

Abstract
Background
Calmodulin (CaM) is encoded by three genes, CALM1, CALM2, and CALM3, all of which harbor pathogenic variants linked to long QT syndrome (LQTS) with early and severe expressivity. These LQTS-causative variants reduce CaM affinity to Ca2+ and alter the properties of the cardiac L-type calcium channel (CaV1.2). CaM also modulates NaV1.5 and the ryanodine receptor, RyR2. All of these interactions may play a role in disease pathogenesis. Here, we determine the spectrum and prevalence of pathogenic CaM variants in a cohort of genetically elusive LQTS, and functionally characterize the novel variants.
Methods and Results
Thirty-nine genetically elusive LQTS cases underwent whole exome sequencing to identify CaM variants. Non-synonymous CaM variants were overrepresented significantly in this heretofore LQTS cohort (15.4%) compared to exome aggregation consortium (0.04%; p<0.0001). When the clinical sequelae of these 6 CaM-positive cases was compared to the 33 CaM-negative cases, CaM-positive cases had a more severe phenotype with an average age of onset of 8 months, an average QTc of 679 ms, and a high prevalence of cardiac arrest. Functional characterization of one novel variant, E141G-CaM, revealed an 11-fold reduction in Ca2+ binding affinity and a functionally-dominant loss of inactivation in CaV1.2, mild accentuation in NaV1.5 late current, but no effect on intracellular RyR2-mediated calcium release.
Conclusions
Overall, 15% of our genetically elusive LQTS cohort harbored non-synonymous variants in CaM. Genetic testing of CALM1-3 should be pursued for individuals with LQTS, especially those with early childhood cardiac arrest, extreme QT prolongation, and a negative family history.
Keywords: calmodulin, L-type calcium channels, long QT syndrome, ryanodine receptor, sodium channels
Introduction
Calmodulin (CaM) is an essential Ca2+ sensing, signal-transducing protein. Calcium-induced activation of CaM regulates many calcium-dependent processes and modulates the function of cardiac ion channels including the long QT syndrome (LQTS)-associated CACNA1C-encoded CaV1.2 calcium channel and SCN5A-encoded NaV1.5 sodium channel, as well as the catecholaminergic polymorphic ventricular tachycardia (CPVT)-associated RYR2-encoded ryanodine receptor.1-3 Interestingly, there are three calmodulin genes, CALM1 (chr14q31), CALM2 (chr2p21), and CALM3 (chr19q13),4 with unique nucleotide sequences that all encode for a completely identical 149 amino acid CaM protein,5, 6 which are expressed differentially in the human heart.7
Variants in all three of the calmodulin genes (CALM1, CALM2, and CALM3) have been described recently in LQTS.7-9 LQTS is a disorder of ventricular myocardial repolarization characterized by the prolongation of the heart-rate corrected QT interval (QTc) on a resting electrocardiogram (ECG), manifesting clinically as syncope, seizures, or sudden death in the setting of a structurally normal heart. In 2013, whole exome sequencing (WES) was utilized on two parent-child trios of severe cases of LQTS presenting during infancy yielding de novovariants in CALM1 and CALM2.7 Follow-up cohort analysis identified two additional LQTS patients with variants in CALM1. Since then, additional CALM2 variants have also been identified in cases of LQTS and LQTS/CPVT overlap phenotypes.8 Finally, a variant was identified in CALM3-encoded CaM in a severe case of LQTS.9
All of the identified LQTS-associated variants in CaM characterized to date have reduced affinity for Ca2+ and attenuated CaV1.2 inactivation through the loss of calcium dependent inactivation (CDI).7, 8, 10, 11 Here, we describe the spectrum, prevalence, and functional consequence of novel CaM variants identified within our cohort of 39 unrelated patients with genetically elusive LQTS.
Methods
Study subjects
The study population consisted of 39 unrelated patients with clinically diagnosed LQTS with a Schwartz score ≥ 3.5 (Table 1) that were referred to the Windland Smith Rice Sudden Death Genomics Laboratory at Mayo Clinic, Rochester, MN for genetic testing. All 39 patients were genotype-negative for all known LQTS-susceptibility genes: AKAP9, ANKB, CACNA1C, CAV3, KCNE1, KCNE2, KCNH2, KCNJ2, KCNJ5, KCNQ1, SCN4B, SCN5A, SNTA1, and TRDN. Thus, the 39 cases tested here represent those patients that would fall within the estimated 20% remnant of patients with a clinically certain diagnosis of LQTS, yet remain without an identified genetic cause following genetic testing of all currently known LQTS susceptibility genes. This study was approved by the Mayo Foundation Institutional Review Board and informed consent was obtained for all patients.
Table 1.
Demographics of our genotype negative LQTS cohort
| Number of Probands | 39 |
| Age at Diagnosis, years ± SD | 20 ± 18 |
| Range | 0-65 |
| < 5 years of age (%) | 10 (26) |
| ≥ 5 years of age (%) | 29 (74) |
| Males (%) | 16 (41) |
| QTc, ms ± SEM | 538 ± 13 |
| Syncope (%) | 15 (38) |
| Cardiac Arrest (%) | 14 (36) |
| Positive Family History (%) | 14 (36) |
Whole Exome Sequencing (WES) with Targeted Calmodulin Gene Analysis
All 39 LQTS patients underwent WES and subsequent calmodulin (CaM) gene (CALM1, CALM2, and CALM3) specific analysis. Briefly, paired-end libraries were prepared following the manufacturer's protocol (Illumina, San Diego, CA and Agilent, Santa Clara, CA) using the Bravo liquid handler from Agilent. Whole exon capture was carried out using the protocol for Agilent's SureSelect Human All Exon v4 + UTRs kit. Exome libraries were loaded onto TruSeq Rapid run paired end flow cells at concentrations of 9 pM to generate cluster densities of 600,000-800,000/mm2 following Illumina's standard protocol using the Illumina cBot and TruSeq Rapid Paired end cluster kit version 1. The flow cells were sequenced as 100 × 2 paired end reads on an Illumina HiSeq 2500 using TruSeq Rapid SBS kit version 1 and HiSeq data collection version 2.0.12.0 software. Base-calling was performed using Illumina's RTA version 1.17.21.3.
The Illumina paired end reads were aligned to the hg19 reference genome using Novoalign 2.08 (http://novocraft.com) followed by the sorting and marking of duplicate reads using Picard (http://picard.sourceforge.net). Local realignment of INDELs and base quality score recalibration were then performed using the Genome Analysis Toolkit 2.7-4 (GATK).12 Single nucleotide variants (SNVs) and insertions/deletions (INDELs) were called across all of the samples simultaneously using GATK's Unified Genotyper with variant quality score recalibration.13
Following the Mayo Clinic bioinformatics pipeline analysis, the data was provided in comprehensive Microsoft Excel (Redmond, WA) spreadsheet with all of the identified variants across WES samples. Using filter functions in Microsoft Excel, we searched for variants within CALM1 (NM_006888), CALM2 (NM_001743), and CALM3 (NM_005184) that had a genotype call quality score of > 20 and a read depth of > 10. To be considered as a putative pathogenic variant, the identified CaM variants had to be i) non-synonymous (amino acid altering), ii) involve a highly conserved amino acid, iii) and absent in the Exome Aggregation Consortium (ExAC , n=60,706) database.14 All CaM variants identified through WES were Sanger sequence verified. Primer sequences and conditions are available upon request.
