

Abstract
Genetic regulatory proteins inducible by small molecules are useful synthetic biology tools as sensors and switches. Bacterial allosteric transcription factors (aTFs) are a major class of regulatory proteins, but few aTFs have been redesigned to respond to new effectors beyond natural aTF-inducer pairs. Altering inducer specificity in these proteins is difficult because substitutions that affect inducer binding may also disrupt allostery. We engineered an aTF, the Escherichia coli lac repressor, LacI, to respond to one of four new inducer molecules: fucose, gentiobiose, lactitol or sucralose. Using computational protein design, single-residue saturation mutagenesis or random mutagenesis, along with multiplex assembly, we identified new variants comparable in specificity and induction to wild-type LacI with its inducer, isopropyl β-D-1-thiogalactopyranoside (IPTG). The ability to create designer aTFs will enable applications including dynamic control of cell metabolism, cell biology and synthetic gene circuits.
Introduction
Allosteric transcription factors (aTFs) encompass several large families of proteins that provide environmental response in bacteria. Upon binding a small molecule, aTFs undergo a conformational change that alters their affinity for an operator DNA sequence that is often found upstream of regulated metabolic operons or transporter genes1–4. Allosteric transcription factors have been co-opted for use as gene expression switches5 that are a cornerstone in synthetic biological applications. For example, aTFs can serve as intracellular metabolite sensors to enable directed evolution of biosynthetic pathways6,7, as devices to control information flow and feedback regulation in synthetic gene networks8, and as switches in metazoan systems to provide synthetic control of cell differentiation and development.
Expanding aTFs to respond to new molecules can greatly increase their utility9,10. Inducer recognition and transcriptional response in aTFs are tightly coupled through allostery, making redesign toward new inducers challenging. Residues mediating allostery are generally unknown and can be distributed throughout the protein structure11; additionally, ligand-binding domain substitutions often disrupt allosteric communication with the DNA-binding domain8,12. High-throughput genetic approaches offer the possibility of understanding allostery at molecular resolution13, but this promise remains unrealized.
Previous work has demonstrated that random or saturation mutagenesis can evolve greater specificity in LuxR, a promiscuous aTF14, and new inducer responses in NahR15, DmpR16, XylR17, TetR18 or AraC19. Notably, saturation of five key positions yielded mevalonate-responsive AraC variants useful for metabolic engineering10. Computational approaches can sample a much larger mutagenic space; for example, homology modeling-based redesign of PobR was used for 3,4-dihydroxybenzoate response20, and mechanistic insights were leveraged to introduce vanillin response to QacR21.
Here we present a general strategy to engineer aTF response to new inducer molecules, using the E. coli lac repressor, LacI, as a test case.
Results
Choice of new inducer molecules
LacI, which natively regulates the lactose catabolism operon, lacZYA, in response to the disaccharide allolactose, also responds to IPTG. As new inducer targets, we chose gentiobiose, fucose, and synthetically derived lactitol and sucralose, four saccharides not metabolizable by E. coli22. These molecules represent targets with increasing apparent chemical difference from known LacI inducers (Supplementary Fig. 1).
Design, synthesis and assembly of variants
To capture the effects of protein residues both proximal and distal to the ligand-binding pocket, we used three methods to create LacI variants: computational design, protein-wide single-amino-acid saturation mutagenesis and error-prone PCR.
Using an adaptation of the Rosetta software suite23,24, we computationally designed LacI variants to bind the three target ligands that are most dissimilar from the native inducer: fucose, lactitol and sucralose (Online Methods and Supplementary Note). Rosetta has been used to design proteins with new ligand-binding interactions25, though it does not account for allostery. We generated DNA oligonucleotides encoding designed variants by microarray-based synthesis26, which specifies a pool of exact oligonucleotide sequences (Online Methods). Because of oligonucleotide length limitations, residues mutable during Rosetta design were confined to three segments of lacI (encoding residues 73–125, 148–197 and 245–296), encompassing the majority of the ligand-binding pocket. We synthesized and cloned LacI libraries encoding each single segment (mean of 4.2 mutations per gene) and combined them through overlap PCR to capture full designs with mutations in each segment (mean of 12.6 mutations per gene).
Substitution of aTF residues distal to the ligand interface can influence induction through long-range effects19,27–29. Thus, we created a variant library encoding all LacI single-amino-acid substitutions using microarray-synthesized DNA) by tiling mutable sequences in windows of 36 residues, totaling 6,800 variants. Sampling by high-throughput sequencing indicated that this library captured ∼88% of all single mutations, with at least 17 of the 19 possible substitutions encoded at ∼74% of positions (Supplementary Fig. 2). Finally, we amplified lacI codons 67–297 by error-prone PCR to generate a library with a mean of five mutations per gene (Online Methods).
A screen to identify LacI variants with new ligand response
Affinity-based screens can evaluate binding, but not allostery, so we developed an in vivo selection-screening method designed to capture aTF variants functional in both allosteric states: DNA-bound in the absence of inducer and allosterically activated by inducer (Fig. 1 and Online Methods). Into the genome of E. coli, we integrated a reporter construct consisting of the genes encoding green fluorescent protein (GFP) and TolC under transcriptional control of the LacI-regulated promoter, pLlacO5; TolC is an E. coli outer membrane porin that mediates the entry of the bacteriocin toxin, colicin E1 (ref. 30). First, we enriched for LacI variants that bind DNA and repress transcription (generally 15–60% per library) by colicin E1 selection (Supplementary Fig. 3a). We verified that colicin E1 selection strongly enriched (>99.5%) for clones encoding full-length lacI variants devoid of frameshift mutations, a common occurrence owing to deletion errors in array-synthesized DNA. Subsequently we collected variants that activated transcription of the GFP gene in response to a target ligand by fluorescence-activated cell sorting (FACS; Supplementary Fig. 3b). We assayed these inducible cells clonally by high-throughput flow cytometry to measure baseline GFP and induction ratios (fluorescence ratio of induced to uninduced cells) for the new inducer (Supplementary Table 1). Wild-type LacI was induced 15-fold by IPTG in this screening system (Supplementary Fig. 4).
Figure 1.
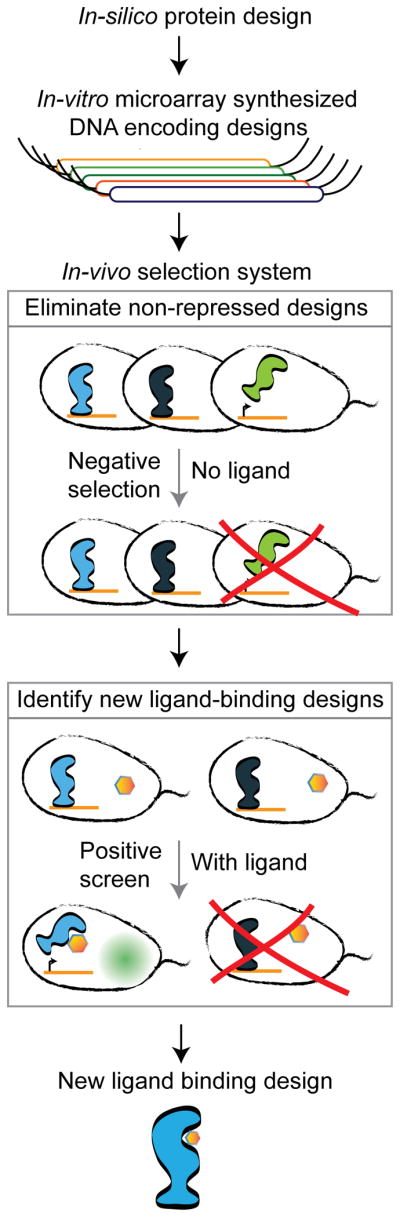
General workflow for designing new ligand binding in an aTF. Schematic diagram showing the steps in the design workflow.
