

ABSTRACT
Phosphatidylserine (PtdSer) receptors that are responsible for the clearance of dying cells have recently been found to mediate enveloped virus entry. Ebola virus (EBOV), a member of the Filoviridae family of viruses, utilizes PtdSer receptors for entry into target cells. The PtdSer receptors human and murine T-cell immunoglobulin mucin (TIM) domain proteins TIM-1 and TIM-4 mediate filovirus entry by binding to PtdSer on the virion surface via a conserved PtdSer binding pocket within the amino-terminal IgV domain. While the residues within the TIM-1 IgV domain that are important for EBOV entry are characterized, the molecular details of virion–TIM-4 interactions have yet to be investigated. As sequences and structural alignments of the TIM proteins suggest distinct differences in the TIM-1 and TIM-4 IgV domain structures, we sought to characterize TIM-4 IgV domain residues required for EBOV entry. Using vesicular stomatitis virus pseudovirions bearing EBOV glycoprotein (EBOV GP/VSVΔG), we evaluated virus binding and entry into cells expressing TIM-4 molecules mutated within the IgV domain, allowing us to identify residues important for entry. Similar to TIM-1, residues in the PtdSer binding pocket of murine and human TIM-4 (mTIM-4 and hTIM-4) were found to be important for EBOV entry. However, additional TIM-4-specific residues were also found to impact EBOV entry, with a total of 8 mTIM-4 and 14 hTIM-4 IgV domain residues being critical for virion binding and internalization. Together, these findings provide a greater understanding of the interaction of TIM-4 with EBOV virions.
IMPORTANCE With more than 28,000 cases and over 11,000 deaths during the largest and most recent Ebola virus (EBOV) outbreak, there has been increased emphasis on the development of therapeutics against filoviruses. Many therapies under investigation target EBOV cell entry. T-cell immunoglobulin mucin (TIM) domain proteins are cell surface factors important for the entry of many enveloped viruses, including EBOV. TIM family member TIM-4 is expressed on macrophages and dendritic cells, which are early cellular targets during EBOV infection. Here, we performed a mutagenesis screening of the IgV domain of murine and human TIM-4 to identify residues that are critical for EBOV entry. Surprisingly, we identified more human than murine TIM-4 IgV domain residues that are required for EBOV entry. Defining the TIM IgV residues needed for EBOV entry clarifies the virus-receptor interactions and paves the way for the development of novel therapeutics targeting virus binding to this cell surface receptor.
INTRODUCTION
Ebolavirus and Marburgvirus, members of the Filoviridae family, are enveloped, negative-sense RNA viruses that cause severe hemorrhagic fever in humans and nonhuman primates. A member of the Ebolavirus genus, Ebola virus (EBOV), is a causative agent of episodic filovirus outbreaks in Africa, including the most recent and deadly West African epidemic that began in December 2013 (1, 2). Contributing to the virulent nature of this virus is the ability of EBOV to infect a broad range of cells, including macrophages and dendritic cells (DCs), that are early targets of infection and are thought to be responsible for virus spread to other cell populations such as hepatocytes, endothelial cells, and fibroblasts (3).
Several protein families have been identified as cell surface receptors mediating filovirus entry into cells. These include C-type lectins and phosphatidylserine (PtdSer) receptors (4). Several different PtdSer receptors can mediate virion uptake, including the Tyro3, Axl, and Mer (TAM) family of receptor tyrosine kinases and the T-cell immunoglobulin mucin (TIM) family of receptors, through interactions with PtdSer on the surface of virions (5–13). PtdSer within the outer leaflet of the viral envelope mimics PtdSer presentation on an apoptotic cell and allows virions to utilize cellular clearance mechanisms for engulfment. This unconventional mechanism utilized by enveloped and some lipid-enclosed nonenveloped viruses for cellular internalization has been termed apoptotic mimicry (14).
The TIM protein family members are type I cell surface glycoproteins (GPs). While there are four functional family members (TIM-1, TIM-2, TIM-3, and TIM-4) in mice, only three (TIM-1, TIM-3, and TIM-4) are present in humans. Murine TIM-1 (mTIM-1) and mTIM-4 and human TIM-1 (hTIM-1) and hTIM-4 can serve as enveloped virus receptors, whereas the other family members do not (11, 13, 15). TIM-1 and -4 are expressed on distinct populations of cells, with TIM-1 found on CD4+ T cells (16, 17) and some B and epithelial cell populations (11, 18–20) and TIM-4 present on some macrophage and DC subsets (16, 21, 22).
The hTIM-1 and -4 and mTIM-1 and -4 ectodomains are relatively conserved and composed of an amino-terminal immunoglobulin variable (IgV)-like domain and a mucin-like domain (MLD) (23). While both domains are required for EBOV internalization into cells (24), virion-associated PtdSer is thought to interact directly with residues within the IgV domain (12). The structures of all of the mTIM and hTIM IgV domains have been solved (25–29). While the IgV domains from different TIMs are similar, they are not identical, with differences in structure found between TIM homologs, as well as between the murine and human orthologs. Nonetheless, all IgV domain structures contain a conserved PtdSer binding pocket composed of the same structural loops.
Ectopic expression of hTIM-4 enhances the entry of many enveloped viruses, including filoviruses, flaviviruses, New World arenaviruses, and alphaviruses, to a level similar to that of hTIM-1 (12, 13), but the details of the virion–TIM-4 interactions that are required for virus internalization have not been studied. Investigations of the binding and uptake of PtdSer liposomes or apoptotic bodies have demonstrated that two central PtdSer binding pocket residues, N121 and D122, are critically important for mTIM-4 or hTIM interactions (21, 25, 27). In addition to these critical residues, two other mTIM-4 IgV domain residues, W119 and F120, have been found to be important for PtdSer liposome or apoptotic body binding (21, 25, 30). The latter residues reside within the FG loop that makes up the upper ridge of the PtdSer binding pocket.
Prior to our present studies, the IgV domain residues required for TIM-4-mediated EBOV entry were not defined. However, structural comparisons of the IgV domains of hTIM-4 and mTIM-4 with other TIMs indicate that there are distinct differences between the different family members within this domain. This suggests that IgV domain residues required for virion interactions may differ for the various family members. Here, we identified IgV domain residues that are critical for mTIM-4- and hTIM-4-mediated filovirus entry, defining residues that are required for optimal virion binding and uptake. Our mutagenesis studies demonstrate that more hTIM-4 than mTIM-4 IgV domain residues affect EBOV entry. Fourteen hTIM-4 but only eight mTIM-4 IgV domain residues were important for EBOV entry. These same residues were found to affect PtdSer liposome binding as well, indicating that it is PtdSer on the virion, and not the viral GP, that mediates virion interactions with TIM-4. Surprisingly, we found that several of the required IgV residues, particularly in hTIM-4, reside outside the PtdSer binding pocket. This is evidence that differences in the murine and human IgV domain structures impact virion PtdSer-receptor interactions.
MATERIALS AND METHODS
Cell lines.
Human embryonic kidney cell lines HEK 293T and HEK 293 were maintained in Dulbecco's modified Eagle's medium (DMEM) with 5% fetal bovine serum (FBS) and 1% penicillin-streptomycin (P/S). For generation of a HEK 293 cell line stably expressing hTIM-4, the hTIM-4 sequence was cloned into a cytomegalovirus (CMV)-driven expression plasmid that also expressed a neomycin (Neo) resistance gene. HEK 293 cells were transfected with the hTIM-4–Neo plasmid by a polyethylenimine (PEI) transfection protocol. Cells were selected by passage in the presence of 1 mg/ml G418 in DMEM with 10% FBS and 1% P/S. To obtain a cell population that expressed high levels of TIM-4, single cells were plated and grown in a 96-well plate. Clonal populations were obtained and screened for hTIM-4 expression. A clonal population with a high hTIM-4 expression level was obtained and maintained under selection with 1 mg/ml G418.
Pseudovirion and recombinant virus production. (i) EBOV VSV GFP pseudovirion generation.
Vesicular stomatitis virus (VSVΔG-GFP) pseudovirions encoding green fluorescent protein (GFP) in place of the GP open reading frame and pseudotyped with EBOV GP (EBOV VSVΔG-GFP) were produced as previously described (11, 31, 32). Briefly, HEK 293T cells were transfected with a CMV-driven expression plasmid containing EBOV (formerly Zaire EBOV) GP lacking the GP1 MLD or with full-length EBOV GP (FL EBOV). EBOV GP lacking the MLD was used for most of these studies because of its ability to confer the same tropism as FL EBOV GP and to produce higher pseudovirus titers (33–35). Twenty-four hours following transfection, the cells were transduced with VSVΔG-GFP pseudovirions bearing Lassa virus GPC. Four hours after transduction, the medium was removed, the cells were washed twice with phosphate-buffered saline (PBS), and fresh medium was added. The EBOV GP-pseudotyped virions were collected at 48 and 72 h following transduction, pooled, and filtered through a 0.45-μm filter. Virus stocks were aliquoted and stored at −80°C until use.
(ii) EBOV GP/rVSV generation.
Recombinant, replication-competent VSV encoding GFP and EBOV GP lacking the GP1 MLD replaced the native VSV GP-encoding gene within the genome (EBOV GP/rVSV). Infectious virus was produced as previously described (12). EBOV GP/rVSV stocks were produced in Vero cells, an African green monkey kidney epithelial cell line, by infection at a low multiplicity of infection (MOI) of ∼0.001. Supernatants were collected 48 h following infection and filtered through a 0.45-μm filter, and aliquots were stored at −80°C until use. The titer of the EBOV GP/rVSV stock was determined by endpoint dilution on Vero cells, and infection was scored 5 days following infection for GFP positivity with an inverted fluorescence microscope. Virus titers were calculated as the 50% tissue culture infective dose per milliliter by the Reed-Muench method (36).
Protein structures.
