

Abstract
Purpose
Radiation (RT) is critical to the treatment of high-grade gliomas (HGGs) but cures remain elusive. The BRAF mutation V600E is critical to the pathogenesis of 10–20% of pediatric gliomas, and a small proportion of adult HGGs. Here we aim to determine whether PLX4720, a specific BRAF V600E inhibitor, enhances the activity of radiation (RT) in human HGGs in vitro and in vivo.
Methods
Patient-derived HGG lines harboring wild-type BRAF or BRAF V600E were assessed in vitro to determine IC50 values, cell cycle arrest, apoptosis and senescence and elucidate mechanisms of combinatorial activity. A BRAF V600E HGG intracranial xenograft mouse model was used to evaluate in vivo combinatorial efficacy of PLX4720+RT. Tumors were harvested for immunohistochemistry to quantify cell cycle arrest and apoptosis.
Results
RT+PLX4720 exhibited greater anti-tumor effects than either monotherapy in BRAF V600E but not in BRAF WT lines. In vitro studies showed increased Annexin V and decreased S phase cells in BRAF V600E gliomas treated with PLX4720+RT, but no significant changes in β-galactosidase levels. In vivo, concurrent and sequential PLX4720+RT each significantly prolonged survival compared to monotherapies, in the BRAF V600E HGG model. Immunohistochemistry of in vivo tumors demonstrated that PLX4720+RT decreased Ki-67 and phospho-MAPK, and increased γH2AX and p21 compared to control mice.
Conclusions
BRAF V600E inhibition enhances radiation-induced cytotoxicity in BRAF V600E-mutated HGGs, in vitro and in vivo, effects likely mediated by apoptosis and cell cycle, but not senescence. These studies provide the pre-clinical rationale for clinical trials of concurrent radiotherapy and BRAF V600E inhibitors.
Keywords: BRAF V600E, radiotherapy, targeted inhibitors, high-grade gliomas
INTRODUCTION
The treatment of high-grade gliomas involves maximal safe surgical resection followed by radiation therapy and concurrent chemotherapy with alkylating agents. These approaches extend the lives of patients with high-grade gliomas by a median of 15 months [1]. While expanding understanding of tumor genetics is paving the way for personalized cancer therapies [2], studies are also demonstrating that tumors, which invariably harbor genetic heterogeneity, develop resistance to molecularly targeted inhibitors when used as monotherapy. New multimodality approaches are desperately needed to improve outcomes, particularly in patients with brain tumors.
RAF may play an important role in brain tumor formation and response to therapy [3]. Genetic studies have revealed the oncogenic role of BRAF V600E in several human tumors, including a small proportion of adult brain tumors [3–5] and 6–25% of grade 2–4 pediatric brain tumors [6, 7]. In vitro studies have also shown that constitutive activation of RAF can promote glioma formation in mice [8]. At least one study suggests that children with BRAF V600E mutated gliomas have poorer outcomes than those with tumors expressing wild-type BRAF [9]. A substantial body of literature suggests constitutive expression of RAF or RAF overexpression may also play a role in radiation response [10, 11].
Therapeutic opportunity is presented by the finding that BRAF V600E is highly druggable. BRAF V600E inhibitors reduce phosphorylation of mitogen-activated protein kinase (MAPK) with subsequent downstream effects on apoptosis and cell cycle inhibition. FDA-approved vemurafenib (PLX4032) targeting BRAF V600E mutated cells has changed the natural history of metastatic melanoma [12]. The most concerning side effect seen with this drug is the development of keratoacanthomoas, a low-grade skin cancer commonly treated by excision. Another BRAF V600E-targeted inhibitor, dabrafenib, has also been FDA-approved for use in the clinic with BRAF V600E-mutated tumors [13, 14]. Additionally, preclinical data using both vemurafenib and its preclinical analogue, PLX4720, show combinatorial activity with radiation in tumor cell lines of diverse origins [15, 16].
In this study, we asked whether the combination of radiation with PLX4720, the preclinical analogue of vemurafenib, would provide superior tumor control in brain tumors harboring the BRAF V600E mutation. Our findings show additive activity between PLX4720 and radiation, both in vitro and in vivo, mediated by cell cycle effects and apoptosis, but not senescence. Furthermore, to our knowledge, this is the first demonstration of survival advantage with the combination of BRAF V600E-targeted inhibitors and radiation in an animal model. Our data provide in vivo validation of the in vitro data on BRAF V600E targeted inhibitors combined with radiation [15, 16], and inform clinical trials with combined modality therapies for patient with BRAF V600E-mutated brain tumors.
MATERIALS AND METHODS
Cell lines and BRAF mutational analysis
Human high-grade glioma cell lines AM-38 and DBTRG-05MG were purchased from the American Type Culture Collection and the Japan Health Sciences Foundation Health Science Research Resources bank. The BRAF WT cell lines GBM6, GBM8, and GBM36 were established from primary patient tumors by the laboratory of C. David James and were propagated according to previously published methods [17]. The BRAF gene in each of these cell lines was sequenced to confirm the presence or absence of the BRAF V600E mutation by the UCSF genomics core according to previously published methods [15].
