

ABSTRACT
The herpes simplex virus (HSV) virion host shutoff (vhs) RNase destabilizes cellular and viral mRNAs, suppresses host protein synthesis, dampens antiviral responses, and stimulates translation of viral mRNAs. vhs mutants display a host range phenotype: translation of viral true late mRNAs is severely impaired and stress granules accumulate in HeLa cells, while translation proceeds normally in Vero cells. We found that vhs-deficient virus activates the double-stranded RNA-activated protein kinase R (PKR) much more strongly than the wild-type virus does in HeLa cells, while PKR is not activated in Vero cells, raising the possibility that PKR might play roles in stress granule induction and/or inhibiting translation in restrictive cells. We tested this possibility by evaluating the effects of inactivating PKR. Eliminating PKR in HeLa cells abolished stress granule formation but had only minor effects on viral true late protein levels. These results document an essential role for PKR in stress granule formation by a nuclear DNA virus, indicate that induction of stress granules is the consequence rather than the cause of the translational defect, and are consistent with our previous suggestion that vhs promotes translation of viral true late mRNAs by preventing mRNA overload rather than by suppressing eIF2α phosphorylation.
IMPORTANCE The herpes simplex virus vhs RNase plays multiple roles during infection, including suppressing PKR activation, inhibiting the formation of stress granules, and promoting translation of viral late mRNAs. A key question is the extent to which these activities are mechanistically connected. Our results demonstrate that PKR is essential for stress granule formation in the absence of vhs, but at best, it plays a secondary role in suppressing translation of viral mRNAs. Thus, the ability of vhs to promote translation of viral mRNAs can be largely uncoupled from PKR suppression, demonstrating that this viral RNase modulates at least two distinct aspects of RNA metabolism.
INTRODUCTION
Viruses deploy diverse strategies to shut off host protein synthesis in order to gain access to the cellular translational machinery and blunt host antiviral responses. Alpha- and gammaherpesviruses, influenza A virus, poxviruses, and the severe acute respiratory syndrome (SARS) coronavirus do so in part by producing proteins that globally destabilize host mRNAs (reviewed in references 1 and 2). Among the best characterized of these viral mRNA destabilizers is the virion host shutoff (vhs) protein (encoded by the UL41 gene) of herpes simplex virus (HSV) and other alphaherpesviruses (3, 4) (reviewed in reference 5). vhs is a Fen-1 family endoribonuclease that is packaged into the virion tegument and delivered into the cytoplasm following fusion of the viral envelope with the host plasma membrane. Although purified vhs degrades essentially any RNA in vitro (6–9), only mRNAs are targeted in vivo (10–12). This selectivity appears to arise through the ability of vhs to bind components of the host mRNA cap-binding complex eIF4F (13), namely, the RNA helicase eIF4AII and the helicase cofactors eIF4H and eIF4B (14–17). Consistent with this hypothesis, vhs-induced mRNA decay initiates at or near regions of translation initiation both in mammalian cell extracts and in infected cells (18–21), and mutations that affect the initiation codon of the target mRNA alter the cleavage pattern (21).
vhs plays important and diverse roles during infection. It is presumed to facilitate access of viral mRNAs to the translational apparatus by lowering the levels of most host mRNAs (2). In addition, it reduces the half-lives of viral mRNAs, thereby sharpening the transitions between the successive phases of viral protein synthesis (10, 11). vhs also dampens certain host antiviral responses, including induction and activity of the type I interferon (IFN) system (22–24), production of other proinflammatory cytokines and chemokines (25), and activation of dendritic cells (26). The effects of vhs on the IFN system in particular contribute to the severe attenuation of vhs mutants in mouse models of HSV infection (22, 27–29).
vhs also directly or indirectly enhances translation of viral mRNAs, likely through at least two distinct mechanisms. First, Sciortino et al. have provided evidence that vhs inhibits activation of the double-stranded RNA (dsRNA)-activated protein kinase PKR (protein kinase R) (31), consistent with an earlier suggestion by Pasieka et al. (28), who observed enhanced levels of phosphorylated eIF2α during infection with a vhs mutant. PKR is one of four host stress-activated kinases that are capable of phosphorylating the translation initiation factor eIF2α on serine residue 51, leading to inhibition of translation of most cellular and viral mRNAs (reviewed in reference 32). Other members of the eIF2α kinase family are PKR-like endoplasmic reticulum kinase (PERK), general control nonrepressed 2 (GCN2), and heme-regulated inhibitor (HRI), which mediate responses to endoplasmic reticulum (ER) stress, amino acid deprivation, and oxidative stress, respectively (reviewed in references 33 and 34). Like many viruses, HSV deploys multiple mechanisms to reduce eIF2α phosphorylation: the viral ICP34.5 protein binds protein phosphatase 1α, redirecting it to dephosphorylate eIF2α (35), the US11 protein binds dsRNA and PKR and blocks PKR activation (36, 37), and glycoprotein B prevents activation of PERK (38). Pasieka et al. (24) proposed that vhs suppresses PKR activation by limiting the production of complementary viral RNA species capable of annealing to form dsRNA, potentially providing an additional mechanism for viral inhibition of PKR activation and eIF2α phosphorylation.
We recently showed that vhs also activates translation of viral mRNAs in an apparently eIF2α phosphorylation-independent fashion in certain cell types (39, 40). In HeLa cells, translation of viral true late mRNAs such as gC, US11, and UL47 is severely impaired independent of eIF2α phosphorylation, while translation of earlier viral mRNAs and cellular mRNAs proceeds normally; in contrast, no translational blockade occurs in Vero cells (39). The translation blockade in HeLa cells is accompanied by the accumulation of stress granules (SGs), aggregates of stalled translation initiation complexes that accumulate when translation initiation is impaired (reviewed in reference 34). The impaired translation of true late mRNAs appears to stem from the timing of mRNA accumulation rather than any feature of late mRNA structure, as US11 mRNA is efficiently translated at late times when it is engineered to accumulate early during infection (40). On the basis of these results, we proposed that the translational block arises as a consequence of “mRNA overload,” whereby excessive amounts of viral late mRNAs titrate one or more limiting translation initiation factors that are stably sequestered by the actively translated mRNAs produced earlier (40). According to this hypothesis, vhs prevents mRNA overload by reducing the levels of cellular and early viral mRNAs, allowing efficient translation of mRNAs produced after the onset of viral DNA replication. This hypothesis implies that one or more translation initiation factors are limiting in HeLa cells compared to Vero cells. Consistent with this suggestion, viral protein synthesis is much more sensitive to eIF4A inhibition by hippuristanol in HeLa cells than in Vero cells (39).
SGs form when translation initiation is stalled through any of several distinct mechanisms, including eIF2α phosphorylation (41), inhibiting eIF4A or eI4FG (42), or depleting eIF4B, eIF4H, or poly(A) binding protein (43). SGs consist of aggregates of the translationally stalled mRNAs and associated 40S ribosomal subunits, translation initiation factors, and a variety of RNA binding proteins including TIA-1 and G3BP1, which play key roles in SG assembly (reviewed in reference 34). They are considered to be storage depots where translationally arrested mRNAs are sorted for reentry into polysomes if stress conditions are alleviated or directed to processing (P) bodies for degradation.
Many viruses activate stress kinases including PKR, potentially triggering SG formation. Some viruses hijack SG components for their own benefit, while others encode proteins that block SG assembly (reviewed in references 44, 45, 46, and 47). For example, West Nile virus minus-strand RNA binds TIA-1 and TIAR to promote virus replication (48). In contrast, the poliovirus 3C protease cleaves G3BP1, causing the SGs that form early during infection to disperse (49). Similarly, influenza A virus genes encode multiple proteins that inhibit SG formation (50, 51). These and other observations have led to the suggestion that SGs have antiviral activity (49). For example, they might reinforce translational arrest by sequestering viral mRNAs (50). In addition, emerging evidence suggests that SGs serve as signaling platforms for RIG-I-dependent signaling and other aspects of innate immunity (52–55; reviewed in reference 44).