Functional Analysis
The novel CaM variant (p.E141G) was characterized functionally using Ca2+ binding assays and patch-clamp electrophysiological recording to assess its pathogenicity.
Generation of recombinant CaM and measurement of Ca2+ binding to CaM
Wildtype and mutant CaM proteins were prepared and the Ca2+ affinities for WT- and E141G-CaM were determined using methods previously described.7
CaM mammalian expression vectors and site-directed mutagenesis
The EGFP-HA-CaM (EGFP: enhanced green florescent protein) vector, kindly provided by Dr. Emanuel Strehler, Mayo Clinic, Rochester, MN, was used for the heterologous expression electrophysiology studies of CaV1.2 and NaV1.5 in TSA201 cells. The p.E141G missense variant was engineered into EGFP-HA-CaM vector using primers containing the missense variant (available upon request) in combination with the Quikchange II XL Site-Directed Mutagenesis Kit (Stratagene, La Jolla, CA). The integrity of all constructs was verified by DNA sequencing.
Heterologous expression and electrophysiological analysis of CaV1.2 co-expressed with WT- or E141G-CaM
The constructs utilized to recapitulate CaV1.2 have been described previously.15 TSA201 cells were cultured in Dulbecco's Modification of Eagle's Medium supplemented with 10% Fetal Bovine Serum (FBS), 1.0% L-glutamine, and 1.2% penicillin/streptomycin solution in a 5% CO2 incubator at 37°C. Heterologous expression of CaV1.2 was accomplished by co-transfecting 1 μg CACNA1C ([(EYFP)Nα1c,77] in pcDNA),15 1 μg CACNB2b (in pIRES2-dsRED2),15 and 1 μg CACNA2D1 (in pcDNA3.1)15 vectors with either 0.5 μg a green fluorescent protein empty vector (GFP-EV; kindly provided by Dr. Gianrico Farrugia, Mayo Clinic, Rochester MN), EGFP-HA-CaM-WT, or EGFP-HA-CaM-E141G vectors using 9 μl Lipofectamine 2000 (Invitrogen, Carlsbad, CA). The media was replaced with fresh OPTI-MEM after 4-6 hours. Transfected TSA201 cells were cultured in OPTI-MEM and incubated for 48 hours and cells exhibiting green, red, and yellow fluorescence were selected for electrophysiological experiments. Standard whole-cell patch clamp technique was used to measure CaV1.2 WT currents co-expressed with WT- or E141G-CaM at room temperature (22-24 °C) as described previously.15 In addition, we also examined persistent ICaL current, which was measured at the end of a 500 ms long depolarization.
Animal use and experiments in ventricular myocytes
The use of animals in this study was approved by the Animal Care and Use Committees of Vanderbilt University, Nashville, TN, USA and performed in accordance with National Institutes of Health guidelines. Single ventricular myocytes from 10- to 16-week-old C57BL/6 mice were isolated by enzymatic digestion using collagenase as previously described.16 Inactivation of CaV1.2 current was studied in freshly isolated murine ventricular myocytes using whole-cell patch clamp technique. CaM (total concentration 6 μM for all experiments) was added to pipette solution and then dialyzed into the cell via patch pipette. Currents were elicited with 500-ms depolarizing pulses to 0 mV from holding potential (HP) of −70 mV applied every 2 minutes to track the effect of CaM over time, as it diffuses into the cell. Usually, the effect of CaM on inactivation reached its maximum 4-6 min after start of dialysis. 15-ms pre-pulses to −40 mV were applied prior to the test pulse to inactivate Na+ currents. Cells were pre-treated with ryanodine (50 μM) and thapsigargin (10 μM) for 30 min prior to experiment to prevent sarcoplasmic reticulum (SR) Ca2+ release. Experiments were conducted at room temperature. For mixing studies, a mixture of 75% of WT CaM and 25% of mutant CaM (total CaM concentration 6 μM) was used. Tau values represent mono-exponential fit of the last 350 ms of the current during depolarizing step.
In addition, Ca2+ spark measurements to examine RyR2 activity were completed as previously described.3
Heterologous expression and electrophysiological analysis of NaV1.5 co-expressed with WT- or E141G-CaM
In order to recapitulate the SCN5A-encoded NaV1.5 sodium channel, 1 μg of the human cardiac voltage-dependent Na+ channel α subunit (H558/Q1077del, Genbank accession no.AY148488) in the pcDNA3 vector (Invitrogen, Carlsbad, CA) was co-transfected with either 0.5 μg GFP-EV, EGFP-HA-CaM-WT, EGFP-HA-CaM-E141G, or 0.25 μg EGFP-HA-CaM-WT + 0.25 μg EGFP-HA-CaM-E141G with the use of 4μl Lipofectamine in TSA201 cells. Transfected TSA201 cells were cultured in OPTI-MEM (Gibco, Carlsbad, CA) and incubated for 24 hours after transfection and cells exhibiting green fluorescence were selected for electrophysiological experiments. Standard whole-cell patch clamp technique was used to measure NaV1.5 WT currents co-expressed with WT-CaM, E141G-CaM, or WT-CaM + E141G-CaM (to mimic the heterozygous state of the patient) at room temperature as previously described.17 Late INa was measured at the end of 700 ms long depolarization.
Statistical analysis
Data are presented as mean values ± standard error of the mean (SEM) or ± 95% confidence intervals. Comparisons were made using one-way ANOVA or Student's t-test where appropriate and p-values < 0.05 were considered significant. Chi square test with Yates correction was utilized to derive the p-value to test for the enrichment of variants in the 39 variants versus ExAC.14 Categorical comparisons were made using a Fisher's exact test and p-values < 0.05 were considered significant.
Results
Genetic Analysis
Our genetically elusive LQTS cohort consisted of 39 individuals (41% males, average age at diagnosis was 20 ± 18 years, 26% were < 5 years-old). The average QTc was 538 ± 13 ms, 38% experienced syncope, 36% cardiac arrest, and 36% had a positive family history of cardiac arrhythmias or sudden unexplained death (Table 1).
WES revealed 6 CaM missense variants, p.D96V (c.287 A>T, CALM2), p.D130G (c.389 A>G, CALM2), p.D130V (c.389 A>T, CALM2), p.E141G (c.422 A>G, CALM1), and two cases with p.F142L (c.426 C>G, CALM1), in a total of 6 of 39 cases (15.4%; Table 2, Figure 1). Both p.D130V and p.E141G represent novel CaM variants. The, p.D96V, p.D130G, and p.F142L CaM variants have been characterized functionally and described previously as LQTS-susceptibility variants (Table 2).7, 10, 11, 18 Our previously discovered CALM3-variant positive child was not included in this cohort of unrelated cases as the child's variant had been discovered by WES and genomic triangulation (proband/parent trio) akin to the previous CALM1 and CALM2 discoveries.9
Table 2.
