

Abstract
Motivation: The goal of deciphering the human glycome has been hindered by the lack of high-throughput sequencing methods for glycans. Although mass spectrometry (MS) is a key technology in glycan sequencing, MS alone provides limited information about the identification of monosaccharide constituents, their anomericity and their linkages. These features of individual, purified glycans can be partly identified using well-defined glycan-binding proteins, such as lectins and antibodies that recognize specific determinants within glycan structures.
Results: We present a novel computational approach to automate the sequencing of glycans using metadata-assisted glycan sequencing, which combines MS analyses with glycan structural information from glycan microarray technology. Success in this approach was aided by the generation of a ‘virtual glycome’ to represent all potential glycan structures that might exist within a metaglycomes based on a set of biosynthetic assumptions using known structural information. We exploited this approach to deduce the structures of soluble glycans within the human milk glycome by matching predicted structures based on experimental data against the virtual glycome. This represents the first meta-glycome to be defined using this method and we provide a publically available web-based application to aid in sequencing milk glycans.
Availability and implementation: http://glycomeseq.emory.edu
Contact: sagravat@bidmc.harvard.edu
Supplementary information: Supplementary data are available at Bioinformatics online.
1 Introduction
Glycans play integral roles in many essential biological functions including cell signaling, molecular recognition, immunity and inflammation generally via their specific interactions with proteins (Varki and Lowe, 2009). Unlike the template driven process for synthesizing linear nucleic acids and proteins, glycans are enzymatically synthesized and are thus products of many genes forming linear and branched sequences of stereospecific monosaccharides that provide unique surfaces for protein interactions (Bertozzi and Rabuka, 2009). Understanding the specificity of glycan-binding proteins (GBPs) provides clues to their functions, and defining specificity is accomplished by comparing the structures of glycans bound by a GBP with related structures that are unbound (Smith et al., 2010). This requires that the glycan ligands be completely defined, which is not a trivial task. In addition, while the human glycome is estimated to be at least 10 times larger than the proteome (Cummings, 2009); to date, no glycome or meta-glycome (partial or sub-glycome) related to a tissue, organ or cell type has been defined. Furthermore no method is currently available with the requisite precision or speed to be incorporated into an automated sequencing platform.
Historically, methods for glycan sequencing were developed to address different aspects of glycan structure and included purification of glycans and application of a variety of chemistries to deduce structure (Mulloy et al., 2009). The predominant Mass Spectrometric (MS) approaches to glycan structure are limited in their ability to fully identify glycan structure that includes sequence, linkage, and anomericity; however, a large amount of information is generated from mixtures of glycans. One example of high-throughput automated annotation of MS peaks was described in the Cartoonist algorithm (Goldberg et al., 2005). The algorithm initially selects annotations from a list of biologically plausible glycans based on a set of archetype cartoons manually derived from a priori knowledge of the biosynthetic pathways known to express certain N-glycans. It then assigns a confidence score to the set of glycan annotations for the most abundant signals. The annotations represent the composition and topology of the structure though portions of the structure contain specific monosaccharides and glycosidic bonds based on constraints imposed by the biosynthetic pathway. A de novo approach, termed STAT (Gaucher et al., 2000), predicts glycan structures by generating all possible topologies of a glycan structure based on the precursor ion mass, charge carrier and product ion mass from the MSn data. A review (von der Lieth et al., 2006) of automated interpretation of MS Spectra for glycan structures provides an overview of various approaches; however, all of the existing approaches fall short of fully characterizing glycan structures including the linkage and anomericity.