HSV vhs mutants induce robust SG accumulation during infection of many cell lines (39, 56, 57), and herpes simplex virus 2 (HSV-2) vhs is able to block SG formation in response to sodium arsenite (which activates HRI) without reducing eIF2α phosphorylation in transient-transfection experiments (57). These data indicate that vhs is able to suppress SG formation downstream of stress kinase activation. The ability of vhs to suppress PKR activation and inhibit SG formation raises interesting questions about the roles of PKR and SGs in mediating the translational defect exhibited by vhs mutants during infection of restrictive HeLa cells. We examined these questions by studying the effect of knocking out PKR on the vhs-null phenotype. We report here that PKR is essential for SG induction during vhs-null virus infection but plays only a secondary role in attenuating viral true late gene expression.
MATERIALS AND METHODS
Cells and viruses.
293T cells, HeLa cells, genetically modified HeLa cells, and BSC40 cells were maintained in Dulbecco's modified Eagle medium (DMEM) supplemented with 10% heat-inactivated fetal bovine serum (FBS), 100 U/ml penicillin, and 100 μg/ml streptomycin. Vero cells were grown in the same medium containing 5% FBS. The wild-type HSV-1 strain used in this study was KOS. The KOS-derived vhs mutant ΔSma (58) was obtained from G. S. Read (University of Missouri—Kansas City). HSV-1 was propagated, and the titers of the virus were determined on Vero cells. Wild-type vaccinia virus strain Copenhagen was provided by G. McFadden (University of Florida, Gainesville), and the E3L-null derivative ΔE3L (59) was obtained from B. Jacobs (Arizona State University). Vaccinia virus was propagated, and the titers of the virus were determined on BSC40 cells.
Plasmids.
pLKO.1-shPKR1379, a lentivirus short hairpin RNA (shRNA) vector from the Sigma-Aldrich human shRNA library that targets human PKR mRNA (sh insert, CCGGTCCTGGCTCATCTCTTTATTCCTCGAGGAATAAAGAGATGAGCCAGGATTTTT) was obtained from the RNA interference (RNAi) core facility of the Li Ka Shing Institute of Virology, University of Alberta. pMD2.G, which expresses vesicular stomatitis virus (VSV) G protein, and psPAX2, a lentivirus packaging plasmid, were obtained from Addgene (plasmids 12259 and 12260, respectively). pEQ1509 and pEQ1510 (60), doxycycline-inducible clustered regularly interspaced short palindromic repeat (CRISPR)-Cas9 gene-editing plasmids that target the first coding exon of the human PKR gene, were generous gifts from Adam Geballe and Stephanie Child, Fred Hutchinson Cancer Research Center, Seattle, WA. Both plasmids were derived from pEQ1508 (60). The PKR-targeting sequences in the guide RNAs are GTCTCTTCCATTGTAGGATA (pEQ1509) and GTTCAGGACCTCCACATGAT (pEQ1510).
Construction of PKR knockdown and knockout HeLa cell derivatives.
A polyclonal PKR knockdown HeLa cell population was generated by transducing HeLa cells with the lentivirus derived from pLKO.1-shPKR1379. Briefly, 293T cells were cotransfected with psPAX2, pMD2.G, and pLKO.1-shPKR1379. The lentivirus-containing supernatant was then used to infect 60-mm dishes of HeLa cells. Transduced cells were initially selected with 2.5 μg/ml puromycin (Sigma) and maintained in 1.5 μg/ml puromycin until the population was established. PKR knockout clones were derived by CRISPR-Cas9-mediated gene editing (61) using pEQ1509 and pEQ1510. HeLa cells were transfected with the two targeting plasmids and pEGFP (included to monitor transfection efficiency and allow sorting of the transfected cells via flow cytometry). The day following transfection, the cells were treated with 1 μg/ml doxycycline (Fluka) for 24 h. The GFP-expressing cells were then isolated using a BD FACSAria III fluorescence activated cell sorter (BD Biosciences) and plated to allow recovery for approximately 1 week. The cells were then diluted and seeded into 96-well plates at 0.5 to 1 cell/well. Candidate PKR knockout clones were identified by screening for loss of PKR by Western blotting. The genotype of the PKR KO1 clone was assessed by PCR amplification of the region surrounding the targeted sites using Platinum Pfx DNA polymerase (Invitrogen) and primers 5′-CTGCATTTATGTGAGACTGA and 5′-TTCTTGCGATAGTTTGCTG.
Induction of stress granules.
Wild-type and modified HeLa cells were scored for their capacity to form stress granules following exposure to 0.5 mM sodium arsenite (Sigma) for 45 min or 6 h after transfection with poly(I·C) (4 μg/six-well plate) (Sigma catalog no. P1530, lot no. 114M4028V) using Lipofectamine 2000 (Thermo Fisher Scientific).
Single-cycle virus growth assay.
Wild-type and modified HeLa cells in 12-well plates were infected at a multiplicity of infection (MOI) of 10 PFU/cell with the indicated vaccinia virus or HSV-1 isolate. Virus absorption was carried out for 1 h in 0.25 ml of serum-free DMEM. For experiments with vaccinia virus, the inoculum was removed, the monolayers were rinsed twice with phosphate-buffered saline (PBS), and DMEM containing 10% FBS was added. Infected cultures were harvested at 24 h postinfection by scraping the infected cells into the medium. Virus yields were determined by plaque titration on BSC40 cells. For experiments with HSV-1, infections, washes, and sample preparation were performed by the methods of Duguay et al. (62) with harvesting occurring at the times indicated in Fig. 6, followed by titration on Vero cells.
FIG 6.

Inactivating PKR does not appreciably promote the growth of ΔSma in HeLa cells. (A to D) HeLa (A), PKR KD (B), PKR KO1 (C), and PKR KO2 (D) cells were infected with 10 PFU/cell of HSV-1 KOS or ΔSma, and viral yields were determined at the indicated times postinfection (hours postinfection [h.p.i]) by plaque assay on Vero cells. Comparable results were obtained in an independent replicate of this experiment.
Western blot analysis.
Cell extracts were prepared and analyzed as previously described (40) using primary antibodies specific for the HSV-1 proteins gC (mouse; catalog no. P1104; Virusys Corporation), gB (mouse; catalog no. P1123; Virusys Corporation), ICP4 (mouse; catalog no. P1101; Virusys Corporation), ICP27 (mouse; catalog no. P1113; Virusys Corporation), ICP34.5 (rabbit; gift from Ian Mohr), US11 (mouse; gift from Ian Mohr), UL47 (rabbit; gift from Gillian Elliott), thymidine kinase (TK) (rabbit; gift from William C. Summers), and VP16 (mouse; LP1; gift from Tony Minson) and the cellular proteins β-actin (mouse; catalog no. A5441; Sigma), phospho-PKR (threonine 446; rabbit; ab13447; Abcam), PKR (mouse; sc-6282; Santa Cruz), phospho-eIF2α (serine 51; rabbit; catalog no. 9721; Cell Signaling), eIF2α (rabbit; catalog no. 9722; Cell Signaling). Primary antibodies were detected using suitable secondary antibodies coupled to Alexa Fluor 680 (Invitrogen) or IRDye800 (Rockland) using the Odyssey infrared imaging system (LI-COR).
Immunofluorescence.