CaM Variants in LQTS and LQTS/CPVT Overlap Phenotypes.
| Study | Variants | Gene | Disease | Sex | Race | QTc (ms) |
Age at diagnosis |
Events | Increased Ca2+ Dissociation C-Domain |
Cav1.2 | Nav1.5 | RyR2 |
|---|---|---|---|---|---|---|---|---|---|---|---|---|
| Crotti | p.D96V | CALM2 | LQTS | F | Hispanic | 690 | prenatal | Cardiac arrest | 14-fold | Loss of CDI | No effect | No effect |
| Boczek | p.D96V | CALM2 | LQTS | F | Hispanic | 690 | prenatal | Cardiac arrest | 14-fold | Loss of CDI | No effect | No effect |
| Makita | p.N98I | CALM2 | LQTS | M | White – England | 555 | 17 months | Cardiac arrest | 7-fold | NA | NA | NA |
| Makita/Nyegaard | p.N98S | CALM2 | LQTS/CPVT | M | Japanese | 478 | 5 years | Syncope during exertion | 1.7-fold decreased affinity | 1.6-fold decrease in CDI | NA | Greater open probability |
| Crotti | p.D130G | CALM1 | LQTS | F | White – Italy | 630 | 6 months | Cardiac arrest | 54-fold | Loss of CDI | 7.5-fold increase in fetal Nav1.5 late current | Lower binding affinity |
| Crotti | p.D130G | CALM1 | LQTS | M | Grecian | 610 | 1 month | Cardiac arrest | 54-fold | Loss of CDI | 7.5-fold increase in fetal Nav1.5 late current | Lower binding affinity |
| Reed | p.D130G | CALM3 | LQTS | M | White | 690 | birth | None | 54-fold | Loss of CDI | 7.5-fold increase in fetal Nav1.5 late current | Lower binding affinity |
| Boczek | p.D130G | CALM2 | LQTS | F | Indian | 740 | birth | Cardiac arrest | 54-fold | Loss of CDI | 7.5-fold increase in fetal Nav1.5 late current | Lower binding affinity |
| Boczek | p.D130V | CALM2 | LQTS | M | White | 800 | birth | Cardiac arrest | NA | NA | NA | NA |
| Makita | p.D132E | CALM2 | LQTS/CPVT | F | White – Germany | 578 | <9 years | Exercise induced syncope | 23-fold | NA | NA | NA |
| Makita | p.D134H | CALM2 | LQTS | F | Japanese | 579 | 6 years | Cardiac arrest | 13-fold | NA | NA | NA |
| Makita | p.Q136P | CALM2 | LQTS/CPVT | F | Morracan | 500 | 8 years | Syncope – SCD age 11 | 9-fold | NA | NA | NA |
| Boczek | p.E141G | CALM1 | LQTS | M | Indian | 610 | 4 years | Cardiac arrest | 11-fold decreased affinity | 8.1-fold increase in Cav1.2 late current | 2.7-fold increase in Nav1.5 late current | No effect |
| Crotti | p.F142L | CALM1 | LQTS | F | White – Italy | 600 | prenatal? | Cardiac arrest | 5-fold | Yes – loss of CDI | No effect | Lower binding affinity |
| Boczek | p.F142L | CALM1 | LQTS | F | Black | 612 | birth | Cardiac arrest – SCD 2 | 5-fold | Yes – loss of CDI | No effect | Lower binding affinity |
| Boczek | p.F142L | CALM1 | LQTS | M | Hispanic | 620 | birth | SCD – 1 year | 5-fold | Yes – loss of CDI | No effect | Lower binding affinity |
Figure 1.
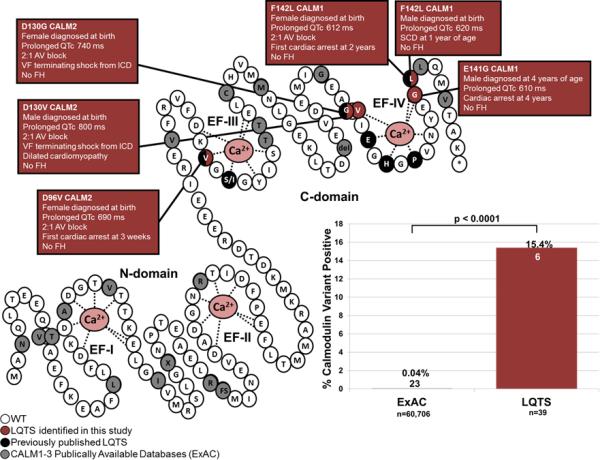
CaM variants identified in individuals with LQTS and the publically available databases. On the right is a schematic rendering of the CaM protein highlighting the N-domain and C-domain, each containing two EF hands (labeled EF-I through EF-IV) with Ca2+ (pink) bound. White circles represent the WT residues, red circles represent the variant residues found in our LQTS cohort, black circles represented previously published CaM variants in LQTS, grey circles represent variant residues found in all three CaM proteins in ExAC. The bar graph on the left compares the frequency of variant positive individuals in the ExAC (23/60,706; 0.04%) to our LQTS cohort (6/39; 15.4%; p<0.0001).
The p.D96V variant was identified in a female who was diagnosed prenatally with LQTS (Figure 1). She was delivered at term and her first ECG demonstrated a QTc of 690 ms, 2:1 AV block, and T-wave alternans (TWA). She was treated with beta-blockers and discharged. At 1-month of age, she experienced her first sudden cardiac arrest (SCA), and a single chamber internal cardiac defibrillator (ICD) was implanted. She developed a brain injury from the SCA and at 2-years-old developed seizures as a result. The family history was negative for arrhythmias and sudden death. Parental DNA was unavailable to test for de novo versus familial inheritance of the variant.
The p.D130G variant was identified in a female born at term that was noted to have bradycardia (Figure 1). An ECG, recorded 12 hours after birth, revealed a QTc of 740 ms and 2:1 AV block. She was treated with beta-blockers, phenytoin, spironolactone, potassium, and a single chamber pacemaker in the first week of life. At 6-years-old, a single chamber ICD was implanted and beta-blocker therapy continued. At 11- and 14-years-old, she experienced appropriate ICD discharges for ventricular fibrillation (VF). The family history was negative. Parental DNA was unavailable.
The p.D130V variant was identified in a male born with 2:1 AV block in utero (Figure 1). QT prolongation (QTc = 800 ms) was documented at birth, macroscopic TWA was observed, and beta-blocker therapy was initiated. Echocardiograms at the first month of life showed restrictive cardiomyopathy with borderline systolic function. In his second month of life, he developed torsades de pointes, and subsequently had a left T2-T4 sympathetic denervation and minimally invasive epicardial ICD. His beta-blocker was combined with sodium channel blockers. At 6-years-old he experienced an appropriate VF-terminating shock after a medication dose change by his parents. His echocardiograms demonstrated dilated cardiomyopathy with hypertrabeculation, and continued QT prolongation. At 7-years-old, he had an additional VF-terminating shock after missing a single beta-blocker dose. The family history was negative. The p.D130V was absent in both parents, thus confirming a de novo occurrence in the child.
The p.E141G variant was identified in a male with his first unwitnessed syncopal event at 3 years of age from which he was found unconscious and spontaneously recovered (Figure 1). He experienced his first SCA at 4 years of age in which CPR was initiated, and after 30 minutes, consciousness was regained. After his SCA, an ECG revealed a QTc of 610 ms, and he was treated subsequently with beta blockers and sodium channel blockers. His QT prolongation has persisted, however the index case is now 11-years-old and has not experienced an episode since the initiation of treatment. An echocardiogram revealed mild LV dilation; however the cardiac valves and structure were normal. In addition, it was noted that he has speech and motor skill delay. He has negative family history. The p.E141G was not identified in either parent confirming a de novo occurrence in the child.