Cell growth, clonogenic, senescence, and cell cycle analyses
Cell growth assays were performed with CellTiter-Glo® Luminescent Cell Viability reagent (Promega, Madison, WI), with at least three replicates per assay, 72 hours after treatment. The IC50 was defined as the concentration leading to 50% viability. Clonogenic survival (colony forming) assays were performed as previously described [18]. First, cells were treated with PLX4720 alone (monotherapy, without radiation) to determine the IC50 (50% maximal inhibitory concentration) and the IC20 (20% maximal inhibitory concentration). Subsequently cells were treated with a combination of 0.5 μM PLX4720 (the IC20) and varying doses of radiation. Cells were pretreated with PLX4720 for 24 hours prior to irradiation, and then incubated in the corresponding media (DMSO as control or PLX4720) until sufficient time had elapsed for colony formation (defined as 50 cells per colony). Cells were irradiated using a cesium source at a dose rate of 1.97 Gy/min. Surviving fractions were normalized to the plating efficiency of each cell line, and cell survival measurements were fitted to a linear quadratic mathematical model using GraphPad Prism 5.0 software [Surviving Fraction = exp(−α*Dose + β*Dose2)]. The dose enhancement ratio (DER) was calculated at 90% survival. The DER is the ratio of the radiation dose required to achieve 90% cell survival using radiation alone and the radiation dose required to achieve the same biological effect (90% cell survival) using radiation plus PLX4720. A DER value >1 indicates cooperativity between the drug and radiation, because a lower dose of radiation is necessary for 90% inhibition when radiation is administered concurrently with the drug.
For cell cycle analyses, cells were harvested at the exponential phase of growth after 24 hours of treatment with 0.05% DMSO, single agent PLX4720 or radiation, or a combination of PLX4720 immediately followed by radiation, and analyzed by flow cytometry (BD FACSCalibur) using the FITC BrdU Flow Kit (BD Pharmingen TM, San Diego, CA) with 7-AAD DNA content stain as previously described [15]. Apoptosis levels were established by flow cytometry 72 hours after treatment using Fluorescein IsoThioCyanate (FITC) Annexin V and propidium iodide (PI) staining (BD Pharmigen TM, San Diego, CA). To quantify proportions of senescent cells, β-galactosidase-based flow cytometry was performed 72 hours after treatment. Cells were incubated for 2 hours with 33 uM C12FDG (Molecular Probes, Waltham, MA) in culture medium prior to measurement of mean C12-fluorescein levels on a log scale [19]. Data were analyzed using FlowJo software (TreeStar), GraphPad Prism (San Diego, CA, USA), and R programming (Vienna, Austria).
Firefly luciferase modification of AM-38 tumor cells
AM-38 cells were modified with a Fluc lentivirus as previously described [17, 20]. The cells were treated with D-luciferin potassium salt (Gold Biotechnology, St. Louis, MO) and the luminescence was analyzed with a Xenogen IVIS System (Xenogen Corporation, Alameda, CA) to determine transformation efficiency.
In vivo experiments with intracranial xenografts of BRAF V600E-mutated glioma
All animal experiments were conducted under the ethical provisions of the UCSF Institutional Animal Care and Use Committee. We implanted cultured AM-38 cells intracranially into the brains of five-week old athymic nude mice [purchased from Taconic (Hudson, NY, USA)]. At the time of tumor implantation, mice were anesthetized with 100 mg/kg ketamine and 10 mg/kg xylazine. The skull was incised and 5 × 105 cells were injected 3 mm to the right of the midline, behind the bregma [21]. Mice were randomized to one of five treatment arms: 1) control (DMSO); 2) PLX4720 alone (10 mg/kg over 14 consecutive days); 3) radiation alone (1 Gy × 3 on alternating days); 4) concurrent PLX4720 (10 mg/kg over 14 consecutive days) and radiation (1 Gy × 3 on alternating days); and 5) sequential XRT (1 Gy × 3 on alternating days), followed by PLX4720 (10 mg/kg over 14 consecutive days).
One mouse from each cohort was sacrificed 6 hours after the final treatment and the brain preserved in paraformaldehyde for immunohistochemistry (see below for methods). Animals were monitored daily for constitutional and neurological symptoms, and were euthanized when significantly symptomatic from burden of disease. Survival analyses were conducted using Kaplan-Meier methods and statistical significance estimated by the log rank test.
Histological staining and immunohistochemistry
For immunohistochemical analyses, BRAF V600E intracranial xenograft (AM-38) mice were randomized to the following treatment groups and sacrificed 6 hours after completion of the last treatment (for Figure 4) and when significantly symptomatic from burden of disease (for Supplemental Figure 3): control (DMSO); PLX4720 alone (10 mg/kg over 14 consecutive days); radiation alone (XRT 1 Gy × 3 on alternating days); or combination of sequential XRT (1 Gy × 3 on alternating days), followed by PLX4720 (10 mg/kg over 14 consecutive days).
Figure 4.