The mTIM-4 (Protein Data Bank [PDB] code 3BI9) (25), hTIM-4 (PDB code 5DZN), and hTIM-1 (PDB code 5DZO) (28) IgV domain crystal structures were obtained from the Research Collaboratory for Structural Bioinformatics (RCSB) PDB. The crystal structures were manipulated and rendered with PyMOL (37).
TIM-4 mutagenesis.
TIM-4 point mutations were introduced into a CMV immediate-early-promoter-driven hTIM-4 or mTIM-4 expression plasmid (OriGene) by the QuikChange (Stratagene) methodology previously described (12). Mutagenesis primers contained targeted nucleotide changes flanked by base pairs of homologous sequence. PCR was performed with Pfu Turbo DNA polymerase (Stratagene) and an S1000 thermal cycler (Bio-Rad) for 17 cycles (95°C for 30 s, 55°C for 1 min, and 68°C for 14 min). The parental plasmid was removed by digesting the PCR product with DpnI (New England BioLabs). The plasmid was transformed into bacteria, and single colonies were selected for plasmid purification/analysis. All mutants were confirmed by DNA sequencing, and the characteristics of these mutants are summarized in Table 1.
TABLE 1.
hTIM-4 and mTIM-4 IgV domain mutationsa
| Mutation(s) | Location | Expression | Mean % of WT transduction ± SEM | Mean % of WT infection ± SEM |
|---|---|---|---|---|
| mTIM-4 | ||||
| R49A | BC loop | WT | 77 ± 4.4 | 128 ± 10.9 |
| N61A | CC′ loop | WT | 108 ± 8.7 | NDb |
| N61D | CC′ loop | WT | 103 ± 6.1 | |
| S62A | CC′ loop | WT | 118 ± 7.4 | ND |
| K63A | CC′ loop | WT | 62 ± 4.1 | 80 ± 8.3 |
| R70A | C′ β-strand | WT | 90 ± 4.4 | ND |
| F92A | DE loop | WT | 92 ± 13.8 | ND |
| G93A | DE loop | WT | 112 ± 24.2 | ND |
| G105A | EF loop | WT | 124 ± 12.3 | ND |
| D106A | EF loop | WT | 116 ± 21.5 | ND |
| R113A | F β-strand | WT | 48 ± 9.8 | 22 ± 1.2 |
| V116A | FG loop | WT | 93 ± 5.0 | ND |
| P117A | FG loop | WT | 118 ± 2.9 | ND |
| P117R | FG loop | WT | 96 ± 8.2 | ND |
| G118A | FG loop | WT | 61 ± 10.2 | 23 ± 6.8 |
| W119A | FG loop | WT | 60 ± 8.8 | 43 ± 8.3 |
| F120A | FG loop | WT | 61 ± 8.7 | 48 ± 10.3 |
| N121A | FG loop | WT | 7 ± 1.8 | 14 ± 2.8 |
| N121D | FG loop | WT | 10 ± 3.0 | 20 ± 7.9 |
| D122A | FG loop | WT | 12 ± 0.8 | 14 ± 4.7 |
| D122N | FG loop | WT | 12 ± 2.1 | 11 ± 5.3 |
| ND121/122DN | FG loop | WT | 4 ± 5.6 | 7.6 ± 2.0 |
| K124A | G β-strand | WT | 117 ± 13 | ND |
| K125A | G β-strand | WT | 110 ± 14.6 | ND |
| hTIM-4 | ||||
| R35A | AB loop | WT | 103 ± 11.1 | 101 ± 8.8 |
| T37A | B β-strand | WT | 81 ± 7.1 | 82 ± 15.5 |
| S46A | BC loop | WT | 87 ± 4.6 | ND |
| H47A | BC loop | WT | 109 ± 18.3 | ND |
| N48A | BC loop | WT | 95.4 ± 8.4 | ND |
| S49A | BC loop | WT | 46 ± 5.5 | 81 ± 4.5 |
| S49R | BC loop | WT | 42 ± 3.6 | ND |
| Y61A | CC′ loop | WT | 64 ± 6.2 | 94 ± 7.5 |
| Y61P | CC′ loop | WT | 70 ± 3.3 | 105 ± 9.8 |
| S62A | CC′ loop | WT | 64 ± 3.6 | 104 ± 4.9 |
| G63A | CC′ loop | WT | 44 ± 6.1 | 47 ± 9 |
| G63K | CC′ loop | WT | 43 ± 2.8 | ND |
| K65A | CC′ loop | WT | 115 ± 5.9 | 109 ± 4.9 |
| R70A | C′ β-strand | WT | 58 ± 9.1 | 42 ± 2.8 |
| M74A | C′C″ loop | WT | 101 ± 6.0 | ND |
| R75A | C′C″ loop | WT | 109 ± 7.4 | ND |
| P91A | DE loop | WT | 126 ± 22.6 | 102 ± 1.2 |
| P91R | DE loop | WT | 78 ± 8.3 | 107 ± 13.9 |
| R92A | DE loop | WT | 124 ± 13.3 | 97 ± 6.7 |
| G93A | DE loop | WT | 1 ± 0.5 | 6 ± 3.9 |
| G93A | DE loop | 50% of WT | 8 ± 1.8 | 7.5 ± 0.9 |
| G93V | DE loop | <10% of WT | 4 ± 3.4 | 10 ± 3.2 |
| R92AG93A | DE loop | >80% of WT | 6 ± 2.3 | 12 ± 3.6 |
| D94A | DE loop | WT | 77 ± 2.7 | 20 ± 10.2 |
| D94E | DE loop | WT | 123 ± 16.9 | ND |
| V95A | DE loop | WT | 85 ± 11.7 | 77 ± 4.4 |
| S96A | DE loop | WT | 81 ± 5.8 | 94 ± 5.0 |
| L97A | E β-strand | WT | 1 ± 0.6 | 19 ± 9.5 |
| T98A | E β-strand | WT | 74 ± 1.8 | 62 ± 9.2 |
| R113A | F β-strand | WT | 8 ± 2.6 | 7.4 ± 1.9 |
| V116A | FG loop | WT | 54 ± 5.8 | 27 ± 5.4 |
| P117R | FG loop | WT | 101 ± 12.4 | 96 ± 5.1 |
| G118A | FG loop | WT | 7.5 ± 1.0 | 10 ± 1.5 |
| W119A | FG loop | WT | 60 ± 7.3 | 29 ± 9.3 |
| F120A | FG loop | WT | 53 ± 13.2 | 27 ± 10.8 |
| N121A | FG loop | WT | 10 ± 6.8 | 7 ± 1.9 |
| N121D | FG loop | WT | 4 ± 2.1 | 5.4 ± 1.6 |
| D122A | FG loop | WT | 4 ± 1.1 | 6.8 ± 1.6 |
| D122N | FG loop | WT | 3 ± 1.0 | 5.4 ± 1.1 |
| ND121/122AA | FG loop | WT | 3 ± 0.9 | ND |
| ND121/122DN | FG loop | WT | 5 ± 0.9 | 5.8 ± 1.1 |
| K124A | G β-strand | WT | 47 ± 5.7 | 42 ± 5.9 |
| I125A | G β-strand | WT | 54 ± 4.2 | 52 ± 7.9 |
Shown are the TIM-4 mutations generated, the location of each mutation in the IgV domain, surface expression compared to that of the WT protein as determined by polyclonal antiserum binding of transfected cells, and the impact of each mutation on TIM-4-mediated EBOV VSVΔG-GFP pseudovirion transduction and EBOV GP/rVSV infection. All mutations were compared to WT TIM-4, which was set at 100%. Bold indicates IgV domain mutants that have WT expression but reduced transduction/infection.
ND, not determined.
Surface expression.
HEK 293T cells were PEI transfected with the empty vector or wild-type (WT) or mutant TIM-4 expression vectors. HEK 293T cells were utilized for our studies because of their ability to be easily transfected with TIM-4-expressing plasmids, their lack of endogenous expression of TIM family members, and their poor permissivity to EBOV infection (11, 12). Twenty-four hours following transfection, cells were lifted with trypsin and a portion of the population was seeded into a 48-well plate while the rest was seeded into a 6-well plate. Forty-eight hours following transfection, the cells in the 6-well plate were lifted with PBS plus 5 mM EDTA and washed with PBS plus 5% FBS for analysis of TIM-4 surface expression. The cells were incubated with hTIM-4-, mTIM-4-, human Mer-, or human integrin αv-specific goat polyclonal primary antiserum (R&D Systems) or control goat IgG for 1 h on ice. Cells were washed and incubated with a DyLight 649-conjugated anti-goat secondary antibody (Jackson ImmunoResearch) for 20 min on ice. The amount of TIM-4 surface expression was determined by the percentage of TIM-4-positive cells in the FL4 channel with a FACSCalibur flow cytometer (BD Bioscience). Mutant hTIM-4 and mTIM-4 expression was normalized to WT hTIM-4 or mTIM-4 expression to determine differences in surface expression that may occur as a result of the mutations. The presence of Mer or integrin was assessed by histogram overlays with the control goat IgG-stained cells.
Transductions and infections.
For the transduction experiments, 48-well plates were seeded with equal numbers of cells transiently transfected with WT and mutant TIM-4. At 48 h following transfection, the cells were transduced with EBOV VSV-GFP pseudovirions (MOI of 0.8 [titer determined on HEK 293T cells]). Twenty-four hours following transduction, cells were lifted with Accutase (Fisher) and assessed for GFP expression in the FL1 channel with a FACSCalibur flow cytometer (BD Biosciences).
For infection studies, 48-well plates were seeded with equal numbers of cells transiently transfected WT or mutant TIM-4. Forty-eight hours following transfection, cells were infected with EBOV GP/rVSV at an inoculum that resulted in ∼25 to 30% of the cells becoming infected (MOI of ∼0.25) or serial dilutions of virus. Twenty-four to 48 h later, cells were lifted with Accutase and fixed in 3.7% formaldehyde for at least 30 min. Cells were then washed with PBS and analyzed for GFP expression by flow cytometry.