We used a genomically integrated single reporter copy to avoid fluorescence artifacts arising from fluctuations in plasmid copy number. Higher reporter copy numbers allow higher total fluorescence, which yields higher fold induction31; we expect that the induction of each LacI variant would scale accordingly in a multicopy reporter system, higher than the values reported here. Although GFP has been used for dual positive and negative reporter screens for AraC19, we preferred a TolC-based negative selection for enriching transcriptionally repressed variants owing to the poorer resolvability of the flow cytometer at low fluorescence levels.
We targeted computational design to three segments of the LacI protein (residues 73–125, 148–197 and 245–296), which form the ligand-binding pocket, but 14 of the 15 highest-ranked Rosetta full-protein designs (five per ligand) did not repress transcription when tested independently (Supplementary Fig. 5), suggesting that a high mutational burden inactivates allostery. We therefore chose to screen the libraries encoding single Rosetta-designed segments for target ligand response. In addition, we screened LacI single-amino-acid substitution and error-prone PCR libraries for induction by each ligand.
New ligand responses by LacI variants
We identified Rosetta-designed LacI variants that responded to fucose, lactitol or sucralose (Fig. 2). The best clones showed induction values similar to wild-type LacI induction by IPTG (15-fold; Supplementary Fig. 4), demonstrating that new ligand binding can be engineered without compromising allosteric regulation. For each target ligand, we found multiple unique variants that resulted in response to the same ligand (Fig. 2, Supplementary Fig. 6a–c and Supplementary Table 1). The diversity of responsive variants differed across the three ligands. For example, sucralose-responsive sequences were the most diverse, with the most responsive clones often containing four or more mutations (Fig. 2e,f and Supplementary Table 1). Fucose response was mediated by independent substitutions to residues in different regions of the binding pocket (Q291T or I79Q; Fig. 2a,b), but lactitol response nearly always required substitution of Ile79 (Fig. 2c,d). Substitutions of Ile79 and Gln291 were frequently present in variants that responded to new inducers (Fig. 2), suggesting that these residues may determine ligand specificity of the binding pocket.
Figure 2. Characterization of Rosetta-designed variants responding to new inducers.

(a–f) Data for fucose (a,b), lactitol (c,d) and sucralose (e,f). Amino-acid substitution profiles with heat maps indicating fold induction are included (a,c,e). We computed these values by normalizing the induction value of each clone by the number of mutations it contained and reporting the highest such value per unique position and amino acid pair. Dose-response curves for variants and wild-type (WT) LacI induced with the target ligand are also shown (b,d,f). Error bars represent s.d. of fold induction from three biological replicates.
The Ile79 substitutions found in the top lactitol-responsive variants tended to be small (glycine or alanine) to accommodate the bulky ligand, or have hydroxyl- or thiol-containing side groups (threonine, tyrosine or cysteine) capable of hydrogen bonding with the sugar alcohol, and were mostly distinct from the substitutions in fucose-responsive Ile79 variants (glutamine, alanine, methionine or leucine). This comparison suggests that Ile79 has an important role in ligand specificity. However, computational alignment studies predicting ligand specificity-determining residues in LacI family proteins had identified neither Ile79 nor Gln291 as key determinants32,33. Thus, our structure-based computational design targeted cryptic determinants of ligand specificity.
Furthermore, we compared our laboratory-evolved fucose-responsive variants to naturally occurring fucose-responsive aTFs in the GalR/S family. We found that three designed substitutions conferring fucose response (I79L, I79M and Y273F; Supplementary Fig. 7a,b) were also significantly differentially conserved between fucose-responsive GalR/S proteins and orthologous LacI sequences (P = 0.0471; Online Methods). This result suggests that our design method recapitulates natural evolutionary solutions to fucose binding.
We tested the utility of error-prone PCR for aTF mutation because this method is simple and widely accessible. Error-prone PCR generated variants responsive to fucose or lactitol but not to sucralose, and was much less effective than computational design as measured by the maximum induction of variants (10.5-fold versus 5.0-fold for fucose and 7.1-fold versus 4.8-fold for lactitol; Supplementary Table 2a) or the proportion of variants after sorting that showed twofold or greater induction (42.7% versus 17.7% for fucose and 27.1% versus 5.2% for lactitol; Supplementary Table 2b). The computationally designed variants with the strongest response to sucralose contained four mutations; this combinatorial complexity of mutations is likely too large to be sufficiently sampled by error-prone PCR.
Distributed substitutions affecting LacI ligand binding
Substitutions distal to the ligand- or DNA-binding domains can influence the affinity of LacI for ligand or DNA through cryptic allosteric networks27–29. We systematically investigated this effect on new ligand binding by assaying 6,000 single-amino-acid substitutions of LacI for response to gentiobiose, a molecule highly similar to the native inducer, allolactose.
From single-amino-acid saturation mutagenesis libraries, we identified many gentiobiose-responsive variants, including some with substitutions far from the ligand-binding site; substitutions primarily clustered in three regions: the binding pocket, the dimerization interface and the DNA-binding domain (Fig. 3a,b). The top variants with a single substitution in the binding pocket (Q291H), dimerization interface (R255H) or DNA-binding domain (V20A) showed similar induction (7.7-fold, 8.4-fold and 6.7-fold, respectively), suggesting that allosteric effects of distal substitutions are as potent as ligand-proximal substitutions in the binding pocket (Fig. 3a–c).
Figure 3. Characterization of gentiobiose-responsive variants from the protein-wide single-amino-acid substitution library.
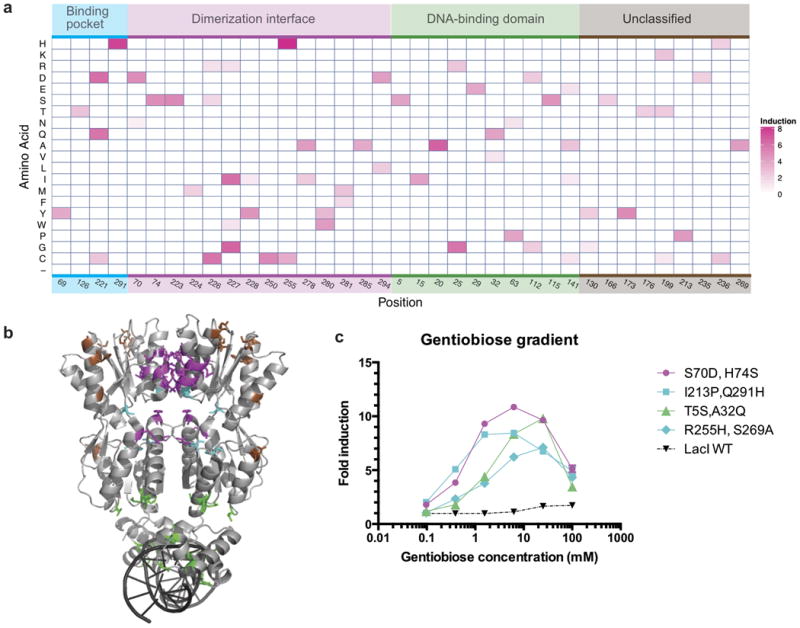
(a) Amino-acid substitution profile with heat map indicating fold induction. Substitutions are classified into four groups on the basis of their location in the protein structure: ligand-binding pocket (cyan), dimerization interface (purple), DNA-binding domain (green), and otherwise unclassified (brown). (b) LacI structure (PDB identifier 1LBG) that includes wild-type side chains with residue substitutions in gentiobiose-responsive variants showing greater than 4.0-fold induction highlighted and colored by classification. (c) Dose-response curves for four gentiobiose-responsive variants (colored by classification of substitutions) and WT LacI. Secondary mutations are due to synthesis errors. Error bars represent s.d. of fold induction from three biological replicates; error bars are not displayed where they overlap plot markers.