In spite of its limitations, MS techniques, especially those that include more laborious multistage MS (Ashline et al., 2014a) have proven extremely useful in doing the deep sequencing required to fully define a glycan structure. However, a method for high throughput, deep sequencing of glycan structure will likely require a combination of techniques. Defined glycan microarrays provide a high-throughput approach for identifying epitopes on individual glycans when interrogated with GBPs that bind known glycan determinants. Although the determinant or epitope alone cannot provide the information needed to fully characterize the glycan structure, it does provide ‘metadata’ that can be applied to reveal the structure. We developed Metadata-Assisted Glycan Sequencing or MAGS as a structural approach that combines MS data with glycan microarray data (Smith and Cummings, 2013), and recently demonstrated its utility in defining over 20 novel structures among the human milk glycans (HMGs) (Ashline et al., 2014b; Yu et al., 2014). The glycan microarray data used in this approach is acquired from libraries of relatively pure glycans, analogous to a shotgun glycan microarray (SGM) (Song et al., 2011) where glycans, representing a selected glycome, are fluorescently derivatized, and separated by multidimensional chromatography to resolve isomeric structures and obtain relatively pure components in a tagged glycan library. The separated glycans are then printed as a SGM comprising the selected glycome. We reasoned that MAGS could be used as a general approach to define any glycome that could be presented as a SGM; however, manual analysis of the data generated from hundreds of glycans in a microarray would be a tedious process. We therefore developed a software package, termed GlycomeSeq, to sequence HMGs through automated meta-analysis of experimental data based on our MAGS (Smith and Cummings, 2013) approach.
We reasoned that a key to more automatable sequencing is to create a ‘virtual glycome’ comprising all theoretical structures, and then test the predicted structures from the obtained metadata against this virtual glycome; we also used a novel algorithm to filter candidate glycan structures through this knowledge base to arrive at a single structure consistent with all available information. For this study, we selected the free glycans of human milk, since the glycans are easily accessible and a large literature is available about them. In addition to the nutritional disaccharide lactose (10–15 g/l), human milk contains a complex mixture of larger, free oligosaccharides or glycans (5–10 g/l) that are not efficiently metabolized in the stomach of the neonate and reach the intestines where they are thought to have probiotic activity (Bode, 2012), as well as to provide protection against pathogens by interfering with adhesion of pathogens to intestinal epithelial cells as ‘decoy receptors’ (Jantscher-Krenn et al., 2012; Kuhn et al., 2008; Ruiz-Palacios et al., 2003; Yu et al., 2014). Milk glycans have also been implicated in having beneficial innate immune and immunomodulatory effects (Duska-McEwen et al., 2014), decreasing colon contractility (Bienenstock et al., 2013) and promoting gut epithelial cell maturation (Holscher et al., 2014; Kuhn et al., 2008; Ruiz-Palacios et al., 2003). Recent studies on the biological functions of human milk have indicated that breast fed infants have soluble milk glycans circulating in blood at detectable levels suggesting potential systemic effects of such glycans (Goehring et al., 2014). The structures and quantities of the free glycans in human milk vary widely among individual mothers based on their genetics, which controls the expression of the human Lewis blood groups and time of lactation (Bode, 2012). A variety of factors influence the repertoire of the human glycome, including genetics, environment, and time (or conditions of synthesis); and like the human genome, the glycome of the human milk free glycans is an ‘average set of structures’ that represents all of the possible structures that could exist at any one time or in any one individual. Thus, there is great interest in defining the human milk metaglycome and its components, since they may differ in many ways between different sources and the overall repertoire of glycans in a milk sample is subject to many variables. The computational approach developed here was successful in identifying glycan structures within the human milk metaglycome and the approach should be applicable to other cellular and tissue metaglycomes.
2 Methods
In prior studies related to the current development we developed a functional glycomics approach using a SGMs of human milk (Yu et al., 2012, 2014), in which the SGMs are interrogated with viruses and antibodies that recognize unique glycan determinants. We also used defined lectins and antibodies before and after specific exoglycosidases to obtain detailed information about the repertoire of glycan determinants in individual glycans within the library. These data generated large quantities of metadata on each glycan; we then used logic to arrive at a structural solution based on the identification of specific determinants by antibody and lectin binding (Yu et al., 2014). However, the manual data processing to arrive at structures was extremely time consuming, and we reasoned that an algorithm could be used to apply the logic generated from the structural information and specificity of glycosyltransferases available from previous studies on milk glycans. If this could be accomplished as an automated, high-throughput method, it could be applicable as a general approach to defining a human metaglycome.