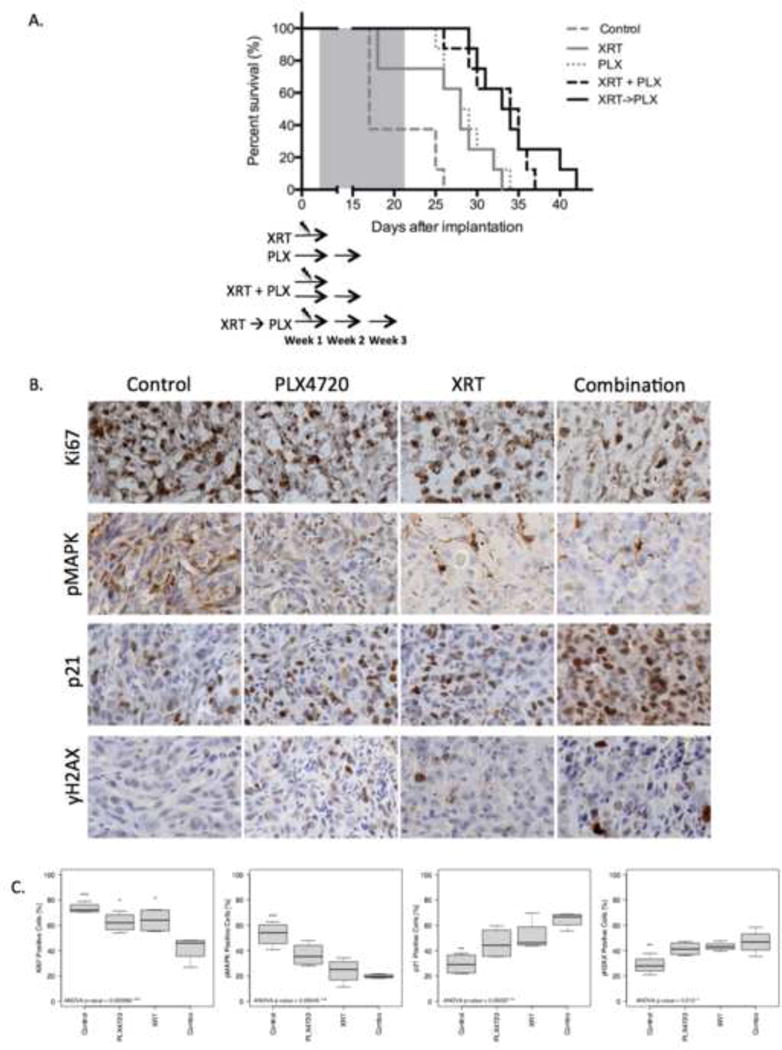
PLX4720 augments radiation antitumor effects in vivo. (A) BRAF V600E, firefly luciferase modified, intracranial xenograft model (AM-38) mice were randomized to control (DMSO); PLX4720 alone (10 mg/kg over 14 consecutive days); radiation alone (XRT 1 Gy × 3 on alternating days); concurrent PLX4720 (10 mg/kg over 14 consecutive days) + radiation (1 Gy × 3 on alternating days); or sequential XRT (1 Gy × 3 on alternating days), followed by PLX4720 (10 mg/kg over 14 consecutive days). Combined PLX4720 and radiation therapy produced statistically significant prolonged survival compared to respective monotherapies. P values: Concurrent treatment vs control p<0.0001; vs PLX4720 p=0.0170; vs radiation p=0.0077. Sequential treatment vs control p<0.0001; vs PLX4720 p=0.0158; vs radiation p=0.0059. There was no difference in survival between concurrent and sequential arms (p=0.57). (B and C) For immunohistochemical analyses, BRAF V600E intracranial xenograft model (AM-38) mice were sacrificed 6 hours after the final treatment. PLX4720 + radiation significantly reduced tumor proliferation in vivo, assessed by Ki-67 staining, compared to control and each monotherapy. Combination therapy significantly decreased pMAPK and increased p21 and γH2AX levels compared to control but not compared to single agents. P values are shown in Supplemental Table 2.
Mouse brains were preserved overnight in 4% paraformaldehyde and processed through ethanol dehydration series for paraffin embedding. Five micron paraffin sections were cut, deparaffinized, rehydrated with decreasing ethanol concentrations. The presence of tumor was confirmed by hematoxylin and eosin staining, and the brain specimens were examined by a neuropathologist. Next, the sections were stained using anti-Ki-67 (Dako, Carpinteria, CA), γH2AX (Bethyl, Montgomery, TX), p21 (Oncogene Science, Cambridge, MA), pMAPK (Cell Signaling, Danvers, MA) and anti-cleaved caspase-3 (Cell Signaling, Danvers, MA) antibodies, detected with DAB substrate (Vector Labs, CA) and counterstained with hematoxylin, as previously described [22].
Image-based quantification was performed for each stain. The data represent measurements from one mouse per group with comparable tumor burden. Five 20× pictures of areas with highest numbers of positive cells were taken per tumor (Leica, DMLS microscope) and both positively and negatively stained cells were quantified. Percentages of positive cells were averaged per tumor. To assess for significant differences among the treatment groups we first performed an analysis of variance (ANOVA) followed by student t-tests for pairwise comparison as previously described [17].
RESULTS
BRAF mutational analysis and in vitro IC50 values for PLX4720
BRAF mutational analysis showed that two of the cell lines used in these studies carried the BRAF V600E mutation (DBTRG-05MG, AM-38), while GBM6, GBM8 and GBM36 expressed wild-type (WT) BRAF. Using a cell viability assay, we determined the IC50 of PLX4720 to be 2 μM and 1.8 μM in the BRAF V600E mutated cell lines DBTRG-05MG and AM-38, respectively; however, in the BRAF WT cell line GBM8, the IC50 was not reached using cell viability studies even with concentrations of PLX4720 up to 20 μM (Supplemental Table 1).
To confirm the IC50 of PLX4720 in these cell lines, we also performed clonogenic assays. As determined by clonogenic assay, the IC50 values of PLX4720 were 2 μM and 0.5 μM in DBTRG-05MG, AM-38, respectively; however, in the BRAF WT cell line, IC50 values were not reached, and in some cells lines growth potentiation was observed (Supplemental Table 1).
Combinatorial efficacy of PLX4720 and radiation in clonogenic assays
To determine the effect of combination treatment with PLX4720 and radiation, glioma cell lines were treated with 0.5 μM PLX4720 and received 0, 2, 4, or 6 Gy of radiation. Dose Enhancement Ratios (DER) calculated at 90% survival were less than 1 for BRAF WT cell line (0.91 for GBM6; Figure 1) reflecting lack of synergy and perhaps antagonism, but greater than 1 for BRAF V600E cell lines (1.42 for AM-38 and 1.80 for DBT-RG) demonstrating sensitizing interactions (Figure 1).
Figure 1.
