

Abstract
Viruses such as influenza suppress host immune function by a variety of methods. This may result in significant morbidity through several pathways, including facilitation of secondary bacterial pneumonia from pathogens such as Streptococcus pneumoniae. PKH26-PCL dye was administered intranasally to label resident alveolar macrophages (AMs) in a well-established murine model prior to influenza infection to determine turnover kinetics during the course of infection. More than 90% of resident AMs were lost in the first week after influenza, while the remaining cells had a necrotic phenotype. To establish the impact of this innate immune defect, influenza-infected mice were challenged with S. pneumoniae. Early AM-mediated bacterial clearance was significantly impaired in influenza-infected mice - about 50% of the initial bacterial inoculum could be harvested from the alveolar airspace 3 hours later. In mock-infected mice, by contrast, more than 95% of inocula up-to-50-fold higher was efficiently cleared. Co-infection during the AM depletion phase caused significant body weight loss and mortality. Two weeks after influenza, the AM population was fully replenished with successful re-establishment of early innate host protection. Local GM-CSF treatment partially restored the impaired early bacterial clearance with efficient protection against secondary pneumococcal pneumonia. We conclude that resident AM depletion occurs during influenza infection. Among other potential effects, this establishes a niche for secondary pneumococcal infection by altering early cellular innate immunity in the lungs resulting in pneumococcal outgrowth and lethal pneumonia. This novel mechanism will inform development of novel therapeutic approaches to restore lung innate immunity against bacterial super-infections.
Keywords: Influenza, Pneumococcal Pneumonia, Super-infections, Alveolar Macrophages, Immunomodulation
Introduction
Influenza and pneumonia are a leading cause of morbidity and mortality in both children and adults in the United States (1). In developing countries, acute lower respiratory infections are the leading cause of death in children younger than 5 years of age (2). Most influenza-related mortality is not due to the viral infection alone. Instead, secondary bacterial pneumonia complicates many severe cases in influenza-infected hosts (3). This results in a tremendous economic burden due to increased hospitalizations, medical costs, and indirect costs during both pandemic and inter-pandemic influenza periods (4). Complicating the picture, treatment of secondary bacterial pneumonia may not be successful even in the antibiotics era. Globally increased rates of antimicrobial resistance among many common respiratory bacterial pathogens and the mechanisms of the drugs themselves can both complicate treatment and cure (5–8). The specter of influenza pandemics makes it crucial to understand how influenza infection alters the host’s local innate immunity to the benefit of establishing secondary bacterial infections.
Lungs are protected against bacterial infections by various components of innate and adaptive immunity (9, 10). Influenza-infected patients are vulnerable to bacterial super-infections, suggesting defects in some or all of these resistance and clearance mechanisms. Certain bacterial pathogens have been commonly isolated from patients with secondary bacterial infections, such as Streptococcus pneumoniae, (4, 11, 12). Immunity to S. pneumoniae is incompletely understood at present in intact hosts, and defects in immunity in compromised hosts are an active area of research. Among host innate immune players, resident alveolar macrophages (AMs) are considered to be the most important first line of defense against respiratory pneumococcal infections (13), through their high phagocytic capacity (14–16). These cells therefore represent an intriguing target for study. However, the strategies for differentiation of resident macrophage subsets in the airways and lung tissues are insufficient to distinguish AMs from lung interstitial macrophages or inflammatory monocytic cells invading the lung in response to infections. In some studies, AMs have been putatively identified based on their surface immunophenotype as CD11chi F4/80hi cells. Indeed, CD11c and F4/80 surface markers are highly expressed on AM surface, but some inflammatory macrophages/dendritic cells can express these markers as well (17–19). Thus, gating on these two markers only will not differentiate the various types of cells during influenza infection, potentially leading to erroneous conclusions about the absolute numbers of resident macrophages.
Previous studies of viral-bacterial synergism in our laboratory have focused on influenza virus virulence factors and their damaging effects on respiratory tract epithelial cells, together with synergistic inflammatory lung injury during co-infection (3, 12). The goal of the present study was to examine the effect of influenza infection on pulmonary innate immune cells, particularly resident macrophages. We reasoned that early escape from the first line of defense in the lung could have profound effects on immunity to a variety of pathogens, including secondary bacterial invaders. Enhanced bacterial growth and replication through this mechanism could allow enhanced expression of virulence factors and the resulting inflammatory response. We used standard murine influenza virus infection and co-infection models (20), in which BALB/c female mice are infected intranasally by a sublethal dose of influenza A virus – this primary infection may then be followed by a sublethal dose of S. pneumoniae at different time points after influenza infection. However, we improved on this model by introducing more specific gating strategies for distinguishing resident macrophage subsets, coupled with in vivo labeling of lung-resident macrophages prior to influenza infection. This allowed accurate validated tracking of resident macrophages during the course of influenza infection. We demonstrate that influenza virus depletes alveolar macrophages. This innate defect impairs early bacterial clearance, supporting development of secondary bacterial pneumonia. These data have implications for understanding virus-induced host immune suppression that may lead to improved prevention and treatment of primary or secondary infections in the lungs.
Materials and Methods
Influenza viruses
We used the St. Jude strain of mouse-adapted influenza virus A/Puerto Rico/8/34 (H1N1), referred to as “PR8”, as well as the human clinical isolate of the pandemic influenza virus A/California/04/09 (H1N1) referred to as “pdm H1N1.” All viruses were passaged once through Madin-Darby canine kidney (MDCK) cells, stocks were grown by a single passage through eggs, and allantoic fluid was stored at −80°C. The viral titers of the stocks were characterized via median tissue culture infective dose (TCID50) assay in MDCK cells.
Bacterial strains
S. pneumoniae A66.1, a type 3 encapsulated strain, was engineered to express luciferase (Kevin Francis and Jun Yu, Xenogen Corporation, Alameda, CA). Pneumococci were grown in Todd Hewitt broth (Difco Laboratories, Detroit, MI) to an OD620 of approximately 0.4 and then frozen at −80°C mixed 2:1 with 5% sterile glycerol. The titers of the frozen stocks were quantitated on tryptic soy agar (Difco Laboratories, Detroit, MI) supplemented with 3% v/v sheep erythrocytes (blood agar). In all instances, the infectious dose administered was confirmed by serial dilution and plating of the bacterial suspension on blood agar plates.
Mice
Six- to 8-week-old female BALB/c mice (Jackson Laboratory, Bar Harbor, ME) were maintained in a Biosafety Level 2 facility in the Animal Resource Center at St. Jude. Animals were given general anesthesia that consisted of 2.5% inhaled isoflurane (Baxter Healthcare Corporation, Deerfield, IL) prior to all interventions, and all studies were approved by the Animal Care and Use Committee at St. Jude.
Infectious model
Infectious agents were diluted in sterile PBS and administered intranasally in a volume of 100 μl (50 μl per nostril) to anesthetized mice held in an upright position. In all experiments of influenza infection, PR8 influenza virus was given at a dose of 25 doses infectious for 50% of tissue culture wells (TCID50) per 100 μl per mouse, which caused about 10% weight loss and no mortality when given alone. In co-infection experiments, PR8 infection was followed at the specified time point by pneumococcal challenge with 200 CFU per mouse. Infected mice were weighed and assessed daily for illness and mortality for 7 days after pneumococcal challenge. The virus infectious dose in the pdm H1N1 experiment was 600 TCID50 per mouse, which caused morbidity and weight loss comparable to PR8 infection (data not shown).