The p.F142L variant was identified in a female. Her ECG shortly after birth showed a QTc of 612 ms and 2:1 AV block (Figure 1). Her initial treatment consisted of beta-blockers and at 19 months a dual chamber pacemaker was implanted. Shortly thereafter, she had SCA, which caused anoxic brain injury and seizure-like syncopal episodes. After the initiation of this genetic investigation, the patient died suddenly. She was brought to the emergency room for altered mental status. Echocardiogram revealed severely diminished left ventricular systolic dysfunction. Soon after arrival she deteriorated to ventricular fibrillation and was unable to be resuscitated. Her autopsy revealed cardiomegaly with dilation and hypertrophy. She had no family history of arrhythmias, and her mother was negative for the p.F142L variant. The father's sample was unavailable.
This p.F142L variant was also identified in a male. He was delivered at term, and bradycardia was noted. This prompted an ECG which revealed a QTc of 620 ms. He was treated with beta-blockers and had a pacemaker placed for atrial pacing. This combination therapy was associated with an attenuation of his QTc down to 540 ms. Shortly after recruitment into our study at 1 year and 3 months of age, this patient died suddenly. He was placed for a nap and was found gasping for air, unconscious and blue, and unfortunately was unable to be resuscitated. Interrogation of his pacemaker showed sinus rhythm with 1:1 conduction just before a period of fast VF which stabilized into VT. He had no prior recorded events on his pacemaker. ECGs were obtained for the parents and siblings, all were normal, and both parents were negative for the p.F142L variant, supporting de novo occurrence.
Over-representation of CaM variants in LQTS versus ExAC
Non-synonymous CaM variants were overrepresented significantly in our LQTS cohort (6/39; 15.4%) compared to ExAC (23/60,706; 0.04%, p<0.0001; Figure 1). The 21 rare non-synonymous CaM variants identified in ExAC are shown in Figure 1 and Supplemental Table 1. Interestingly, all 8 of the pathogenic CaM residues (D96, N98, D130, D132, D134, Q136, E141, and F142)7-9 that are affected in LQTS patients reside within one of the four EF-hands, where Ca2+ binds to CaM. Specifically, these pathogenic variants localize to either the third (D96 and N98) or fourth (D130, D134, Q136, E141, and F142) EF-hands located in the C-domain (Figure 1). Moreover, the side chains of 7 of these 8 residues interact directly with Ca2+. In contrast, only 4 of the 21 unique variants in ExAC (G24, I28, A103, and A104) reside within an EF-hand calcium binding loop, however none of them involve a residue whose side chain interacts directly with calcium (Figure 1). In addition, there are two extremely rare protein truncating variants (G42* and D51Gfs*5) present in the ExAC browser that were identified in 2/60,706 individuals. However, given the severity of CaM-related LQTS, this would suggest that having a non-functioning CaM protein from one of six CaM-generating alleles is not pathogenic.14
Demographic and clinical characteristics of patients harboring CaM variants
Compared to our original LQTS referral cohort that comprised 541 patients in 2005 where the average age at diagnosis was 24 ± 16 years, the average QTc was 482 ± 57 ms, and 12% had cardiac arrest,19 Table 3 details the differences between LQT1-3 positive and LQT1-3 negative cases in this original cohort,19 the CaM-positive cases herein, and the CaM-negative cases that still remain genetically elusive. The average age of CaM-positive patients was significantly younger (0.67 years) compared to CaM-negative (23 years; p<0.01; Figure 2A; Table 3).19 In addition, the yield of CaM variants was significantly higher in patients < 5-years-old (6/10, 60%) compared to patients ≥ 5 years-old (0/29, 0%, p<0.0001; Figure 2B). The average QTc was significantly longer in CaM- positive patients (679 ± 32 ms) compared to CaM-negative patients (514 ± 9 ms; p<0.0001; Figure 2C; Table 3).19 In addition, the occurrence of cardiac arrest was significantly higher in CaM-positive patients (6/6; 100%) compared to CaM-negative patients (8/33; 24%, p<0.001; Figure 2D; Table 3).19
Table 3.
Demographics of CaM-positive, CaM-negative compared to a previously published cohort of 541 cases of LQTS
| CaM-Positive (n=6) | CaM-Negative (n=33) | LQT1-3 Positive19 (n=272)* | LQT1-3 Negative19 (n=269)† | |
|---|---|---|---|---|
| Male/Female | 3/3 | 13/20 | 94/178 | 89/180 |
| Age at Diagnosis years ± SD | 0.67 ± 2 | 23 ± 18 | 23 ± 16 | 25 ± 16 |
| QTc, ms ± SEM | 679 ± 32 | 514 ± 9 | 494 ± 51 | 470 ± 60 |
| Cardiac Arrest (%) | 100 | 29 | 13 | 12 |
LQT1-3 Positive represents individuals who were positive for putative pathogenic variants in KCNQ1, KCNH2, or SCN5A from a larger cohort of 541 unrelated individuals with LQTS from a previously published study.19
LQT1-3 Negative represents individuals who were negative for putative pathogenic variants in KCNQ1, KCNH2, or SCN5A from a larger cohort of 541 unrelated individuals with LQT from a previously published study.19
Figure 2.
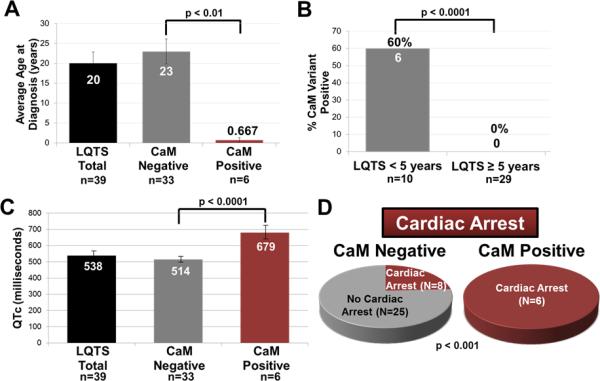
Demographic and clinical characteristics of the CaM-positive patients. (A) Bar graph comparing average age of diagnosis for our CaM-negative cases (23 ± 3 years), and CaM-positive cases (0.67 ± 0.7 years; p < 0.01). (B) Bar graph comparing the percent CaM-positive patients < 5 years of age (6/10; 60%) to patients ≥ 5 years of age (0/29; 0%; p < 0.0001). (C) Bar graph comparing the QTc of CaM negative patients (514 ± 9 ms) to CaM-positive patients (679 ± 32 ms; p < 0.0001). (D) Pie charts comparing the number of patients who had experienced cardiac arrest in our CaM-negative (8/33; 24%) versus CaM-positive patients (6/6; 100%; p < 0.001). Data in (A) and (B) are shown as mean ± SEM.