Many gentiobiose-responsive variants had substitutions at the dimerization interface of the ligand-binding domain (Fig. 3b). Despite the fact that we designed single mutations, the most responsive variants each contained an additional substitution arising from DNA synthesis errors; a double mutant encoding two dimerization interface substitutions (S70D,H74S) showed the highest induction (>10-fold; Fig. 3c). Library sequencing before and after negative selection revealed that many substitutions in the dimerization interface ablate DNA binding (Supplementary Fig. 8). For example, at residue Ala250, 12 of 19 possible substitutions were not tolerated, but the permissible A250C substitution generated a gentiobiose response (Supplementary Fig. 8). These results are consistent with mutational and biophysical studies showing that allosteric signal propagation in wild-type LacI upon IPTG binding involves communication between monomers via the dimer interface34.
It was more surprising that substitutions in the DNA-binding domain (for example, T5S, V15I, V20A, N25G and H29E) or near this domain (for example, H112D, H112G and L115S), about 40–50 Å from the ligand-binding pocket, retained DNA binding and yielded a strong gentiobiose response (Fig. 3a,b). We measured the dose-dependent response of four top variants carrying mutations within distinct regions: the binding pocket (Q291H,H173Y), N-terminal dimerization interface (S70D,H74S), C-terminal dimerization interface (R255H,S269A) and the DNA-binding domain (T5S,A32Q; Fig. 3c). This positional diversity underscores the whole-protein phenomenon of allostery, suggesting that many distal substitutions can subtly rearrange the conformation of the ligand-binding domain to alter ligand specificity.
Ligand promiscuity of LacI variants
We found that nearly all variants responsive to a new inducer retained a strong response to IPTG. To assess ligand promiscuity, we measured the induction of select variants against all five ligands: fucose, lactitol, sucralose, gentiobiose and IPTG (Fig. 4 and Supplementary Fig. 9). Ligand promiscuity was widespread, as most variants showed some reactivity to more than one new inducer.
Figure 4. Ligand cross-reactivity of LacI variants.

(a–d) For top variants displayed in Figures 2 and 3, a dose response was determined for indicated ligands. Values displayed represent the highest fold induction at any ligand concentration. Variants displayed were designed for binding to gentiobiose (a), fucose (b), lactitol (c) and sucralose (d). Error bars represent s.d. of fold induction from three biological replicates.
Fucose- and gentiobiose-responsive variants showed cross-reactivity with each other (Fig. 4a,b). However, substitutions I79G, I79S and I79T were specific to lactitol (Fig. 4c and Supplementary Fig. 9c), again highlighting the previously uncharacterized role of Ile79 in ligand-specificity determination32,33.
Variants responsive to lactitol and sucralose were overall less promiscuous (Fig. 4c,d and Supplementary Fig. 9); in particular, the N246D,Y273H variant was highly specific to sucralose (Fig. 4d). The pervasive response to IPTG by nearly all engineered variants shows that response to the native inducer is robust to many substitutions, and highlights the need for activity maturation or negative design against IPTG binding.
Activity maturation to improve specificity and induction
We used two approaches to mature the activity of variants: for greater ligand specificity, we individually shuffled promiscuous hits with mutations that ablate off-target binding, and to increase induction, we combined multiple beneficial mutations for the same ligand Using FACS, we collected 44 variants that reduced IPTG induction, called Is variants12, with substitutions near the binding pocket (Supplementary Table 3). We combined these Is variants with the gentiobiose-responsive promiscuous variant Q291H, and via screening we uncovered chimeras that not only completely lost IPTG induction (Fig. 5a), but also showed greater induction with gentiobiose.
Figure 5. Activity maturation of LacI.

(a) Induction response of WT LacI and three variants toward gentiobiose and IPTG. Q291H is a promiscuous variant found during the initial screen. Activity-matured variants Q291H,A266L,T276I and Q291H,T276L,S279G were found after shuffling with Is variants. (b) Induction response of WT LacI and three LacI variants toward sucralose and IPTG. Quadruple mutant I160S,H163W,S191A,L196R was uncovered in the initial sucralose-response screen. Activity-matured variants N125S,I160S,H163W,S191A,L196R,R303L and N125S,I160S,H163W,S191A,L196R were found after shuffling of a library of sucralose-responsive variants. Error bars represent s.d. of fold induction from three biological replicates.
To increase induction values, we shuffled together the genes encoding 31 sucralose-responsive variants (Supplementary Table 1) via PCR reassembly of DNase I gene fragments (Online Methods). We identified several clones with improved induction by sucralose, up to 22-fold (Fig. 5b), which exceeds the wild-type LacI response to IPTG (15-fold). These variants also showed a dramatic decrease in IPTG induction compared to their parent sequence (threefold versus 14-fold, respectively; Fig. 5b). The activity maturation goals of increased specificity and induction appear to be coupled, and may simultaneously improve as the binding pocket adjusts to better fit a new ligand. These results show that simple combinatorial mutational strategies can substantially improve the specificity and fold induction of initial hits.
Crystal structure of a sucralose-responsive variant
To understand the molecular details of how a computationally designed LacI variant binds a bulkier inducer such as sucralose, we crystallized apo and sucralose-bound forms of a sucralose-responsive quadruple mutant (Fig. 6 and Supplementary Table 4).
Figure 6. Crystal structure and GFP induction with ligand of sucralose-binding LacI design variant (D149T,S193D,V150A,I156L).
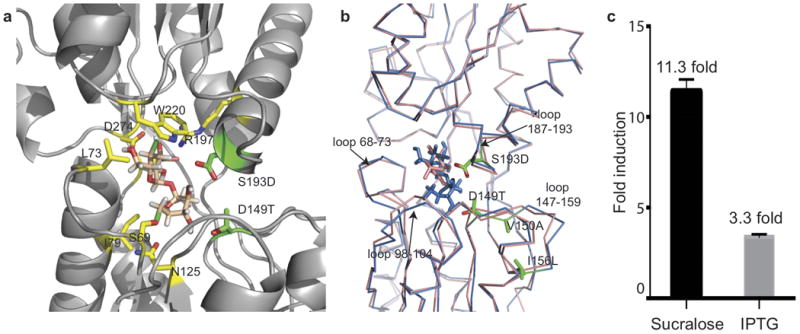
(a) Zoomed-in view of sucralose bound to LacI quadruple mutant. Designed residues D149T and S193D are shown in green; V150A and I156L are outside the field of view. Other key interactions of native residues are shown in yellow. (b) Backbone C-α structural superposition of WT LacI (pink) and sucralose-binding LacI variant (blue). Designed residues are shown in green, and loops undergoing substantial conformational change are marked. (c) Fold induction response of the sucralose-binding variant with sucralose and IPTG at 100 mM ligand concentration. PDB identifiers of the LacI variant in apo and sucralose-bound forms are 4RZS and 4RZT, respectively.
The chlorines are stabilized either by π -bond interactions with aromatic residues or by electron acceptor groups, involving residues Trp220, Phe161 and Phe293 in π interactions, and Ser69 and Asn125 in electron withdrawal (Fig. 6a). Sucralose displaces the binding-pocket loops (Fig. 6b) and makes more optimal hydrogen bonds with the protein through substitutions that alleviate steric clash, D149T and S193D, and substitutions that improve side-chain packing in the loop segment 148–159, V150A and I156L (Fig. 6b).