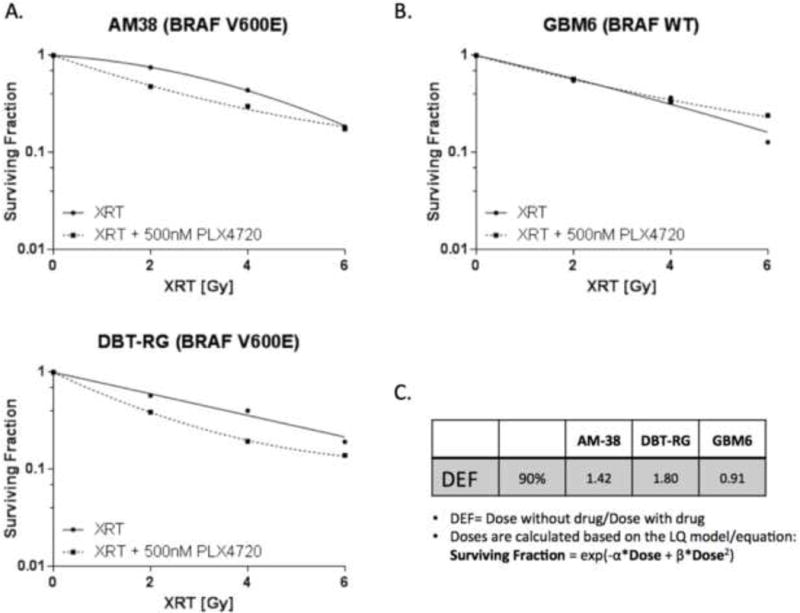
Radiation cooperates with PLX4720 to reduce clonogenic survival in BRAF V600E-mutant but not BRAF WT glioma cells. (A) BRAF V600E glioma cell lines; (B) BRAF WT glioma cell line; (C) Dose Enhancement Ratio (DER) for each cell line calculated at 90% survival level. PLX4720 (500 nM) was administered 24 hour before radiation (0, 2, 4, 6 Gy). Surviving fractions, shown as mean + SE (quadruplicate samples), were normalized to plating efficiency for each cell line. Radiation alone: solid line. Radiation + PLX4720: dashed line. XRT: radiation therapy. Clonogenic survival data points were fitted to a linear quadratic mathematical model.
Combinatorial treatment with PLX4720 and radiation decreased the proportion of cells in S phase and increased the proportion of apoptotic but not senescent cells in BRAF V600E-mutated glioma cells
We investigated the mechanisms underling the combinatorial cytotoxicity of PLX4720 and radiation. We began by examining cell cycle modulation and analyzed the proportion of glioma cells in various phases of the cell cycle after each treatment using flow cytometry. In AM-38 and DBTRG-05MG cells (BRAF V600E), treatment with PLX4720 reduced S phase mostly through a G1 arrest, radiation alone reduced S phase mostly through a G2 arrest and combined treatment of radiation + PLX4720 showed the most pronounced reduction in S phase due to both G1 and G2 arrests. Minimal cell cycle effects were seen in GBM8 and GBM36 cells (BRAF WT) although mild reductions in S-phase were seen with all three treatments (Figure 2, Supplemental Figure 1A).
Figure 2.
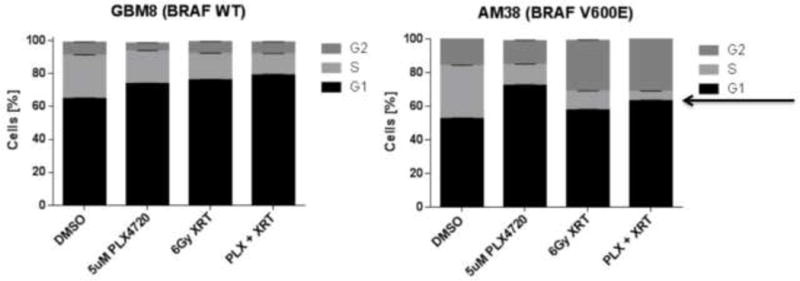
Radiation cooperates with PLX4720 to reduce S phase in BRAF V600E glioma cells. Bar graphs display distribution of cells within each cell cycle phase for GBM8 (BRAF WT) and AM-38 (BRAF V600E) treated with either radiation therapy (XRT; 6 Gy), PLX4720 (5 μM), PLX4720 + radiation (5 μM PLX4720 followed 24 hours later by 6 Gy radiation), or DMSO control. Cell cycle analyses were performed 24 hours after the last treatment. An arrow indicates S phase content.
We next examined the induction of apoptosis after each treatment modality. In BRAF V600E cell lines, treatment with PLX4720 or radiation alone increased the number of apoptotic cells as measured by Annexin V. Combination treatment with PLX4720 and radiation showed a reproducible and significant increase in Annexin V when compared to treatment with either single modality (Figure 3, Supplemental Figure 1B). The proportions of senescent cells, as measured by β-galactosidase levels, were not significantly altered by the combinatorial treatment regimen in BRAF WT or BRAF V600E mutated cells at the same time point (Supplemental Figure 2).
Figure 3.
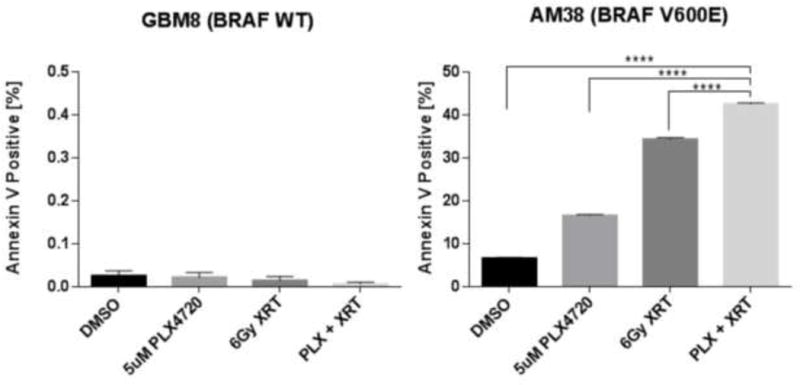
Radiation cooperates with PLX4720 to induce apoptosis in BRAF V600E but not in BRAF WT glioma cells. Bar graphs quantitate apoptosis (mean + SE), as measured by Annexin V expression for GBM8 (BRAF WT) and AM-38 (BRAF V600E) treated with either radiation therapy (XRT; 6 Gy), PLX4720 (5 μM), PLX4720 + radiation (5 μM PLX4720 followed 24 hours later by 6 Gy radiation), or DMSO control. Annexin V expression was measured 72 hours after the last treatment.