In vivo labeling of lung-resident macrophages
We intranasally administered 100 μl of 10 mM PKH26-PCL dye (Sigma-Aldrich, St. Louis, MO) into anesthetized mice 5 days before influenza or mock infection, as previously described (21).
Imaging of live mice
Mice were then imaged for 60 seconds using an IVIS CCD camera (Caliper Life Sciences, Alameda, CA) daily after pneumococcal challenge to monitor in vivo pneumococcal pneumonia development. Total photon emission from selected and defined areas within the images of each mouse was quantified using LivingImage software (Caliper Life Sciences, Alameda, CA) as described previously (22) and expressed as the flux of relative light units per minute. Pneumonia was defined as visible bioluminescence within the thorax and detection of a flux of > 11,000 relative light units per minute.
Flow cytometric analysis of immune cells in BALF, post-lavage lungs, and mediastinal lymph nodes
Following euthanasia by CO2 inhalation, the trachea was exposed and cannulated with a 24-gauge plastic catheter (Becton Dickinson Infusion Therapy Systems, Inc., Sandy, UT). Lungs were lavaged 4 times with 1 ml of cold sterile Hank’s buffered salt solution (HBSS) supplemented with 0.1 mM ethylenediaminetetraacetic acid (EDTA).The whole lungs after lavage or mediastinal lymph nodes were harvested and physically homogenized by syringe plunger against a 40-μm cell strainer and washed in FACS buffer consisting of HBSS, 0.1 mM EDTA, and 1% heat-inactivated fetal bovine serum. Cell suspension of BALF, post-lavage lung homogenate, or mediastinal lymph nodes homogenate were centrifuged at 4°C, 350 × g for 7 min, and the BALF supernatant was stored at –80°C. Flow cytometry (LSRII, and LSRII Fortessa, Becton Dickinson, San Jose, CA) was performed on the cell pellets after incubation with 75 μl of 1:200 dilution of Fc block (anti-mouse CD16/CD32, BD Bioscience Inc., San Jose, CA) on ice for 10 min, followed by surface marker staining with anti-mouse antibodies: CD11c (eFluor 450), F4/80 (FITC), Ly6G (PerCp-Cy5.5), Ly6C (APC), and CD11b (APC-eFluor 780; eBioscience Inc., San Diego, CA). The data were analyzed using FlowJo 8.8.6 (Tree Star, Ashland, OR) where viable cells were gated from an FSC/SSC plot. First, we gated out neutrophils (CD11bhi Ly6Ghi) then sub-gated macrophages (CD11chi F4/80hi) based on CD11b surface expression into AMs (CD11b−ve), IMs (CD11blow-int), and recruited exudate macrophages (CD11bhi). Viable and non-viable cells were counted before surface marker staining, and the percentage viability was counted via the trypan blue exclusion method using a Cell Countess System (Invitrogen, Grand Island, NY). The absolute numbers of different cell types were calculated based on the proportion of viable events analyzed by flow cytometry as related to the total number of viable cells per sample. Live/dead aqua dye was added during flow cytometric surface staining to determine the total numbers of dead AMs and IMs. This dye binds to free amines after penetrating the impaired cell membrane of dead cells. First, AMs and IMs were gated as mentioned above but without prior gating for viable events based on an FSC/SSC dot plot. Live/dead aqua-positively stained AMs and IMs were gated as dead cells, and their numbers were calculated based on the proportion of dead events from the total events, analyzed by flow cytometry. Average mean fluorescence intensity of PKH26-PCL was measured for different subsets of macrophages in the PE channel.
Confocal laser scanning microscopy of PKH26-labeled Lungs
Five days after in vivo PKH26-PCL labeling of lung macrophages, euthanasia by CO2 inhalation was performed, and the trachea was exposed and cannulated with a 24-gauge plastic catheter (Becton Dickinson Infusion Therapy Systems, Inc., Sandy, UT). Lungs were harvested after instillation of 1.2 ml of 4% freshly prepared formaldehyde with PBS. Harvested lungs were fixed in 4% formaldehyde with PBS at room temperature for 1 hour. Then every lung was washed with PBS and cut into 4 pieces before mounting into PBS in 4 wells of Nunc Lab-Tek chambered cover glass (Thermo Scientific, Rochester, NY) before microscopic examination. Fluorescence was visualized with a Nikon Eclipse TE2000-E inverted microscope equipped with C2 confocal system and a 40X/1.3NA numeric aperature oil objective (Nikon Instruments, Melville, NY). Image collection and analysis were performed with Nikon NIS-Elements software (Version 4.13).
Cytospin slides preparation
After harvesting BALF as described above, and before staining cells for flow cytometry analysis, BALF cells were resuspended in PBS, cytospun (Thermo Scientific, Ashville, NC) onto glass slides, and stained with Diff-Quick (Quik-Dip stain; Mercedes Medical, Sarasota, FL). Neutrophils, monocytes/macrophages, and lymphocytes were identified by morphology and images were taken from different representative fields of stained macrophages under high power fields.
Early pneumococcal clearance determination
Early pneumococcal clearance was determined by measuring the pneumococcal count remaining within alveolar airspace. Briefly, mice were euthanized by CO2 inhalation 3 h after pneumococcal inoculation. BALF was harvested using sterile HBSS supplemented with 0.1 mM EDTA (lavage by 1 ml twice), and then half the BALF was spread on blood agar plates supplemented with 0.4 mg kanamycin/ml blood agar to select for kanamycin-resistant luciferase-expressing pneumococci (22) and incubated at 37°C overnight.
Local GM-CSF treatment regimen
We treated anesthetized mice intranasally with 25 μg of recombinant mouse GM-CSF (Invitrogen, Grand Island, NY) in 100 μl of sterile PBS on days –1 and +1 before and after PR8 infection. We treated control mice with vehicle (PBS) only.
Statistical Analysis
Comparison of survival and pneumonia development between groups of mice was done with the log-rank chi-squared test on the Kaplan-Meier survival data. Comparison of bacterial titers, cell counts, and mean fluorescence intensity of PKH26-PCL in BALF, and post-lavage lung homogenate between groups were compared using analysis of variance (ANOVA). Comparison of weight loss between groups of mice was done using Student’s t-test for pair-wise comparisons. A p-value of < 0.05 was considered significant for these comparisons. Prism 4 for Windows (GraphPad Software, Inc., V 4.03) was used for all statistical analyses.