This variant showed strong sucralose and much-attenuated IPTG responses (Fig. 6c) with an uninduced baseline comparable to wild-type LacI, showing that allostery was not disrupted by the four substitutions. The malleability of the binding pocket to accommodate a large, chemically divergent inducer highlights the evolvability of natural aTFs to bind diverse ligands.
Discussion
Understanding and modifying the allosteric regulation of proteins is of considerable interest in the fields of biotechnology and medicine, given the prevalence of allostery in enzyme regulation, protein drug activity, and small-molecule sensing. However, designing a protein to alter allosteric regulation is more challenging than designing for binding alone.
Our results suggest a general strategy for engineering aTF ligand responses (Supplementary Fig. 10). Single mutations or error-prone PCR may be sufficient for target inducers that closely resemble a known inducer, but for more dissimilar target ligands, computational design is preferred to produce complex mutational combinations required for response. The plasticity of each aTF may vary considerably, so related aTFs should be engineered if no responsive variants are initially found, with the caveats that the aTF structure, operator DNA sequence, and inducer identity must be available; each new aTF that is characterized becomes a potential starting point for biosensor design. Once initial hits are found, their activity can be matured for greater induction or specificity as required.
Besides aTFs, other biosensor paradigms include riboswitches35, reporter domain-coupled allosteric proteins36, ligand-dependent protein dimerization37 and ligand-conditional protein stability38 in which proteins are engineered to be stabilized through ligand binding and degraded otherwise. Riboswitches, which are also allosteric gene regulators, have proven surprisingly hard to engineer.
Designer aTFs should find utility in many applications. Metabolic engineering approaches increasingly rely on high-throughput screens and selections to identify productive cells6,7. Alleviating the reliance of this approach on natural sensory proteins opens new opportunities for biosynthesis of many valuable chemicals, including the identification of novel biosynthetic pathways in metagenomic libraries derived from microbes and plants. For instance, the gentiobiose-responsive LacI variant could be used to screen for β-glucosidases that carry out transglycosylation of glucose to produce gentiobiose39.
New aTFs can also be powerful cell-biological discovery tools. The dynamic composition of metabolites in a cell is a signature of the phenotypic state of the cell. Engineered aTFs that respond to key metabolites could expand single-cell interrogation beyond the genome and transcriptome to report on metabolic dynamics of each cell at high temporal resolution. The widely used TetOn-Off system for mammalian gene regulation40 relies on a bacterial aTF (TetR) adapted for mammalian cells. An expanded repertoire of similarly adapted engineered aTFs with non-interacting inducers would enable tunable, independent control of multiple genes to exquisitely regulate signaling, development and differentiation pathways. We expect the ability to create aTFs responsive to new target molecules to have wide-reaching benefits for synthetic biology.
Online Methods
lacI expression vector and screening strain construction
The E. coli strain K12 MG1655 derivative EcNR2 (ref. 41) was modified by lambda Red recombineering42 to replace the native lacI gene with a zeocin-resistance cassette. The tetR and bla genes found on the lambda prophage were similarly replaced with a tetracycline-resistance cassette. We then used recombineering to create the final screening strain by replacing the native promoters 5′ to the tolC gene with a linear PCR product encoding the following: one copy of promoter pLlacO5 controlling transcription of a cocistron of superfolder GFP43 and a kanamycin-resistance cassette, and a second copy of pLlacO in a divergent orientation controlling transcription of tolC. All modifications were verified by sequencing.
A copy of the E. coli lacI gene that had been recoded to facilitate cloning site sequences (Supplementary Table 5) was cloned into a low-copy plasmid backbone (SC101 origin of replication) carrying a spectinomycin resistance, to create plasmid pSC101_lacI_specR (Supplementary Table 5). The lacI variant gene was expressed from the strong pLtetO promoter5, which is unregulated in the screening strain because of the deletion of tetR. Colonies of the screening strain fluoresce visibly under blue light, but fluorescence was no longer visible after transformation with plasmid pSC101_lacI_specR. Observed reversions of the repressed phenotype were low and did not necessitate restoration of the MutS+ phenotype in this strain.
Rosetta computational design of LacI proteins
Computationally designed LacI variant candidates were generated using the Rosetta software suite23,24 for fucose, lactitol and sucralose. For each ligand, we generated a library of hundreds of allowable conformational isomers, or conformers, using OpenEye Omega software44,45. These conformers sample discrete states along rotatable torsions of various bonds. Each conformer was docked into the ligand-binding pocket of a high-resolution crystal structure of LacI34 (PDB identifier 2P9H). The design protocol consists of multiple rounds of rigid-body perturbation of the ligand position followed by combinatorial mutagenesis and backbone minimization to optimize the interaction of the selected conformer within the pocket. The mutagenesis included exhaustive sampling of the rotameric states of each amino acid in a backbone context–dependent manner. All designs were loosely filtered on the basis of a standard set of energy terms that ensures diversity of solutions while eliminating poor designs (the Supplementary Note includes full method details). Rosetta is distributed under a free academic license (http://www.rosettacommons.org/).
Construction of LacI variant libraries
We constructed the lacI gene variant libraries by cloning oligonucleotides encoding the desired mutations into plasmid pSC101_lacI_specR amplified by PCR with primers (Supplementary Table 6) to appropriately linearize, add BsaI recognition sequence (5′-GGTCTCN) and remove the wild-type lacI coding sequence segment to be replaced. Oligonucleotide pools encoding Rosetta-designed sequences were obtained from Agilent Technologies, and each encoded substitutions within one of the following sets of lacI codons: 73–125, 148–197 and 245–296. Constructing the single-residue replacement library involved replacing lacI codons 3–359 with one missense codon encoding each of the remaining 19 natural amino acids (6,802 sequences). Oligonucleotides encoding these mutations were synthesized on a B3 Synthesizer (CustomArray), and were organized into the following tiles spanning the lacI gene: 3–38, 39–74, 75–110, 111–146, 147–182, 183–218, 219–254, 255–290, 291–326 and 327–359.
Oligonucleotides in each pool were encoded as a concatamer of the forward priming sequence, a BsaI restriction site (5′-GGTCTCN), appropriate four-base upstream overhang, lacI mutant sequence segment, appropriate four-base downstream overhang, the reverse complement of the BsaI restriction site (5′-NGAGACC-3′) and the reverse complement of the reverse priming sequence. Subpools were amplified using primers specific to each subpool (Supplementary Table 6) from each oligonucleotide pool by means of quantitative PCR (SYBR qPCR master mix, KAPA Biosystems; 20 μl reaction volume; 0.1–1 ng oligonucleotide pool template) until the second inflection point on a real-time plot of cycle number versus well fluorescence indicated amplification saturation was beginning, following ref. 26.
We constructed error-prone PCR libraries by amplifying the lacI codons 67–297 with GeneMorf II polymerase (Agilent Technologies) using primers including BsaI recognition sites (Supplementary Table 6) and a 15-ng gene fragment template, which resulted in 10 μg of PCR product for 670-fold amplification and a calculated mean of 5.3 coding mutations per lacI gene. Subpool amplification PCR products and error-prone PCR products were digested with BsaI-HF enzyme (New England Biolabs (NEB)), and appropriate plasmid backbones were digested with BsaI-HF and DpnI enzymes (NEB). Backbone termini were dephosphorylated with Antarctic phosphatase enzyme (NEB). All digested nucleotides were cleaned up with Agencourt AMPure XP beads (Beckman Coulter; 1:1 ratio of beads to DNA), fragments were ligated into backbones with T4 DNA ligase (NEB), and ligation products were purified with AMPure XP beads and transformed into electrocompetent E. coli DH10B cells (NEB). After 1 h, a 1 μl aliquot of transformed cells was plated onto LB spectinomycin selective medium for estimation of the transformed library size.