To address the human milk meta-glycome and to develop an automated system for MAGS, we used data available from the SGM from 10 different donors with mixed blood groups (Yu et al., 2014), and selected data from the analysis of 42 HMGs and 14 standards (Supplementary Table S1). This included 33 glycans whose structures were predicted by manual analysis of MALDI-TOF data, antibody and lectin binding data, and MSn analysis (Ashline, et al., 2014b; Yu et al., 2014). In this article, we describe the approach to developing an automated system for MAGS, and we show how automated analysis of multiple modalities can enrich the set of predicted structures for an unknown glycan target by introducing the concept of a ‘virtual glycome’ of human milk free glycans as a knowledge base and an algorithm for filtering candidate structures from the virtual glycome.
2.1 Generating the virtual human milk soluble glycome
Based on previous studies of human milk soluble glycan structures and the specificities of enzymes involved in their synthesis, we established a set of biologically plausible rules that can be used to define all of the possible structures synthesized as free glycans in human milk without regard to differential genetics or stage of lactation (see Supplementary Material A). These rules can then be used to computationally generate and store all possible glycan structures including structural isomers into a database. Seeking to establish a general method to generate a virtual glycome, we developed a novel approach using regular expressions (REs) to represent the biosynthetic rules for a particular metaglycome. For our initial attempt, we focused solely on the human milk soluble glycome and we describe the method below.
The Virtual Glycome Generator algorithm is initialized with the core lactose structure, a threshold parameter for the maximum core size of the glycome, and the patterns that represent the extensions and terminal modifications of HMG biosynthesis. The actual representation of the extension and terminal modification patterns is declared using REs as described in Supplementary Figure S1. A RE is a sequence of symbols or characters (also known as a string) that represent a set of patterns that describe regular languages from Formal Language Theory. Common applications of REs include validating email addresses in a web form, replacing or extracting values in a text file, UNIX shell commands such as ls or grep etc. REs can use operators and meta-characters to express non-trivial patterns of strings including repeating characters, optional characters and classes of characters. In our Virtual Glycome Generator algorithm, we are interested in the class of regular languages that are star-free, such that the language described is finite but also supports the union, disjunction and finite repetition of patterns. For example, rather than using the Kleene Star operator (i.e. ‘*’), the RE must be bounded by a minimum and maximum range (i.e. ‘{0,1}’).
An RE is typically used to check for a match between an input string and a pattern. In our case, we are interested in generating all the input strings that can possibly represent a pattern. This turns our problem into the question of how can we generate all the possible strings (or glycan structures) that match a given RE? From the field of Automata Theory, we know we can use a Finite-State Automaton (FSA) to implement a RE. An FSA recognizer starts in an initial state and transitions to other states based on the sequence of symbols from the input, and if the automaton is in an accepting state when the input terminates, then it is said to accept the input, thus representing a match against the RE.
Since we are actually interested in generating output from the FSA, we reasoned we could simulate the automaton to output all possible strings that would be accepted by the FSA. We used a non-recursive backtracking algorithm to find all possible accepting states. The algorithm accumulates the input characters for each transition until reaching an accepting state; when it outputs the resulting string to an array. We used the Linear Code nomenclature (Banin et al., 2002) to represent the set of symbols for the monosaccharides, anomericity, linkages and branches to define the REs. We describe the full algorithm to generate the virtual glycome in Supplementary Material B.
The set of glycan structures generated by this algorithm represents the ‘virtual glycome’ for human milk free glycans and is used as the knowledge base for glycan structure prediction. Since glycans commonly form branched structures, we utilized Extensible Markup Language (XML) as the format to store generated glycan structures in the virtual glycome database. XML has the advantage of supporting hierarchical queries using the XML Path Language (XPath). XPath is analogous to a SQL query used to query records from a relational database though XPath provides a mechanism to query and navigate through hierarchical representations of data, a feature that is well suited for branched glycan structures and utilized extensively in the candidate glycan filtering step described in the ‘Glycan Structure Prediction’ method.