Transduction and infection studies of mutant TIM-4-expressing HEK 293T cells were normalized to WT transduction or infection by dividing the sample mean from replicates by the WT mean in the same experiment. The normalized transduction or infection ratios were then normalized to TIM-4 expression by dividing mutant TIM-4 expression by WT TIM-4 expression.
Production and quantification of soluble TIM-4 proteins.
Hemagglutinin (HA)-tagged soluble hTIM-4 and mTIM-4 proteins used in the enzyme-linked immunosorbent assay (ELISA) studies were generated by replacing the transmembrane domain and cytoplasmic tail with an HA tag sequence followed by a stop codon. Point mutations in soluble hTIM-4 and mTIM-4 were created by the QuikChange mutagenesis protocol described above. The expression plasmids were transfected into HEK 293T cells, and secreted proteins were collected 48 h following transfection in Opti-MEM with 1% P/S. Supernatants were filtered through a 0.2-μm filter, aliquoted, and stored at −80°C. Recombinant soluble TIM-4 protein expression was determined by dot blot and Western blot assays with rabbit polyclonal anti-HA antiserum (Sigma). Signals were quantified and protein amounts were normalized with the Odyssey CLx Imaging system and Image Studio software (LI-COR).
Virion/PtdSer–TIM-4 ELISAs.
EBOV VSVΔG-GFP pseudovirion and PtdSer/PtdC liposome binding ELISAs were performed as described previously (12). Briefly, plates were precoated with EBOV VSVΔG-GFP pseudovirions (∼5 × 106 transducing units as determined by titration on Vero cells), 50 μM PtdSer, or 50 μM phosphatidylcholine (PtdC) liposomes in 1× TBS+ (150 mM NaCl, 25 mM Tris, and 10 mM CaCl2, pH 7.2) overnight. All experiments included wells incubated with 1× TBS+ alone as a background binding control for each protein examined. Plates were blocked for 2 h at 4°C with 1× TBS plus 2% bovine serum albumin and then incubated with HA-tagged soluble hTIM-4 protein for 2 h. The plates were probed with rabbit polyclonal anti-HA antiserum (Sigma) for 1 h and horseradish peroxidase-conjugated secondary anti-rabbit antiserum for 1 h and developed with Ultra TMB (Thermo Scientific). Absorbance was read at 450 nm.
Virion internalization experiments.
HEK 293T cells were PEI transfected with WT TIM-4, mutant TIM-4, or the empty vector. Forty-eight hours following transfection, cells were lifted with PBS plus 5 mM EDTA and resuspended in 1× Hanks' balanced salt solution with CaCl2 and MgCl2 with 30 mM HEPES (pH 7.2). Fluorescein isothiocyanate (FITC)-labeled EBOV VSVΔG-GFP pseudovirions produced as previously described (12) were added (MOI of ∼6.5) and bound to transfected cells for 1 h on ice. A portion of each cell population was shifted to 37°C for 1 or 4 h to allow internalization. At the time points indicated following internalization, cells were treated with trypsin-EDTA (Gibco) for 10 min at 37°C. After the 10-min treatment, medium was added to the cell-trypsin mixture and cells were pelleted. The supernatant was removed, and the cells were washed twice with PBS and once with PBS plus 5% FBS. Fluorescence was determined by flow cytometry. Data were analyzed with FlowJo cytometry analysis software to gate the percentage of GFP-positive cells in the FL1 channel compared to that of empty-vector control cells.
Internalization was normalized to internalization of virus from empty-vector-transfected HEK 293T cells equaling 100%, and relative internalization was determined by dividing the amount of GFP-positive TIM-4-expressing cells by the empty-vector-expressing cells and multiplying by 100.
EBOV entry inhibition studies with RGD peptide and 3.47.
Prior to transduction, the hTIM-4-expresssing HEK 293 stable cell line was incubated at 37°C for 30 min with increasing concentrations of arginyl-glycyl-aspartic acid (RGD) peptide (Abbiotec) or 3.47 inhibitor (a kind gift from Kartik Chandran, Albert Einstein College of Medicine). Concentrations were based on amounts that have been previously demonstrated to inhibit integrin binding (RGD peptide) or EBOV entry (3.47) (38–40). Cells in the presence of the inhibitor were transduced with FL EBOV VSV-GFP pseudovirions (MOI of 0.7 [titer determined on Vero cells]). Transduction was quantified 24 h later by flow cytometry as described above, and percent entry was normalized to transduction of cells in medium alone.
Statistics.
All experiments were performed at least three independent times with at least two replicates per cell type. Data from the independent experiments were pooled to generate means and standard deviations of the means. One-sample t tests were performed to compare transduction, infection, or internalization values to 100%, which represents normalized WT TIM-4 transduction or infection or empty-vector-transfected HEK 293T cell virus internalization. Student t tests were performed to compare pseudovirion and liposome binding to WT and mutant TIM-4 proteins.
RESULTS
Expression of hTIM-4 and mTIM-4 enhances filovirus entry.
Previous studies have demonstrated that cell surface expression of hTIM-4 enhances filovirus entry into host cells (10, 12, 13). However, the ability of mTIM-4 to mediate EBOV entry has not been shown, although previous work has utilized mTIM-4 to define TIM-4 structure and function as a receptor for PtdSer liposomes and apoptotic bodies (21, 22, 25). Our initial studies assessed the ability of increasing concentrations of transfected mTIM-4 or hTIM-4 in HEK 293T cells to enhance EBOV entry. For these studies, we transduced the cells with VSV that expresses GFP in place of the native GP (VSVΔG-GFP) that is pseudotyped with the EBOV GP (EBOV VSVΔG-GFP). Ectopic expression of either hTIM-4 or mTIM-4 increased EBOV entry into HEK 293T cells in a dose-dependent manner compared to that into empty-vector-transfected cells (Fig. 1A).
FIG 1.

Expression of human or murine TIM-4 enhances EBOV entry. (A) EBOV entry was enhanced when increasing amounts of hTIM-4 and mTIM-4 were transfected into HEK 293T cells. Forty-eight hours following transfection, cells were transduced with EBOV VSVΔG-GFP pseudovirions. Twenty-four hours later, transduction was quantified by assessing GFP expression by flow cytometry. Background transduction of empty-vector-transfected HEK 293T is indicated by the dotted line (∼1.5% of cells). (B) Amino acid sequence alignment of the mTIM-4 and hTIM-4 IgV domains generated with the Clustal W software program. β-Sheets are indicated by a line and a corresponding letter above the alignment, as previously assigned by Santiago et al. (25). Residue numbers are noted below the amino acid sequences. Asterisks represent conserved sequences, colons represent conservative mutations, and periods represent semiconservative mutations. (C) Overlay of the IgV domain structures of mTIM-4 (blue) and hTIM-4 (green). IgV domain structures of mTIM-4 (PDB code 3BI9) (25) and hTIM-4 (PDB code 5DZN) (28) were obtained from the RCSB PDB and aligned in PyMOL. (D) Residues found in this study to impact EBOV entry are identified in the structures of the mTIM-4 IgV domain (left) and the hTIM-4 IgV domain (right). β-Sheets (mTIM-4, blue; hTIM-4, green) are labeled with the corresponding letters. Red residues are located in the PtdSer binding pocket, and teal residues are located outside the PtdSer binding pocket.
Sequence and structural differences are present in IgV domains of mTIM-4 and hTIM-4.
To compare the mTIM-4 and hTIM-4 IgV domains, we aligned the amino acid sequences of these domains, revealing 63.5% sequence homology between the two domains with high conservation within the FG loop, where many of the PtdSer binding pocket residues reside (Fig. 1B). However, the sequence of the CC′ loop that forms the bottom of the PtdSer binding pocket was not as highly conserved between mTIM-4 and hTIM-4. Next, we aligned the structures of the mTIM-4 IgV domain and the recently elucidated hTIM-4 IgV domain (25, 28). In general, the hTIM-4 and mTIM-4 IgV structures were similar (Fig. 1C). Differences in the AB loop were evident, with the hTIM-4 loop extending further away from the core of the IgV domain than the mTIM-4 loop. However, the most distinct difference found from the overlay resided in the CC′ loop that makes up the bottom ridge of the PtdSer binding pocket (Fig. 1C). Specifically, the defined C′ β-sheet of hTIM-4 was smaller, increasing the length of the CC′ loop and resulting in the bottom loop occluding a portion of the unoccupied PtdSer binding pocket.
From the amino acid and structural differences found in the CC′ loop of the PtdSer binding pocket, we predicted that IgV residues required for mTIM-4- and hTIM-4-dependent virus entry would differ. Hence, TIM-4 alanine-scanning mutagenesis was performed to identify IgV domain residues that are critical for hTIM-4- and mTIM-4-mediated filovirus entry. Alanine-scanning mutagenesis of 31 hTIM-4 and 19 mTIM-4 IgV domain residues was performed, and the TIM-4 constructs were tested for their impact on filovirus entry (Fig. 1D and Table 1).
PtdSer binding pocket residues N121 and D122 are necessary for EBOV entry.
EBOV binding and entry were assessed in cells transfected with plasmids expressing either WT or IgV domain-mutated hTIM-4 or mTIM-4. Others have demonstrated that the asparagine 121 (N121) and aspartic acid 122 (D122) residues within the IgV domain PtdSer binding pocket coordinate the binding of a Ca2+ ion in the pocket and mutagenesis of these residues abrogates PtdSer liposome or apoptotic body interactions (21, 25, 30). To confirm and extend this work to EBOV entry studies, we generated a series of mutant mTIM-4 and hTIM-4 constructs with N121 and D122 altered singly or in combination to either alanine (ND121/122AA) or the reciprocal residue (ND121/122DN). Upon the transient transfection of these mutant mTIM-4 or hTIM-4 constructs into HEK293T cells, levels of surface expression the mutant proteins were indistinguishable from those of the WT protein (Table 1). Along with transduction studies with VSV pseudovirions, infectivity studies with recombinant VSV that contained both the EBOV GP gene and GFP in place of the G-encoding gene in the VSV genome (EBOV GP/rVSV) were performed (11, 12, 31, 32). Expression of WT mTIM-4 or hTIM-4 mediated robust levels of virus entry, as measured by pseudovirion transduction (Fig. 1A and 2A) or virus infection (Fig. 2B), and these values were set to 100% for comparison. Similar to previous findings with hTIM-1, N121 and D122 mutations singly or in combination profoundly inhibited virus transduction and infection mediated by mTIM-4 or hTIM-4 (12, 28) (Fig. 2A to C). These mutant constructs provided no better EBOV entry than the empty-vector control. Additionally, we observed that these same mutations abrogated mTIM-4- or hTIM-1-mediated entry of VSV virions pseudotyped with FL EBOV or Marburg virus GP (data not shown).