Selection and screening protocols for ligand response
For library transformations, we made the the screening strain electrocompetent by harvesting early log phase cells (10 ml per transformation at OD600 nm = 0.15–0.25), removing salt through two washes with ice-cold 10% glycerol, and resuspended the cells in 50 μl cold 10% glycerol. 10 ng of library plasmid were electroporated into the competent cells, which were recovered for 1 h in 1 ml of SOC medium. To estimate the number of transformants, we plated 1 μl of recovered cells on selective LB spectinomycin medium, and we added the remainder of the recovered cells to 10 ml of LB spectinomycin medium for overnight selection.
Screening-strain cells expressing LacI variants that do not bind to operator DNA constitutively express tolC and GFP genes; these were eliminated through negative selection by overnight selection with colicin E1 protein. We added 5 μl of saturated library transformation culture to 150 μl of LB spectinomycin medium supplemented with tenfold serial dilutions of purified colicin E1 protein (2.73 mg/ml), in the range from 1:100 to 1:1,000,000; a control population of the same library was grown overnight without colicin E1. Enrichment of DNA-bound lacI variants was verified by flow cytometry the next day by measuring the fraction of GFP+ cells in the colicin E1 incubated and control populations. A 1:100,000 dilution of colicin E1 (20.7 ng/ml) was generally found to be optimal.
After negative selection, the colicin E1 selected cells were washed twice with LB and grown for 1 h in LB spectinomycin lacking colicin E1. These cells were then subjected to a ligand-response test. To carry out a response test, 1.5 μl of saturated culture was added to 150 μl of LB supplemented with spectinomycin and 3 mM concentration of the target ligand; the tested library grown in identical conditions but without ligand was used as a negative control.
Cells with GFP signal greater than the ligand-free control were collected using FACS on an Avalon S3 Sorter (Propel Labs). Because cells expressing a LacI variant responsive to the target ligand often presented a subtle signal, we used the uninduced control to set sorting gates per library-inducer pair. After observing 100,000 cells for the induced and uninduced conditions, the sorting gate was set to maximize the difference in cells falling above the gate between the induced and uninduced conditions. This generally resulted in collecting the top 0.1–1% of the induced library, except where a larger proportion of cells clearly fell above the uninduced condition (for example, for gentiobiose inductions). FACS-isolated cells were immediately recovered in LB and plated on LB-agar containing spectinomycin at a several dilutions to yield a plate with hundreds of clearly separated colonies. Depending on the library, we picked 48–192 colonies into a 96-well plate to clonally test induction response. Each clone was incubated overnight with and without the target ligand (3 mM) concentration. After 16–20 h, the GFP response of each clone was measured with and without the ligand on a flow cytometer with high-throughput sampler (LSRFortressa, Beckton-Dickenson; Supplementary Table 1). The sequence of each lacI variant in ligand-responsive clones was determined using Sanger sequencing (Supplementary Table 1).
Expression and purification of sucralose-responsive LacI variant
Expression strain construction
To overexpress the sucralose-binding variant (carrying substitutions D149T, V150A, I156L and S193D), we cloned into a pET14b vector (Novagen via EMD Millipore) with a constitutive T7 promoter. The lacI variant gene was cloned downstream of His and thrombin tags of the vector. We used the arabinose-inducible T7 expression host BL21-AI (Life Technologies, Inc.) to avoid inducing protein expression with IPTG, which could lead to binding artifacts. We also modified the commercially available E. coli BL21-AI strain by deleting the wild-type copy of lacI gene to avoid heterodimer formation with the substituted variant. We transformed BL21-AI with the pKD46 plasmid42, containing the lambda-Red recombineering machinery on a temperature-sensitive origin of replication (plasmid lost above 37 °C). We replaced the wild-type lacI gene through homologous recombination by transforming the pKD46-containing BL21-AI with a zeocin-resistance cassette flanked by homology arms targeting the lacI endogenous locus. We induced lambda-Red expression with 1% arabinose 1 h before transformation with donor cassette DNA. After recovery, the transformed cells were plated on zeocin-containing LB-agar plates, and colonies were screened to identify wild-type lacI gene deleted strain. The pKD46 plasmid was subsequently removed by growing the cells at 37 °C overnight. The pET14b vector with sucralose-binding variant was transformed into BL21-AI lacI(x02237)zeo for overexpression.
LacI variant protein overexpression and purification
Several colonies of the expression host containing the sucralose-binding LacI variant were used to inoculate a 350 ml LB culture supplemented with 100 μg/ml ampicillin and grown at 37 °C with 230 r.p.m. shaking overnight to an OD600 of 4.6. 35 ml of the overnight inoculum was added to each of the six 2.5-l shake flasks containing 1 l Terrific broth medium with 100 μg/ml ampicillin and grown at 37 °C with 230 r.p.m. shaking. The culture temperature was equilibrated to 18 °C before expression was induced using 0.5 mM IPTG when the OD600 nm was 2.8. The induced cultures were grown at 18 °C for 19 h before they were harvested.
The cell pellet was resuspended at a 2 ml/g ratio in buffer A (20 mM Tris-HCl, pH 8.0, 0.3 M NaCl and10% glycerol), supplemented with 10 mM imidazole, 2 mM βME, 2 μg/ml DNase I, 0.1 mg/ml lysozyme, 1 mM PMSF, 1 tablet/100 ml lysate cOmplete protease inhibitor cocktail (Roche) and 5 mM MgCl. Lysis was done by sonication and the lysate was centrifuged at 35,000g at 4 °C for 30 min. Affinity chromatography was carried out by nutating 1 ml of HisPur Ni-NTA resin (Thermo Scientific) with the cleared lysate at 4 °C for 1 h. The Ni-NTA resin was packed onto a gravity column and then washed twice by 10 column volumes (CV) of buffer A with 10 mM imidazole, once by 10 CV of buffer A with 50 mM imidazole, and then eluted twice by 10 CV of buffer A with 0.3 M imidazole. The fractions containing predominantly target protein were pool and concentrated using a 10 kDa molecular weight cutoff (MWCO) Amicon Ultra-15 concentrator (EMD Millipore).
Size-exclusion chromatography was subsequently performed on a HiLoad 16/60 Superdex 200 PG column (GE Healthcare) at a flow rate of 1 ml/min in buffer A. Pure peak fractions were pooled and concentrated to 12.8 mg/ml using the same type of concentrator mentioned above. This concentrate was subject to a three-step (555-fold dilution factor each step) dialysis in a buffer containing 0.2 M Tris, pH 7.4, 0.2 KCl, 1 mM EDTA, 0.3 mM DTT in 6 kDa MWCO D-Tube Dialyzer Mini dialysis devices (Novagen, 71504-3). The dialyzed protein solution was centrifuged at 16,000g at 4 °C for 5 min, and the concentration was measured to be 12.2 mg/ml via Bradford assay.
Crystallography, X-ray data collection and structure solution
The crystal that led to the unliganded LacI variant structure was grown using hanging-drop vapor-diffusion method in a 24-well VDX plate (Hampton Research). The crystal drop of 0.4 μl of the reservoir solution plus 1.6 μl 12.1 mg/ml protein concentrate was set up against 500 μl of reservoir solution, which was composed of 16% polyethylene glycol 3,350 and 200 mM ammonium nitrate. After 5 weeks of growth, the crystal grew to approximately 600 nm × 200 nm by 200 nm and was harvested by flash-freezing in liquid nitrogen with 23% glycerol as cryoprotectant.