2.2 Glycan structure prediction
The GlycomeSeq algorithm requires a spreadsheet input file (Supplementary Material C) that contains the composition and glycan array binding data for each glycan target. The composition, determined by MS, of the glycans are reported based on the number of hexose (H) residues, which are the single reducing terminal Glc plus Gal (the only hexoses found in human milk free glycans); N-acetylhexosamine (N), which is only GlcNAc; deoxyhexose (F), which is only Fucose; and Sialic Acid (S), which is only Neu5Ac. For example, the composition of the glycan, Neu5Acα2-3Galβ1-3(Fucα1-4)GlcNAcβ1-3Galβ1-4Glc, is H3N1F1S1; and the core structure (without Fucose and Sialic acid additions) would be H3N1.We proceed through the steps of the algorithm below.
As illustrated in Figure 1, the algorithm initially selects all structures from the database that match the composition of the unknown target. We then evaluate the positive binders and select the intersection of candidate structures from the remaining set of structures that contain the determinant for the positive binding GBPs. The selection of the candidate structures that contain the determinant is based on an XPath query defined for the GBP. Each of the XPath queries is defined in Supplementary Material C.
Fig. 1.

GlycomeSeq Algorithm. Combines the metadata from MS and glycan array binding data to select and filter out candidate structures from the virtual glycome
Next, for each non-binding GBP, we filter out candidate structures that contain the determinant for each non-binding GBP using an XPath query. This step is completed for all negative binders to eliminate candidate structures for that target. Finally, if we have additional determinants as identified by MSn reporter ions (e.g. linear lactose, branched lactose, terminal fucosylated LacNAc etc.) we select the intersection of candidate structures in our final set of predicted structures.
3 Results
We use our Virtual Glycome Generator to store the virtual human milk soluble glycan glycome into a database. Table 1 shows the theoretical number of glycans that can exist for each composition based on the biosynthetic rules of HMGs (Supplementary Material A) up to a dodecaose core structure (H7N5).
Table 1.
The virtual glycome of human milk free glycans
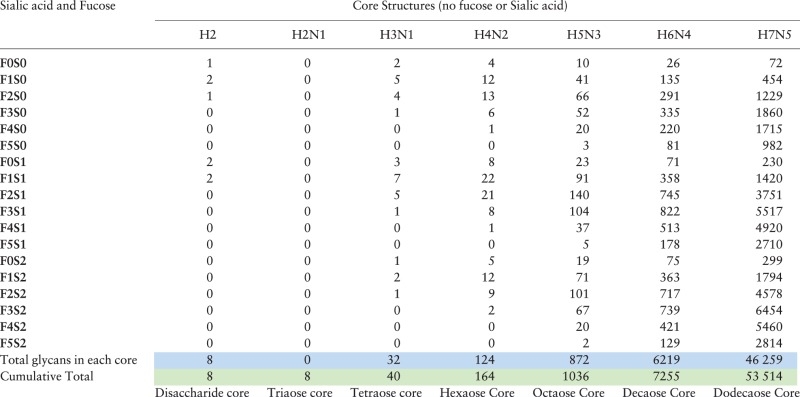 |
Core Structures, which are unsubstituted with fucose or sialic acid as indicated by the designation F0S0 in the top row, are designated by composition where H represents hexose (a single reducing terminal glucose and galactose residues) followed by the number of residues; i.e. H2 is the lactose core structure (Galβ1-4Glc), and N represents GlcNAc residues i.e. H3N1 represents the isomers comprised of a single reducing terminal glucose, 2 galactose residues and a single GlcNAc. The composition H2N1 was included to be comprehensive, but this structure is not found as a free glycan in human milk (see Supplementary Material A). The numbers for each composition indicate the number of isomers with the indicated composition that can be biosynthetically generated in human milk based on the rules described in Supplementary Material A. For example, the composition H5N3F2S1 is shared among 140 isomeric structures in the virtual glycome.