Results
In vivo labeling of lung-resident macrophages can distinguish AMs from interstitial macrophages
Before examining any alterations in the lung-resident innate immune cells in influenza-infected hosts, we determined critical techniques for the differential analysis of the heterogeneous population of macrophages in both alveolar airspace and post-lavage lung tissue. First, we refined a flow cytometry technique for gating resident macrophages so that clear differentiation between different cells types could be accomplished. We used the common markers CD11chi and F4/80hi and added gating by another marker, CD11b (β2-integrin). CD11b has been shown to be weakly expressed on AM cell surface (23–25) while highly expressed on granulocytes, exudate macrophages, monocytes, and some dendritic cells (26–29). Therefore, AMs are better gated as CD11chi F4/80hi CD11bdim in BAL fluid as shown recently (25, 30). This allowed differentiation in our flow cytometric analysis of the two major subsets of lung-resident macrophages in mock-infected mice. Thus, AMs could be gated as CD11chi F4/80hi CD11b−ve and interstitial macrophages (IMs) as CD11chi F4/80hi CD11blo-int in both bronchoalveolar lavage fluid (BALF) and post-lavage lung homogenate (Fig. 1A).
Figure 1. In vivo labeling of lung-resident macrophages confirms validity of flow cytometric gating strategy.

(A) Flow cytometry dot plots show the gating strategy of alveolar macrophages (AMs, R4 gate) and interstitial macrophages (IMs, R5 gate) of mock-infected mice in BALF (top plots) and post-lavage lung homogenate (bottom plots).
(B) In vivo labeling of lung-resident macrophages using PKH26-PCL dye before influenza infection can distinguish AMs (solid red bars) from IMs (solid black bars) based on MFI of PKH26-PCL dye in both BALF and post-lavage lung homogenate (n = 4).
***P < 0.001 Tukey’s multiple comparison test (ANOVA). The bar graphs show the average ± SD
To confirm our gating strategy for both subsets of lung-resident macrophages and to differentiate them from recruited macrophages, we did in vivo labeling of lung-resident macrophages before influenza infection using intranasally administered PKH26-Phagocytic Cell Labeling (PKH26-PCL) dye. Both subsets of lung-resident macrophages were intensely labeled by the dye, showing high means of the fluorescence intensity of PKH26-PCL dye (MFI-PKH26). Nonetheless, significant differences in MFI-PKH26 were observed between AMs and IMs in both BALF and post-lavage lung homogenates, with IMs showing significantly lower MFI-PKH26 than AMs (Fig. 1B). Selective labeling of lung-resident macrophages was also confirmed by confocal microscopy of fixed naïve PKH26-labeled lungs showing the same pattern of difference in PKH26-MFI (Supplemental Fig. 1). These differences reflect the spatial and functional differences between the two major subsets of lung-resident macrophages, with IMs having less accessibility to the intranasally administered dye and lower phagocytic capacity than AMs (14, 24, 31). Thus, in vivo labeling method confirmed the validity of our flow cytometric gating strategy for the lung-resident macrophage subsets.
Influenza virus depletes alveolar macrophages
To determine if influenza infection causes any alteration in the numbers of lung-resident macrophages, we determined the percentages of lung-resident macrophages in flow cytometric dot plots and calculated their absolute numbers. We infected BALB/c female mice intranasally with the mouse-adapted H1N1 influenza virus strain A/Puerto Rico8/34 (PR8) using a sublethal dose (25 doses infectious for 50% of tissue culture wells (TCID50) per 100 μl). Seven days after influenza virus infection, more than 90% of the AM pool was depleted (Fig. 2A).To exclude the possibility that the observed depletion of AMs is unique to the use of a mouse-adapted influenza virus strain (PR8), we examined changes in the AM pool size using a human clinical influenza isolate from the 2009 influenza pandemic, the A/California/04/09 H1N1 strain (pdm H1N1). We found that AMs were significantly depleted within 7 days after pdm H1N1 infection to levels comparable to PR8 infection (Fig. 2B).
Figure 2. AMs are depleted during influenza infection.

Absolute numbers of AMs in bronchoalveolar lavage fluid (solid bars) and post-lavage lung homogenates (open bars) of PR8-infected (A), or 2009 pandemic H1N1-infected mice (B) 7 days after influenza infection are significantly lower than in mock-infected (naive) mice (n ≥5 each). ***P < 0.001 compared with mock-infected (naïve) mice. The bar graphs show the average ± SEM.
Tracking of AMs and IMs dynamic changes during influenza infection
To track depletion kinetics of AMs and changes of the IM pool size during influenza infection, we again did in vivo labeling of lung-resident macrophages followed by sublethal PR8 influenza infection and studied different time points in the first 2 weeks after influenza virus infection. We found that AMs were significantly depleted in the alveolar airspace shortly after PR8 infection starting on the first day p.i. (Fig. 3A). Considering that some AMs may not be completely harvested in BALF and still adhere to the respiratory tract lining, we also analyzed post-lavage lungs and found that higher numbers of AMs remained in the post-lavage lung homogenate. These remaining AMs were significantly depleted starting 3 days p.i. compared with those in mock-infected mice (Fig. 3B). Consequently, 3 days after PR8 influenza infection was identified as the earliest time point of significant whole lung AM depletion. The AM pool was partially replenished 9 days p.i. (in BALF only), while complete replenishment appeared 11 days p.i. (Fig. 3A, 3B). Conversely, the IM pool was not significantly depleted during the course of influenza infection; indeed, it was expanded at later time points (Fig. 3A, 3B).
Figure 3. Tracking of AMs and IMs dynamic changes during influenza infection.

Absolute numbers of AMs and IMs in BALF (A) and post-lavage lung homogenate (B) of PR8-infected mice harvested 1, 3, 5, 7, 9, 11, and 14 days p.i. Tracking the changes in MFI of PKH26-PCL dye that distinguishes resident macrophages in BALF (C) and post-lavage lungs (D) during influenza infection. *P < 0.05,**P < 0.01,***P < 0.001 compared with mock-infected (naïve) mice (Fig. A, B) or influenza-infected mice 1 day p.i. (Fig. C, D), n ≥4. Dunnett’s multiple comparison test (ANOVA). The bar graphs show the average ± SD
To confirm the validity of the tracking process of lung-resident macrophages during the course of influenza infection, dynamic changes in their MFI-PKH26 were measured. At any time point after influenza infection, AMs showed the highest MFI-PKH26 compared to IMs or recruited exudates macrophages (Supplemental Fig. 2), confirming our gating strategy that differentiates between AM and IM subsets. The MFI-PKH26 of AMs was significantly decreased during the full replenishment period starting 11 days p.i. (Fig. 3C, 3D). Nonetheless, AMs still had significantly higher MFI-PKH26 than that of IMs during the course of infection (Supplemental Fig. 2). The MFI-PKH26 of IMs was significantly decreased shortly after influenza infection. However, no significant changes in their absolute numbers were observed in post-lavage lungs in the first week of infection (Fig. 3B, 3D). In the second week, the IM pool significantly expanded with a significant increase in absolute numbers harvested from BALF (Fig. 3A).
To test whether these observed changes in PKH26-MFI of lung-resident macrophages were due to changes in their absolute numbers or due to instability of the dye, we measured the stability of PKH26-PCL dye in vivo in mock-infected lungs. PKH26-PCL dye demonstrated efficient labeling and stability inside the lung-resident macrophages for at least 14 days after mock-infection (i.e. 19 days after labeling), thereby covering the tracking period during which we did the kinetics analyses (Supplemental Fig. 3).