Combination treatment of PLX4720 and radiation prolonged survival in an intracranial xenograft model of BRAF V600E-mutated malignant astrocytoma
To determine if combination treatment with radiation and PLX4720 in BRAF V600E-mutated cell lines impacted survival in an in vivo animal model, we further tested this combination in an intracranial xenograft model of the BRAFV600E-mutated glioma cell line AM-38. This combination was tested using two different regimens: concurrent PLX4720 with radiation, and sequential radiation followed by PLX4720. There was a statistically significant prolonged survival in mice that received concurrent PLX4720 and radiation, compared to treatment with DMSO control (p<0.0001), PLX4720 alone (p=0.0170) or radiation alone (p=0.0077). This survival benefit remained significant with sequential treatment with PLX4720 and radiation, when compared to treatment with control (p<0.0001), PLX4720 alone (p=0.0158) or radiation alone (p=0.0059). There was no difference in survival between the concurrent and sequential arms (p=0.57) (Figure 4A).
Analysis of mice tumor tissues showed reduced proliferation and increased cleaved caspase 3 in tumor tissues treated with PLX4720 and radiation
Immunohistochemical analyses of tumor tissues upon completion of treatment showed that the group treated with a combination of PLX4720 and radiation had significantly lower proliferation index (by Ki-67 staining) in the combination group when compared to PLX4720 alone (p=0.002) and XRT alone (p=0.01; ANOVA p<0.01; Figure 4B,C). Combination treatment also decreased pMAPK levels (Figure 4B,C) when compared to control (p<0.01), and PLX4720 alone (a trend of p=0.06) but not radiation alone (p>0.05; ANOVA p<0.01). In assessing γH2AX foci formation, combination therapy increased double-stranded DNA damage breaks compared to control (p=0.02; ANOVA p=0.01) but these increases were not significant when compared to single agents (p>0.05). Levels of p21, a marker of senescence as well as a G1/S checkpoint regulator, were significantly increased in combination treatment when compared to control (p=0.02, ANOVA p<0.01) but not when compared to single agents. The detailed pairwise t-test comparisons are shown in Supplemental Table 2. In mice followed to the end of the survival study, the group treated with a combination of PLX4720 and radiation had retained a lower proliferation index (by Ki-67 staining) in the combination group when compared to control group (p=0.01), PLX4720 alone (p<0.01) but not XRT alone (p=0.2) (Supplemental Figure 3A,B). Moreover, we documented increased cleaved caspase-3 levels in combination treatment (Supplemental Figure 3A,B) that were significant when compared to control (p<0.01), but not when compared to radiation alone (p=0.2222) or PLX4720 alone (p=0.2222).
DISCUSSION
As advances in scientific understanding illuminate the molecular pathways driving gliomagenesis, targeted therapies for brain tumors have increasingly become the focus of clinical trials. Prior studies have suggested that constitutive activation of the RAF oncogene plays a role in the radiation response [10, 11]. Several novel targeted BRAF V600E inhibitors have been tested in the clinic, and at least one (dabrafenib) has been shown to cross the blood brain barrier and demonstrate efficacy against BRAF V600E-mutated brain metastases [12, 14]. As radiation therapy plays an integral role in the treatment of high-grade and recurrent gliomas; as in vitro data suggest combinatorial activity between radiation and BRAF V600E-targeted inhibitors; and as there is growing evidence of resistance to therapy by targeted inhibitors, studying the combination of RAF inhibitors with radiation in an intracranial xenograft model of a BRAF V600E-mutated glioma has important clinical implications.
In this study, we show that PLX4720, a BRAF V600E inhibitor, cooperates with radiation in vitro in BRAF V600E-mutated glioma cell lines, even when a small concentration of the inhibitor (substantially lower than the IC50) is used. Akin to data from BRAF V600E-mutated melanoma [23], growth potentiation is observed in BRAF WT cell lines treated with PLX4720 (Supplemental Table 1). In our intracranial xenograft model of BRAF V600E-mutated gliomas, combination treatment with radiation plus PLX4720 leads to a survival advantage and lower tumor proliferation index when compared to treatment by either monotherapy (radiation or BRAF V600E inhibitor). This survival advantage persists regardless of whether PLX4720 is administered sequentially or concurrently with radiation, suggesting that this combination is additive in vivo as well. Our in vitro data suggest that combination treatment with PLX4720 and radiation increases the rate of double stranded DNA breaks, thereby increasing the proportion of cells undergoing apoptosis, and the frequency of cells arrested at cell cycle checkpoints in the BRAF V600E background. In vivo we did not observe any skin toxicities in these mice corresponding to reports from adults treated with vemurafenib and radiotherapy [24], perhaps due to differing mechanisms of combination in humans compared to murine models.
The establishment of a BRAF V600E animal model in which survival differences can be demonstrated may help resolve conflicting data from in vitro studies about RAF inhibitors. To our knowledge, this is the first demonstration of survival advantage with the combination of radiation and BRAF V600E-targeted inhibitors in an animal model of BRAF-mutant cancer. In a prior study with in vitro clonogenic assays, our group demonstrated additive activity between BRAF V600E inhibitors and radiation in colon cancer and melanoma cell lines [15]. In contrast, a detailed clonogenic study of several melanoma cell lines treated with radiation and PLX4032 displayed heterogeneity in the radiation enhancement ratio [16]. These differing results suggest that the BRAF V600E mutation may not be the only molecular aberration driving the favorable combination of radiation with a BRAF V600E inhibitor. This report also reproduces and extends the results of a prior study in which treatment with PLX4720 enhanced survival compared to vector in BRAF V600E mutated brain tumors [25].