Although core structures (without fucose or sialic acid) as large as H10N8 have been reported, the amounts of glycans with core structures greater than H5N3 are vanishingly small. Nevertheless, the virtual glycome of human milk soluble glycans is certainly >50 000 different possible structures. The web-based software tool, GlycomeSeq, provides the number of isomers within each composition and will display the structures of all isomers within each composition (https://glycomeseq.emory.edu/).
In an effort to further our understanding of the functional and structural roles of HMGs we registered accession numbers in GlyTouCan (Aoki-Kinoshita et al., 2015) for the predicted structures that were verified by independent structural methods. We also shared our virtual HMG database with UniCarbKB (Campbell et al., 2014), which can be found at http://unicarbkb.org/milk. The UniCarbKB platform is a knowledgebase that allows public access to a curated database of glycan structures and associated metadata including publications and glycan structural classification by taxonomy, tissue, protein etc. In addition to sharing the glycan structures, GlycomeSeq also provides link outs to the UniCarbKB website for each of the predicted structures where the user can view any associated metadata for the glycan structure.
3.1 Identification of determinants within the structures of HMGs
The simple compilation of glycan structures is considered to have limited value since little information on function can be generated from lists of structures. However, having a compilation of all of the possible structures within a particular glycome may permit some useful predictions regarding the relationship of structure and function. Although the total number of possible free milk glycans with disaccharide to dodecasaccharide core structures is estimated to be 53 514 (Table 1), the region of a complex carbohydrate molecule that is required for the specific recognition of a biologically relevant GBP has been termed the glycan determinant (Cummings, 2009), which is comprised of di- to pentasaccharides; the number of determinants is significantly less than the total number of structures. As an exercise to test this hypothesis, we identified the number of non-reducing, terminal determinants from di- to pentasaccharide determinants in the human milk metaglycome as a function of increasing core structure size. Examples of determinants identified by the defined lectins and antibodies used in this study are shown in Supplementary Figure S2. The results of this analysis are shown in Figure 2.
Fig. 2.
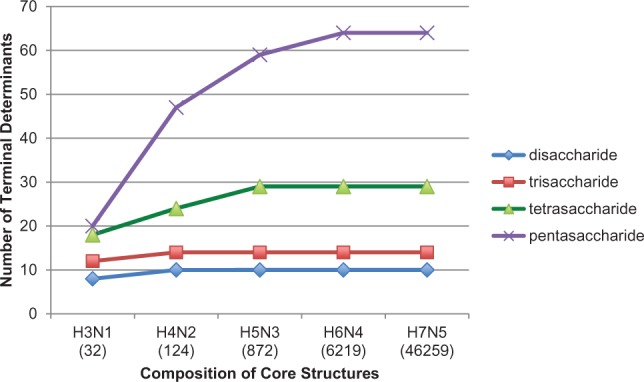
Number of terminal determinants in HMG glycome as a function of increasing core structure size. The composition of the core structures in indicated by number of hexoses (H, galactose and reducing terminal glucose) and the number of N-acetylglucosamines (N) in each core structure. The number of unique glycan structures including 0–5 frucose residues and 0–2 sialic acid residues in indicated in parentheses under each core composition. The data show the number of di- (diamond), tri- (square), tetra- (triangle) and pentasaccharide (cross) determinants found among the glycans comprising each
Although the total number of structures in each core size increases dramatically as shown by the number of glycans indicated for each core size (Fig. 2, parentheses), the number of terminal determinants increases to an apparent asymptotic value; i.e. 10 terminal disaccharide determinants for all human milk free glycans, 14 terminal trisaccharide determinants, 28 terminal tetrasaccharide determinants and 64 terminal pentasaccharide determinants. Thus, while the number of possible free glycan structures in any human milk sample may be enormous, there appear to be a limited number of potentially relevant biologically active terminal determinants that we would predict to be recognized by GBPs and other glycan recognition molecules. Interestingly, over 90% of the determinants in each determinant size are represented in the glycans with a core composition of H5N3, while glycans with larger core structures are found in vanishingly small amounts. These observations address the questions of how many possible free glycans can exist in human milk, and how many of these structures may be biologically relevant. Although the number of possible structures of HMGs may be well over 50 000 and depend on individual genetics, time since initiation of lactation, diet and possible time of day, the number of biologically relevant terminal determinants may be <100. The free glycans of human milk may, therefore, present a cluster or bouquet of thousands of individual structures where these biosynthetic pathways may have evolved to support a relatively limited number of biologically relevant determinants. By this unique biological process the microheterogeneity within a metaglycome is less important than the total number of relevant determinants expressed.