We did further characterization of the different macrophage subsets in the lungs during influenza infection based on their surface expression levels of Ly6C antigen that can be used to differentiate between resident and recruited inflammatory macrophage subsets, in addition to their maturation stages (27, 28). We found that resident AMs didn’t express surface Ly6C, while resident lung IMs express intermediate levels, reflecting their intermediate stage of maturation and supporting the previous studies suggesting them as precursors for AMs (24, 32). In contrast, the recruited exudates macrophages with CD11bhigh phenotype showed significantly high expression levels of Ly6C (Supplemental Fig. 4).
Influenza infection induces cell death of AMs
To determine whether AM depletion is due to a cell death process, we measured the total numbers of dead AM cells during PR8 infection using a cell viability dye (live/dead aqua). The total numbers of dead AMs were significantly higher in both BALF and post-lavage lungs of influenza-infected mice than in mock-infected mice (Fig. 4A). In contrast, there was no significant difference between the total numbers of dead IMs in influenza-infected and mock-infected mice (Fig. 4B). To determine the type of AM cell death process, we examined alveolar airspace macrophages for any morphologic changes associated with influenza infection. Diff-Quick -stained cytospin slides of BALF cells harvested from influenza-infected mice 3 days p.i. showed many macrophages with cellular damage manifestations. They were characterized by distorted nuclei and more vacuoles in cytoplasm than in mock-infected mice (Fig. 4C). This suggests that cell death was due to a secondary necrotic process.
Figure 4. Influenza infection induces significant AM death.

Total numbers of dead AMs (A) and IMs (B) are calculated during influenza infection. (C) Cytospin of Diff quick-stained BALF cells from mock-infected and PR8-infected mice 3 days p.i at magnification × 500. *P < 0.05, **P < 0.01 Dunnett’s multiple comparison test (ANOVA) compared with mock-infected naive mice. The bar graphs show the average ± SD.
To determine if AMs were lost due to recruitment to the lung-draining lymph nodes, we examined the mediastinal lymph node 7 days after PR8 influenza infection and didn’t observe significant recruitment of PKH26-PCL– labeled AMs to it (data not shown).
Influenza infection enhances susceptibility to secondary pneumococcal pneumonia
Influenza-mediated death of AMs is likely to have significant effects on primary and secondary immunity. To explore one potential defect, we studied the permissiveness of influenza-infected hosts to secondary respiratory bacterial infections. We first infected BALB/c female mice intranasally with PR8 influenza virus using a sublethal dose (25 TCID50). Influenza-infected mice showed mild morbidity manifested as loss of ~ 10% of their original body weight within 7 days after influenza infection (Fig. 5A). At that timepoint, we induced secondary bacterial infection via intranasal administration of a small inoculum (200 CFU) of the serotype 3 clinical isolate of S. pneumoniae, A66.1. Influenza-infected mice showed high susceptibility to secondary pneumococcal infection with a significant increase in morbidity and continuous body weight loss (Fig. 5A). All co-infected mice died within 3 to 5 days after pneumococcal inoculation (Fig. 5B). In contrast, all single influenza- or single pneumococcus-infected mice recovered quickly with no mortality (Fig. 5A, 5B). To determine whether the co-infected mice died due to pneumococcal pneumonia, we monitored pneumococcal growth in vivo through bioluminescent imaging of the lungs (22). Only co-infected mice developed serious pneumococcal infections; pneumonia occurred within 48–72 h after pneumococcal inoculation (Fig. 5C).
Figure 5. Murine influenza-pneumococcal co-infection model.

(A) Body weight loss of single influenza-infected (influenza and PBS 7 days p.i.), single pneumococcal-infected (PBS and S. pneumoniae), and co-infected (influenza and S. pneumoniae) mice (n > 5 each). (B) Survival rate of single influenza-infected, single pneumococcus-infected, and co-infected mice (n > 5 each). (C) Thorax bioluminescence of luciferase-expressing A66.1 pneumococcus in co-infected mice showing development of pneumococcal pneumonia. The bar graphs show the average ± SD. *** P < 0.001 Student’s t-test at each timpoint compared with single influenza-infected mice group (Fig. A), *** P < 0.001 log-rank test on the Kaplan Meier survival data (Fig. B). (D) Pneumococcal titers harvested 3 h after bacterial inoculation (inoculum of 200 or more CFUs) from alveolar airspace of mock-infected and influenza-infected mice 7 days p.i. are shown as percentage of inoculum. ***P < 0.001 compared with mock-infected (naïve) mice.
AM depletion during influenza infection impairs early pneumococcal clearance
To determine whether the observed depletion of the AM pool contributes to enhanced susceptibility to secondary pneumococcal infection in influenza-infected hosts, AM phagocytic function was assessed in vivo by measuring early pneumococcal clearance with or without influenza infection. First, we determined the earliest time point at which a small pneumococcal inoculum (200 CFU) could be efficiently cleared within the alveolar airspaces of naive mice. Pneumococcal clearance was tested at different time points (1, 2, 3, and 4 h) after bacterial inoculation. Complete pneumococcal clearance was observed within 3 h after pneumococcal inoculation in naïve mice (data not shown). Then, we tested the ability of influenza-infected lungs to clear this small dose of pneumococcus at this early time point. This experiment demonstrated that early pneumococcal clearance was significantly impaired in the alveolar airspace of influenza-infected mice compared with mock-infected controls. Mock-infected mice could efficiently clear more than 95% of up to 50-fold higher doses of pneumococcus, while influenza virus-infected mice were unable to clear the basal inoculum (Fig. 5D).
Influenza-infected mice demonstrate increased susceptibility to secondary pneumococcal pneumonia during the AM depletion phase
To determine whether AM depletion during influenza infection correlates with susceptibility to secondary pneumococcal infection, we examined the early pneumococcal clearance at different time points after influenza infection. Early pneumococcal clearance was significantly impaired during PR8 infection from 3 until 9 days p.i. (Fig. 6A). This period of early pneumococcal clearance impairment closely mirrors the phase of the AM pool depletion observed during PR8 infection (Fig. 3A, 3B). Later, the impaired early pneumococcal clearance was restored (Fig. 6A).
Figure 6. Early pneumococcal clearance from alveolar airspace is impaired during the AM depletion phase in influenza-infected mice.