Cells growing on glass coverslips were fixed with 4% paraformaldehyde for 15 min at room temperature and then permeabilized with ice-cold methanol for 20 min. Coverslips were blocked in 5% bovine serum albumin (BSA) in PBS for 1 h before antibody staining. Goat polyclonal antiserum against human TIA-1 (Santa Cruz Biotechnology, Santa Cruz, CA) was used at a dilution of 1:100, and the Alexa Fluor 488-labeled chicken anti-goat secondary antiserum (Invitrogen) was used at a dilution of 1:400. All antibodies were diluted in blocking buffer. After washing, coverslips were mounted using Vectashield (Vector Laboratories Inc.) supplemented with 1 μg/ml 4′,6′-diamidino-2-phenylindole (DAPI). Digital images were acquired sequentially using a spinning disk confocal microscope (Wave FX; Quorum Technologies, Guelph, Ontario, Canada) integrated on an Olympus IX-81 inverted microscope base (Olympus, Richmond Hill, Ontario, Canada) using a 40×/1.3-numerical-aperture objective lens. Images were captured on an EM-CCD (ImageEM X2; Hamamatsu Photonics, Hamamatsu City, Japan) through standard fluorescence filter sets (Semrock, Inc., Rochester, NY). Composites of representative images were prepared using Adobe Photoshop software.
RESULTS
vhs-deficient HSV-1 activates PKR in HeLa cells without inducing enhanced levels of eIF2α phosphorylation.
We sought to evaluate the role of PKR in determining the phenotype of HSV-1 vhs mutants in primate cells. As a first step, we compared the ability of wild-type HSV-1 strain KOS and the vhs-deficient ΔSma derivative to activate PKR in HeLa cells, where translation of viral true late mRNAs is severely impaired and SGs accumulate in the absence of vhs function, and in Vero cells, where viral translation proceeds normally and SGs do not form (39). For controls, these cells were also infected with wild-type vaccinia virus (VV) strain Copenhagen and a VV derivative lacking the PKR antagonist E3L (63) (ΔE3L [59]). In HeLa cells, HSV-1 KOS and ΔSma both induced phosphorylation of PKR on residue T446, a hallmark of PKR activation (Fig. 1A). Consistent with previous work (31), ΔSma induced substantially higher levels of PKR phosphorylation than KOS did. In contrast, neither virus detectably activated PKR in Vero cells. As expected, VV ΔE3L provoked more robust PKR activation than wild-type VV in HeLa cells. Surprisingly, both VV isolates displayed similar activity in Vero cells. While it remains unclear why ΔE3L strain does not exhibit enhanced activity relative to wild-type VV in Vero cells, these results demonstrate that PKR is susceptible to activation by virus infection in this cell line. We therefore suspect that the failure of HSV-1 to activate PKR in Vero cells reflects the activity of one or more HSV-1-encoded PKR inhibitors such as US11 (36, 37).
FIG 1.

HSV-1 activates PKR in HeLa cells but not in Vero cells. (A) HeLa and Vero cells were infected with 10 PFU/cell of wild-type HSV-1 KOS (KOS), the vhs mutant HSV-1 KOS ΔSma (ΔSma), wild-type vaccinia virus (VV), or E3L mutant vaccinia virus (ΔE3L). Cell extracts prepared 12 h postinfection were then analyzed by Western blotting using antibodies directed against phosphorylated PKR (P-PKR), total PKR, phosphorylated eIF2α (P-eIF2α), total eIF2α, β-actin, and HSV-1 ICP27. (B) HeLa cells were infected with KOS or ΔSma in the presence (+) or absence (−) of 300 μg/ml phosphonoacetic acid (PAA), and cell extracts prepared 12 h postinfection were analyzed for phosphorylated PKR by Western blotting. Glyceraldehyde-3-phosphate dehydrogenase (GAPDH) served as the loading control.
Although ΔSma strongly activated PKR in HeLa cells, the levels of phosphorylated eIF2α were not obviously increased compared to cells infected with wild-type KOS (Fig. 1A) (39, 40). In contrast, E3L-null VV provoked a large increase. Perhaps the failure of ΔSma to induce increased levels of eIF2α phosphorylation despite more robust PKR activation stems from the ability of the viral ICP34.5 protein to reverse eIF2α phosphorylation (35) (see Discussion).
SGs fail to form during vhs-null virus infection of HeLa cells when viral DNA replication is blocked by phosphonoacetic acid (PAA) (39). PAA similarly prevented PKR activation by KOS and ΔSma (Fig. 1B). One interpretation of this result is that complementary viral transcripts capable of forming double-stranded RNA accumulate to levels sufficient to activate PKR after the onset of viral DNA replication, as suggested by Pasieka et al. (24).
Overall, the results outlined in this section indicate that HSV-1 infection activates PKR in a viral DNA replication-dependent fashion in restrictive HeLa cells, but not in permissive Vero cells, and that vhs-deficient HSV-1 induces higher levels of PKR activation than wild-type virus does. These findings raised the possibility that PKR plays important roles in SG formation and/or inhibiting translation of viral true late mRNAs during infection of HeLa cells with vhs mutant HSV-1. We tested this possibility by determining the effects of knocking down and knocking out PKR on SG formation and the expression of selected viral true late proteins.
Construction and characterization of PKR knockdown and knockout HeLa cell derivatives.
We transduced HeLa cells with a lentivirus vector expressing an shRNA targeting PKR mRNA to generate a polyclonal knockdown cell population (PKR KD) in which PKR levels were reduced by ca. 80% relative to control HeLa cells (Fig. 2A and additional data not shown). In addition, we used CRISPR-Cas9-mediated gene editing to generate PKR mutant HeLa cell clones. Here, we cotransfected cells with two Cas9 plasmids expressing guide RNAs that target two sites in the first coding exon of the PKR gene. One of these sites spans the 5′ splice acceptor sequence, and the other site is located in the 3′ portion of the exon (Fig. 2B; Materials and Methods). Candidate clones were screened for PKR expression by Western blotting, and two independent PKR-deficient clones (PKR KO1 and PKR KO2) were identified (Fig. 2A and additional data not shown). We confirmed by PCR that the PKR KO1 clone lacks an intact PKR gene; instead, two mutant alleles were detected. One of these alleles lacks all of the sequences between the two targeted sites, while the other allele bears two separate deletions, one at each of the two targeted sites. Both alleles lack the 5′ splice acceptor site (summarized in Fig. 2B). We have not yet analyzed the genotype of the KO2 cell line.
FIG 2.