3.2 GlycomeSeq algorithm
The GlycomeSeq algorithm has been implemented in a software package that executes within a web application (http://glycomeseq.emory.edu). It takes an input file in a format described in Supplementary Material C; however, the example on the website is preloaded with the data from Supplementary Material C and the web application is available at http://glycomeseq.emory.edu. In our initial study, we have focused only on the neutral free glycans of human milk. The data used for demonstrating our automated sequencing approach were obtained from the validation of the HM-SGM reported previously (Yu et al., 2014). The data from 11 GBPs with known specificities binding to 42 purified HMGs and 14 standards of known structure before and after treatment with the specific exoglycosidases, β1-3-galactosidase, β1-4-galactosidase and α1-2-fucosidase and endo-β1-4-galactosidase are shown in Supplementary Table S1. The determinants (subset of a glycan structure containing di- to pentasaccharides) identified by defined GBP binding are shown in Supplementary Figure 2.
The application outputs a table with the predictions for each target and the associated metadata (see http://glycomeseq.emory.edu). To assist with the interpretation and derivation of the results, the application includes explanations for predicted structures and candidate structures that were ruled out as predicted structures due to filtering from the negative binders. Each predicted structure includes the metadata that was used to include it in the final result. For example, Figure 3, which is an excerpt from the application output of http://glycomeseq.emory.edu, shows the results of GlycomeSeq analysis of HMG-20 where the prediction is a single structure and three ‘Ruled out Candidates’ are listed.
Fig. 3.

GlycomeSeq display for HMG-20. (A) The display shows the Sample ID (HMG-20), the composition and the number of isomers in the virtual glycome with that composition (Virtual Candidates). The positive metadata supporting the single prediction is shown and the structures of the Candidate glycans ruled out by non-binding lectins or antibodies are presented as ‘Ruled out Candidates’. Finally the single prediction is shown. All structures are presented in a linear code (Banin et al., 2002). (B) Clicking on the linear code under ‘Prediction’ in 4A., displays the candidate structure and the positive metadata. If one clicks on the sample ID in 4B (HMG-20), the link to the structure in the Virtual Milk Glycome in the UniCarbKB database is displayed (not shown). (C–E) are displayed when the linear codes under Ruled out Candidates (3A.) are clicked on from top to bottom, respectively. Here the logic for ruling out the structure from the database is presented for each
3.3 Determination of structures using GlycomeSeq
The results of the structural analysis of the glycan targets on the microarray comprised of 42 unknown glycans and 14 standards can be displayed using the uploaded sample in the GlycomeSeq web site (http://glycomeseq.emory.edu), or observed in the summary provided in Supplementary Table S2. We applied the algorithm to 33 pure glycans. Using manual application of the MAGS logic, we limited the application of the algorithm to glycan targets that we determined to be single structures. The mass calculation limits the selection of only one composition for each glycan target, and the presence of more than one structure is obvious since the GBP-binding data will discover an excess of determinants for a single composition. Using these criteria, we determined that nine targets were mixtures of two to five glycans. The algorithm returned single results for 20 of the glycans. Of the remaining 13 targets analyzed, three glycans (HMG-38, -39 and -42) were not found in the database of all human milk free glycans (virtual glycome) and could, therefore, not be returned as single structures. The structures with no predictions are a result of the metadata identifying a positive ‘anti-H type 2’ determinant (Fucα1-2Galβ1-4GlcNAc) and a positive Lewis Y determinant [Fucα1-2Galβ1-4(Fucα1-3)GlcNAc] in their structures, which would indicate that the unknown target contains a substructure that is inconsistent with the biosynthetic rules (absence of H-type 2 glycans) we used to generate the virtual milk glycome (Supplementary Material A). Thus, the algorithm as currently written permits us to identify exceptions to the biosynthetic rules and raises questions for further experimentation.