(A) Pneumococcal CFUs harvested 3 h after bacterial inoculation (inoculum of 200 CFUs) from the alveolar airspace of mock-infected and influenza-infected mice 1, 3, 5, 7, 9, 11, and 14 days p.i. Secondary pneumococcal pneumonia development (B) and mortality (C) are manifested in influenza-infected mice that are secondarily pneumococcal-infected during the AM depletion phase (n ≥4). *P < 0.05,***P < 0.001 compared with mock-infected mice, Dunnett’s multiple comparison test (ANOVA) (Fig. A). *P < 0.05,** P < 0.01 *** P < 0.001 compared with Day 1 or Day 14 co-infection groups, log-rank test on the Kaplan Meier survival data (Fig. B, C)
To determine whether the impaired early pneumococcal clearance during influenza infection enhances susceptibility to secondary pneumococcal pneumonia development, we monitored mouse lungs via bioluminescence imaging 24 h after bacterial inoculation. Among the influenza-infected mice, 100% developed secondary pneumococcal pneumonia when they were secondarily infected by pneumococcus 3, 5, or 7 days after influenza infection. In contrast, all influenza-infected mice that were secondarily infected either 1 days or 14 days p.i. cleared the bacterial dose efficiently and did not develop secondary pneumococcal pneumonia (Fig. 6B). Secondary pneumococcal pneumonia development was less frequent in influenza-infected mice that were secondarily infected by pneumococcus on 9 or 11 days p.i., with only 60% or 40% of pneumonic mice, respectively (Fig. 6B). As expected in this model, all co-infected mice that developed secondary pneumococcal pneumonia died within few days due to bacterial pneumonia (Fig. 6C).
Local GM-CSF treatment decreases secondary pneumococcal pneumonia development in influenza-infected mice
To restore the early bacterial clearance efficiency that was impaired during the AM depletion phase, we tested local recombinant GM-CSF treatment as a means to accelerate replenishment of the depleted AM pool in influenza-infected mice. Recombinant GM-CSF was intranasally administered in 2 doses on days −1 and +1 before and after PR8 infection (Fig. 7A). First, we analyzed the effect of this GM-CSF treatment regimen on the size of the AM and IM pools. GM-CSF treatment in influenza-infected mice resulted in a significant expansion of the IM pool with partial replenishment of the AM pool (Fig. 7B).
Figure 7. Local recombinant GM-CSF treatment improved secondary pneumococcal pneumonia development in influenza-infected mice.
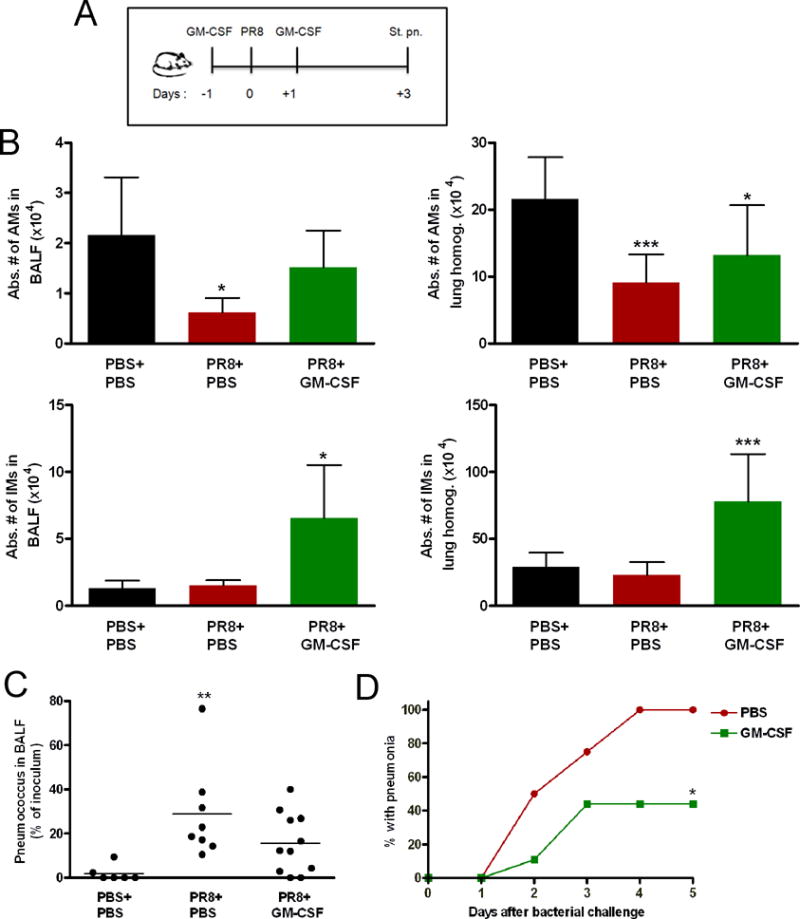
(A) Intranasal administration of recombinant GM-CSF into PR8-infected mice on days −1 and +1 before and after infection, followed by 200 CFUs of pneumococcus on day 3 improved early pneumococcal clearance from the alveolar airspace (C) and secondary pneumococcal pneumonia development (n = 9) (D) compared with mock-treated co-infected mice (n = 4). (B) The absolute numbers of AMs and IMs increased in GM-CSF-treated influenza-infected mice analyzed 3 days p.i (n ≥4). *P < 0.05,***P < 0.001 Dunnett’s multiple comparison test (ANOVA), compared with mock-treated mock-infected mice (Fig. B, C). The bar graphs show the average ± SD. *P < 0.05 compared with Day 1 or Day 14 co-infection groups, log-rank test on the Kaplan Meier survival data (Fig. D).
To evaluate the efficacy of local GM-CSF treatment in the co-infection model, pneumococcus was administered 3 days after PR8 infection into GM-CSF-treated and mock-treated mice groups (Fig. 7A). Local GM-CSF treatment led to better early pneumococcal clearance in influenza-infected mice than in mock-treated mock-infected mice. Conversely, early pneumococcal clearance remained impaired in mock-treated, influenza-infected mice (Fig. 7C). Interestingly, some of the GM-CSF-treated influenza-infected mice could efficiently clear the bacterial inoculum, while others could not. Next, we tested the ability of local GM-CSF treatment to prevent secondary pneumococcal pneumonia development after bacterial inoculation 3 days p.i. Local GM-CSF treatment protected more than 50% of co-infected mice against secondary pneumococcal pneumonia. Meanwhile, all mock-treated co-infected mice developed pneumococcal pneumonia (Fig. 7D).
Discussion
Influenza is well known to increase susceptibility to secondary bacterial infections, such as secondary pneumococcal pneumonia. The increased permissiveness of influenza-infected lungs to pneumococcal outgrowth suggests a defect in the host innate immune defenses that establishes a niche for bacterial infections. In this study, we examined a novel mechanism of influenza-mediated immune suppression – depletion of alveolar macrophages. The resulting immune defect is likely to have pleiotropic effects on primary and secondary immunity. We examined one potential effect, bacterial escape from early innate immunity contributing to enhanced vulnerability to respiratory bacterial super-infections. Resident AMs are essential for early bacterial clearance and protection against bacterial infections (13, 21). We found that influenza infection induces depletion of AM pool for certain period during which early clearance of small pneumococcal inocula is significantly impaired. This influenza-associated damaging effect on AMs is correlated with enhanced vulnerability to secondary pneumococcal pneumonia. It is likely that other effects on immunity may be seen based on the lack of AMs at this crucial period of viral infection and clearance.