FIG 2.

TIM-4 IgV domain PtdSer binding pocket residues N121 and D122 are critical for EBOV entry. HEK 293T cells were transfected with WT or N121/D122 TIM-4 IgV domain mutant constructs and assessed for their impact on EBOV VSVΔG-GFP pseudovirion transduction (A) and EBOV GP/rVSV infection (B). GFP expression was quantified by flow cytometry 24 h following transduction or infection. Relative transduction and infection were normalized to TIM-4 surface expression for each construct as described in Materials and Methods. WT TIM-4 represents 100% transduction and infection in panels A and B, respectively. Significance was determined by one-sample t test. **, P < 0.01; ***, P < 0.001. (C) ND121/122DN (DN) mutant TIM-4 does not enhance EBOV GP/rVSV infection. Forty-eight hours following transfection with TIM-4 plasmids or the empty vector, cells were infected with serial dilutions of EBOV GP/rVSV. Infection was quantified by flow cytometry 48 h following infection. (D) ND mutant TIM-4 constructs have reduced binding to EBOV VSVΔG-GFP pseudovirions. HEK 293T supernatants containing HA-tagged WT or mutant TIM-4 protein were incubated on ELISA plates prebound with EBOV VSVΔG-GFP pseudovirions. Relative protein amounts present in supernatants in a representative immunoblot assay are shown at the bottom. The dotted line represents TIM-4 binding to plates not coated with pseudovirions. Soluble TIM-4 proteins were detected with anti-HA antiserum. Significance was determined by Student t test compared to WT TIM-4 binding to EBOV pseudovirions. ***, P < 0.001. (E) WT TIM-4, not the DN PtdSer binding pocket mutant, increases internalization of FITC-labeled EBOV VSVΔG-GFP pseudovirions. HEK 293T cells were transfected with WT TIM-4 or ND121/122DN mutant TIM-4. Pseudovirions were bound at 4°C for 1 h and then shifted to 37°C for the time indicated to allow virus internalization. Cells were treated with trypsin-EDTA to remove cell surface-bound, noninternalized virus. FITC expression was quantified by flow cytometry following trypsin-EDTA treatment and washing. Internalization into empty-vector-transfected cells represents 100%. Significance was determined by one-sample t test. **, P < 0.01; ***, P < 0.001.
We further examined the impact of mutating these TIM-4 residues on individual steps in the entry process. We examined TIM-4 binding to EBOV pseudovirions via an ELISA with HA-tagged soluble WT or mutant mTIM-4 and hTIM-4 proteins. The recombinant proteins in cell supernatants were normalized for expression by immunoblotting for HA (Fig. 2D, bottom) and added to virion-bound plates. WT mTIM-4 and hTIM-4 bound well to the pseudovirion-coated plates but not the untreated ELISA plates; however, the N121/D122 PtdSer binding pocket mutant constructs had significantly reduced binding that was similar to background levels (Fig. 2D). Consistent with the inability of mutant constructs to bind to EBOV pseudovirions, we did not detect internalization of FITC-labeled EBOV pseudovirions into cells expressing ND121/122DN double mutant hTIM-4 or mTIM-4 but observed time-dependent internalization of virions into cells expressing the WT TIM-4 proteins (Fig. 2E).
Additional PtdSer binding pocket residues affect optimal TIM-4-mediated entry.
We next examined the importance of other PtdSer binding pocket residues for virion binding and infection. Alanine mutagenesis of mTIM-4 residues N61 and S62, which make up the outer bottom ridge of the pocket within the CC′ loop, had no impact on EBOV pseudovirion entry (Fig. 3A). However, alanine substitution of residues located more deeply within the pocket, K63 and R113, affected mTIM-4-mediated EBOV transduction. These two mutations were also assessed for their impact on EBOV GP/rVSV infection (Fig. 3B). The K63A mutation did not affect infection at an MOI of 0.25, whereas R113A did. Consistent with the R113A entry findings, R113A mutant mTIM-4 also had reduced binding of EBOV pseudovirions (Fig. 3D). The F120A mutation, which is located on the upper FG loop of the mTIM4 PtdSer binding pocket, also decreased virion binding and EBOV entry (Fig. 3). The impact of these three mutations on binding and transduction or infection was more variable and, in general, more modest than that observed upon the mutation of N121 or D122, with EBOV binding reduced by ∼50 to 85% and transduction or infection reduced by ∼20 to 80%.
FIG 3.

Additional PtdSer binding pocket hTIM-4 IgV domain residues affect EBOV entry. HEK 293T cells were transfected with WT or mutant TIM-4 constructs and assessed for their impact on EBOV VSVΔG-GFP pseudovirion transduction (A) or EBOV GP/rVSV infection (B). GFP expression was quantified by flow cytometry 24 h following transduction or infection. Relative transduction and infection were normalized to TIM-4 surface expression for each construct as described in Materials and Methods. WT TIM-4 represents 100% transduction and infection in panels A and B, respectively. The significance of differences from WT TIM-4 was determined by one-sample t test. *, P < 0.05; **, P < 0.01; ***, P < 0.001. (C) Y61A and S62A mutant hTIM-4 CC″ loop constructs demonstrated about 4-fold less infectivity than WT hTIM-4. Forty-eight hours following transfection with TIM-4 plasmids or the empty vector, cells were infected with serial dilutions of EBOV GP/rVSV. Infection was quantified by flow cytometry 48 h following infection. The significance of differences from WT TIM-4 infection was determined by Student t test. **, P < 0.01. (D) Identification of TIM-4 PtdSer binding pocket mutant constructs with reduced binding to EBOV VSVΔG-GFP pseudovirions. HEK 293T supernatants containing HA-tagged WT or mutant TIM-4 proteins were incubated on ELISA plates prebound with EBOV VSVΔG-GFP pseudovirions. The dashed line represents binding of WT pseudovirions to an uncoated plate. Relative soluble TIM-4 expression present in supernatants is shown below in a representative immunoblot assay with anti-HA antiserum. The significance of differences from WT TIM-4 binding to EBOV pseudovirions was determined by Student t test. *, P < 0.05; **, P < 0.01; ***, P < 0.001.
In contrast, mutagenesis of these same five amino acids (residues 61, 62, 63, 113, and 120) in the hTIM-4 PtdSer binding pocket significantly reduced transduction of EBOV GP pseudovirions (Fig. 3A), with G63A, R113A, and F120A also demonstrating a reduced ability to support EBOV GP/rVSV infection at an MOI of ∼0.25 (Fig. 3B). While the alanine substitutions at positions 61 and 62 had a significant impact on transduction, these mutations supported WT levels of EBOV GP/rVSV infection at this relatively high MOI (Fig. 3B). To look more closely at the ability of these two mutant constructs to support infection, serial dilutions of EBOV GP/rVSV were assessed in WT, Y61A, and S62A hTIM-4-transfected cells at 48 h of infection. At low input virus concentrations, both of the mutant constructs were ∼4-fold less infectious than the WT; however, that deficiency was overcome with the addition of larger amounts of virus (Fig. 3C). Our other mutant TIM constructs that were found to be significantly different from the WT with transducing virus but not infectious virus, such as mTIM-4 mutant K63A, may have similar infectivity curves. Consistent with our transduction or infection findings, all five mutant hTIM-4 constructs studied had reduced binding to virus (Fig. 3D).
IgV domain residues near the upper loop of the PtdSer binding pocket are important for TIM-4-mediated EBOV entry.
Previous hTIM-1 IgV domain mutagenesis studies identified several residues adjacent to the PtdSer binding pocket that affect hTIM-1-mediated virus entry (12). In a similar manner, additional TIM-4 IgV residues might be critical for EBOV entry. To explore this possibility, we targeted additional TIM-4 residues that corresponded either to positions found to be important in hTIM-1 or to residues that were surface exposed as determined by structural analysis. Alanine substitution of a number of IgV residues had little to no impact on EBOV uptake (Table 1); however, we did identify several additional residues that influenced virus binding and entry. As with hTIM-1, mutation of G118 and W119, which sit along the top of the FG loop, altered virus binding and entry mediated by either mTIM-4 or hTIM-4 (Fig. 4A to C), suggesting that structural alteration of this loop affects the ability of PtdSer to bind within the PtdSer binding pocket. Interestingly, mutagenesis of V116 decreased EBOV binding and entry mediated by hTIM-4 but did not impact mTIM-4-mediated EBOV entry. In a similar manner, an alanine substitution at I125 on the G β-strand of hTIM-4 altered hTIM-4-dependent entry, but a K125A mutation in mTIM-4 had no effect on virus uptake.
FIG 4.