In 10 instances the algorithm generated multiple candidate structures. In these cases, there was no indication of excess determinants indicating that these targets were mixtures. Reporting two or more possible structures indicates that metadata to distinguish between two structures are missing. For example, in HMG-23 (see Supplementary Table S2) the internal Lewis x determinant in the actual structure (Yu et al., 2014) cannot be detected by any currently known antibody or lectin, and the algorithm has not been sufficiently refined to identify fragment ions indicating the location of the internal Lex determinant in the structure, and in addition there is no known, specific GBP that can distinguish between a linear and a branched glycan. Therefore, using the available binding data and the knowledge base, the algorithm returned the three possible structures (two linear and one branched) among all isomers of that composition that were consistent with available data. One of the predicted structures was the correct one. If such GBPs were available or if an appropriate fragment ion were identified and added to the knowledge base, the algorithm would be able to return the correct prediction
In most cases, we used sequential MSn analysis to confirm all of the structures in the array including targets comprised of multiple glycans as shown in Supplementary Table S2, which summarizes the predictions for each glycan based on the data from the analysis of 42 HMGs and 14 standards selected for analysis. The structures of glycan targets HMG-9, -19, -36, -38, -39, -40, -42, -46, -59 and -64 were confirmed for this report and a description of the structural analysis is provided in Supplementary Material D and Supplementary Table S2. The others had been confirmed in a previous description of HMGs that bound rotavirus adhesion proteins (Ashline et al., 2014a; Yu et al., 2014).
4 Discussion
In the current studies, we developed a novel computational approach to address the fundamental question regarding the size of the human milk free glycan meta-glycome and if structural information and metadata can be combined to facilitate high-throughput sequencing of glycans. Predictions as to the number of different glycans that can exist in human milk have varied from a few hundred to many thousands. In our study, we applied a novel approach to address that question as shown in Table 1 where we estimated that number of HMGs limited to a core structure no larger that a dodecasaccharide could be ∼53 000 structures. Obviously, no single individual will produce all possible glycans because the structures synthesized will depend on many factors including, the genotype, time after initiation of lactation, time of day, and nutrition state of the donors. Nevertheless, it is clear that many thousands of different glycans are synthesized and secreted into the milk of all human mothers. Thus, modern computational approaches as developed here are essential to help identify and characterize the complex metaglycomes of human milk.
We hypothesized that a bioinformatics approach for structural analysis that combines the knowledge of a database of fully characterized glycan structures and experimental metadata from glycan microarrays and MS analysis would be able to automate the sequencing for the human milk meta-glycome. This approach is summarized in the algorithm GlycomeSeq (Fig. 2), and as shown in Supplementary Table S2, GlycomeSeq was able to identify all of the standards on the array that were found in the database of all human milk free glycans (virtual glycome). The algorithm correctly predicted no structures for ‘Agal LNT’, ‘LNFP IV’ (H2) and ‘Ley-Lex’ since they do not occur in human milk. GlycomeSeq was able to identify a single prediction in the unknown glycan targets in 20 out of 33 cases, and in all cases drastically reduced the number of candidate glycans from the large numbers of possible isomers in each composition. Structures of the glycan targets were confirmed or determined by independent structural methods and were in agreement with the predicted structures from GlycomeSeq.
Generally, where the algorithm produced multiple predictions for a target, there may not be enough meta-data available to eliminate candidate structures or the data indicates a binding motif should be present but is actually missing in a candidate structure. Such instances may occur due to weak binding, cross-reactivity of GBP for a target glycan, steric effects that prevent detection of a determinant that actually exists in the target, or insufficient data quality. For example, glycan HMG-76 with composition H6N4F2S0 has 291 possible structures in the virtual glycome. GlycomeSeq predicted two structures and all those contained determinants for the GBPs and the MSn reporter ions. Neither of the two predicted structures was ruled out during the negative binder filtering-step. In spite of not being able to generate a single predicted structure, the algorithm was able to eliminate all but 2 candidate structures from the 291 possible isomers with this composition. Such information is invaluable to analysts using mass spectrometry (MS) to define this glycan target and facilitates more targeted MS techniques to distinguish isomers.