Functional Characterization
Since p.D96V, p.D130G, and p.F142L are disruptive functionally (Table 2),7, 10, 11 we confidently conclude that these variants are responsible for the LQTS phenotype observed in our patients. Although p.D130V is novel, we believe that because glycine (G) and valine (V) are replacing a critical negatively charged aspartic acid (D) known to interact with the positively charged calcium ion (Figure 1), p.D130V will functionally and clinically mimic p.D130G. The p.E141G variant was the only variant affecting a novel residue and we therefore characterized it functionally in this study.
E141G-CaM impairs Ca2+ binding by CaM
In line with previous observations, the p.E141G variant, located in EF-hand IV of the C-domain (Figure 1), decreased the Ca2+ affinity of the C-domain (by 11-fold) but did not significantly alter the Ca2+ affinity of the N-domain (Figure 3A).
Figure 3.
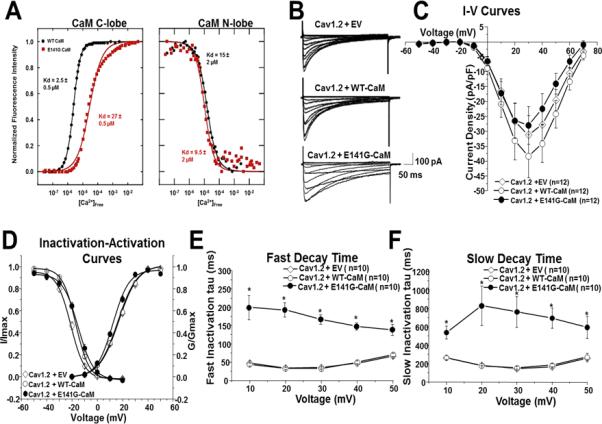
Ca2+ titration curves for WT- and E141G-CaM and patch clamp analysis in TSA201 cells. (A) Data were used to derive dissociation constants (Kd, in μM) for the each domain. E141G-CaM led to an 11-fold reduction in Ca2+ affinity of CaM C-domain compared to WT-CaM, whereas N-domain Ca2+ binding was not statistically different. Values are averages of 3 experiments, and error was determined by analysis of the curve fits. (B) Representative tracings of whole cell CaV1.2 current from TSA201 cells expressing CaV1.2+EV, CaV1.2+WT-CaM and CaV1.2+E141G-CaM determined from a holding potential −90 mV to testing potential of +70 mV in 10 mV increments with 500 ms duration. (C) Current-voltage relationship for CaV1.2+EV, CaV1.2+WT-CaM, and CaV1.2+E141G-CaM. All values represent mean ± SEM. (D) Inactivation-activation curves of CaV1.2+EV, CaV1.2+WT-CaM and CaV1.2+E141G-CaM (n=8-12). Steady-state inactivation was determined from a holding potential of −90 mV to pre-pulse of 20 mV in 10 mV increments with 10 s duration followed by a test pulse of 30 mV with 500 ms duration. I/Imax represents normalized calcium current and G/Gmax represents normalized conductance. Fast (E) and slow (F) decay time of CaV1.2+EV, CaV1.2+WT-CaM and CaV1.2+E141G-CaM. *P<0.05 vs. CaV1.2+WT-CaM.
E141G-CaM disrupts CaV1.2's CDI
The previously published LQTS-associated CaM variants significantly impaired CaV1.2 calcium dependent inactivation (CDI) leading to a loss of inactivation.11,10 Therefore, we examined the effects of p.E141G-CaM on CaV1.2 in a heterologous expression system. Typical CaV1.2 tracings of voltage-dependent activation from CaV1.2 with GFP-EV, WT- and E141G-CaM are shown in Figure 3B with holding potential at −90 mV to various depolarization potentials (see figure legend). Current-voltage relationship shows that p.E141G-CaM did not change CaV1.2 peak current density (Figure 3C). However, p.E141G-CaM shifted CaV1.2 V1/2 of steady-state inactivation 1.9 mV from −16.8 ± 0.23 mV (CaV1.2+WT-CaM; n=11) to −14.9 ± 0.27 mV (CaV1.2+E141G-CaM; n=9). The p.E141G-CaM also shifted CaV1.2 V1/2 of activation −3.9 mV from 16.4 ± 0.97 mV (CaV1.2+WT-CaM, n=12) to 12.5 ± 0.76 mV (CaV1.2+E141G-CaM; n=12; p < 0.05; Figure 3D).
CaV1.2+E141G-CaM exhibited much slower fast and slow decay time across the voltages +10 mV to +50 mV compared with CaV1.2+WT-CaM (n=10 for each group, p<0.05) (Figure 3E-F). Typical CaV1.2 current tracings from CaV1.2+EV, WT-CaM, and E141G-CaM are shown in Figure 4A. E141G-CaM increased CaV1.2's persistent current 7.1 fold from 2.1 ± 0.5% (CaV1.2 + WT-CaM; n=9) to 17.0 ± 3.0% (CaV1.2 + E141G-CaM; n=10, p<0.05; Figure 4B).
Figure 4.
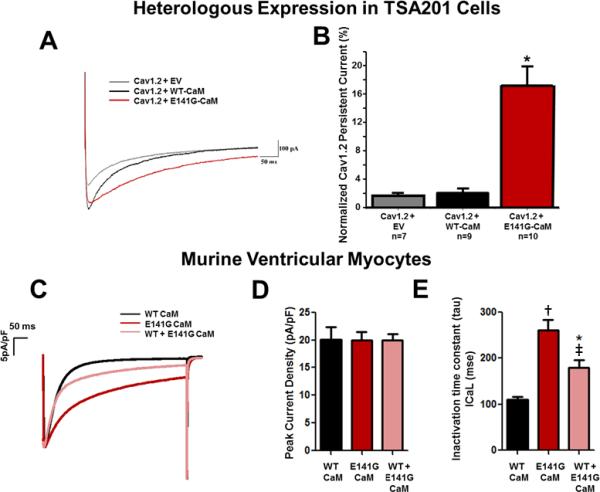
E141G-CaM leads to increased CaV1.2 persistent current and alters current inactivation without affecting peak current density in murine ventricular myocytes. (A) Representative tracings of persistent CaV1.2 current from CaV1.2+EV, CaV1.2+WT-CaM and CaV1.2+E141G-CaM determined from a holding potential of −90 mV to +30 mV with 500ms duration in TSA201 cells. (B) Group data showing CaV1.2 late current normalized to peak (%) for CaV1.2+EV, CaV1.2+WT-CaM, and CaV1.2+E141G-CaM in TSA201 cells. *P<0.05 vs. CaV1.2+WT-CaM. (C) Representative examples of traces for each experimental group in murine ventricular myocytes. (D) Average current densities (pA/pF) obtained in cells dialyzed with WT- or E141G-CaM in murine ventricular myocytes. (E) Effect of E141G-CaM alone or mixed with 75% WT CaM on inactivation time constant of the CaV1.2 compared to WT-CaM in murine ventricular myocytes. Data are mean ± SD. (n=7 *P<0.05; †P<0.001 vs. WT-CaM. ‡P<0.01 vs. E141G-CaM alone).
We next tested the effect of E141G-CaM in native murine ventricular myocytes. As in our heterologous expression system, E141G-CaM drastically impaired CaV1.2 current inactivation without affecting peak currents (Figure 4C-E). Even a 25% fraction of E141G-CaM in the presence of 75% WT-CaM was sufficient to significantly impair the CaV1.2 inactivation, which is consistent with a dominant negative effect.