For co-crystallization experiments, sucralose was dissolved in the dialysis buffer to a concentration of 0.5 M. a small amount of the 0.5 M sucralose solution was added to the unliganded protein concentrate at a 1:150 (v/v) ratio, giving a final sucralose concentration of 3 mM and final protein concentration of 12.1 mg/ml (equivalent of 0.3 mM). Crystallization was carried out using hanging-drop vapor-diffusion method. A Mosquito liquid handler (TTP LabTech) set up a drop of 140 nl sucralose containing protein concentrate with 70 nl of crystallization reagent against 100 μl of the same reagent in the reservoir that was composed of 0.1 M HEPES, pH 7.5, 10% polyethylene glycol 6,000 and 5% 2-methyl-2,4-pentanediol (MPD). After 4 weeks, the structure-producing crystal grew to ∼150 nm × 150 nm × 20 nm, and was harvested with 20% glycerol as cryo-protectant.
X-ray data for both the unliganded and sucralose co-crystal structures was collected at the Advanced Photon Source (APS) beam line 24-ID-C at the Argonne National Laboratory. The data were processed using XDS46, followed by anisotropic data removal47. Structure solution was found by molecular replacement using the program Phaser44, using the core domain (residues 62–332) of an existing LacI structure (PDB ID: 1JYE) as the search model. Model was built using the program Coot48 and refined using the program REFMAC49. Translation/libration/screw50 (TLS) vibrational motion analysis and noncrystallographic symmetry (NCS) restraints were used during model refinement.
LacI ortholog/paralog identification and alignment methods
We accessed a database of complete bacterial genomes from ftp://ftp.ncbi.nlm.nih.gov/genomes/archive/old_refseq/Bacteria/ (29 January 2013) and formatted it for BLAST searching. We used BLASTP 2.2.21 (ref. 51) to search the bacterial genome database using full-length protein queries of E. coli LacI and full-length E. coli LacZ, separately, using default settings. Our LacI ortholog set contained 41 sequences from 13,591 total matches meeting these criteria: the top hit in each species, ignoring subspecies; occurred within 10 kb of any E. coli LacZ hit from the same species, accounting for subspecies; and had an E value of less than 0.01. The remainder of the matches were called E. coli LacI paralogs.
Alignments of all sequence matches from the E. coli LacI BLASTP query were done with CLUSTAL 2.0.12 (ref. 52) using default settings and fast pairwise alignment. From the resulting alignment, non-gap E. coli LacI sequence positions were used as a positional reference. Heat-map grids depicting conservation values show the percentage of utilization for the amino-acid at the E. coli LacI position indicated. Gaps are shown as “–” on the y axis. Heat-map grids of induction values are the maximum weighted induction value, which scales the total induction found equally across all co-occurring mutations. Structure diagrams indicate positions with greater than twofold induction over no ligand, colored by the frequency the position was found in the screen.
Comparison of LacI fucose-responsive variants to GalR/S fucose-responsive proteins
To compare our designed substitutions to naturally evolved aTF sequences, we relied on known fucose-responsive GalR/S proteins. We computed whether 17 LacI ligand-proximal substitutions (<5 Å from IPTG in PDB structure 2P9H) conferring greater than twofold response to fucose were enriched among five experimentally characterized fucose-responsive GalR/S orthologs53,54. After alignment, we independently calculated the frequency of amino acids at each position for 41 high-confidence natural LacI orthologs (Supplementary Fig. 7a), and for the five GalR/S orthologs (Supplementary Fig. 7b). We subtracted the LacI ortholog frequencies from GalR/S frequencies at every aligned position and identity, and defined differentially conserved identities to be in the top 5% of this subtracted frequency set. Within these differentially conserved identities, we identified three (I79L, I79M and Y273F) of the 17 ligand-proximal fucose-responsive variants. By Fisher's exact test, this result is significant (P = 0.0471).
Analysis of negative selection via high-throughput sequencing
Single amino-acid libraries were prepared for amplicon sequencing by nested PCR amplification using a first round of PCR with primers annealing within the lacI gene (Supplementary Table 6) and a second round of PCR with primers annealing within the first set of primers and containing i5 or i7 indexing sequences and adapters for sequencing on the MiSeq instrument (Illumina) using 300-base paired-end reads. Paired sequencing reads were collapsed and filtered for sequencing errors using FLASH v. 1.2.11 with a maximum overlap of 300 (ref. 55). Collapsed reads were then translated in three frames, aligned to wild-type protein LacI with BLAT v.35 using default settings, and the best translated alignment by percentage match was retained56. Protein sequences were then trimmed according to the amplicon number and known flanking sequence from the library designs. Sequences with mismatches in the fixed flanking sequences or different than the expected length (containing insertions or deletions) were discarded. Protein sequences were then counted for pre- and post-selection, respectively, for each amplicon. Only sequences harboring a single amino-acid change found in either the pre- or postselection were retained for further analysis. We assembled the single amino-acid sequences as rows in a table with counts for pre- and postselection values as columns. A psuedo-count of one was added to each column. Counts between pre- and postselection were then quantile normalized using the “normalize.quantiles” function from preprocessCore in R v.3.1.1. Log2 fold-changes were computed from the ratio of preselection divided by postselection quantile-normalized counts57. Each protein sequence was positioned and shown with respect to the wild-type LacI position. Final heat-map grids, associated line plots and histograms were created in ggplot2 (ref. 58).
Shuffling with Is variants for enhanced specificity
LacI variants that do not respond to IPTG are called Is clones59. We sorted cells from the single-amino-acid mutant library that showed no GFP signal when incubated with IPTG overnight. After recovery, growth and plating, we picked about 200 colonies for Sanger sequencing. This Is set contained both variants that are allosterically broken and variants that do not recognize IPTG but retain allosteric regulation. Because we were interested in the latter type, we picked only variants with substitutions near the binding pocket, reasoning that these were more likely to reduce IPTG binding without affecting allostery. Although some of the binding-pocket variants may also be allosterically ‘broken’, this set is also likely to contain variants that do not bind to IPTG. We picked about 44 variants (Supplementary Table 3) to form our curated Is set. To switch the specificity of gentiobiose hit Q291H (Fig. 5a), we amplified each of the 44 Is variants including the backbone using PCR primers encoding a Q291H substitution. This gave us 44 chimeras containing Q291H and the Is substitutions. We carried out isothermal assembly of all chimeras together in a single tube. After transformation, recovery and plating, we picked about 100 clones for testing induction with IPTG and gentiobiose.
Supplementary Material
Acknowledgments
We thank B. Turczyk and D. Weigand for synthesizing the single-amino-acid substitution library on the Custom Array synthesizer, and G. Cuneo and V. Toxavidis for assistance with flow cytometry and FACS. We thank Rosetta@ home participants for providing the computing resources necessary for this work. This work was supported by the US Department of Energy (DOE) (DE-FG02-02ER63445 to G.M.C.), a Wyss Technology Development Fellowship (to S.R.), grant T32 HG002295 from the National Human Genome Research Institute, NIH (supporting A.S.G.), and the US National Institute of General Medical Sciences (grant 1P41 GM103533 to S.F.). The sucralose-responsive LacI mutant was purified and crystallized with assistance from the UCLA-DOE Protein Expression Technology Center, the UCLA-DOE X-ray Crystallography Core Facility (both supported by DOE grant DE-FC02-02ER63421) and the UCLA Crystallization Core Facility; in particular we thank M. Collazo for help with protein crystallization. X-ray data collection was facilitated by M. Capel, K. Rajashankar, N. Sukumar, F. Murphy and I. Kourinov of the Northeastern Collaborative Access Team beamline 24-ID-C at the Advanced Photon Source of Argonne National Laboratory, which is supported by US National Institutes of Health grants P41 RR015301 and P41 GM103403. Use of the Advanced Photon Source is supported by the DOE under contract DE-AC02-06CH11357.