One observation stemming from this study is that examination of lung-resident macrophages in the setting of influenza infection should be cautiously performed. During influenza infection, diverse chemokines are upregulated in lungs, activating an influx of heterogeneous populations of innate immune cells, such as monocytes, inflammatory macrophages, and monocyte-derived dendritic cells. The dynamic changes in the phagocyte populations in influenza-infected lungs require critical methods to distinguish resident macrophages from recruited ones. Based on the surface phenotype of lung-resident macrophages in naïve mice, we used an extensive gating strategy to analyze and differentiate between resident and recruited macrophages. Thus, AMs were gated as CD11chi F4/80hi CD11b−ve, while IMs and recruited macrophages were gated as CD11chi F4/80hi CD11blo-int and CD11chi F4/80hi CD11bhigh, respectively. Further characterization showed the ability of Ly6C marker to differentiate between resident and recruited phagocytes and their maturation stage in influenza-infected lungs. Ly6C antigen is expressed by circulating blood monocytes and macrophages which can be recruited to tissues under inflammation conditions (27). However, Ly6C expression is down-regulated during differentiation of blood monocytes or macrophages into tissue resident macrophages after migration to tissue. Therefore, it can be added as a suitable marker to differentiate macrophage subsets and their stages of maturation into tissue resident phenotype (28). Moreover, in vivo labeling of lung-resident macrophages was done to validate our gating strategy and to distinguish between resident AMs and recruited macrophages. The PKH26-PCL dye used to label macrophages forms fluorescent microparticles, which can be taken up by resident phagocytes in lungs and remain stable for more than 21 days (21), and emitting high fluorescence intensity for at least 19 days based on our findings.
Using this to track lung-resident macrophages during influenza infection, we found a significant depletion of the AM pool across a limited time period after infection. About 70% of AMs were depleted after 3 days, reaching a nadir (> 90% depletion) 7 days after PR8 influenza infection. This matches the previously established boundaries for maximum synergism in the secondary bacterial infection model (33), which parallel the typical order and timing of infections in humans (34). Interestingly, in another influenza infection model using a different strain, percentage of resident AMs – gated as CD11chigh Mac-1−ve– decreased on day 3 p.i (35), which is consistent with our findings. In contrast, they didn’t analyze absolute numbers of cells.
In most studies of primary or secondary pneumococcal pneumonia in animal models, AM-mediated protection has not been rigorously studied (21). This appears mainly to be because of the use of high doses of pneumococcus (≥ 1×106 CFU) to induce a robust and reproducible pneumonia, doses which overwhelm the phagocytic capacity of resident AMs. Instead, an influx of neutrophils in the setting of development of type-specific antibody, the second line of innate immune defense coupled with the early adaptive response, was found in these models to be important for host protection against primary pneumococcal pneumonia (36). However, in the perhaps more physiologically relevant setting of a relatively small dose of pneumococcus (200 CFU) after sublethal mild influenza infection, we can demonstrate a strong effect mediated by AMs. This low bacterial inoculum successfully caused lethal secondary pneumococcal pneumonia within 48–72 h after bacterial inoculation, pneumonia that was dependent on an absence of AMs.
In vivo labeling of lung-resident macrophages in this study was a novel approach that allowed successful differentiation of AMs from recruited macrophages. Furthermore, monitoring changes of MFI-PKH26 for lung-resident macrophages reflected the dynamic changes in their absolute numbers during influenza infection. For instance, expansion of the AM pool during the recovery phase was accompanied by significant decrease in their MFI-PKH26. Nevertheless, the AM population showed the highest MFI-PKH26 at various times during the first 2 weeks after influenza infection. Interestingly, this significantly high MFI-PKH26 of AMs, even during their replenishment phase, implies that the AM pool was replenished mainly via differentiation of the PKH26-labeled IMs into an AM surface phenotype rather than maturation of PKH26-unlabeled recruited blood macrophages. These results support the findings that the origin of the AM pool is mainly through differentiation of intermediate lung macrophages rather than self-proliferation (24). In contrast, the quick drop in MFI-PKH26 of IMs in the first week after influenza infection, without apparent changes in their absolute cell numbers, suggests that influenza infection may induce partial depletion of the IM pool. However, this partial depletion can be quickly re-compensated by the proliferative capacity of IMs, maturation of the recruited macrophages, or both (14, 24).
Other studies were not able to identify depletion of resident AMs; because of some limitations in their analyses. Mostly they used gating strategies not sufficient to distinguish between resident AMs, IMs and recruited exudate macrophages. For example, gating for a single marker like F4/80 antigen or macrophage-specific esterase staining of macrophages isolated from BAL fluid during influenza infection is targeting mixed populations of resident and recruited macrophages whose total cell numbers increase during infection (37). In addition, although the same in vivo labeling method was used before, AM depletion couldn’t be identified during influenza (21). Based on their analysis, they gated AMs as CD11c+ve PKH26+ve which may be mixed with IMs. They analyzed AM numbers on day 9 after influenza infection in BAL fluid only. Based on our model, we showed certain period for AM depletion. Therefore, analyzing AMs at single time point (day 9 p.i) is not conclusive regarding AM depletion event during influenza infection. Furthermore, analyzing cells in BAL fluid only may dismiss significant numbers of AMs remaining adherent in the lungs after lavage leading to underestimation of resident AM pool size during comparison between influenza-infected and mock-infected mice. Likewise, in another study they didn’t show AM depletion; because of missing CD11b marker in their AM gating, as well as harvesting only BAL fluid for kinetics analysis (17). Taken together, missing important marker that distinguishes between different lung-resident macrophage subsets and recruited monocytic cells or using BAL fluid only for analyzing resident AMs may lead to erroneous conclusions regarding their kinetics during infections.
GM-CSF is a cytokine with diverse functions critical for effective innate immunity in lungs. Among these activities, it regulates AM differentiation and activation (23, 38), enhances proliferation of resident pulmonary macrophages (28), and expands the pool of resident AMs (39). It also has an important role in pulmonary surfactant homeostasis (40). Recent studies showed that GM-CSF over expression in lungs has prophylactic activity against lethal influenza and pneumococcal pneumonias (39, 41, 42). We found that local recombinant GM-CSF treatment in influenza-infected mice induced significant expansion of the IM pool. In addition, absolute numbers of AMs increased under this treatment regimen in influenza-infected mice. This increase was not to the degree as that of IMs, possibly because they were analyzed at an early time point (3 days p.i). Thus, our data support the previous studies showing GM-CSF enhances the proliferation capacity of lung-resident macrophages and maturation of AMs. Considering the higher proliferative capacity of IMs over AMs, our short treatment regimen could significantly expand the pool of IMs more than AMs. As a result of partial replenishment of the AM pool with pulmonary GM-CSF treatment, influenza-infected mice had partial restoration of efficient early pneumococcal clearance. Furthermore, they manifested improved protection against secondary pneumococcal pneumonia. Although pulmonary GM-CSF treatment has some drawbacks such as inflammatory activity (43) that can render its use in humans problematic by exacerbating inflammatory lung injury, the results of our study are promising. They suggest that strategies seeking to balance the protective AM replenishment effects and the adverse effect of exuberant inflammation induction via combining an adjunctive anti-inflammatory therapy with GM-CSF treatment might be possible.