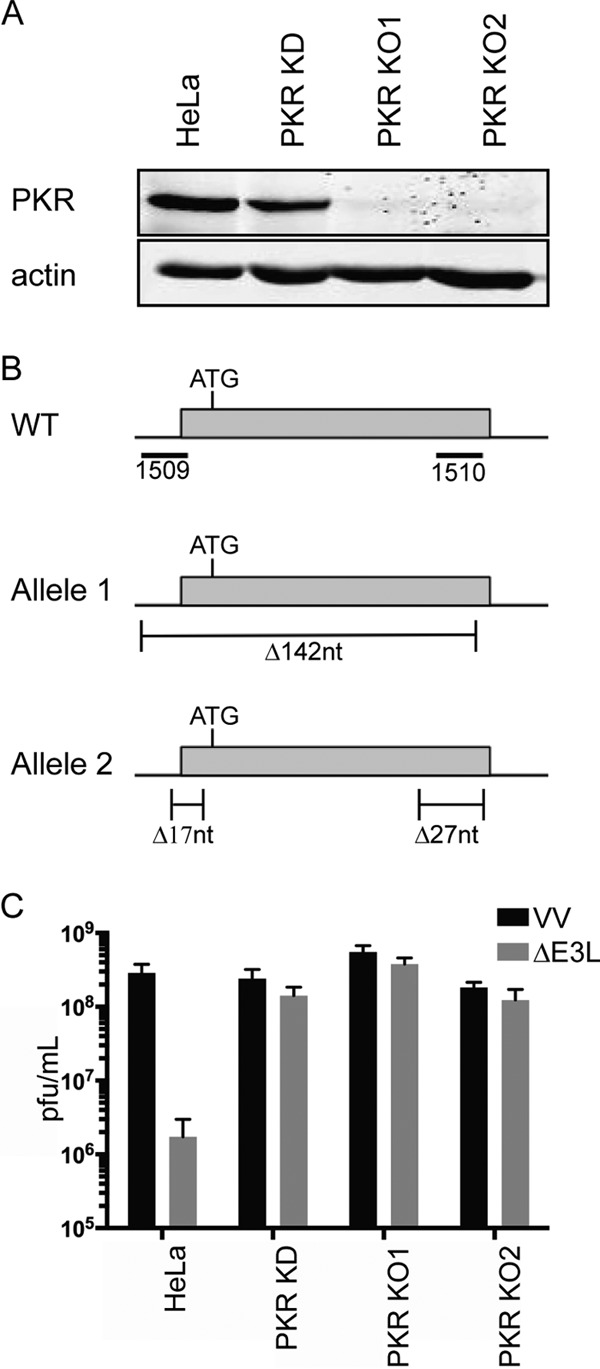
Characterization of PKR knockdown and knockout HeLa cells. (A) Extracts of wild-type HeLa cells and HeLa PKR knockdown (PKR KD), knockout 1 (KO1) and knockout 2 (KO2) cells were analyzed by Western blotting for PKR. β-Actin served as the loading control. (B) Diagram of the PKR mutations present in the PKR KO1 cell line. For the wild type (WT), the locations of the sequences targeted by the pEQ1509 and pEQ1510 Cas9 plasmids are indicated relative to the first coding exon of the human PKR gene. The position of the translational initiation codon is shown. For allele 1 and allele 2, the locations and sizes (in nucleotides [nt]) of the deletions present in the two mutant PKR alleles detected in the PKR KO1 cell line are shown. (C) The PKR KD and KO cells complement E3L mutant vaccinia virus. The indicated cell lines were infected with 10 PFU/cell of wild-type or E3L-null vaccinia virus (VV and ΔE3L), and viral yields were determined 24 h postinfection by plaque assay. The means plus standard deviations (error bars) for data obtained in three independent experiments are displayed.
We tested for functional inactivation of PKR by scoring the KD and KO cells for their ability to support replication of E3L-null vaccinia virus, which is severely restricted by PKR in human cells. As reported previously (64), replication of ΔE3L VV was reduced ca. 100-fold relative to wild-type VV in unmodified HeLa cells (Fig. 2C); in contrast, it replicated to essentially wild-type levels in the KD and KO derivatives.
PKR mediates eIF2α phosphorylation and SG formation in response to dsRNA, while other eIF2α kinases mediate the response to other stresses (reviewed in reference 34). We therefore asked whether the HeLa PKR KD and KO derivatives are competent to form SGs in response to transfected poly(I·C), a dsRNA mimic, and sodium arsenite, which activates the HRI stress kinase (Fig. 3). Untreated cells showed diffuse nuclear and cytoplasmic staining of TIA-1, a SG marker. In wild-type HeLa cells, poly(I·C) triggered the formation of prominent cytoplasmic foci of TIA-1 characteristic of SGs in ca. 70% of the cells. In contrast, no SGs were observed in poly(I·C)-transfected PKR KO1 and KO2 cells. The KD cell population displayed an intermediate phenotype, with ca. 9% of the cells showing large SG-like aggregates. Notably, the cells displaying these aggregates also displayed diffuse TIA-1 staining. In contrast, all of the cell lines showed abundant SGs in response to sodium arsenite. These results provide additional evidence that PKR function is severely impaired in the KD cell population and eliminated in the KO clones.
FIG 3.
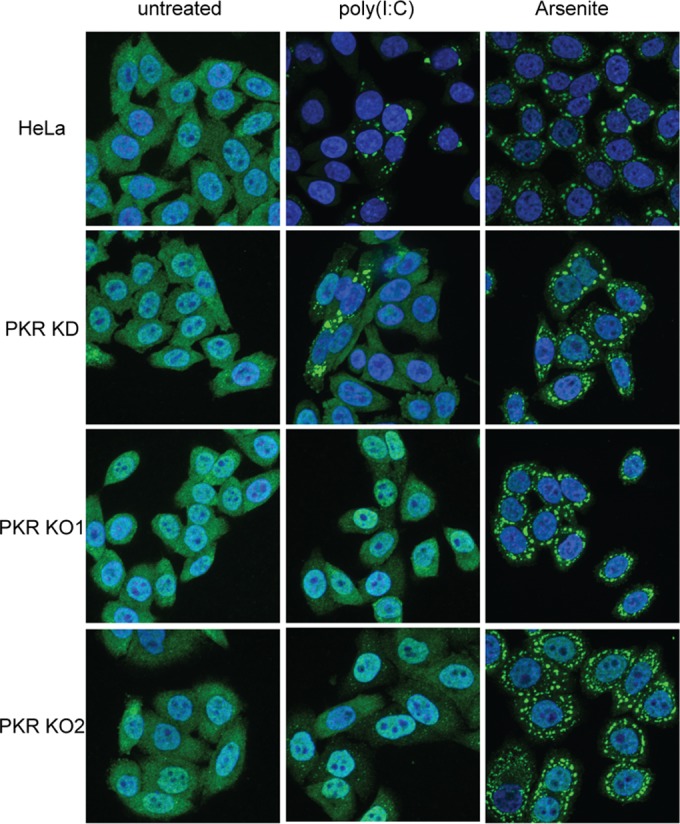
The PKR KD and KO cell lines fail to form SGs in response to poly(I·C). The indicated cells were left untreated, transfected with poly(I·C) for 6 h, or exposed to 0.5 mM sodium arsenite for 45 min. The cells were then scored for stress granules by immunofluorescence using an antibody directed to TIA-1. The results shown are representative of those obtained in four independent experiments.
PKR is required for stress granule formation during infection in the absence of functional vhs.
We evaluated the role of PKR in SG formation following infection with ΔSma virus. Wild-type, PKR KD, PKR KO1 and PKR KO2 HeLa cells were infected with wild-type KOS and ΔSma and examined 12 h postinfection for the presence of SGs by staining for TIA-1 (Fig. 4). We consistently observed a reduction in the intensity of the TIA-1 signal after infection. As reported previously (39), ΔSma induced formation of prominent cytoplasmic SGs in a subset of infected HeLa cells (76% in this experiment). In contrast, KOS failed to trigger SG formation, but instead it induced nuclear TIA-1 foci, as previously reported for wild-type HSV-2 (65). These nuclear foci do not contain the SG markers G3BP1 or G3BP2 (data not shown), and hence do not represent SGs. SGs were markedly reduced in the KD cell population (12% of the cells scored positive) and absent in the KO1 and KO2 cell lines. Similar results were obtained using eIF4A as a SG marker (data not shown). These data establish that PKR is essential for SG formation during vhs-null virus infection.
FIG 4.