Footnotes
Accession codes. Protein Data Bank (PDB): sucralose-responsive LacI variant (D149T,V150A,I156L,S193D) crystallized in apo and sucralose-bound forms, 4RZS and 4RZT, respectively. Gene Expression Ombibus (GEO): library sequencing read data, GSE75009.
Author Contributions: N.D.T., F.J.I., G.M.C. and S.R. conceived the study. N.D.T., S.F., G.M.C. and S.R. designed experiments. N.D.T., A.S.G. and S.R. performed experiments and carried out bioinformatic studies. R.M. and D.B. generated computational protein design candidates. S.C., D.C., M.A. and S.K. solved the crystal structure of a sucralose-binding variant. S.K. helped with Agilent OLS chip library design. J.R. helped with optimizing screening protocols. N.D.T., A.S.G., S.F., G.M.C. and S.R. analyzed the data. N.D.T., A.S.G., S.F., G.M.C. and S.R. wrote the paper.
Competing Financial Interests: S.R., N.D.T. and G.M.C. have filed a patent application (PCT/US15/16868) covering biosensor design methods.
References
- 1.Weickert MJ, Adhya S. A family of bacterial regulators homologous to Gal and Lac repressors. J Biol Chem. 1992;267:15869–15874. [PubMed] [Google Scholar]
- 2.Schell MA. Molecular biology of the LysR family of transcriptional regulators. Annu Rev Microbiol. 1993;47:597–626. doi: 10.1146/annurev.mi.47.100193.003121. [DOI] [PubMed] [Google Scholar]
- 3.Gallegos MT, Schleif R, Bairoch A, Hofmann K, Ramos JL. Arac/XylS family of transcriptional regulators. Microbiol Mol Biol Rev. 1997;61:393–410. doi: 10.1128/mmbr.61.4.393-410.1997. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Ramos JL, et al. The TetR family of transcriptional repressors. Microbiol Mol Biol Rev. 2005;69:326–356. doi: 10.1128/MMBR.69.2.326-356.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Lutz R, Bujard H. Independent and tight regulation of transcriptional units in Escherichia coli via the LacR/O, the TetR/O and AraC/I1-I2 regulatory elements. Nucleic Acids Res. 1997;25:1203–1210. doi: 10.1093/nar/25.6.1203. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Dietrich JA, Shis DL, Alikhani A, Keasling JD. Transcription factor-based screens and synthetic selections for microbial small-molecule biosynthesis. ACS Synth Biol. 2013;2:47–58. doi: 10.1021/sb300091d. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Raman S, Rogers JK, Taylor ND, Church GM. Evolution-guided optimization of biosynthetic pathways. Proc Natl Acad Sci USA. 2014;111:17803–17808. doi: 10.1073/pnas.1409523111. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Lu TK, Khalil AS, Collins JJ. Next-generation synthetic gene networks. Nat Biotechnol. 2009;27:1139–1150. doi: 10.1038/nbt.1591. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Dietrich JA, McKee AE, Keasling JD. High-throughput metabolic engineering: advances in small-molecule screening and selection. Annu Rev Biochem. 2010;79:563–590. doi: 10.1146/annurev-biochem-062608-095938. [DOI] [PubMed] [Google Scholar]
- 10.Tang SY, Cirino PC. Design and application of a mevalonate-responsive regulatory protein. Angew Chem Int Edn Engl. 2011;50:1084–1086. doi: 10.1002/anie.201006083. [DOI] [PubMed] [Google Scholar]
- 11.Süel GM, Lockless SW, Wall MA, Ranganathan R. Evolutionarily conserved networks of residues mediate allosteric communication in proteins. Nat Struct Biol. 2003;10:59–69. doi: 10.1038/nsb881. [DOI] [PubMed] [Google Scholar]
- 12.Markiewicz P, Kleina LG, Cruz C, Ehret S, Miller JH. Genetic studies of the lac repressor. XIV Analysis of 4000 altered Escherichia coli lac repressors reveals essential and non-essential residues, as well as “spacers” which do not require a specific sequence. J Mol Biol. 1994;240:421–433. doi: 10.1006/jmbi.1994.1458. [DOI] [PubMed] [Google Scholar]
- 13.Raman S, Taylor N, Genuth N, Fields S, Church GM. Engineering allostery. Trends Genet. 2014;30:521–528. doi: 10.1016/j.tig.2014.09.004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Collins CH, Arnold FH, Leadbetter JR. Directed evolution of Vibrio fischeri LuxR for increased sensitivity to a broad spectrum of acyl-homoserine lactones. Mol Microbiol. 2005;55:712–723. doi: 10.1111/j.1365-2958.2004.04437.x. [DOI] [PubMed] [Google Scholar]
- 15.Cebolla A, Sousa C, de Lorenzo V. Effector specificity mutants of the transcriptional activator NahR of naphthalene degrading Pseudomonas define protein sites involved in binding of aromatic inducers. J Biol Chem. 1997;272:3986–3992. doi: 10.1074/jbc.272.7.3986. [DOI] [PubMed] [Google Scholar]
- 16.Wise AA, Kuske CR. Generation of novel bacterial regulatory proteins that detect priority pollutant phenols. Appl Environ Microbiol. 2000;66:163–169. doi: 10.1128/aem.66.1.163-169.2000. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Galvão TC, Mencía M, de Lorenzo V. Emergence of novel functions in transcriptional regulators by regression to stem protein types. Mol Microbiol. 2007;65:907–919. doi: 10.1111/j.1365-2958.2007.05832.x. [DOI] [PubMed] [Google Scholar]
- 18.Scholz O, Köstner M, Reich M, Gastiger S, Hillen W. Teaching TetR to recognize a new inducer. J Mol Biol. 2003;329:217–227. doi: 10.1016/s0022-2836(03)00427-3. [DOI] [PubMed] [Google Scholar]
- 19.Tang SY, Fazelinia H, Cirino PC. AraC regulatory protein mutants with altered effector specificity. J Am Chem Soc. 2008;130:5267–5271. doi: 10.1021/ja7109053. [DOI] [PubMed] [Google Scholar]
- 20.Jha RK, Chakraborti S, Kern TL, Fox DT, Strauss CEM. Rosetta comparative modeling for library design: engineering alternative inducer specificity in a transcription factor. Proteins. 2015 May 13; doi: 10.1002/prot.24828. [DOI] [PubMed] [Google Scholar]
- 21.de Los Santos ELC, Meyerowitz JT, Mayo SL, Murray RM. Engineering transcriptional regulator effector specificity using computational design and in vitro rapid prototyping: developing a vanillin sensor. ACS Synth Biol. 2015 Aug 19; doi: 10.1021/acssynbio.5b00090. [DOI] [PubMed] [Google Scholar]
- 22.AbuOun M, et al. Genome scale reconstruction of a Salmonella metabolic model: comparison of similarity and differences with a commensal Escherichia coli strain. J Biol Chem. 2009;284:29480–29488. doi: 10.1074/jbc.M109.005868. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Jiang L, et al. De novo computational design of retro-aldol enzymes. Science. 2008;319:1387–1391. doi: 10.1126/science.1152692. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Röthlisberger D, et al. Kemp elimination catalysts by computational enzyme design. Nature. 2008;453:190–195. doi: 10.1038/nature06879. [DOI] [PubMed] [Google Scholar]
- 25.Tinberg CE, et al. Computational design of ligand-binding proteins with high affinity and selectivity. Nature. 2013;501:212–216. doi: 10.1038/nature12443. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Kosuri S, et al. Scalable gene synthesis by selective amplification of DNA pools from high-fidelity microchips. Nat Biotechnol. 2010;28:1295–1299. doi: 10.1038/nbt.1716. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Swint-Kruse L, Elam CR, Lin JW, Wycuff DR, Shive Matthews K. Plasticity of quaternary structure: twenty-two ways to form a LacI dimer. Protein Sci. 2001;10:262–276. doi: 10.1110/ps.35801. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Swint-Kruse L, Zhan H, Fairbanks BM, Maheshwari A, Matthews KS. Perturbation from a distance: mutations that alter LacI function through long-range effects. Biochemistry. 2003;42:14004–14016. doi: 10.1021/bi035116x. [DOI] [PubMed] [Google Scholar]