Overall, this study suggests a novel mechanism of influenza-mediated immune suppression that resulted in increased vulnerability of influenza-infected hosts to bacterial super-infections. Resident AM depletion during influenza infection establishes a niche for secondary pneumococcal infection by altering early cellular innate immunity in the lungs, thereby allowing pneumococcal outgrowth causing lethal pneumococcal pneumonia. The precise functional characterization of this novel finding can change the way researchers look at the alteration of pulmonary cellular innate immunity during sublethal influenza infections. There are likely to be important effects on immunity beyond the bacterial escape studied here. Furthermore, these findings should open avenues for novel immunomodulating therapeutic interventions to prevent respiratory bacterial super-infections by quick replenishment of the critical innate immune effectors during both pandemic and seasonal influenza.
Supplementary Material
Acknowledgments
We thank Richard Webby, Amali Samarasinghe, and Irina Alymova for helpful discussions.
This work was supported by the American Lebanese Syrian Associated Charities (ALSAC). Confocal microscopy images were acquired at the Cell & Tissue Imaging Center which is supported by SJCRH and NCI P30 CA021765-34.
Footnotes
Disclosures
The authors have no financial conflicts of interest.
References
- 1.Minino AM, Heron MP, Murphy SL, Kochanek KD. Deaths: final data for 2004. Natl Vital Stat Rep. 2007;55:1–119. [PubMed] [Google Scholar]
- 2.Liu L, Johnson HL, Cousens S, Perin J, Scott S, Lawn JE, Rudan I, Campbell H, Cibulskis R, Li M, Mathers C, Black RE. Global, regional, and national causes of child mortality: an updated systematic analysis for 2010 with time trends since 2000. Lancet. 2012;379:2151–2161. doi: 10.1016/S0140-6736(12)60560-1. [DOI] [PubMed] [Google Scholar]
- 3.McCullers JA. Preventing and treating secondary bacterial infections with antiviral agents. Antivir Ther. 2011;16:123–135. doi: 10.3851/IMP1730. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Morens DM, Taubenberger JK, Fauci AS. Predominant role of bacterial pneumonia as a cause of death in pandemic influenza: implications for pandemic influenza preparedness. J Infect Dis. 2008;198:962–970. doi: 10.1086/591708. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Musher DM, Dowell ME, Shortridge VD, Flamm RK, Jorgensen JH, Le Magueres P, Krause KL. Emergence of macrolide resistance during treatment of pneumococcal pneumonia. N Engl J Med. 2002;346:630–631. doi: 10.1056/NEJM200202213460820. [DOI] [PubMed] [Google Scholar]
- 6.Linares J, Ardanuy C, Pallares R, Fenoll A. Changes in antimicrobial resistance, serotypes and genotypes in Streptococcus pneumoniae over a 30-year period. Clinical microbiology and infection : the official publication of the European Society of Clinical Microbiology and Infectious Diseases. 2010;16:402–410. doi: 10.1111/j.1469-0691.2010.03182.x. [DOI] [PubMed] [Google Scholar]
- 7.Song JH, Thamlikitkul V, Hsueh PR. Clinical and economic burden of community-acquired pneumonia amongst adults in the Asia-Pacific region. International journal of antimicrobial agents. 2011;38:108–117. doi: 10.1016/j.ijantimicag.2011.02.017. [DOI] [PubMed] [Google Scholar]
- 8.McCullers JA, English BK. Improving therapeutic strategies for secondary bacterial pneumonia following influenza. Future microbiology. 2008;3:397–404. doi: 10.2217/17460913.3.4.397. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Kohlmeier JE, Woodland DL. Immunity to respiratory viruses. Annu Rev Immunol. 2009;27:61–82. doi: 10.1146/annurev.immunol.021908.132625. [DOI] [PubMed] [Google Scholar]
- 10.Lipscomb MF, Hutt J, Lovchik J, Wu T, Lyons CR. The pathogenesis of acute pulmonary viral and bacterial infections: investigations in animal models. Annu Rev Pathol. 2010;5:223–252. doi: 10.1146/annurev-pathol-121808-102153. [DOI] [PubMed] [Google Scholar]
- 11.Wang XY, Kilgore PE, Lim KA, Wang SM, Lee J, Deng W, Mo MQ, Nyambat B, Ma JC, Favorov MO, Clemens JD. Influenza and bacterial pathogen coinfections in the 20th century. Interdiscip Perspect Infect Dis. 2011;2011:146376. doi: 10.1155/2011/146376. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.McCullers JA. Insights into the interaction between influenza virus and pneumococcus. Clin Microbiol Rev. 2006;19:571–582. doi: 10.1128/CMR.00058-05. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Knapp S, Schultz MJ, van der Poll T. Pneumonia models and innate immunity to respiratory bacterial pathogens. Shock. 2005;24(Suppl 1):12–18. doi: 10.1097/01.shk.0000191385.41689.f3. [DOI] [PubMed] [Google Scholar]
- 14.Franke-Ullmann G, Pfortner C, Walter P, Steinmuller C, Lohmann-Matthes ML, Kobzik L. Characterization of murine lung interstitial macrophages in comparison with alveolar macrophages in vitro. J Immunol. 1996;157:3097–3104. [PubMed] [Google Scholar]
- 15.Dockrell DH, Marriott HM, Prince LR, Ridger VC, Ince PG, Hellewell PG, Whyte MK. Alveolar macrophage apoptosis contributes to pneumococcal clearance in a resolving model of pulmonary infection. J Immunol. 2003;171:5380–5388. doi: 10.4049/jimmunol.171.10.5380. [DOI] [PubMed] [Google Scholar]
- 16.Marriott HM, Dockrell DH. The role of the macrophage in lung disease mediated by bacteria. Exp Lung Res. 2007;33:493–505. doi: 10.1080/01902140701756562. [DOI] [PubMed] [Google Scholar]
- 17.Herold S, Steinmueller M, von Wulffen W, Cakarova L, Pinto R, Pleschka S, Mack M, Kuziel WA, Corazza N, Brunner T, Seeger W, Lohmeyer J. Lung epithelial apoptosis in influenza virus pneumonia: the role of macrophage-expressed TNF-related apoptosis-inducing ligand. J Exp Med. 2008;205:3065–3077. doi: 10.1084/jem.20080201. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Bradley LM, Douglass MF, Chatterjee D, Akira S, Baaten BJ. Matrix metalloprotease 9 mediates neutrophil migration into the airways in response to influenza virus-induced toll-like receptor signaling. PLoS Pathog. 2012;8:e1002641. doi: 10.1371/journal.ppat.1002641. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Bedoret D, Wallemacq H, Marichal T, Desmet C, Quesada Calvo F, Henry E, Closset R, Dewals B, Thielen C, Gustin P, de Leval L, Van Rooijen N, Le Moine A, Vanderplasschen A, Cataldo D, Drion PV, Moser M, Lekeux P, Bureau F. Lung interstitial macrophages alter dendritic cell functions to prevent airway allergy in mice. The Journal of clinical investigation. 2009;119:3723–3738. doi: 10.1172/JCI39717. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Karlstrom A, Boyd KL, English BK, McCullers JA. Treatment with protein synthesis inhibitors improves outcomes of secondary bacterial pneumonia after influenza. J Infect Dis. 2009;199:311–319. doi: 10.1086/596051. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Sun K, Metzger DW. Inhibition of pulmonary antibacterial defense by interferon-gamma during recovery from influenza infection. Nat Med. 2008;14:558–564. doi: 10.1038/nm1765. [DOI] [PubMed] [Google Scholar]