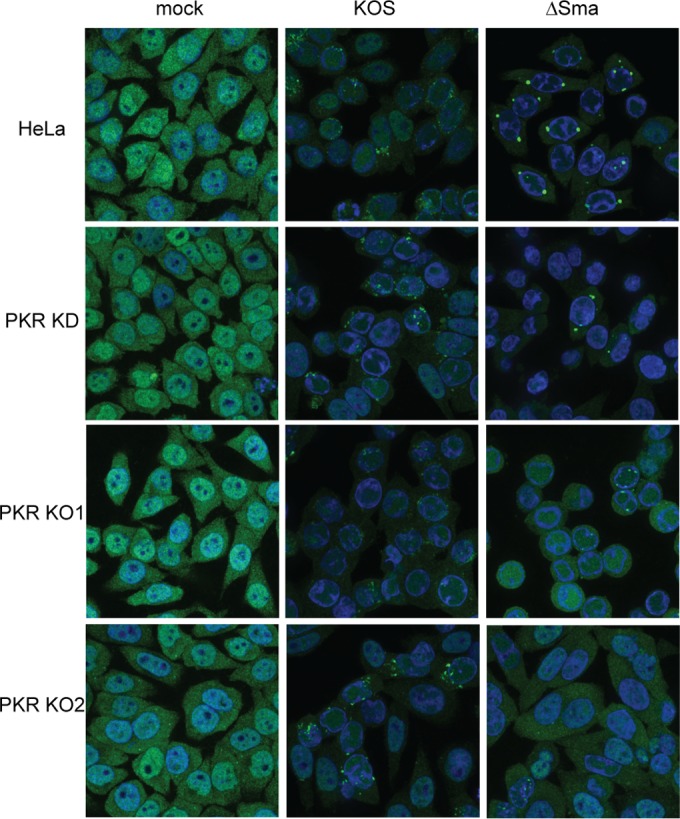
The PKR KO cell lines fail to form SGs following infection with vhs mutant HSV-1. The indicated cell lines were infected with 10 PFU/cell of HSV-1 KOS or ΔSma and then scored at 12 h postinfection for stress granules by immunofluorescence using an antibody directed to TIA-1. The results shown are representative of two independent experiments.
Inactivating PKR does not fully rescue viral true late protein expression or viral replication in the absence of vhs.
We next asked whether inactivating PKR alleviates the defect in the accumulation of viral true late proteins in the absence of vhs. Cells were infected with 10 PFU/cell of KOS or ΔSma and harvested 12 h postinfection, and extracts were analyzed by Western blotting using antibodies directed against selected viral proteins (Fig. 5). As previously reported (39, 40), ΔSma displayed severely reduced levels of the true late proteins gC, US11, and UL47 compared to KOS in HeLa cells: the results obtained from three independent experiments were quantified and are presented in Table 1. Leaky late proteins such as VP16 and gB exhibited a less pronounced reduction, and no effect was observed for TK (early) or ICP27 (immediate early). Knocking down or knocking out PKR detectably enhanced the expression of gC, US11, and UL47, but the levels remained well below those found in KOS-infected cells (Table 1). Thus, inactivating PKR does not fully restore translation of true late mRNAs during vhs-null virus infection of HeLa cells; the levels of gC and UL47 in particular remained severely compromised. These results suggest that PKR (and likely eIF2α phosphorylation) are not the primary drivers of the block to translation of viral true late mRNAs in the absence of vhs. This conclusion is in accord with the finding that eIF2α is phosphorylated to similar extents in the presence and absence of vhs in HeLa cells and the PKR KD and KO derivatives (Fig. 1A and 5) (39, 40).
FIG 5.

Effects of knocking down and knocking out PKR on viral protein accumulation. The indicated cell lines were infected with 10 PFU/cell of HSV-1 KOS or ΔSma, and cell extracts prepared 12 h postinfection were analyzed by Western blotting for the levels of HSV-1 gC, US11, UL47, gB, VP16, TK, ICP4, ICP27, ICP34.5, cellular β-actin, and phosphorylated PKR.
TABLE 1.
Expression of the true late proteins during infection with HSV-1a
| Protein | Protein expression in the following cells: |
|||
|---|---|---|---|---|
| HeLa | KD | KO1 | KO2 | |
| gC | 0.02, 0.03, 0.01 | 0.12, 0.12, 0.05 | 0.18, 0.13, 0.09 | 0.13, 0.10, 0.04 |
| US11 | 0.13, 0.17, 0.05 | 0.58, 0.43, 0.14 | 0.59, 0.56, 0.26 | 0.42, 0.41, 0.15 |
| UL47 | 0.03, 0.10, 0.07 | 0.15, 0.12, 0.13 | 0.11, 0.11, 0.16 | 0.18, 0.12, 0.09 |
The levels of gC, US11, and UL47 protein observed during infection with HSV-1 ΔSma are expressed relative to those in cells infected with wild-type HSV-1 KOS. The results of three independent experiments are presented. The Western blot data obtained in experiment 3 are displayed in Fig. 5.
We also evaluated the effect of knocking out PKR on the replication of ΔSma in a one-step growth experiment (Fig. 6). Cells were infected with 10 PFU/cell of KOS and ΔSma, and viral yields were determined by plaque assay on Vero cells. As previously reported (39), yields of ΔSma were 50- to 100-fold lower than those of KOS in HeLa cells. A similar reduction was observed in the PKR KD and KO cells (Fig. 6). Thus, inactivating PKR had little or no effect on the growth defect of ΔSma in HeLa cells.
DISCUSSION
We have drawn three major conclusions based on the results reported in this study. First, we confirm a previous report that HSV-1 vhs mutants induce higher levels of PKR activation than wild-type virus does (31). Second, we show that PKR is essential for SG formation during HSV-1 infection in the absence of vhs. Third, notwithstanding these two findings, PKR activation and SGs are not primary drivers of the translational defect exhibited by viral true late mRNAs in HeLa cells in the absence of vhs function.
Our finding that ΔSma induces substantially higher levels of PKR activation than wild-type KOS agrees with the results of Sciortino et al. (31). This finding suggests that vhs directly or indirectly inhibits PKR activation or limits the production of dsRNA or another activator of PKR during infection. Pasieka et al. (24) suggested that vhs inhibits PKR activation by reducing the levels of complementary viral RNAs capable of annealing to form dsRNA at late times postinfection. This hypothesis is consistent with the ability of vhs to block PKR activation in response to certain mammalian cDNA expression plasmids in transient-transfection experiments (31; our unpublished data) and our finding that PKR activation requires viral DNA replication (and hence presumably viral late RNA accumulation; Fig. 1B). It will be important to determine the origin and identity of the PKR inducer that accumulates after the onset of viral DNA replication. Alternatively, it is possible that vhs acts indirectly, for example by stimulating the synthesis of a viral PKR inhibitor such as US11. Consistent with this hypothesis, US11 protein levels are greatly reduced in HeLa cells in the absence of vhs (39, 40). Further studies are required to test these and other possibilities, which are not mutually exclusive.
We found that the strong PKR activation that occurs in the absence of vhs does not lead to increased eIF2α phosphorylation relative to wild-type virus in HeLa cells (Fig. 1 and 5) (39, 40). This finding contrasts with previous results from the Leib and Morrison groups, who demonstrated enhanced levels of phosphorylated eIF2α in mouse embryo fibroblasts infected with HSV-1 and HSV-2 vhs mutants (24, 30). Similarly, we have previously observed enhanced eIF2α phosphorylation in human fibroblasts and U2OS cells infected with ΔSma virus (39). These different outcomes may stem at least in part from cell type-specific variation in the effects of vhs mutations on the expression of ICP34.5, which reverses eIF2α phosphorylation (35): Wylie et al. (30) showed that HSV-2 vhs mutants display reduced levels of ICP34.5 in mouse fibroblasts, whereas we observed no such reduction in HeLa cells (Fig. 5).
PKR is essential for SG formation during infection with certain RNA viruses (51, 66–68) and for accumulation of SG-like structures in vaccinia virus-infected cells (69). Our data establish that PKR is also required for SG formation during infection with vhs-deficient HSV-1, a nuclear DNA virus. HSV-1 fails to detectably activate PKR in Vero cells (Fig. 1), likely explaining why HSV-1 fails to induce SGs in this cell line (39). SG formation in response to stress kinase activation is thought to stem solely from eIF2α phosphorylation and the consequent block to translation initiation (70). However, this view is difficult to reconcile with our findings, suggesting that the situation is more complex, at least in cells infected with an HSV-1 vhs mutant. SGs can also arise in an eIF2α-independent fashion when translation initiation is blocked by inhibiting eIF4A or eIF4G (42) or reducing the levels of eIF4B, eIF4H, or poly(A) binding protein (43). We have previously suggested that excessive amounts of viral and cellular mRNA accumulate during vhs mutant infection, titrating one or more limiting translation initiation factors and leading to impaired translational initiation on viral true late mRNAs through mRNA overload (40). It is possible that mRNA overload renders SG formation exquisitely sensitive to small or highly localized changes in the levels of phosphorylated eIF2α. Alternatively, one or more PKR substrates other than eIF2α may drive SG formation in response to mRNA overload. Further studies are required to distinguish between these possibilities.