E141G-CaM accentuates NaV1.5 late current
Next, we determined if E141G-CaM affected the electrophysiological characteristics of NaV1.5. Typical INa tracings of voltage-dependent activation from NaV1.5 with GFP-EV, WT- and E141G-CaM are shown in Figure 5A with holding potential at −100 mV to various depolarization potentials (see figure legend). Current-voltage relationship shows that WT- and E141G-CaM did not change NaV1.5 peak current density, voltage-dependent inactivation or activation, or decay time (Figure 5B-D).
Figure 5.
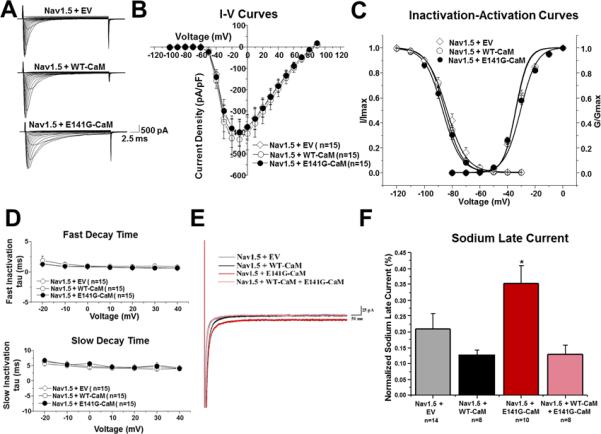
E141G-CaM leads to increased NaV1.5 late current. (A) Representative tracings of whole cell NaV1.5 current from TSA201 cells expressing NaV1.5+EV, NaV1.5+WT-CaM, and NaV1.5+E141G-CaM determined from a holding potential of −100 mV to testing potential of +90 mV in 10 mV increments with 24 ms duration. (B) Current-voltage relationship for NaV1.5+EV, NaV1.5+WT-CaM, and NaV1.5+E141G-CaM. All values represent mean ± SEM. (C) Inactivation-activation curves of NaV1.5+EV, NaV1.5+WT-CaM and NaV1.5+E141G-CaM (n=13-15). Steady-state inactivation obtained from a holding potential of −120 mV to pre-pulse of 0 mV in 10 mV increments with 976 ms duration followed by a test pulse of 0 mV with 24 ms duration. I/Imax represents normalized sodium current, G/Gmax represents normalized conductance. (D) Fast and slow decay time of NaV1.5+EV, NaV1.5+WT-CaM, and NaV1.5+E141G-CaM. (E) Representative tracings of NaV1.5 late current from NaV1.5+EV, NaV1.5+WT-CaM, NaV1.5+E141G-CaM, and NaV1.5+WT-CaM+E141G-CaM determined from a holding potential of −120 mV to −20 mV with 700ms duration. (F) Group data showing NaV1.5 late current normalized to peak (%) for NaV1.5+EV, NaV1.5+WT-CaM, NaV1.5+E141G-CaM, and NaV1.5+WT-CaM+E141G-CaM. *P<0.05 vs. NaV1.5+WT-CaM.
Typical NaV1.5 late current tracings from NaV1.5 with EV, WT-, E141G-CaM, and WT-CaM+E141G-CaM are shown in Figure 5E. E141G-CaM increased NaV1.5's late current 1.7-fold from 0.13 ± 0.01% (NaV1.5+WT-CaM; n=8) to 0.35 ± 0.06% (NaV1.5+E141G-CaM; n=10; p<0.05; Figure 5F). However, when WT-CaM was co-expressed with E141G-CaM, there was no longer an effect on the NaV1.5 peak late current (Figure 5E-F). This result suggests that unlike for Cav1.2, there was no functional dominance of E141G-CaM in the regulation of NaV1.5 late currents.
E141G-CaM has no effect on RyR2 Ca2+ release channels and SR Ca2+ handling
CaM variants have also been identified in patients with CPVT,18 leading to activation of RyR2 SR Ca2+ release channels.3 However, to date, the known LQTS-associated CaM variants have exerted no effect or only slightly reduced RyR2 Ca2+ release channel activity.20 Therefore, we determined the effect of E141G-CaM on intracellular Ca2+ handling by measuring Ca2+ sparks (a measure of RyR2 Ca2+ release channel activity) and found that E141G-CaM had no effect on Ca2+ sparks and SR Ca2+ content (Figure 6).
Figure 6.
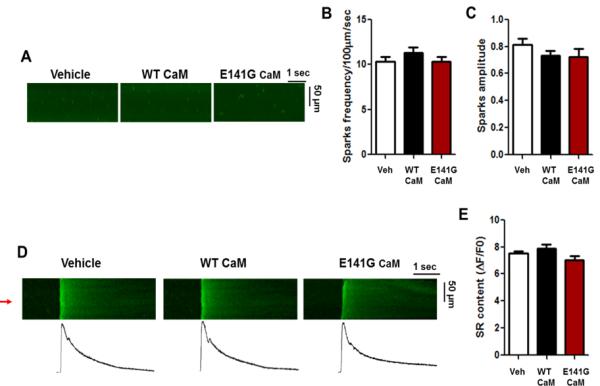
E141G-CaM has no effect on Ca2+ sparks and SR Ca2+ content. (A) Representative line-scan images of Ca2+ sparks in permeabilized mouse ventricular myocytes in CaM-free (vehicle), WT-CaM, and E141G-CaM in the presence of AIP2 (1μM). (B) Average Ca2+ spark frequency and (C) Ca2+ spark amplitude. Data are mean ± SD (n=20). (D) Line scan (top) and (bottom) line plot (red arrow) examples of SR Ca2+ content evaluated by 10 mM caffeine-evoked Ca2+ transient in CaM-free (vehicle), WT-CaM, and E141G-CaM in the presence of AIP2 (1μM). (E) Average SR Ca2+ content. Data are mean ± SD (n=4).
Discussion
Our examination of 39 patients with genetically elusive LQTS identified 6 individuals (15.4%) that harbored variants in either CALM1 or CALM2. Three of the identified variants, p.D96V, p.D130G, and p.F142L, had been published previously in other cases of LQTS,7 one variant represented a novel amino acid change at a previously described position, p.D130V, and the final variant, p.E141G, was novel. Within our cohort, CaM variants were overrepresented significantly when compared to the publically available databases in essentially a modified case:control analysis, which provides additional evidence that single amino acid substitutions within CaM, particularly those involving calcium-interacting residues in the C-domain, are not well tolerated.