More FG loop residues are required for optimal hTIM-4-mediated entry than for mTIM-4-mediated entry. HEK 293T cells were transfected with WT TIM-4 or FG loop mutant constructs and assessed for their impact on EBOV VSVΔG-GFP pseudovirion transduction (A) and EBOV GP/rVSV infection (B). GFP expression was quantified by flow cytometry 24 h following transduction or infection. Relative transduction and infection were normalized to TIM-4 surface expression for each construct as described in Materials and Methods. WT TIM-4 represents 100% transduction and infection in panels A and B, respectively. The significance of differences from WT TIM-4 was determined by one-sample t test. *, P < 0.05; **, P < 0.01; ***, P < 0.001. (C) FG loop mutant TIM-4 constructs with reduced transduction and infection also have reduced binding to EBOV VSVΔG-GFP pseudovirions. HEK 293T supernatants containing HA-tagged WT or mutant TIM-4 protein were incubated with ELISA plates prebound with EBOV VSVΔG-GFP pseudovirions. The dashed line represents the binding of WT pseudovirions to an uncoated plate. Relative amounts of soluble TIM-4 present in supernatants in a representative immunoblot assay with anti-HA antiserum are shown at the bottom. The significance of differences from WT TIM-4 binding to EBOV pseudovirions was determined by Student t test. *, P < 0.05; **, P < 0.01; ***, P < 0.001.
Additional IgV residues previously found to interact with PtdSer in membranes are important for EBOV uptake.
Interestingly, mTIM-1 and mTIM-4 have similar binding affinities for PtdSer (27) but mTIM-4 binding to PtdSer-containing artificial membranes displays significant cooperativity compared to the other TIM proteins (27, 41). Tietjen et al. recently proposed that mTIM-4 binds in a cooperative manner to PtdSer in membranes through ionic interactions with additional IgV domain residues (41). Four mTIM-4 basic IgV domain residues were found (R49, K63, R70, and K124) in that study to mediate direct interactions with membrane-bound PtdSer. These residues are thought to bind independently of the central residues within the PtdSer binding pocket that most robustly bind PtdSer in a Ca2+-dependent manner (41). Therefore, the impact of these basic residues on viral entry was assessed. In mTIM-4, one of these residues, K63, resides within the PtdSer binding pocket and, as shown above, modestly decreases EBOV pseudovirion transduction but not EBOV GP/rVSV infection (Fig. 3A and B). Surprisingly, mTIM-4 residues R70 and K124 had no impact on EBOV entry, whereas residue R49, which resides on the BC loop, also modestly inhibited EBOV transduction but had no effect on EBOV GP/rVSV infection when the virus was added at an MOI of ∼0.25 (Fig. 5A and B).
FIG 5.

TIM-4 IgV domain residues outside the PtdSer binding pocket are important for EBOV entry. HEK 293T cells were transfected with WT or IgV domain mutant TIM-4 constructs outside the PtdSer binding pocket and assessed for their impact on EBOV VSVΔG-GFP pseudovirion transduction (A) and EBOV GP/rVSV infection (B). GFP expression was quantified by flow cytometry 24 h following transduction or infection. Relative transduction and infection were normalized to TIM-4 surface expression for each construct as described in Materials and Methods. WT TIM-4 represents 100% transduction and infection in panels A and B, respectively. The significance of differences from WT TIM-4 was determined by one-sample t test. *, P < 0.05; **, P < 0.01; ***, P < 0.001. (C) hTIM-4 residues outside the PtdSer binding pocket with reduced binding to EBOV VSVΔG-GFP pseudovirions were identified. HEK 293T supernatants containing HA-tagged WT or mutant TIM-4 proteins were incubated on ELISA plates prebound with EBOV VSVΔG-GFP pseudovirions. The dashed line represents the binding of WT pseudovirions to an uncoated plate. Relative protein amounts present in supernatants in a representative immunoblot assay with anti-HA antiserum are shown at the bottom. The significance of differences from WT TIM-4 binding to EBOV pseudovirions was determined by Student t test. *, P < 0.05; **, P < 0.01; ***, P < 0.001.
We also mutated the hTIM-4 IgV domain residues that align with the four mTIM-4 basic residues. While the residues located at positions 70 and 124 are conserved between mTIM-4 and hTIM-4, hTIM-4 contains a serine and a glycine at positions 49 and 63, respectively. Interestingly, in hTIM-4, basic residues are located near these residues (H47 and K65) and we assessed the role of these residues in EBOV entry as well. Mutagenesis of hTIM-4 residues at positions 49, 63, 70, and 124 significantly reduced EBOV binding and entry (Fig. 3 and 5). However, alanine substitution of adjacent basic residues H47 and K65 had no impact on hTIM-4-mediated EBOV entry (Fig. 5A). Because of the lack of conservation between mTIM-4 and hTIM-4 at residues 49 and 63, we also altered the serine at hTIM-4 position 49 to an arginine and the glycine at hTIM-4 position 63 to a lysine to mimic the mTIM-4 residues and assessed transduction. The hTIM-4 S49R and G63K mutant constructs still demonstrated reduced EBOV entry similar to alanine mutant constructs (Table 1). So, of the residues originally identified by Tietjen et al., two basic mTIM-4 IgV domain residues (R49 and K63) and four of the corresponding hTIM-4 IgV domain residues (S49, G63, R70, and K124) influenced EBOV entry.
The same TIM-4 residues that impact EBOV entry also have decreased PtdSer liposome binding.
A recent study by Yuan et al. suggests that hTIM-1 interacts not only with virion-associated PtdSer but also with the EBOV GP to facilitate cellular virus entry (28). This recent observation stands in contrast to a series of other studies providing evidence that TIM proteins interact not with EBOV GP but solely with PtdSer on the virion (12, 13, 29). To determine whether the residues we have identified as important for EBOV entry interact with PtdSer or EBOV GP, we tested the abilities of our WT and mutant mTIM-4 and hTIM-4 proteins to bind to PtdSer and PtdC liposomes in an ELISA. We found that the mutations that reduced EBOV binding and entry similarly reduced or eliminated the ability of mTIM-4 and hTIM-4 to bind to PtdSer liposomes (Fig. 6). Neither TIM molecule bound to PtdC liposomes. These findings suggest that the TIM-4 residues we identified here are important for EBOV entry because of their ability to bind PtdSer on the surface of EBOV virions.
FIG 6.
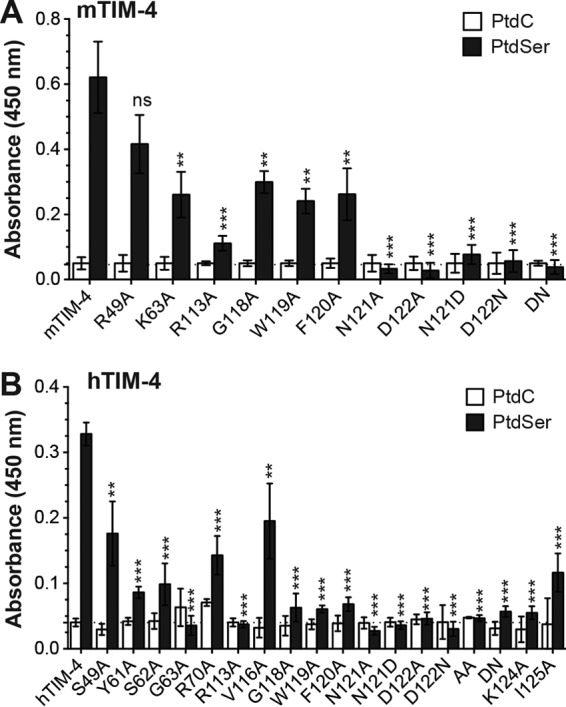
mTIM-4 and hTIM-4 IgV domain mutants found to inhibit EBOV entry also have reduced binding to PtdSer liposomes. HEK 293T supernatants containing HA-tagged mouse (A) or human (B) WT or mutant TIM-4 proteins were incubated on ELISA plates prebound with 50 μM PtdSer or PtdC liposomes. Relative protein amounts present in supernatants were normalized in an immunoblot assay with anti-HA antiserum prior to ELISA analysis. The significance of differences from WT TIM-4 protein PtdSer binding was determined by Student t test. *, P < 0.05; **, P < 0.01; ***, P < 0.001; ns, not significant.
Integrin–TIM-4 interactions are not required for TIM-4-mediated EBOV entry.
Work by others suggests that TIM-4 does not directly mediate internalization of PtdSer liposomes or apoptotic bodies (30, 42, 43). Instead, it is proposed that TIM-4 passes the PtdSer-containing cargo to other PtdSer receptor complexes such as Gas6/Mer and/or MFG-E8 complexed with integrin αVβ3 or αVβ5 (42–45). Since HEK 293T cells express integrin αV but not Mer on their surface (Fig. 7A), we investigated the importance of TIM-4 interactions with integrins for EBOV entry. Both mTIM-4 and hTIM-4 have an integrin binding RGD motif within their IgV domains, and RGD motifs are known to interact with integrins; however, the mTIM-4 and hTIM-4 RGD motifs are located in different positions within the IgV domain (Fig. 1B and Table 1). We sought to determine if mutagenesis of the residues composing this motif significantly reduced TIM-4-mediated entry. We found that alanine substitutions of mTIM-4 G105 and D106 had no impact on the ability of mTIM-4 to mediate EBOV entry into cells, indicating that integrin interactions through this motif are not required for mTIM-4-dependent EBOV entry (Fig. 7B). Mutagenesis of the RGD motif of hTIM-4 residues resulted in a change in the overall IgV domain structure, reducing hTIM-4 detection by a panel of anti-hTIM-4 IgV monoclonal antibodies or hTIM-4 polyclonal antiserum (data not shown). Our evidence suggests that alanine substitutions of these DE loop residues structurally alter the IgV domain, thereby disrupting protein structure and/or function and significantly reducing hTIM-4-mediated virus entry (Table 1).
FIG 7.