The ability of vhs to limit PKR activation likely contributes to suppression of SG formation during infection with wild-type HSV-1. However, vhs is also able to block SG assembly in response to sodium arsenite, which activates the HRI stress kinase (57, 65). In this case, vhs acts downstream of eIF2α phosphorylation (65). Perhaps vhs can also attenuate SG formation by globally lowering mRNA levels; consistent with this possibility, the influenza virus A host shutoff RNase PA-X also strongly inhibits SG formation (50). Alternatively, vhs may perturb SG formation or stability through its interactions with components of the eIF4F complex (13-15, 17) and/or other SG components.
Our data establish that PKR is not the primary driver of the barrier to translation of viral true late mRNAs during infection in the absence of vhs. This finding is consistent with our previous proposal that the translational blockade stems from mRNA overload (39, 40). We currently seek to further test the mRNA overload hypothesis and identify the limiting translation factor(s). Inactivating PKR did however have a modest stimulatory effect on viral true late protein levels. Perhaps PKR-dependent sequestration of viral true late mRNAs into SGs in a subset of infected cells plays a secondary role in reinforcing the barrier to viral protein synthesis triggered by mRNA overload. Consistent with this view, our previous data demonstrate that the translational blockade arises before SGs are evident in the majority of infected cells (39).
As reviewed in the introduction, SGs have been proposed to serve one or more antiviral functions. Our data demonstrate that they play at best a secondary role in attenuating HSV-1 gene expression or virus replication in the absence of vhs; consistent with this observation, Wylie et al. (30) found that eliminating SG formation did not restore wild-type levels of virus replication to vhs mutants. These observations raise the question of what, if any, antiviral effects SGs have against HSV. An intriguing emerging hypothesis is that SGs serve as platforms for RIG-1-dependent signaling (44, 53), PKR activation (54, 71, 72), and other aspects of innate immunity (55), in addition to their roles as sorting depots for translationally stalled mRNAs. It will therefore be of great interest to determine whether SG suppression contributes to the ability of vhs to dampen the type IFN system (22–24), reduce proinflammatory cytokine production (25), and suppress NF-κB activation (73).
ACKNOWLEDGMENTS
We thank Adam Geballe and Stephanie Child for pEQ1509 and pEQ1510, Dorothy Kratochwil-Otto for help with cell sorting, and Stephen Ogg for advice on confocal microscopy.
This research was supported by operating grants from the Canadian Institutes for Health Research (MOP 37995 and MOP 37990) and Alberta Innovates - Health Solutions. J.R.S. is a Canada Research Chair in Molecular Virology (Tier I).
Funding Statement
The funders had no role in study design, data collection and interpretation, or the decision to submit the work for publication.
REFERENCES
- 1.Abernathy E, Glaunsinger B. 2015. Emerging roles for RNA degradation in viral replication and antiviral defense. Virology 479–480:600–608. doi: 10.1016/j.virol.2015.02.007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Read GS. 2013. Virus-encoded endonucleases: expected and novel functions. Wiley Interdiscip Rev RNA 4:693–708. doi: 10.1002/wrna.1188. [DOI] [PubMed] [Google Scholar]
- 3.Read GS, Frenkel N. 1983. Herpes simplex virus mutants defective in the virion-associated shutoff of host polypeptide synthesis and exhibiting abnormal synthesis of alpha (immediate early) viral polypeptides. J Virol 46:498–512. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Kwong AD, Kruper JA, Frenkel N. 1988. Herpes simplex virus virion host shutoff function. J Virol 62:912–921. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Smiley JR. 2004. Herpes simplex virus virion host shutoff protein: immune evasion mediated by a viral RNase? J Virol 78:1063–1068. doi: 10.1128/JVI.78.3.1063-1068.2004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Everly J, David N, Feng P, Mian IS, Read GS. 2002. mRNA degradation by the virion host shutoff (Vhs) protein of herpes simplex virus: genetic and biochemical evidence that Vhs is a nuclease. J Virol 76:8560–8571. doi: 10.1128/JVI.76.17.8560-8571.2002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Taddeo B, Roizman B. 2006. The virion host shutoff protein (UL41) of herpes simplex virus 1 is an endoribonuclease with a substrate specificity similar to that of RNase A. J Virol 80:9341–9345. doi: 10.1128/JVI.01008-06. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Taddeo B, Zhang W, Roizman B. 2006. The UL41 protein of herpes simplex virus 1 degrades RNA by endonucleolytic cleavage in absence of other cellular or viral proteins. Proc Natl Acad Sci U S A 103:2827–2832. doi: 10.1073/pnas.0510712103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Zelus BD, Stewart RS, Ross J. 1996. The virion host shutoff protein of herpes simplex virus type 1: messenger ribonucleolytic activity in vitro. J Virol 70:2411–2419. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Kwong AD, Frenkel N. 1987. Herpes simplex virus-infected cells contain a function(s) that destabilizes both host and viral mRNAs. Proc Natl Acad Sci U S A 84:1926–1930. doi: 10.1073/pnas.84.7.1926. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Oroskar AA, Read GS. 1989. Control of mRNA stability by the virion host shutoff function of herpes simplex virus. J Virol 63:1897–1906. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Gaglia MM, Covarrubias S, Wong W, Glaunsinger BA. 2012. A common strategy for host RNA degradation by divergent viruses. J Virol 86:9527–9530. doi: 10.1128/JVI.01230-12. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Page HG, Read GS. 2010. The virion host shutoff endonuclease (UL41) of herpes simplex virus interacts with the cellular cap-binding complex eIF4F. J Virol 84:6886–6890. doi: 10.1128/JVI.00166-10. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Doepker RC, Hsu W-L, Saffran HA, Smiley JR. 2004. Herpes simplex virus virion host shutoff protein is stimulated by translation initiation factors eIF4B and eIF4H. J Virol 78:4684–4699. doi: 10.1128/JVI.78.9.4684-4699.2004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Feng P, Everly DN Jr, Read GS. 2001. mRNA decay during herpesvirus infections: interaction between a putative viral nuclease and a cellular translation factor. J Virol 75:10272–10280. doi: 10.1128/JVI.75.21.10272-10280.2001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Sarma N, Agarwal D, Shiflett LA, Read GS. 2008. Small interfering RNAs that deplete the cellular translation factor eIF4H impede mRNA degradation by the virion host shutoff protein of herpes simplex virus. J Virol 82:6600–6609. doi: 10.1128/JVI.00137-08. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Feng P, Everly DN, Read GS. 2005. mRNA decay during herpes simplex virus (HSV) infections: protein-protein interactions involving the HSV virion host shutoff protein and translation factors eIF4H and eIF4A. J Virol 79:9651–9664. doi: 10.1128/JVI.79.15.9651-9664.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Elgadi MM, Hayes CE, Smiley JR. 1999. The herpes simplex virus vhs protein induces endoribonucleolytic cleavage of target RNAs in cell extracts. J Virol 73:7153–7164. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Elgadi MM, Smiley JR. 1999. Picornavirus internal ribosome entry site elements target RNA cleavage events induced by the herpes simplex virus virion host shutoff protein. J Virol 73:9222–9231. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Karr BM, Read GS. 1999. The virion host shutoff function of herpes simplex virus degrades the 5′ end of a target mRNA before the 3′ end. Virology 264:195–204. doi: 10.1006/viro.1999.9986. [DOI] [PubMed] [Google Scholar]