When examining the clinical characteristics of these six patients harboring CaM variants, we found that they had an early age at onset with the average age of onset at 8 months, average QTc of 679 ± 32 ms, and all experienced cardiac arrest (Figure 2, Table 3).19 Moreover, all CaM variants were shown to occur as de novo when parental DNA was available for testing, thus supporting the malignant nature of LQTS-related CaM variants. These clinical characteristics seemed even more severe than those of the previously published LQT1-3 genotype-positive and LQT1-3 genotype-negative LQTS (Table 3),19 and the previously described LQTS7 and LQTS/CPVT8 patients with CaM variants (n=10), where the average age at diagnosis was 3 years, the average QTc was 591 ± 22 ms, and 60% experienced cardiac arrest (Table 2). However, if we separate the previously described CaM-positive patients into two categories, LQTS/CPVT overlap phenotypes and a pure LQTS phenotype, it becomes more apparent that LQTS/CPVT patients (n=3)8 may be less severe, as the average age at diagnosis was 7 years, the average QTc was 519 ± 53 ms, with only 33% experiencing cardiac arrest. In contrast, the patients with a pure LQTS phenotype (n=7) were similar to our LQTS CaM-positive patients, with an average age at diagnosis of 14 months, an average QTc of 622 ± 20 ms, and with 86% experiencing cardiac arrest. This provides evidence that CaM-mediated LQTS is very severe and it may be important to delineate these phenotypes, as they may present with different disease severities, and the phenotypic presentation may be regulated by distinct underlying mechanisms. Therefore, continued studies will be necessary to elucidate the key differences between CaM variants and their associated phenotypes. In addition, children with CaM-mediated LQTS who survive will need ongoing surveillance for the potential development of a cardiomyopathy as two of the CaM variant-positive children who have survived past 5 years of age, and one of the deceased patients on autopsy, had developed dilated cardiomyopathy. It is possible that the CaV1.2's impaired CDI may contribute to diastolic calcium excess that could precipitate cardiomyopathic structural remodeling.
CaM modulates the activity of several different ion channels by sensing and transducing Ca2+ signals. When Ca2+ levels are raised, CaM binds up to 4 calcium ions, which stabilize an open conformation within each domain that mediates interactions with CaM's targets. Hence, we first examined the effects of the E141G variant on the Ca2+ affinity of CaM, and found that this mutant resulted in an 11-fold reduction in the Ca2+ affinity of the C-domain. These results are comparable to the Ca2+ binding impairments caused by the 8 variants previously associated with LQTS or LQTS/CPVT overlap phenotypes (5-54 fold increased dissociation of Ca2+ in the C-domain, Table 2), 7, 10, 18 suggesting that impaired Ca2+ binding properties in the C-domain is a common underlying pathomechanism of LQTS-associated CaM variants.
These disrupted Ca2+ affinities have direct implications on CaV1.2's CDI. Typically, CaV1.2 is inactivated with increasing Ca2+ concentrations via a mechanism mediated by the binding of CaM. The altered Ca2+ binding properties of CaM, such as with CaM-E141G, would therefore be expected to impair CaV1.2's CDI. Using whole-cell patch clamp experiments, we found that CaM-E141G shifted inactivation 1.9 mV. These findings were replicated in native ventricular myocytes where CaM-E141G again reduced CaV1.2 inactivation, which closely parallels what has been noted previously with the other LQTS-associated CaM variants.10, 11
Because there are three calmodulin genes expressed in the heart, which each encode for an identical protein, our heterozygous variant only affects 1/6 of the calmodulin alleles. Therefore, we examined the CaM-E141G variant at reduced levels (25:75; mutant:WT), which also maintained a significant effect on inactivation. This suggests that p.E141G has a dominant negative effect and is capable of causing the disease phenotype.
In addition to CaV1.2, it has been established previously that CaM modulates cardiac sodium channel inactivation.21 Previously, Yin and colleagues found that LQTS-associated CaM variants (D96V, D130G, and F142L) did not affect NaV1.5; however D130G led to a 7.5-fold increase in fetal NaV1.5 late current.11 They hypothesized that because CaM-mediated LQTS has an early age of onset, the variants may only affect the fetal isoform of the sodium channel. However, because there have been associations of CaM with the adult isoform of NaV1.5, we wanted to determine whether CaM-E141G may have an effect on channel gating function. Our studies showed that the E141G variant led to a 1.7-fold increase in NaVl.5 late current when expressed as a homozygote. This makes our CaM-E141G variant the first CaM variant shown to affect the adult isoform of the cardiac sodium channel. Interestingly, however, when E141G was expressed with wild-type CaM – mimicking a heterozygote patient – the NaV1.5 late current was normalized. This differs from the effect of E141G-CaM on CaV1.2, where even a 3-fold excess of WT CaM failed to normalize CaV1.2 inactivation.
It has been established that CPVT-associated CaM variants18 affect the functioning of RYR2- encoded ryanodine receptor (RyR2), leading to greater open probability of the channel.3 Because one previously reported LQTS associated CaM variant (N98S) also activates RyR2 and can cause CPVT as well as LQTS,3 we examined the effect of p.E141G on RyR2 Ca2+ release channel activity. The p.E141G variant had no effect on Ca2+ sparks, a measure of RyR2 Ca2+ release channel activity, and did not affect SR Ca2+ content. Hence, our results continue to support the emerging evidence that LQTS-associated CaM variants do not affect RyR2, whereas CPVT-associated CaM variants perturb RyR2 function.
Conclusion
We have identified a novel CaM variant p.E141G, which like other LQTS-associated CaM variants, disrupts Ca2+ binding and leads to increased CaV1.2 window/persistent current. In addition, p.E141G is the first CaM variant identified to also affect the adult NaV1.5 isoform, leading to increased sodium late current. Overall, we found that 15% of genetically elusive LQTS patients harbored a functionally significant CaM variant. The phenotypic characteristics of the CaM-positive individuals from our cohort, combined with the other published cases of LQTS-associated CaM variants, suggest that LQTS patients harboring variants in either CALM1-, CALM2-, or CALM3-encoded CaM present early in life with profound QT prolongation and have a high predilection for cardiac arrest and sudden death. The existing gene panels for LQTS genetic testing should now be expanded to include CALM1, CALM2, and CALM3.
Supplementary Material
Clinical Perspective.
Recently, pathological variants in three genes (CALM1, CALM2, and CALM3) encoding for 100% identical calmodulin (CaM) protein have been associated with early onset long QT syndrome (LQTS). This report sought to characterize the spectrum and prevalence of CaM variants in an unrelated cohort of 39 patients with heretofore genetically elusive LQTS. Overall, six of these cases harbored rare pathogenic variants in one of the three CaM encoding genes. Three previously published variants (p.D96V, p.D130G, p.F142L identified twice) and two novel (p.D130V and p.E141G) variants were identified. Because p.D130V was allelic with a previously characterized pathogenic variant in CaM (p.D130G), we elected to functionally characterize the other novel variant. This variant, p.E141G, disrupted Ca2+ binding and led to altered CaV1.2 window current, similar to previously characterized CaM variants, consistent with LQTS pathogenicity. Closer examination of the phenotypic sequelae of these 6 patients with CaM-mediated LQTS highlighted that such individuals present early in life, have profound QT prolongation, and have a high predilection for cardiac arrest and sudden death when compared to LQTS patients without pathogenic CaM variants. These findings replicated what has been previously described in the literature, and there are now at least 10 described variants in one of the three calmodulin genes associated with LQTS. Taken altogether, patients with calmodulin-associated LQTS are more severe phenotypically than typical LQTS and genetic testing of all three calmodulin genes is warranted when there is evidence of severe, early onset LQTS.
Acknowledgments
We would like to acknowledge gratefully Dr. Emanuel Strehler at the Mayo Clinic for sharing his expertise of the calmodulins and for sharing his calmodulin DNA constructs.