TIM-4 IgV RGD motifs are not important for EBOV entry. (A) HEK 293T cells express integrin αv but not Mer. Polyclonal antiserum (black histogram) against human integrin αv or Mer and a control goat IgG (gray histogram) were used to stain the surface of HEK 293T cells. Surface expression was analyzed by flow cytometry. (B) Mutations in the mTIM-4 IgV domain RGD motif do not reduce EBOV VSVΔG-GFP pseudovirion transduction. HEK 293T cells were transfected with WT or mutant TIM-4 constructs. Transfected cells were transduced with EBOV VSVΔG-GFP pseudovirions at 24 h. GFP expression was quantified 24 h following transduction by flow cytometry. WT mTIM-4 represents 100% transduction, and significance was determined by one-sample t test. ***, P < 0.001. (B) hTIM-4-mediated EBOV entry is not blocked by increasing concentrations of RGD peptide. HEK 293 cells stably expressing hTIM-4 were preincubated with increasing concentrations of RGD peptide or the EBOV entry inhibitor 3.47 at 37°C for 30 min. Cells were transduced in the presence of the peptide or inhibitor with FL EBOV VSVΔG-GFP pseudovirions, and GFP expression was quantified by flow cytometry 24 h following transduction. Transduction of cells in medium alone represents 100%.
To address the contribution of integrins to hTIM-4-mediated EBOV entry in another way, we utilized an RGD peptide competition assay. Initially, we verified that commercially purchased RGD peptide inhibited fibronectin-Vero cell interactions in a dose-dependent manner, as previously reported (46, 47) (data not shown). Upon verification, HEK 293 cells that stably express hTIM-4 were incubated with increasing concentrations of RGD peptide or, as a control, a well-established EBOV entry inhibitor, 3.47, that blocks late endosomal events required for EBOV entry (39, 40). The cells were transduced with EBOV pseudovirions and analyzed 24 h later for GFP expression. We found that the RGD peptide did not compete for EBOV entry into hTIM-4-expressing cells, in contrast to robust inhibition with 3.47 (Fig. 7C). Together, these findings suggest that TIM-4–integrin interactions are not essential for TIM-4-mediated EBOV entry.
DISCUSSION
Here, we identify TIM-4 IgV domain residues critical for TIM-4-mediated filovirus entry. We found that eight mTIM-4 IgV domain residues, five residues in and three residues outside the PtdSer binding pocket, are important for mTIM-4-mediated entry. In contrast, we found that fourteen hTIM-4 IgV domain residues, eight residues in and six residues surrounding or outside the PtdSer binding pocket, are important for hTIM-4-mediated entry. Additionally, we demonstrate that those hTIM-4 and mTIM-4 residues found to affect EBOV binding and entry also affected PtdSer liposome interactions, emphasizing that PtdSer interactions, rather than EBOV GP interactions, are important for TIM-4-dependent entry. Finally, we show that RGD-dependent interactions of TIM-4 with integrins are not required for EBOV entry into HEK 293T cells.
Our results provide evidence that the mTIM-4 and hTIM-4 IgV domains have structurally distinct requirements for PtdSer interactions. This may be due to differences in direct IgV domain residue/PtdSer binding properties or in the IgV domain structures that influence the ability of the pocket to bind to PtdSer. Our studies are unable to distinguish which of these mechanistic possibilities is responsible for our disparate findings with mTIM-4 and hTIM-4.
The structures of mTIM-1 and -4 and hTIM-1 and -4 IgV domains have been solved, showing that residues of the CC′ and FG loops that are important for PtdSer binding make up the PtdSer binding pocket (25, 26, 28, 29). Using those structures, as well as a linear sequence alignment, we summarize the mTIM-4 and hTIM-4 IgV domain residues important for filovirus entry and compare those residues to hTIM-1 IgV domain residues previously found to be important (12) (Fig. 8). Previously, our lab identified eight hTIM-1 PtdSer binding pocket or adjacent residues that are important for hTIM-1-mediated EBOV entry (12). The present study found that six of these eight residues also located within the TIM-4 IgV domain (R113, G118, W119, F120, N121, and D122) to be important for mTIM-4- and hTIM-4-mediated entry. All of these residues either reside in the PtdSer binding pocket or sit along the outside of the FG loop that defines the upper ridge of the pocket. Structural comparisons of the mTIM-1, hTIM-1, mTIM-4, and hTIM-4 IgV domains indicate that the FG loops composing the upper lip of the PtdSer binding pocket are similar in all four proteins (Fig. 1C and 8 and data not shown) and highlight the importance of these residues for TIM-PtdSer interactions. Additional residues located on the lower CC′ loop of the PtdSer binding pocket were also found to be important for PtdSer-TIM interactions. In hTIM-1 and mTIM-4, a single residue in the CC′ loop affected PtdSer binding and EBOV entry (reference 12 and this study). In contrast, CC′ loop residues Y61, S62, and G63 are important for hTIM-4-mediated EBOV entry. The mTIM-4 and hTIM-4 structural comparison in Fig. 1C demonstrates that the CC′ loop of the hTIM-4 pocket is structurally distinct from mTIM-4. Structural differences between the hTIM-4 and hTIM-1 CC′ loops are also apparent in Fig. 8. Perhaps the altered structure of hTIM-4 requires additional lower-loop interactions that are not required for mTIM-4 or hTIM-1 functionality. It should be noted that several other residues near or perhaps even within the lower portion of the PtdSer binding pocket were also found to be important for hTIM-4, but not mTIM-4. These include V116 and I125, which, along with Y61, S62, and G63, may help to maintain the structural integrity of the lower PtdSer binding pocket. Since none of these residues is positively charged, they are not likely to interact directly with PtdSer.
FIG 8.

Identification of TIM-1 and TIM-4 residues important for filovirus entry. (A) Amino acid sequence alignment of the hTIM-1, hTIM-4, and mTIM-4 IgV domains generated with the Clustal W software program. Each β-sheet is indicated above the alignment by a line and a corresponding letter, as previously assigned by Santiago et al. (25). Residue numbers are below the amino acid sequences. Asterisks represent conserved sequences, colons represent conservative mutations, and periods represent semiconservative mutations. (B) Overlay of the IgV domain structures of hTIM-1 (gray) and hTIM-4 (black). The IgV domain structures of hTIM-1 (PDB code 5DZO) and hTIM-4 (PDB code 5DZN) (28) were obtained from the RCSB PDB and aligned in PyMOL. Highlighted residues indicate those found in this study and that of Moller-Tank et al. (12) to impact EBOV entry. Purple (hTIM-1) and cyan (hTIM-4) residues are important for both TIM molecules. Green residues are uniquely important for hTIM-4.
Not all of the mTIM-4 residues found by Tietjen et al. to enhance mTIM-4 cooperative binding to PtdSer-containing membranes also affected virus entry (41). The four basic residues that were identified were proposed to bind directly to lipid head groups of the membrane bilayer and mediate cooperative binding to PtdSer independently of the PtdSer binding pocket residues (41). We demonstrate that two of these residues (R49 and K63), but not R70 or K124, modestly reduce EBOV entry when mutated to alanine. Interestingly, we found that all four hTIM-4 residues present at the same amino acid positions (S49, G63, R70, and K124) are important for hTIM-4-dependent virus entry. However, one characteristic feature of PtdSer binding proteins is the presence of positively charged residues that interact with the membrane surface (48). Since hTIM-4 S49 and G63 are not positively charged residues, we examined adjacent positively charged amino acids residues H47 and K65. Neither of these basic residues was found to be important for hTIM-4-mediated EBOV entry. In addition, as noted above, G63, K70, and K124 sit within or near the PtdSer binding pocket and may impact critical structural constraints of the pocket. Thus, alternative roles for these residues other than direct PtdSer interactions are likely. Future studies need to compare the sensitivity of mTIM-4 and hTIM-4 to PtdSer surface density to determine if both proteins exhibit similar lipid recognition sensitivity.
For several of the mutant TIM-4 constructs studied, an entry defect was noted in the transduction studies that was not detected in the infection studies. Our detailed dose-response curves of two of the mutant hTIM-4 constructs suggested that the relatively high MOIs used may have compensated for any entry deficit. However, it is possible that TIM-mediated viral entry may be influenced by the amount of surface PtdSer on viral particles. To begin to address if there are distinct differences between PtdSer on the surface of our transducing and infectious VSV stocks, we assessed PtdSer levels in three different stocks of each by binding annexin V to virions normalized for VSV matrix levels in ELISAs. We observed modestly higher levels of PtdSer on the surface of our infectious virus stocks than on our pseudovirions (data not shown). This slightly larger amount of PtdSer on infectious virions may contribute to their ability to more effectively use some of the mutant TIM molecules. Differences in PtdSer levels in our pseudovirion stocks versus our infectious stocks may be due to pseudovirion production in HEK 293T cells and infectious virion production in Vero cells.
Nevertheless, it remains unknown how much virion-associated PtdSer is needed for productive TIM interactions. Further, studies have yet to assess if differences in surface PtdSer exposure occur on different viral particles (e.g., VSV pseudovirions verse authentic filoviruses). Recent studies have demonstrated that EBOV matrix protein VP40 selectively induces budding from membranes enriched for PtdSer and that PtdSer regulates the localization and assembly of VP40 at the plasma membrane (49, 50). Additionally, it has been proposed that filovirus budding occurs at sites of lipid rafts, which are rich in PtdSer (51, 52). Therefore, it is possible that filovirus membranes selectively acquire increased amounts of PtdSer while budding from the host cell.
Unlike TIM-1 and TIM-3, TIM-4 does not have a known signaling motif in its cytoplasmic tail (30, 53–56). Additionally, TIM-4-mediated engulfment of apoptotic cells has been shown not to involve the ELMO1/DOCK180/Rac or GULP signaling pathway (30). Consequently, it has been suggested that TIM-4 serves as a cell surface attachment factor for apoptotic bodies by tethering the bodies to the cell surface. Other PtdSer receptor complexes, like integrin αVβ3 and MFG-E8, act to engulf the apoptotic cell (42). Further, others have postulated that TIM-4–integrin complex interactions occur through RGD motifs within hTIM-4 and mTIM-4 IgV domains (57–59). However, alanine substitution of G105 and D106 of mTIM-4 had no effect on EBOV entry, suggesting that these interactions are not required for mTIM-4-dependent EBOV entry (Fig. 7B). Additionally, we demonstrate that an RGD peptide does not inhibit EBOV entry in hTIM-4-expressing cells. Future studies need to investigate if signaling pathways are important for TIM-4-dependent viral entry and, if so, which pathways are required. Studies also need to determine whether TIM-4 itself initiates virus engulfment or whether other PtdSer receptors or cellular factors are potentially important interacting partners for TIM-4-mediated viral entry.