- 21.Shiflett LA, Read GS. 2013. mRNA decay during herpes simplex virus (HSV) infections: mutations that affect translation of an mRNA influence the sites at which it is cleaved by the HSV virion host shutoff (Vhs) protein. J Virol 87:94–109. doi: 10.1128/JVI.01557-12. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Duerst RJ, Morrison LA. 2004. Herpes simplex virus 2 virion host shutoff protein interferes with type I interferon production and responsiveness. Virology 322:158–167. doi: 10.1016/j.virol.2004.01.019. [DOI] [PubMed] [Google Scholar]
- 23.Murphy JA, Duerst RJ, Smith TJ, Morrison LA. 2003. Herpes simplex virus type 2 virion host shutoff protein regulates alpha/beta interferon but not adaptive immune responses during primary infection in vivo. J Virol 77:9337–9345. doi: 10.1128/JVI.77.17.9337-9345.2003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Pasieka TJ, Lu B, Crosby SD, Wylie KM, Morrison LA, Alexander DE, Menachery VD, Leib DA. 2008. Herpes simplex virus virion host shutoff attenuates establishment of the antiviral state. J Virol 82:5527–5535. doi: 10.1128/JVI.02047-07. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Suzutani T, Nagamine M, Shibaki T, Ogasawara M, Yoshida I, Daikoku T, Nishiyama Y, Azuma M. 2000. The role of the UL41 gene of herpes simplex virus type 1 in evasion of non-specific host defence mechanisms during primary infection. J Gen Virol 81:1763–1771. doi: 10.1099/0022-1317-81-7-1763. [DOI] [PubMed] [Google Scholar]
- 26.Samady L, Costigliola E, MacCormac L, McGrath Y, Cleverley S, Lilley CE, Smith J, Latchman DS, Chain B, Coffin RS. 2003. Deletion of the virion host shutoff protein (vhs) from herpes simplex virus (HSV) relieves the viral block to dendritic cell activation: potential of vhs− HSV vectors for dendritic cell-mediated immunotherapy. J Virol 77:3768–3776. doi: 10.1128/JVI.77.6.3768-3776.2003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Leib DA, Harrison TE, Laslo KM, Machalek MA, Moorman NJ, Virgin HW. 1999. Interferons regulate the phenotype of wild-type and mutant herpes simplex viruses in vivo. J Exp Med 189:663–672. doi: 10.1084/jem.189.4.663. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Pasieka TJ, Cilloniz C, Lu B, Teal TH, Proll SC, Katze MG, Leib DA. 2009. Host responses to wild-type and attenuated herpes simplex virus infection in the absence of Stat1. J Virol 83:2075–2087. doi: 10.1128/JVI.02007-08. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Pasieka TJ, Lu B, Leib DA. 2008. Enhanced pathogenesis of an attenuated herpes simplex virus for mice lacking Stat1. J Virol 82:6052–6055. doi: 10.1128/JVI.00297-08. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Wylie KM, Schrimpf JE, Morrison LA. 2009. Increased eIF2α phosphorylation attenuates replication of herpes simplex virus 2 vhs mutants in mouse embryonic fibroblasts and correlates with reduced accumulation of the PKR antagonist ICP34.5. J Virol 83:9151–9162. doi: 10.1128/JVI.00886-09. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Sciortino MT, Parisi T, Siracusano G, Mastino A, Taddeo B, Roizman B. 2013. The virion host shutoff RNase plays a key role in blocking the activation of protein kinase R in cells infected with herpes simplex virus 1. J Virol 87:3271–3276. doi: 10.1128/JVI.03049-12. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Garcia MA, Gil J, Ventoso I, Guerra S, Domingo E, Rivas C, Esteban M. 2006. Impact of protein kinase PKR in cell biology: from antiviral to antiproliferative action. Microbiol Mol Biol Rev 70:1032–1060. doi: 10.1128/MMBR.00027-06. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Samuel CE. 1993. The eIF-2 alpha protein kinases, regulators of translation in eukaryotes from yeasts to humans. J Biol Chem 268:7603–7606. [PubMed] [Google Scholar]
- 34.Anderson P, Kedersha N. 2008. Stress granules: the Tao of RNA triage. Trends Biochem Sci 33:141–150. doi: 10.1016/j.tibs.2007.12.003. [DOI] [PubMed] [Google Scholar]
- 35.He B, Gross M, Roizman B. 1997. The γ134.5 protein of herpes simplex virus 1 complexes with protein phosphatase 1α to dephosphorylate the α subunit of the eukaryotic translation initiation factor 2 and preclude the shutoff of protein synthesis by double-stranded RNA-activated protein kinase. Proc Natl Acad Sci U S A 94:843–848. doi: 10.1073/pnas.94.3.843. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Mulvey M, Poppers J, Ladd A, Mohr I. 1999. A herpesvirus ribosome-associated, RNA-binding protein confers a growth advantage upon mutants deficient in a GADD34-related function. J Virol 73:3375–3385. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Cassady KA, Gross M, Roizman B. 1998. The herpes simplex virus US11 protein effectively compensates for the gamma1(34.5) gene if present before activation of protein kinase R by precluding its phosphorylation and that of the alpha subunit of eukaryotic translation initiation factor 2. J Virol 72:8620–8626. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Mulvey M, Arias C, Mohr I. 2007. Maintenance of endoplasmic reticulum (ER) homeostasis in herpes simplex virus type 1-infected cells through the association of a viral glycoprotein with PERK, a cellular ER stress sensor. J Virol 81:3377–3390. doi: 10.1128/JVI.02191-06. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Dauber B, Pelletier J, Smiley JR. 2011. The herpes simplex virus 1 vhs protein enhances translation of viral true late mRNAs and virus production in a cell type-dependent manner. J Virol 85:5363–5373. doi: 10.1128/JVI.00115-11. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Dauber B, Saffran HA, Smiley JR. 2014. The herpes simplex virus 1 virion host shutoff protein enhances translation of viral late mRNAs by preventing mRNA overload. J Virol 88:9624–9632. doi: 10.1128/JVI.01350-14. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Kedersha NL, Gupta M, Li W, Miller I, Anderson P. 1999. RNA-binding proteins Tia-1 and Tiar link the phosphorylation of eIF-2α to the assembly of mammalian stress granules. J Cell Biol 147:1431–1442. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Mazroui R, Sukarieh R, Bordeleau M-E, Kaufman RJ, Northcote P, Tanaka J, Gallouzi I, Pelletier J. 2006. Inhibition of ribosome recruitment induces stress granule formation independently of eukaryotic initiation factor 2α phosphorylation. Mol Biol Cell 17:4212–4219. doi: 10.1091/mbc.E06-04-0318. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Mokas S, Mills JR, Garreau C, Fournier MJ, Robert F, Arya P, Kaufman RJ, Pelletier J, Mazroui R. 2009. Uncoupling stress granule assembly and translation initiation inhibition. Mol Biol Cell 20:2673–2683. doi: 10.1091/mbc.E08-10-1061. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Onomoto K, Yoneyama M, Fung G, Kato H, Fujita T. 2014. Antiviral innate immunity and stress granule responses. Trends Immunol 35:420–428. doi: 10.1016/j.it.2014.07.006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Reineke LC, Lloyd RE. 2013. Diversion of stress granules and P-bodies during viral infection. Virology 436:255–267. doi: 10.1016/j.virol.2012.11.017. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Valiente-Echeverría F, Melnychuk L, Mouland AJ. 2012. Viral modulation of stress granules. Virus Res 169:430–437. doi: 10.1016/j.virusres.2012.06.004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.White JP, Lloyd RE. 2012. Regulation of stress granules in virus systems. Trends Microbiol 20:175–183. doi: 10.1016/j.tim.2012.02.001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Li W, Li Y, Kedersha N, Anderson P, Emara M, Swiderek KM, Moreno GT, Brinton MA. 2002. Cell proteins TIA-1 and TIAR interact with the 3′ stem-loop of the West Nile virus complementary minus-strand RNA and facilitate virus replication. J Virol 76:11989–12000. doi: 10.1128/JVI.76.23.11989-12000.2002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.White JP, Cardenas AM, Marissen WE, Lloyd RE. 2007. Inhibition of cytoplasmic mRNA stress granule formation by a viral proteinase. Cell Host Microbe 2:295–305. doi: 10.1016/j.chom.2007.08.006. [DOI] [PubMed] [Google Scholar]