Is there a clinical role for agents with anti-neoplastic effects that are additive rather than synergistic with radiation? While classical radiosensitizers are ‘synergistic’ in combination with radiation, PLX4720 shows additive activity with radiation. Cetuximab is another targeted inhibitor whose clinical effectiveness in combination with radiation relies on additive rather than synergistic effects as seen in clonogenic assays [26]. An additive inhibitor may target alternative pathways, helping to overcome drug resistance and increasing the therapeutic index of radiation. In turn, the addition of radiation may decrease the dose of inhibitor needed for efficacy. The latter is an important consideration for dose-limiting toxicities. In BRAF V600E inhibitors, with preclinical evidence of growth activation in BRAF WT tissues and keratoacanthoma development in clinical trials, decreasing normal tissue toxicity may be of particular importance [27].
The photosensitivity of BRAF inhibitors like vemurafenib may increase the toxicity of patients receiving BRAF inhibitors with radiotherapy, even with sequential administration. There are case reports that have demonstrated increased incidence of BRAF-inhibitor related dermatitis in patients who have previously received radiation therapy. Increased radio-dermatitis was also seen in melanoma patients receiving brain radiation, and vemurafenib was associated with a higher risk of dermatitis than dabrafenib [28]. Such cases of cutaneous toxicity or radiation recall dermatitis can be managed with topical corticosteroids, and generally should not necessitate a decreased dose of BRAF inhibitor [29]. Certain centers have reported brain radionecrosis or steroid dependence with sequential administration of radiation after BRAF inhibitors [30, 31]. There are less data on the toxicity of sequential administration or radiation and BRAF inhibitors; however, several case reports have pointed to radiation recall dermatitis and even radiation recall pneumonitis [32] and proctitis [30] when BRAF inhibitors were administered in patients who previously received radiation.
Future experiments are underway to address the limitations of this study. Clonogenic assays were initially designed to address radiobiological questions, and not the efficacy of continuous medical therapies like chemotherapeutics. We have addressed this concern by performing both cell viability (Supplemental Figure 1) and clonogenic assays, with good agreement between the results. In vivo, our study shows only a modest increase in survival with combination treatment with PLX4720 and radiation. The goal in combinatorial experiments is to investigate interactions between drug and radiation and therefore doses and dosing schedules that only have modest effects as single agents are used, allowing details of their combinatorial effects to be interrogated and established. As such, we specifically did not wish to administer a drug alone regimen that has dramatic effects as monotherapy. At the time this study was performed, the only BRAF V600E cell line that could be established as an in vivo xenograft was AM-38, which is known to have a rapid doubling time and swift animal demise. Survival differences based on different treatment conditions are difficult to establish with rapidly proliferating xenografts, so even these modest survival differences are encouraging. Variation in cell lines, drug dosing schedules, and vehicle used for drug administration have been known to affect in vivo tumor growth in xenografts treated by agents including temozolomide, cetuximab and cisplatin, and in certain cell lines, even these chemotherapeutic agents appear to have only modest efficacy [33, 34]. Further in vivo experiments are underway to determine the effects of combining PLX4720 and radiation with an alternative continuous dosing schedule of PLX4720, with a higher dose of PLX4720, with a low-grade glioma cell line expressing BRAF V600E, or with the addition of an inhibitor with complementary activity (e.g. a MEK inhibitor). Additionally, our data suggest a mechanism involving DNA damage may explain the favorable combination of PLX4720 and radiation; therefore, detailed mechanistic questions of which DNA damage response elements are involved in the response to BRAF inhibition and the combination of radiation and PLX4720 are also the subject of future experiments.
In summary, these studies show cooperative activity of PLX4720 and radiation in vitro and in vivo, with a survival advantage of combination treatment in a high-grade glioma xenograft model. Already, these data informed an ongoing trial of concurrent BRAF V600E inhibitor with stereotactic radiosurgery in BRAF-mutant melanomas that metastasized to the brain (http://clinicaltrials.gov/show/NCT01721603), and these data may further guide the next generation of multimodality clinical trials in BRAF V600E-mutated high-grade gliomas. The Pacific Neuro-Oncology Consortium is already conducting a trial of PLX4032 monotherapy in children with BRAF V600E-mutated brain tumors (http://clinicaltrials.gov/show/NCT01748149); the next generation of trials may investigate the combination of BRAF-inhibition with concurrent radiation. Moving forward, for patients with high-grade BRAF-mutant gliomas, this combination of radiation with PLX4720 must be compared to the existing post-operative standard of care (concurrent radiation with temozolomide), with respect to outcome, normal tissue toxicity, and as a modality to overcome resistance to targeted inhibitors in BRAF-mutant cancers.
Supplementary Material
Supplemental Figure 1. Radiation cooperates with PLX4720 to reduce S phase and induce apoptosis in BRAF V600E glioma cells. Flow cytometry analyses of (A–B) cell cycle using BrdU and 7-AAD staining; and (C–D) apoptosis using Annexin V staining. Cells analyzed include (A, C) BRAF WT GBM36 and (B, D) BRAF V600E DBTRG-05MG cells. These additional cell lines complement the data shown in Figures 2 and 3 and utilize identical methods to those delineated in Figures 2 and 3.
Supplemental Figure 2. Flow cytometric in vitro measurement of β-galactosidase levels in (A) GBM36 (BRAF WT) and (B) AM-38 (BRAF V600E) cells reveals higher background levels of senescence in BRAF V600E cells compared to BRAF WT cells but no pronounced effects of PLX4720, radiation or their combination on levels of senescent cells.