Our analysis of the molecular characteristics of both mTIM-4 and hTIM-4 provides a better understanding of the regions of the TIM-4 IgV domain critical for EBOV entry. Unlike TIM-1, which is expressed on epithelial cells, mast cells, B-cells, and activated CD4+ T cells (11, 16–20), TIM-4 is expressed on antigen-presenting cells, including macrophages and DCs, which are important early targets during EBOV pathogenesis (16, 21–23). Thus, the cell expression profiles of these family members do not overlap. Small-molecule inhibitors that bind in the relatively conserved TIM PtdSer binding pocket might be effective against both TIM-1 and TIM-4, providing novel therapeutics to reduce EBOV infection and pathogenesis in vivo.
ACKNOWLEDGMENTS
We thank Stanley Perlman and Anthony Fehr for providing helpful comments on the manuscript.
Funding Statement
The funders had no role in study design, data collection and analysis, the decision to publish, or preparation of the manuscript.
REFERENCES
- 1.Murray MJ. 2015. Ebola virus disease: a review of its past and present. Anesth Analg 121:798–809. doi: 10.1213/ANE.0000000000000866. [DOI] [PubMed] [Google Scholar]
- 2.WHO. 2015. Ebola situation reports. World Health Organization, Geneva, Switzerland: http://apps.who.int/ebola/ebola-situation-reports Accessed 9 September 2015. [Google Scholar]
- 3.Martines RB, Ng DL, Greer PW, Rollin PE, Zaki SR. 2015. Tissue and cellular tropism, pathology and pathogenesis of Ebola and Marburg viruses. J Pathol 235:153–174. doi: 10.1002/path.4456. [DOI] [PubMed] [Google Scholar]
- 4.Rhein B, Maury W. 2015. Ebola virus entry into host cells: identifying therapeutic strategies. Curr Clin Microbiol Rep 2:115–124. doi: 10.1007/s40588-015-0021-3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Alvarez CP, Lasala F, Carrillo J, Muniz O, Corbi AL, Delgado R. 2002. C-type lectins DC-SIGN and L-SIGN mediate cellular entry by Ebola virus in cis and in trans. J Virol 76:6841–6844. doi: 10.1128/JVI.76.13.6841-6844.2002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Takada A, Fujioka K, Tsuiji M, Morikawa A, Higashi N, Ebihara H, Kobasa D, Feldmann H, Irimura T, Kawaoka Y. 2004. Human macrophage C-type lectin specific for galactose and N-acetylgalactosamine promotes filovirus entry. J Virol 78:2943–2947. doi: 10.1128/JVI.78.6.2943-2947.2004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Lennemann NJ, Rhein BA, Ndungo E, Chandran K, Qiu X, Maury W. 2014. Comprehensive functional analysis of N-linked glycans on Ebola virus GP1. mBio 5:e00862-13. doi: 10.1128/mBio.00862-13. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Shimojima M, Takada A, Ebihara H, Neumann G, Fujioka K, Irimura T, Jones S, Feldmann H, Kawaoka Y. 2006. Tyro3 family-mediated cell entry of Ebola and Marburg viruses. J Virol 80:10109–10116. doi: 10.1128/JVI.01157-06. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Hunt CL, Kolokoltsov AA, Davey RA, Maury W. 2011. The Tyro3 receptor kinase Axl enhances macropinocytosis of Zaire ebolavirus. J Virol 85:334–347. doi: 10.1128/JVI.01278-09. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Morizono K, Chen IS. 2014. Role of phosphatidylserine receptors in enveloped virus infection. J Virol 88:4275–4290. doi: 10.1128/JVI.03287-13. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Kondratowicz AS, Lennemann NJ, Sinn PL, Davey RA, Hunt CL, Moller-Tank S, Meyerholz DK, Rennert P, Mullins RF, Brindley M, Sandersfeld LM, Quinn K, Weller M, McCray PB Jr, Chiorini J, Maury W. 2011. T-cell immunoglobulin and mucin domain 1 (TIM-1) is a receptor for Zaire Ebolavirus and Lake Victoria Marburgvirus. Proc Natl Acad Sci U S A 108:8426–8431. doi: 10.1073/pnas.1019030108. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Moller-Tank S, Kondratowicz AS, Davey RA, Rennert PD, Maury W. 2013. Role of the phosphatidylserine receptor TIM-1 in enveloped-virus entry. J Virol 87:8327–8341. doi: 10.1128/JVI.01025-13. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Jemielity S, Wang JJ, Chan YK, Ahmed AA, Li W, Monahan S, Bu X, Farzan M, Freeman GJ, Umetsu DT, Dekruyff RH, Choe H. 2013. TIM-family proteins promote infection of multiple enveloped viruses through virion-associated phosphatidylserine. PLoS Pathog 9:e1003232. doi: 10.1371/journal.ppat.1003232. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Amara A, Mercer J. 2015. Viral apoptotic mimicry. Nat Rev Microbiol 13:461–469. doi: 10.1038/nrmicro3469. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Meertens L, Carnec X, Lecoin MP, Ramdasi R, Guivel-Benhassine F, Lew E, Lemke G, Schwartz O, Amara A. 2012. The TIM and TAM families of phosphatidylserine receptors mediate dengue virus entry. Cell Host Microbe 12:544–557. doi: 10.1016/j.chom.2012.08.009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Meyers JH, Chakravarti S, Schlesinger D, Illes Z, Waldner H, Umetsu SE, Kenny J, Zheng XX, Umetsu DT, DeKruyff RH, Strom TB, Kuchroo VK. 2005. TIM-4 is the ligand for TIM-1, and the TIM-1–TIM-4 interaction regulates T cell proliferation. Nat Immunol 6:455–464. doi: 10.1038/ni1185. [DOI] [PubMed] [Google Scholar]
- 17.Umetsu SE, Lee WL, McIntire JJ, Downey L, Sanjanwala B, Akbari O, Berry GJ, Nagumo H, Freeman GJ, Umetsu DT, DeKruyff RH. 2005. TIM-1 induces T cell activation and inhibits the development of peripheral tolerance. Nat Immunol 6:447–454. doi: 10.1038/ni1186. [DOI] [PubMed] [Google Scholar]
- 18.Ichimura T, Bonventre JV, Bailly V, Wei H, Hession CA, Cate RL, Sanicola M. 1998. Kidney injury molecule-1 (KIM-1), a putative epithelial cell adhesion molecule containing a novel immunoglobulin domain, is up-regulated in renal cells after injury. J Biol Chem 273:4135–4142. doi: 10.1074/jbc.273.7.4135. [DOI] [PubMed] [Google Scholar]
- 19.Uchida Y, Ke B, Freitas MC, Ji H, Zhao D, Benjamin ER, Najafian N, Yagita H, Akiba H, Busuttil RW, Kupiec-Weglinski JW. 2010. The emerging role of T cell immunoglobulin mucin-1 in the mechanism of liver ischemia and reperfusion injury in the mouse. Hepatology 51:1363–1372. doi: 10.1002/hep.23442. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Rodriguez-Manzanet R, Sanjuan MA, Wu HY, Quintana FJ, Xiao S, Anderson AC, Weiner HL, Green DR, Kuchroo VK. 2010. T and B cell hyperactivity and autoimmunity associated with niche-specific defects in apoptotic body clearance in TIM-4-deficient mice. Proc Natl Acad Sci U S A 107:8706–8711. doi: 10.1073/pnas.0910359107. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Kobayashi N, Karisola P, Pena-Cruz V, Dorfman DM, Jinushi M, Umetsu SE, Butte MJ, Nagumo H, Chernova I, Zhu B, Sharpe AH, Ito S, Dranoff G, Kaplan GG, Casasnovas JM, Umetsu DT, Dekruyff RH, Freeman GJ. 2007. TIM-1 and TIM-4 glycoproteins bind phosphatidylserine and mediate uptake of apoptotic cells. Immunity 27:927–940. doi: 10.1016/j.immuni.2007.11.011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Miyanishi M, Tada K, Koike M, Uchiyama Y, Kitamura T, Nagata S. 2007. Identification of Tim4 as a phosphatidylserine receptor. Nature 450:435–439. doi: 10.1038/nature06307. [DOI] [PubMed] [Google Scholar]
- 23.Freeman GJ, Casasnovas JM, Umetsu DT, DeKruyff RH. 2010. TIM genes: a family of cell surface phosphatidylserine receptors that regulate innate and adaptive immunity. Immunol Rev 235:172–189. doi: 10.1111/j.0105-2896.2010.00903.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Moller-Tank S, Albritton LM, Rennert PD, Maury W. 2014. Characterizing functional domains for TIM-mediated enveloped virus entry. J Virol 88:6702–6713. doi: 10.1128/JVI.00300-14. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Santiago C, Ballesteros A, Martínez-Muñoz L, Mellado M, Kaplan GG, Freeman GJ, Casasnovas JM. 2007. Structures of T cell immunoglobulin mucin protein 4 show a metal-ion-dependent ligand binding site where phosphatidylserine binds. Immunity 27:941–951. doi: 10.1016/j.immuni.2007.11.008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Santiago C, Ballesteros A, Tami C, Martinez-Munoz L, Kaplan GG, Casasnovas JM. 2007. Structures of T cell immunoglobulin mucin receptors 1 and 2 reveal mechanisms for regulation of immune responses by the TIM receptor family. Immunity 26:299–310. doi: 10.1016/j.immuni.2007.01.014. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.DeKruyff RH, Bu X, Ballesteros A, Santiago C, Chim YL, Lee HH, Karisola P, Pichavant M, Kaplan GG, Umetsu DT, Freeman GJ, Casasnovas JM. 2010. T cell/transmembrane, Ig, and mucin-3 allelic variants differentially recognize phosphatidylserine and mediate phagocytosis of apoptotic cells. J Immunol 184:1918–1930. doi: 10.4049/jimmunol.0903059. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Yuan S, Cao L, Ling H, Dang M, Sun Y, Zhang X, Chen Y, Zhang L, Su D, Wang X, Rao Z. 2015. TIM-1 acts a dual-attachment receptor for ebolavirus by interacting directly with viral GP and the PS on the viral envelope. Protein Cell 6:814–824. doi: 10.1007/s13238-015-0220-y. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Wang H, Qi JX, Liu NN, Li Y, Gao J, Zhang TH, Chai Y, Gao F, Zhang H, Li XD, Ye X, Yan JH, Lu GW, Gao GF. 2015. Crystal structures of human TIM members: ebolavirus entry-enhancing receptors. Chin Sci Bull 60:3438–3453. doi: 10.1360/N972015-01255. [DOI] [Google Scholar]