- 50.Khaperskyy DA, Emara MM, Johnston BP, Anderson P, Hatchette TF, McCormick C. 2014. Influenza A virus host shutoff disables antiviral stress-induced translation arrest. PLoS Pathog 10:e1004217. doi: 10.1371/journal.ppat.1004217. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Khaperskyy DA, Hatchette TF, McCormick C. 2012. Influenza A virus inhibits cytoplasmic stress granule formation. FASEB J 26:1629–1639. doi: 10.1096/fj.11-196915. [DOI] [PubMed] [Google Scholar]
- 52.Ng CS, Jogi M, Yoo JS, Onomoto K, Koike S, Iwasaki T, Yoneyama M, Kato H, Fujita T. 2013. Encephalomyocarditis virus disrupts stress granules, the critical platform for triggering antiviral innate immune responses. J Virol 87:9511–9522. doi: 10.1128/JVI.03248-12. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Onomoto K, Jogi M, Yoo JS, Narita R, Morimoto S, Takemura A, Sambhara S, Kawaguchi A, Osari S, Nagata K, Matsumiya T, Namiki H, Yoneyama M, Fujita T. 2012. Critical role of an antiviral stress granule containing RIG-I and PKR in viral detection and innate immunity. PLoS One 7:e43031. doi: 10.1371/journal.pone.0043031. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Reineke LC, Kedersha N, Langereis MA, van Kuppeveld FJ, Lloyd RE. 2015. Stress granules regulate double-stranded RNA-dependent protein kinase activation through a complex containing G3BP1 and Caprin1. mBio 6:e02486. doi: 10.1128/mBio.02486-14. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Reineke LC, Lloyd RE. 2015. The stress granule protein G3BP1 recruits protein kinase R to promote multiple innate immune antiviral responses. J Virol 89:2575–2589. doi: 10.1128/JVI.02791-14. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Esclatine A, Taddeo B, Roizman B. 2004. Herpes simplex virus 1 induces cytoplasmic accumulation of TIA-1/TIAR and both synthesis and cytoplasmic accumulation of tristetraprolin, two cellular proteins that bind and destabilize AU-rich RNAs. J Virol 78:8582–8592. doi: 10.1128/JVI.78.16.8582-8592.2004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Finnen RL, Hay TJ, Dauber B, Smiley JR, Banfield BW. 2014. The herpes simplex virus 2 virion-associated ribonuclease vhs interferes with stress granule formation. J Virol 88:12727–12739. doi: 10.1128/JVI.01554-14. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Read GS, Karr BM, Knight K. 1993. Isolation of a herpes simplex virus type 1 mutant with a deletion in the virion host shutoff gene and identification of multiple forms of the vhs (UL41) polypeptide. J Virol 67:7149–7160. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.Beattie E, Denzler KL, Tartaglia J, Perkus ME, Paoletti E, Jacobs BL. 1995. Reversal of the interferon-sensitive phenotype of a vaccinia virus lacking E3L by expression of the reovirus S4 gene. J Virol 69:499–505. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60.Braggin JE, Child SJ, Geballe AP. 2016. Essential role of protein kinase R antagonism by TR1 in human cytomegalovirus replication. Virology 489:75–85. doi: 10.1016/j.virol.2015.11.032. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61.Doudna JA, Charpentier E. 2014. Genome editing. The new frontier of genome engineering with CRISPR-Cas9. Science 346:1258096. doi: 10.1126/science.1258096. [DOI] [PubMed] [Google Scholar]
- 62.Duguay BA, Saffran HA, Ponomarev A, Duley SA, Eaton HE, Smiley JR. 2014. Elimination of mitochondrial DNA is not required for herpes simplex virus 1 replication. J Virol 88:2967–2976. doi: 10.1128/JVI.03129-13. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63.Chang HW, Watson JC, Jacobs BL. 1992. The E3L gene of vaccinia virus encodes an inhibitor of the interferon-induced, double-stranded RNA-dependent protein kinase. Proc Natl Acad Sci U S A 89:4825–4829. doi: 10.1073/pnas.89.11.4825. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64.Zhang P, Jacobs BL, Samuel CE. 2008. Loss of protein kinase PKR expression in human HeLa cells complements the vaccinia virus E3L deletion mutant phenotype by restoration of viral protein synthesis. J Virol 82:840–848. doi: 10.1128/JVI.01891-07. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65.Finnen RL, Pangka KR, Banfield BW. 2012. Herpes simplex virus 2 infection impacts stress granule accumulation. J Virol 86:8119–8130. doi: 10.1128/JVI.00313-12. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66.Okonski KM, Samuel CE. 2013. Stress granule formation induced by measles virus is protein kinase PKR dependent and impaired by RNA adenosine deaminase ADAR1. J Virol 87:756–766. doi: 10.1128/JVI.02270-12. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 67.Lindquist ME, Mainou BA, Dermody TS, Crowe JE Jr. 2011. Activation of protein kinase R is required for induction of stress granules by respiratory syncytial virus but dispensable for viral replication. Virology 413:103–110. doi: 10.1016/j.virol.2011.02.009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 68.Garaigorta U, Heim MH, Boyd B, Wieland S, Chisari FV. 2012. Hepatitis C virus (HCV) induces formation of stress granules whose proteins regulate HCV RNA replication and virus assembly and egress. J Virol 86:11043–11056. doi: 10.1128/JVI.07101-11. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 69.Simpson-Holley M, Kedersha N, Dower K, Rubins KH, Anderson P, Hensley LE, Connor JH. 2011. Formation of antiviral cytoplasmic granules during orthopoxvirus infection. J Virol 85:1581–1593. doi: 10.1128/JVI.02247-10. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 70.Anderson P, Kedersha N. 2006. RNA granules. J Cell Biol 172:803–808. doi: 10.1083/jcb.200512082. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 71.Yoo JS, Takahasi K, Ng CS, Ouda R, Onomoto K, Yoneyama M, Lai JC, Lattmann S, Nagamine Y, Matsui T, Iwabuchi K, Kato H, Fujita T. 2014. DHX36 enhances RIG-I signaling by facilitating PKR-mediated antiviral stress granule formation. PLoS Pathog 10:e1004012. doi: 10.1371/journal.ppat.1004012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 72.Wen X, Huang X, Mok BW, Chen Y, Zheng M, Lau SY, Wang P, Song W, Jin DY, Yuen KY, Chen H. 2014. NF90 exerts antiviral activity through regulation of PKR phosphorylation and stress granules in infected cells. J Immunol 192:3753–3764. doi: 10.4049/jimmunol.1302813. [DOI] [PubMed] [Google Scholar]
- 73.Cotter CR, Kim WK, Nguyen ML, Yount JS, Lopez CB, Blaho JA, Moran TM. 2011. The virion host shutoff protein of herpes simplex virus 1 blocks the replication-independent activation of NF-kappaB in dendritic cells in the absence of type I interferon signaling. J Virol 85:12662–12672. doi: 10.1128/JVI.05557-11. [DOI] [PMC free article] [PubMed] [Google Scholar]