Supplemental Figure 3. Immunohistochemical analyses of survival study mice reveals combination (concurrent) therapy of PLX4720 and radiation significantly decreases tumor proliferation (Ki-67 staining) and increases cleaved caspase 3 (CC3) compared to control mice. Comparisons of combination therapy to each monotherapy were not statistically significant.
Supplemental Table 1. BRAF V600E inhibition by PLX4720 leads to growth inhibition in BRAF V600E cell lines, but IC50 values were not reached in BRAF WT glioma lines.
Supplemental Table 2. T-test comparisons of Ki67, pMAPK, p21 and γH2AX immunohistochemical staining of BRAF V600E tumor xenograft sections treated with PLX4720, radiation or combination (sequential) therapy assessed 6 hours after completion of treatment.
Acknowledgments
The authors acknowledge a Young Investigator Award to TD from the American Society of Clinical Oncology, NIH R01NS091620 (DHK, WAW), Grand Philanthropic Fund (DHK), and University of California Cancer Research Coordinating Committee (DHK).
References
- 1.Stupp R, et al. Radiotherapy plus concomitant and adjuvant temozolomide for glioblastoma. The New England journal of medicine. 2005;352(10):987–96. doi: 10.1056/NEJMoa043330. [DOI] [PubMed] [Google Scholar]
- 2.Bamford S, et al. The COSMIC (Catalogue of Somatic Mutations in Cancer) database and website. British journal of cancer. 2004;91(2):355–8. doi: 10.1038/sj.bjc.6601894. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Basto D, et al. Mutation analysis of B-RAF gene in human gliomas. Acta neuropathologica. 2005;109(2):207–10. doi: 10.1007/s00401-004-0936-x. [DOI] [PubMed] [Google Scholar]
- 4.Knobbe CB, Reifenberger J, Reifenberger G. Mutation analysis of the Ras pathway genes NRAS, HRAS, KRAS and BRAF in glioblastomas. Acta neuropathologica. 2004;108(6):467–70. doi: 10.1007/s00401-004-0929-9. [DOI] [PubMed] [Google Scholar]
- 5.Myung JK, et al. Analysis of the BRAF(V600E) Mutation in Central Nervous System Tumors. Translational oncology. 2012;5:430–6. doi: 10.1593/tlo.12328. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Schindler G, et al. Analysis of BRAF V600E mutation in 1,320 nervous system tumors reveals high mutation frequencies in pleomorphic xanthoastrocytoma, ganglioglioma and extra-cerebellar pilocytic astrocytoma. Acta neuropathologica. 2011;121(3):397–405. doi: 10.1007/s00401-011-0802-6. [DOI] [PubMed] [Google Scholar]
- 7.Schiffman JD, et al. Oncogenic BRAF mutation with CDKN2A inactivation is characteristic of a subset of pediatric malignant astrocytomas. Cancer research. 2010;70(2):512–9. doi: 10.1158/0008-5472.CAN-09-1851. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Lyustikman Y, et al. Constitutive activation of Raf-1 induces glioma formation in mice. Neoplasia. 2008;10(5):501–10. doi: 10.1593/neo.08206. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Horbinski C, et al. Interplay among BRAF, p16, p53, and MIB1 in pediatric low-grade gliomas. Neuro-oncology. 2012;14(6):777–89. doi: 10.1093/neuonc/nos077. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Kasid U, Dritschilo A. RAF antisense oligonucleotide as a tumor radiosensitizer. Oncogene. 2003;22(37):5876–84. doi: 10.1038/sj.onc.1206700. [DOI] [PubMed] [Google Scholar]
- 11.Weidhaas JB, et al. A conserved RAS/mitogen-activated protein kinase pathway regulates DNA damage-induced cell death postirradiation in Radelegans. Cancer research. 2006;66(21):10434–8. doi: 10.1158/0008-5472.CAN-06-2182. [DOI] [PubMed] [Google Scholar]
- 12.Chapman PB, et al. Improved survival with vemurafenib in melanoma with BRAF V600E mutation. The New England journal of medicine. 2011;364(26):2507–16. doi: 10.1056/NEJMoa1103782. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Hauschild A, et al. Dabrafenib in BRAF-mutated metastatic melanoma: a multicentre, open-label, phase 3 randomised controlled trial. Lancet. 2012;380(9839):358–65. doi: 10.1016/S0140-6736(12)60868-X. [DOI] [PubMed] [Google Scholar]
- 14.Long GV, et al. Dabrafenib in patients with Val600Glu or Val600Lys BRAF-mutant melanoma metastatic to the brain (BREAK-MB): a multicentre, open-label, phase 2 trial. The lancet oncology. 2012;13(11):1087–95. doi: 10.1016/S1470-2045(12)70431-X. [DOI] [PubMed] [Google Scholar]
- 15.Dasgupta T, et al. Genotype-dependent cooperation of ionizing radiation with BRAF inhibition in BRAF V600E-mutated carcinomas. Investigational new drugs. 2013 doi: 10.1007/s10637-013-9928-9. [DOI] [PubMed] [Google Scholar]
- 16.Sambade MJ, et al. Melanoma cells show a heterogeneous range of sensitivity to ionizing radiation and are radiosensitized by inhibition of B-RAF with PLX-4032. Radiotherapy and oncology : journal of the European Society for Therapeutic Radiology and Oncology. 2011;98(3):394–9. doi: 10.1016/j.radonc.2010.12.017. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Michaud K, et al. Pharmacologic inhibition of cyclin-dependent kinases 4 and 6 arrests the growth of glioblastoma multiforme intracranial xenografts. Cancer research. 2010;70(8):3228–38. doi: 10.1158/0008-5472.CAN-09-4559. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Franken NA, et al. Clonogenic assay of cells in vitro. Nature protocols. 2006;1(5):2315–9. doi: 10.1038/nprot.2006.339. [DOI] [PubMed] [Google Scholar]