Funding Sources: This work was supported by the Mayo Clinic Windland Smith Rice Comprehensive Sudden Cardiac Death Program, the Sheikh Zayed Saif Mohammed Al Nahyan Fund in Pediatric Cardiology Research, the Dr. Scholl Fund, and the Hannah M. Wernke Memorial Fund. This project was also supported in part by funding from Mayo Clinic's Center for Individualized Medicine (CIM). CNJ was supported by fellowships from the American Heart Association (13POST14380036) and the National Institutes of Health (5 F32 HL117612-02). Research on calcium binding proteins in the Chazin laboratory is supported by an endowed chair. This work was also partly supported by the United States National Institutes of Health (HL88635, HL71670 & HL124935 to BCK)
Footnotes
Conflict of Interest Disclosures: MJA is a consultant for Boston Scientific, Gilead Sciences, Medtronic, and St. Jude Medical. In addition, MJA, DJT, and Mayo Clinic receive sales based royalties from Transgenomic for their FAMILION-LQTS and FAMILION-CPVT genetic tests. However, none of these entities contributed to this study in any manner. NJB, NGH, DY, MLC, DK, HSH, CNJ, WJC, CGL, MS, ALP, YRL, RK and BCK have no conflicts of interest.
References
- 1.Levitan IB. It is calmodulin after all! Mediator of the calcium modulation of multiple ion channels. Neuron. 1999;22:645–648. doi: 10.1016/s0896-6273(00)80722-9. [DOI] [PubMed] [Google Scholar]
- 2.Gabelli SB, Boto A, Kuhns VH, Bianchet MA, Farinelli F, Aripirala S,Y. Regulation of the Nav1.5 cytoplasmic domain by calmodulin. Nat Commun. 2014;5:5126. doi: 10.1038/ncomms6126. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Hwang HS, Nitu FR, Yang Y, Walweel K, Pereira L, Johnson CN, et al. Divergent regulation of ryanodine receptor 2 calcium release channels by arrhythmogenic human calmodulin missense mutants. Circ Res. 2014;114:1114–1124. doi: 10.1161/CIRCRESAHA.114.303391. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Berchtold M, Egli R, Rhyner J, Hameister H, Strehler E. Localization of the human bona fide calmodulin genes calm1, calm2, and calm3 to chromosomes 14q24-q31, 2p21.1-p21.3, and 19q13.2-q13.3. Genomics. 1993;16:461–465. doi: 10.1006/geno.1993.1211. [DOI] [PubMed] [Google Scholar]
- 5.Fischer R, Koller M, Flura M, Mathews S, Strehler-Page M, Krebs J, et al. Multiple divergent mrnas code for a single human calmodulin. J Biol Chem. 1998;263:17055–17062. [PubMed] [Google Scholar]
- 6.SenGupta B, Friedberg F, Detera-Wadleigh S. Molecular analysis of human and rat calmodulin complementary DNA clones. Evidence for additional active genes in these species. J Biol Chem. 1987;262:16663–16670. [PubMed] [Google Scholar]
- 7.Crotti L, Johnson CN, Graf E, De Ferrari GM, Cuneo BF, Ovadia M, et al. Calmodulin mutations associated with recurrent cardiac arrest in infants. Circulation. 2013;127:1009–1017. doi: 10.1161/CIRCULATIONAHA.112.001216. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Makita N, Yagihara N, Crotti L, Johnson CN, Beckmann BM, Roh MS, et al. Novel calmodulin mutations associated with congenital arrhythmia susceptibility. Circ Cardiovasc Genet. 2014;7:466–474. doi: 10.1161/CIRCGENETICS.113.000459. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Reed GJ, Boczek NJ, Etheridge SP, Ackerman MJ. CALM3 mutation associated with long QT syndrome. Heart Rhythm. 2015;12:419–422. doi: 10.1016/j.hrthm.2014.10.035. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Limpitikul WB, Dick IE, Joshi-Mukherjee R, Overgaard MT, George AL, Jr., Yue DT. Calmodulin mutations associated with long qt syndrome prevent inactivation of cardiac l-type ca(2+) currents and promote proarrhythmic behavior in ventricular myocytes. J Mol Cell Cardiol. 2014;74:115–124. doi: 10.1016/j.yjmcc.2014.04.022. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Yin G, Hassan F, Haroun AR, Murphy LL, Crotti L, Schwartz PJ, et al. Arrhythmogenic calmodulin mutations disrupt intracellular cardiomyocyte Ca2+ regulation by distinct mechanisms. J Am Heart Assoc. 2014;3:e000996. doi: 10.1161/JAHA.114.000996. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.McKenna A, Hanna M, Banks E, Sivachenko A, Cibulskis K, Kernytsky A, et al. The genome analysis toolkit: A mapreduce framework for analyzing next-generation DNA sequencing data. Genome Res. 2010;20:1297–1303. doi: 10.1101/gr.107524.110. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.DePristo MA, Banks E, Poplin R, Garimella KV, Maguire JR, Hartl C,P. A framework for variation discovery and genotyping using next-generation DNA sequencing data. Nat Genet. 2011;43:491–498. doi: 10.1038/ng.806. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Exome aggregation consortium (exac) cambridge, ma: [November, 2015]. (url: Http://exac.Broadinstitute.Org) [Google Scholar]
- 15.Boczek NJ, Miller EM, Ye D, Nesterenko VV, Tester DJ, Antzelevitch C, et al. Novel timothy syndrome mutation leading to increase in cacna1c window current. Heart Rhythm. 2015;12:211–219. doi: 10.1016/j.hrthm.2014.09.051. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Knollmann BC, Chopra N, Hlaing T, Akin B, Yang T, Ettensohn K, et al. Casq2 deletion causes sarcoplasmic reticulum volume increase, premature Ca2+ release, and catecholaminergic polymorphic ventricular tachycardia. J Clin Invest. 2006;116:2510–2520. doi: 10.1172/JCI29128. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Van Norstrand DW, Valdivia CR, Tester DJ, Ueda K, London B, Makielski JC, et al. Molecular and functional characterization of novel glycerol-3-phosphate dehydrogenase 1 like gene (GPD1-L) mutations in sudden infant death syndrome. Circulation. 2007;116:2253–2259. doi: 10.1161/CIRCULATIONAHA.107.704627. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Nyegaard M, Overgaard Michael T, Sondergaard Mads T, Vranas M, Behr Elijah R, Hildebrandt Lasse L, et al. Mutations in calmodulin cause ventricular tachycardia and sudden cardiac death. Am J Hum Genet. 2012;91:703–712. doi: 10.1016/j.ajhg.2012.08.015. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Tester DJ, Will ML, Haglund CM, Ackerman MJ. Compendium of cardiac channel mutations in 541 consecutive unrelated patients referred for long qt syndrome genetic testing. Heart Rhythm. 2005;2:507–517. doi: 10.1016/j.hrthm.2005.01.020. [DOI] [PubMed] [Google Scholar]
- 20.Watanabe H, Knollmann BC. Mechanism underlying catecholaminergic polymorphic ventricular tachycardia and approaches to therapy. J Electrocardiol. 2011;44:650–655. doi: 10.1016/j.jelectrocard.2011.07.025. [DOI] [PubMed] [Google Scholar]
- 21.George AL., Jr. Inherited disorders of voltage-gated sodium channels. J Clin Invest. 2005;115:1990–1999. doi: 10.1172/JCI25505. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.