- 22.Francis KP, Yu J, Bellinger-Kawahara C, Joh D, Hawkinson MJ, Xiao G, Purchio TF, Caparon MG, Lipsitch M, Contag PR. Visualizing pneumococcal infections in the lungs of live mice using bioluminescent Streptococcus pneumoniae transformed with a novel gram-positive lux transposon. Infect Immun. 2001;69:3350–3358. doi: 10.1128/IAI.69.5.3350-3358.2001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Guth AM, Janssen WJ, Bosio CM, Crouch EC, Henson PM, Dow SW. Lung environment determines unique phenotype of alveolar macrophages. Am J Physiol Lung Cell Mol Physiol. 2009;296:L936–946. doi: 10.1152/ajplung.90625.2008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Landsman L, Jung S. Lung macrophages serve as obligatory intermediate between blood monocytes and alveolar macrophages. J Immunol. 2007;179:3488–3494. doi: 10.4049/jimmunol.179.6.3488. [DOI] [PubMed] [Google Scholar]
- 25.Janssen WJ, Barthel L, Muldrow A, Oberley-Deegan RE, Kearns MT, Jakubzick C, Henson PM. Fas determines differential fates of resident and recruited macrophages during resolution of acute lung injury. American journal of respiratory and critical care medicine. 2011;184:547–560. doi: 10.1164/rccm.201011-1891OC. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Gonzalez-Juarrero M, Shim TS, Kipnis A, Junqueira-Kipnis AP, Orme IM. Dynamics of macrophage cell populations during murine pulmonary tuberculosis. J Immunol. 2003;171:3128–3135. doi: 10.4049/jimmunol.171.6.3128. [DOI] [PubMed] [Google Scholar]
- 27.Auffray C, Sieweke MH, Geissmann F. Blood monocytes: development, heterogeneity, and relationship with dendritic cells. Annu Rev Immunol. 2009;27:669–692. doi: 10.1146/annurev.immunol.021908.132557. [DOI] [PubMed] [Google Scholar]
- 28.Gordon S, Taylor PR. Monocyte and macrophage heterogeneity. Nat Rev Immunol. 2005;5:953–964. doi: 10.1038/nri1733. [DOI] [PubMed] [Google Scholar]
- 29.Murray PJ, Wynn TA. Protective and pathogenic functions of macrophage subsets. Nat Rev Immunol. 2011;11:723–737. doi: 10.1038/nri3073. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Chamoto K, Gibney BC, Ackermann M, Lee GS, Lin M, Konerding MA, Tsuda A, Mentzer SJ. Alveolar macrophage dynamics in murine lung regeneration. J Cell Physiol. 2012;227:3208–3215. doi: 10.1002/jcp.24009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Zaslona Z, Wilhelm J, Cakarova L, Marsh LM, Seeger W, Lohmeyer J, von Wulffen W. Transcriptome profiling of primary murine monocytes, lung macrophages and lung dendritic cells reveals a distinct expression of genes involved in cell trafficking. Respiratory research. 2009;10:2. doi: 10.1186/1465-9921-10-2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Lehnert BE. Pulmonary and thoracic macrophage subpopulations and clearance of particles from the lung. Environ Health Perspect. 1992;97:17–46. doi: 10.1289/ehp.929717. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.McCullers JA, Rehg JE. Lethal synergism between influenza virus and Streptococcus pneumoniae: characterization of a mouse model and the role of platelet-activating factor receptor. J Infect Dis. 2002;186:341–350. doi: 10.1086/341462. [DOI] [PubMed] [Google Scholar]
- 34.Brundage JF, Shanks GD. Deaths from bacterial pneumonia during 1918–19 influenza pandemic. Emerging infectious diseases. 2008;14:1193–1199. doi: 10.3201/eid1408.071313. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Duan M, Li WC, Vlahos R, Maxwell MJ, Anderson GP, Hibbs ML. Distinct macrophage subpopulations characterize acute infection and chronic inflammatory lung disease. J Immunol. 2012;189:946–955. doi: 10.4049/jimmunol.1200660. [DOI] [PubMed] [Google Scholar]
- 36.van der Sluijs KF, van Elden LJ, Nijhuis M, Schuurman R, Pater JM, Florquin S, Goldman M, Jansen HM, Lutter R, van der Poll T. IL-10 is an important mediator of the enhanced susceptibility to pneumococcal pneumonia after influenza infection. J Immunol. 2004;172:7603–7609. doi: 10.4049/jimmunol.172.12.7603. [DOI] [PubMed] [Google Scholar]
- 37.Hashimoto Y, Moki T, Takizawa T, Shiratsuchi A, Nakanishi Y. Evidence for phagocytosis of influenza virus-infected, apoptotic cells by neutrophils and macrophages in mice. J Immunol. 2007;178:2448–2457. doi: 10.4049/jimmunol.178.4.2448. [DOI] [PubMed] [Google Scholar]
- 38.Shibata Y, Berclaz PY, Chroneos ZC, Yoshida M, Whitsett JA, Trapnell BC. GM-CSF regulates alveolar macrophage differentiation and innate immunity in the lung through PU.1. Immunity. 2001;15:557–567. doi: 10.1016/s1074-7613(01)00218-7. [DOI] [PubMed] [Google Scholar]
- 39.Huang FF, Barnes PF, Feng Y, Donis R, Chroneos ZC, Idell S, Allen T, Perez DR, Whitsett JA, Dunussi-Joannopoulos K, Shams H. GM-CSF in the lung protects against lethal influenza infection. American journal of respiratory and critical care medicine. 2011;184:259–268. doi: 10.1164/rccm.201012-2036OC. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Yoshida M, Ikegami M, Reed JA, Chroneos ZC, Whitsett JA. GM-CSF regulates protein and lipid catabolism by alveolar macrophages. Am J Physiol Lung Cell Mol Physiol. 2001;280:L379–386. doi: 10.1152/ajplung.2001.280.3.L379. [DOI] [PubMed] [Google Scholar]
- 41.Sever-Chroneos Z, Murthy A, Davis J, Florence JM, Kurdowska A, Krupa A, Tichelaar JW, White MR, Hartshorn KL, Kobzik L, Whitsett JA, Chroneos ZC. GM-CSF modulates pulmonary resistance to influenza A infection. Antiviral Res. 2011;92:319–328. doi: 10.1016/j.antiviral.2011.08.022. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Steinwede K, Tempelhof O, Bolte K, Maus R, Bohling J, Ueberberg B, Langer F, Christman JW, Paton JC, Ask K, Maharaj S, Kolb M, Gauldie J, Welte T, Maus UA. Local delivery of GM-CSF protects mice from lethal pneumococcal pneumonia. J Immunol. 2011;187:5346–5356. doi: 10.4049/jimmunol.1101413. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Hamilton JA. Colony-stimulating factors in inflammation and autoimmunity. Nat Rev Immunol. 2008;8:533–544. doi: 10.1038/nri2356. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.