106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Vanden Wymelenberg A, Gaskell J, Mozuch M, BonDurant SS, Sabat G, Ralph J, Skyba O, Mansfield SD, Blanchette RA, Grigoriev IV, Kersten PJ, Cullen D. 2011. Significant alteration of gene expression in wood decay fungi Postia placenta and Phanerochaete chrysosporium by plant species. Appl Environ Microbiol 77:4499–4507. doi: 10.1128/AEM.00508-11. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Tanaka H, Itakura S, Enoki A. 1999. Hydroxyl radical generation by an extracellular low-molecular weight substance and phenol oxidase activity during wood degradation by the white-rot basidiomycete Phanerochaete chrysosporium. Holzforschung 52:21–28. [DOI] [PubMed] [Google Scholar]
- 34.Tanaka H, Yoshida G, Baba Y, Matsumura K, Wasada H, Murata J, Agawa M, Itakura S, Enoki A. 2007. Characterization of a hydroxyl-radical-producing glycoprotein and its presumptive genes from the white-rot basidiomycete Phanerochaete chrysosporium. J Biotechnol 128:500–511. doi: 10.1016/j.jbiotec.2006.12.010. [DOI] [PubMed] [Google Scholar]
- 35.Wei D, Houtman CJ, Kapich AN, Hunt CG, Cullen D, Hammel KE. 2010. Laccase and its role in production of extracellular reactive oxygen species during wood decay by the brown rot basidiomycete Postia placenta. Appl Environ Microbiol 76:2091–2097. doi: 10.1128/AEM.02929-09. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Daniel G, Volc J, Filonova L, Plihal O, Kubatova E, Halada P. 2007. Characteristics of Gloeophyllum trabeum alcohol oxidase, an extracellular source of H2O2 in brown rot decay of wood. Appl Environ Microbiol 73:6241–6253. doi: 10.1128/AEM.00977-07. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Vanden Wymelenberg A, Sabat G, Mozuch MD, Kersten P, Cullen D, Blanchette RA. 2006. Structure, organization, and transcriptional regulation of a family of copper radical oxidase genes in the lignin-degrading basidiomycete Phanerochaete chrysosporium. Appl Environ Microbiol 72:4871–4877. doi: 10.1128/AEM.00375-06. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Pan X, Xie D, Yu RW, Saddler JN. 2008. The bioconversion of mountain pine beetle-killed lodgepole pine to fuel ethanol using the organosolv process. Biotechnol Bioeng 101:39–48. doi: 10.1002/bit.21883. [DOI] [PubMed] [Google Scholar]
- 39.Adler E. 1977. Lignin chemistry. Past, present and future. Wood Sci Technol 11:169–218. [Google Scholar]
- 40.Binder M, Justo A, Riley R, Salamov A, Lopez-Giraldez F, Sjokvist E, Copeland A, Foster B, Sun H, Larsson E, Larsson KH, Townsend J, Grigoriev IV, Hibbett DS. 2013. Phylogenetic and phylogenomic overview of the Polyporales. Mycologia 105:1350–1373. doi: 10.3852/13-003. [DOI] [PubMed] [Google Scholar]
- 41.Kido R, Takeeda M, Manabe M, Miyagawa Y, Itakura S, Tanaka H. 2015. Presence of extracellular NAD(+) and NADH in cultures of wood-degrading fungi. Biocontrol Sci 20:105–113. doi: 10.4265/bio.20.105. [DOI] [PubMed] [Google Scholar]
- 42.Arantes V, Qian Y, Milagres AM, Jellison J, Goodell B. 2009. Effect of pH and oxalic acid on reduction of Fe3+ by a biomimetic chelator and on Fe3+ desorption/adsorption onto wood: Implications for brown-rot decay. Int Biodeter Biodegradation 63:478–483. doi: 10.1016/j.ibiod.2009.01.004. [DOI] [Google Scholar]
- 43.Kersten PJ. 1990. Glyoxal oxidase of Phanerochaete chrysosporium: its characterization and activation by lignin peroxidase. Proc Natl Acad Sci U S A 87:2936–2940. doi: 10.1073/pnas.87.8.2936. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Grigoriev IV, Nikitin R, Haridas S, Kuo A, Ohm R, Otillar R, Riley R, Salamov A, Zhao X, Korzeniewski F, Smirnova T, Nordberg H, Dubchak I, Shabalov I. 2014. MycoCosm portal: gearing up for 1000 fungal genomes. Nucleic Acids Res 42:D699–D704. doi: 10.1093/nar/gkt1183. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Aspeborg H, Coutinho PM, Wang Y, Brumer H III, Henrissat B. 2012. Evolution, substrate specificity and subfamily classification of glycoside hydrolase family 5 (GH5). BMC Evol Biol 12:186. doi: 10.1186/1471-2148-12-186. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Park J, Lee S, Choi J, Ahn K, Park B, Park J, Kang S, Lee YH. 2008. Fungal cytochrome P450 database. BMC Genomics 9:402. doi: 10.1186/1471-2164-9-402. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Skyba O, Cullen D, Douglas CJ, Mansfield SD. Gene expression patterns of wood decay fungi Postia placenta and Phanerochaete chrysosporium are influenced by wood substrate composition during degradation. Appl Environ Microbiol, in press. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.