- 29.Xu J, Matthews KS. Flexibility in the inducer binding region is crucial for allostery in the Escherichia coli lactose repressor. Biochemistry. 2009;48:4988–4998. doi: 10.1021/bi9002343. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.DeVito JA. Recombineering with tolC as a selectable/counter-selectable marker: remodeling the rRNA operons of Escherichia coli. Nucleic Acids Res. 2008;36:e4. doi: 10.1093/nar/gkm1084. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Rogers JK, et al. Synthetic biosensors for precise gene control and real-time monitoring of metabolites. Nucleic Acids Res. 2015;43:7648–7660. doi: 10.1093/nar/gkv616. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Mirny LA, Gelfand MS. Using orthologous and paralogous proteins to identify specificity-determining residues in bacterial transcription factors. J Mol Biol. 2002;321:7–20. doi: 10.1016/s0022-2836(02)00587-9. [DOI] [PubMed] [Google Scholar]
- 33.Pei J, Cai W, Kinch LN, Grishin NV. Prediction of functional specificity determinants from protein sequences using log-likelihood ratios. Bioinformatics. 2006;22:164–171. doi: 10.1093/bioinformatics/bti766. [DOI] [PubMed] [Google Scholar]
- 34.Bell CE, Lewis M. A closer view of the conformation of the Lac repressor bound to operator. Nat Struct Biol. 2000;7:209–214. doi: 10.1038/73317. [DOI] [PubMed] [Google Scholar]
- 35.Werstuck G, Green MR. Controlling gene expression in living cells through small molecule-RNA interactions. Science. 1998;282:296–298. doi: 10.1126/science.282.5387.296. [DOI] [PubMed] [Google Scholar]
- 36.Guntas G, Mansell TJ, Kim JR, Ostermeier M. Directed evolution of protein switches and their application to the creation of ligand-binding proteins. Proc Natl Acad Sci USA. 2005;102:11224–11229. doi: 10.1073/pnas.0502673102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Licitra EJ, Liu JO. A three-hybrid system for detecting small ligand-protein receptor interactions. Proc Natl Acad Sci USA. 1996;93:12817–12821. doi: 10.1073/pnas.93.23.12817. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Maynard-Smith LA, Chen LC, Banaszynski LA, Ooi AGL, Wandless TJ. A directed approach for engineering conditional protein stability using biologically silent small molecules. J Biol Chem. 2007;282:24866–24872. doi: 10.1074/jbc.M703902200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Qin Y, et al. Screening and identification of a fungal β-glucosidase and the enzymatic synthesis of gentiooligosaccharide. Appl Biochem Biotechnol. 2011;163:1012–1019. doi: 10.1007/s12010-010-9105-y. [DOI] [PubMed] [Google Scholar]
- 40.Gossen M, Bujard H. Tight control of gene expression in mammalian cells by tetracycline-responsive promoters. Proc Natl Acad Sci USA. 1992;89:5547–5551. doi: 10.1073/pnas.89.12.5547. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Wang HH, et al. Programming cells by multiplex genome engineering and accelerated evolution. Nature. 2009;460:894–898. doi: 10.1038/nature08187. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Datsenko KA, Wanner BL. One-step inactivation of chromosomal genes in Escherichia coli K-12 using PCR products. Proc Natl Acad Sci USA. 2000;97:6640–6645. doi: 10.1073/pnas.120163297. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Pédelacq JD, Cabantous S, Tran T, Terwilliger TC, Waldo GS. Engineering and characterization of a superfolder green fluorescent protein. Nat Biotechnol. 2006;24:79–88. doi: 10.1038/nbt1172. [DOI] [PubMed] [Google Scholar]
- 44.Hawkins PCD, et al. Conformer Generation with OMEGA: Algorithm and Validation Using High Quality Structures from the Protein Databank and the Cambridge Structural Database. J Chem Inf Model. 2010;50:572–584. doi: 10.1021/ci100031x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Hawkins PCD, Nicholls A. Conformer generation with OMEGA: learning from the data set and the analysis of failures. J Chem Inf Model. 2012;52:2919–2936. doi: 10.1021/ci300314k. [DOI] [PubMed] [Google Scholar]
- 46.Kabsch W. XDS. Acta Crystallogr D Biol Crystallogr. 2010;66:125–132. doi: 10.1107/S0907444909047337. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Strong M, et al. Toward the structural genomics of complexes: crystal structure of a PE/PPE protein complex from Mycobacterium tuberculosis. Proc Natl Acad Sci USA. 2006;103:8060–8065. doi: 10.1073/pnas.0602606103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Emsley P, Lohkamp B, Scott WG, Cowtan K. Features and development of Coot. Acta Crystallogr D Biol Crystallogr. 2010;66:486–501. doi: 10.1107/S0907444910007493. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Murshudov GN, Vagin AA, Dodson EJ. Refinement of macromolecular structures by the maximum-likelihood method. Acta Crystallogr D Biol Crystallogr. 1997;53:240–255. doi: 10.1107/S0907444996012255. [DOI] [PubMed] [Google Scholar]
- 50.Winn MD, Isupov MN, Murshudov GN. Use of TLS parameters to model anisotropic displacements in macromolecular refinement. Acta Crystallogr D Biol Crystallogr. 2001;57:122–133. doi: 10.1107/s0907444900014736. [DOI] [PubMed] [Google Scholar]
- 51.Altschul SF, et al. Gapped BLAST and PSI-BLAST: a new generation of protein database search programs. Nucleic Acids Res. 1997;25:3389–3402. doi: 10.1093/nar/25.17.3389. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Larkin MA, et al. Clustal W and Clustal X version 2.0. Bioinformatics. 2007;23:2947–2948. doi: 10.1093/bioinformatics/btm404. [DOI] [PubMed] [Google Scholar]
- 53.Majumdar A, Rudikoff S, Adhya S. Purification and properties of Gal repressor:pL-galR fusion in pKC31 plasmid vector. J Biol Chem. 1987;262:2326–2331. [PubMed] [Google Scholar]
- 54.Meinhardt S, et al. Novel insights from hybrid LacI/GalR proteins: family-wide functional attributes and biologically significant variation in transcription repression. Nucleic Acids Res. 2012;40:11139–11154. doi: 10.1093/nar/gks806. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Magoč T, Salzberg SL. FLASH: fast length adjustment of short reads to improve genome assemblies. Bioinformatics. 2011;27:2957–2963. doi: 10.1093/bioinformatics/btr507. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Kent WJ. BLAT--the BLAST-like alignment tool. Genome Res. 2002;12:656–664. doi: 10.1101/gr.229202. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Bolstad BM, Irizarry RA, Astrand M, Speed TP. A comparison of normalization methods for high density oligonucleotide array data based on variance and bias. Bioinformatics. 2003;19:185–193. doi: 10.1093/bioinformatics/19.2.185. [DOI] [PubMed] [Google Scholar]
- 58.Hadley W. ggplot2: Elegant Graphics for Data Analysis. Springer; 2009. [Google Scholar]
- 59.Suckow J, et al. Genetic studies of the Lac repressor. XV: 4000 single amino acid substitutions and analysis of the resulting phenotypes on the basis of the protein structure. J Mol Biol. 1996;261:509–523. doi: 10.1006/jmbi.1996.0479. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.