- 30.Park D, Hochreiter-Hufford A, Ravichandran KS. 2009. The phosphatidylserine receptor TIM-4 does not mediate direct signaling. Curr Biol 19:346–351. doi: 10.1016/j.cub.2009.01.042. [DOI] [PubMed] [Google Scholar]
- 31.Brindley MA, Hunt CL, Kondratowicz AS, Bowman J, Sinn PL, McCray PB Jr, Quinn K, Weller ML, Chiorini JA, Maury W. 2011. Tyrosine kinase receptor Axl enhances entry of Zaire ebolavirus without direct interactions with the viral glycoprotein. Virology 415:83–94. doi: 10.1016/j.virol.2011.04.002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Takada A, Robison C, Goto H, Sanchez A, Murti KG, Whitt MA, Kawaoka Y. 1997. A system for functional analysis of Ebola virus glycoprotein. Proc Natl Acad Sci U S A 94:14764–14769. doi: 10.1073/pnas.94.26.14764. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Yang ZY, Duckers HJ, Sullivan NJ, Sanchez A, Nabel EG, Nabel GJ. 2000. Identification of the Ebola virus glycoprotein as the main viral determinant of vascular cell cytotoxicity and injury. Nat Med 6:886–889. doi: 10.1038/78645. [DOI] [PubMed] [Google Scholar]
- 34.Jeffers SA, Sanders DA, Sanchez A. 2002. Covalent modifications of the Ebola virus glycoprotein. J Virol 76:12463–12472. doi: 10.1128/JVI.76.24.12463-12472.2002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Sinn PL, Hickey MA, Staber PD, Dylla DE, Jeffers SA, Davidson BL, Sanders DA, McCray PB Jr. 2003. Lentivirus vectors pseudotyped with filoviral envelope glycoproteins transduce airway epithelia from the apical surface independently of folate receptor alpha. J Virol 77:5902–5910. doi: 10.1128/JVI.77.10.5902-5910.2003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Reed LJ, Muench H. 1938. A simple method of estimating fifty percent endpoints. Am J Epidemiol 27:493–497. [Google Scholar]
- 37.Schrödinger LLC. 2010. The PyMOL molecular graphics system, version 1.3r1. http://www.pymol.org Schrödinger LLC, New York, NY. [Google Scholar]
- 38.Bedal KB, Grassel S, Oefner PJ, Reinders J, Reichert TE, Bauer R. 2014. Collagen XVI induces expression of MMP9 via modulation of AP-1 transcription factors and facilitates invasion of oral squamous cell carcinoma. PLoS One 9:e86777. doi: 10.1371/journal.pone.0086777. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Côté M, Misasi J, Ren T, Bruchez A, Lee K, Filone CM, Hensley L, Li Q, Ory D, Chandran K, Cunningham J. 2011. Small molecule inhibitors reveal Niemann-Pick C1 is essential for Ebola virus infection. Nature 477:344–348. doi: 10.1038/nature10380. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Shoemaker CJ, Schornberg KL, Delos SE, Scully C, Pajouhesh H, Olinger GG, Johansen LM, White JM. 2013. Multiple cationic amphiphiles induce a Niemann-Pick C phenotype and inhibit Ebola virus entry and infection. PLoS One 8:e56265. doi: 10.1371/journal.pone.0056265. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Tietjen GT, Gong Z, Chen CH, Vargas E, Crooks JE, Cao KD, Heffern CT, Henderson JM, Meron M, Lin B, Roux B, Schlossman ML, Steck TL, Lee KY, Adams EJ. 2014. Molecular mechanism for differential recognition of membrane phosphatidylserine by the immune regulatory receptor Tim4. Proc Natl Acad Sci U S A 111:E1463–E1472. doi: 10.1073/pnas.1320174111. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Toda S, Hanayama R, Nagata S. 2012. Two-step engulfment of apoptotic cells. Mol Cell Biol 32:118–125. doi: 10.1128/MCB.05993-11. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Nishi C, Toda S, Segawa K, Nagata S. 2014. Tim4- and MerTK-mediated engulfment of apoptotic cells by mouse resident peritoneal macrophages. Mol Cell Biol 34:1512–1520. doi: 10.1128/MCB.01394-13. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Andersen MH, Graversen H, Fedosov SN, Petersen TE, Rasmussen JT. 2000. Functional analyses of two cellular binding domains of bovine lactadherin. Biochemistry 39:6200–6206. doi: 10.1021/bi992221r. [DOI] [PubMed] [Google Scholar]
- 45.Hanayama R, Tanaka M, Miwa K, Shinohara A, Iwamatsu A, Nagata S. 2002. Identification of a factor that links apoptotic cells to phagocytes. Nature 417:182–187. doi: 10.1038/417182a. [DOI] [PubMed] [Google Scholar]
- 46.Pierschbacher MD, Ruoslahti E. 1984. Variants of the cell recognition site of fibronectin that retain attachment-promoting activity. Proc Natl Acad Sci U S A 81:5985–5988. doi: 10.1073/pnas.81.19.5985. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Sottile J, Hocking DC, Swiatek PJ. 1998. Fibronectin matrix assembly enhances adhesion-dependent cell growth. J Cell Sci 111(Pt 19):2933–2943. [DOI] [PubMed] [Google Scholar]
- 48.Stace CL, Ktistakis NT. 2006. Phosphatidic acid- and phosphatidylserine-binding proteins. Biochim Biophys Acta 1761:913–926. doi: 10.1016/j.bbalip.2006.03.006. [DOI] [PubMed] [Google Scholar]
- 49.Soni SP, Stahelin RV. 2014. The Ebola virus matrix protein VP40 selectively induces vesiculation from phosphatidylserine-enriched membranes. J Biol Chem 289:33590–33597. doi: 10.1074/jbc.M114.586396. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Adu-Gyamfi E, Johnson KA, Fraser ME, Scott JL, Soni SP, Jones KR, Digman MA, Gratton E, Tessier CR, Stahelin RV. 2015. Host cell plasma membrane phosphatidylserine regulates the assembly and budding of Ebola virus. J Virol 89:9440–9453. doi: 10.1128/JVI.01087-15. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Pike LJ, Han X, Chung K-N, Gross RW. 2002. Lipid rafts are enriched in arachidonic acid and plasmenylethanolamine and their composition is independent of caveolin-1 expression: a quantitative electrospray ionization/mass spectrometric analysis. Biochemistry 41:2075–2088. doi: 10.1021/bi0156557. [DOI] [PubMed] [Google Scholar]
- 52.Pike LJ, Han X, Gross RW. 2005. Epidermal growth factor receptors are localized to lipid rafts that contain a balance of inner and outer leaflet lipids: a shotgun lipidomics study. J Biol Chem 280:26796–26804. doi: 10.1074/jbc.M503805200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Binné LL, Scott ML, Rennert PD. 2007. Human TIM-1 associates with the TCR complex and up-regulates T cell activation signals. J Immunol 178:4342–4350. doi: 10.4049/jimmunol.178.7.4342. [DOI] [PubMed] [Google Scholar]
- 54.Lee J, Su EW, Zhu C, Hainline S, Phuah J, Moroco JA, Smithgall TE, Kuchroo VK, Kane LP. 2011. Phosphotyrosine-dependent coupling of Tim-3 to T-cell receptor signaling pathways. Mol Cell Biol 31:3963–3974. doi: 10.1128/MCB.05297-11. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.de Souza AJ, Oriss TB, O'Malley KJ, Ray A, Kane LP. 2005. T cell Ig and mucin 1 (TIM-1) is expressed on in vivo-activated T cells and provides a costimulatory signal for T cell activation. Proc Natl Acad Sci U S A 102:17113–17118. doi: 10.1073/pnas.0508643102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.de Souza AJ, Oak JS, Jordanhazy R, DeKruyff RH, Fruman DA, Kane LP. 2008. T cell Ig and mucin domain-1-mediated T cell activation requires recruitment and activation of phosphoinositide 3-kinase. J Immunol 180:6518–6526. doi: 10.4049/jimmunol.180.10.6518. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.D'Souza SE, Ginsberg MH, Plow EF. 1991. Arginyl-glycyl-aspartic acid (RGD): a cell adhesion motif. Trends Biochem Sci 16:246–250. doi: 10.1016/0968-0004(91)90096-E. [DOI] [PubMed] [Google Scholar]
- 58.Flannagan RS, Canton J, Furuya W, Glogauer M, Grinstein S. 2014. The phosphatidylserine receptor TIM4 utilizes integrins as coreceptors to effect phagocytosis. Mol Biol Cell 25:1511–1522. doi: 10.1091/mbc.E13-04-0212. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.Zhang Q, Wang H, Wu X, Liu B, Liu W, Wang R, Liang X, Ma C, Gao L. 2015. TIM-4 promotes the growth of non-small-cell lung cancer in a RGD motif-dependent manner. Br J Cancer 113:1484–1492. doi: 10.1038/bjc.2015.323. [DOI] [PMC free article] [PubMed] [Google Scholar]