- 19.Debacq-Chainiaux F, et al. Protocols to detect senescence-associated beta-galactosidase (SA-betagal) activity, a biomarker of senescent cells in culture and in vivo. Nature protocols. 2009;4(12):1798–806. doi: 10.1038/nprot.2009.191. [DOI] [PubMed] [Google Scholar]
- 20.Sarkaria JN, et al. Identification of molecular characteristics correlated with glioblastoma sensitivity to EGFR kinase inhibition through use of an intracranial xenograft test panel. Molecular cancer therapeutics. 2007;6(3):1167–74. doi: 10.1158/1535-7163.MCT-06-0691. [DOI] [PubMed] [Google Scholar]
- 21.Hashizume R, et al. Morphologic and molecular characterization of ATRT xenografts adapted for orthotopic therapeutic testing. Neuro-oncology. 2010;12(4):366–76. doi: 10.1093/neuonc/nop033. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Wang J, et al. A reproducible brain tumour model established from human glioblastoma biopsies. BMC cancer. 2009;9:465. doi: 10.1186/1471-2407-9-465. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Poulikakos PI, et al. RAF inhibitors transactivate RAF dimers and ERK signalling in cells with wild-type BRAF. Nature. 2010;464(7287):427–30. doi: 10.1038/nature08902. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Merten R, et al. Increased skin and mucosal toxicity in the combination of vemurafenib with radiation therapy. Strahlentherapie und Onkologie : Organ der Deutschen Rontgengesellschaft … [et al] 2014;190(12):1169–72. doi: 10.1007/s00066-014-0698-x. [DOI] [PubMed] [Google Scholar]
- 25.Nicolaides TP, et al. Targeted therapy for BRAFV600E malignant astrocytoma. Clinical cancer research : an official journal of the American Association for Cancer Research. 2011;17(24):7595–604. doi: 10.1158/1078-0432.CCR-11-1456. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Hidalgo M. Erlotinib: preclinical investigations. Oncology. 2003;17(11 Suppl 12):11–6. [PubMed] [Google Scholar]
- 27.Seiwert TY, Salama JK, Vokes EE. The concurrent chemoradiation paradigm–general principles. Nature clinical practice. Oncology. 2007;4(2):86–100. doi: 10.1038/ncponc0714. [DOI] [PubMed] [Google Scholar]
- 28.Hecht M, et al. Radiosensitization by BRAF inhibitor therapy-mechanism and frequency of toxicity in melanoma patients. Annals of oncology : official journal of the European Society for Medical Oncology / ESMO. 2015;26(6):1238–44. doi: 10.1093/annonc/mdv139. [DOI] [PubMed] [Google Scholar]
- 29.Boussemart L, et al. Vemurafenib and radiosensitization. JAMA dermatology. 2013;149(7):855–7. doi: 10.1001/jamadermatol.2013.4200. [DOI] [PubMed] [Google Scholar]
- 30.Peuvrel L, et al. Severe radiotherapy-induced extracutaneous toxicity under vemurafenib. European journal of dermatology : EJD. 2013;23(6):879–81. doi: 10.1684/ejd.2013.2193. [DOI] [PubMed] [Google Scholar]
- 31.Narayana A, et al. Vemurafenib and radiation therapy in melanoma brain metastases. Journal of neuro-oncology. 2013;113(3):411–6. doi: 10.1007/s11060-013-1127-1. [DOI] [PubMed] [Google Scholar]
- 32.Forschner A, et al. Radiation recall dermatitis and radiation pneumonitis during treatment with vemurafenib. Melanoma research. 2014;24(5):512–6. doi: 10.1097/CMR.0000000000000078. [DOI] [PubMed] [Google Scholar]
- 33.Stein AP, et al. Xenograft assessment of predictive biomarkers for standard head and neck cancer therapies. Cancer medicine. 2015 doi: 10.1002/cam4.387. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Ryu CH, et al. Valproic acid downregulates the expression of MGMT and sensitizes temozolomide-resistant glioma cells. Journal of biomedicine & biotechnology. 2012;2012:987495. doi: 10.1155/2012/987495. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Supplemental Figure 1. Radiation cooperates with PLX4720 to reduce S phase and induce apoptosis in BRAF V600E glioma cells. Flow cytometry analyses of (A–B) cell cycle using BrdU and 7-AAD staining; and (C–D) apoptosis using Annexin V staining. Cells analyzed include (A, C) BRAF WT GBM36 and (B, D) BRAF V600E DBTRG-05MG cells. These additional cell lines complement the data shown in Figures 2 and 3 and utilize identical methods to those delineated in Figures 2 and 3.
Supplemental Figure 2. Flow cytometric in vitro measurement of β-galactosidase levels in (A) GBM36 (BRAF WT) and (B) AM-38 (BRAF V600E) cells reveals higher background levels of senescence in BRAF V600E cells compared to BRAF WT cells but no pronounced effects of PLX4720, radiation or their combination on levels of senescent cells.
Supplemental Figure 3. Immunohistochemical analyses of survival study mice reveals combination (concurrent) therapy of PLX4720 and radiation significantly decreases tumor proliferation (Ki-67 staining) and increases cleaved caspase 3 (CC3) compared to control mice. Comparisons of combination therapy to each monotherapy were not statistically significant.
Supplemental Table 1. BRAF V600E inhibition by PLX4720 leads to growth inhibition in BRAF V600E cell lines, but IC50 values were not reached in BRAF WT glioma lines.
Supplemental Table 2. T-test comparisons of Ki67, pMAPK, p21 and γH2AX immunohistochemical staining of BRAF V600E tumor xenograft sections treated with PLX4720, radiation or combination (sequential) therapy assessed 6 hours after completion of treatment.