The current state of the art approach for automated glycan sequencing is through techniques that automatically interpret MS data. However, these methods and tools are still evolving. Methods vary from (i) matching theoretical peak lists to the mass spectra (Joshi et al., 2004), (ii) matching mass spectra to a database of experimentally determined spectra (Kameyama et al., 2005) (iii) de novo sequencing approaches that match mass spectra to theoretical peak lists from structures that are constrained by biosynthetic pathways (Gaucher et al., 2000; Goldberg et al., 2005; Hu et al., 2014; Lapadula et al., 2005) and (iv) the GlycoWorkbench and Glyco-Peakfinder, a semi-automated annotation tool to assist the manual interpretation of MS data (Ceroni et al., 2008; Maass et al., 2007). All of these approaches are limited by their ability to perform complete structural characterizations that detect linkage position and anomeric configurations, meaning that the predicted glycan structures are ambiguous in certain aspects. Based on our current review of the literature, GlycomeSeq is the only automated high-throughput sequencing method that can predict fully characterized glycan structures including topology, linkages and anomeric configurations.
We also found that the number of terminal determinants that are found in human milk free glycans increase sub-linearly as a function of core structure size. This raises the possibility that the large numbers of isomeric structures represent a type of scaffold upon which specific determinants are created to provide necessary biological functions. From the analysis in Figure 2 we observe that as the number of monosaccharides in the core structures of the glycans increases, the number of terminal determinants increases, which is consistent with the greater branching that can occur in the larger glycans. Interestingly, the number of tetra- and pentasaccharide determinants seems to reach a constant number at a core structure of an octasaccharide, suggesting that free milk glycans up to octasaccharides may represent the biologically relevant set of glycans in human milk. These observations also suggest that the free glycans in human milk present a repertoire of structures that present biologically relevant determinants, and that individual structures are less important that the ‘bouquet’ of determinants.
Adding orthogonal methods as metadata to GlycomeSeq enhances the predictive power of our method. The reporter ions from MSn analysis provide conclusive structural information for fragments in the unknown target structure. For example, HMG-76 has two reporter ions inferred from the MSn analysis; if we eliminate the fragments from our input then the algorithm predicts 47 structures from the binding data alone. Similarly, if we only use the reporter ion fragments from MS, GlycomeSeq predicts 10 structures. By combining the GBP binding data with the MS fragment data; the algorithm generates 2 predicted structures out of 291 possible structures.
5 Conclusion
We describe herein an approach to define a virtual meta-glycome and use it as a knowledge base to predict fully characterized glycan structures using data from MS and glycan microarray-binding experiments. This approach to computational sequencing of the unknown glycans requires (i) determination of the glycan composition using MALDI-TOF analysis, (ii) interrogation of the glycans with lectins and antibodies that bind known determinants, (iii) determination of the set of predicted structures based on automated meta-analysis of the experimental data from the virtual glycome database given the constraints of the rules for the biosynthetic pathway of the glycans. Although several methods have been aimed at glycan annotation of mass spectrometric analysis, to our knowledge no other method has been developed that attempts to solve the glycan structure to the level of GlycomeSeq. This approach has the potential to be a significant breakthrough in glycomics analysis that has thus far been hindered by the complexity and ambiguity of MS analysis. We seek to make improvements and have planned future work for our approach including (i) automate the analysis of the MS spectra to identify the MS reporter ion determinants, (ii) develop a library of XPath queries for the determinants so the user does not have to manually specify them in the spreadsheet, (iii) include exo- and endoglycosidases to yield finer specificity and (iv) apply this method on HMGs with Sialic Acid to further validate this approach before moving onto a much larger meta-glycome.
Supplementary Material
Acknowledgements
The authors would like to acknowledge Dr Ying Yu, who developed the human milk SGM with the excellent technical support of Ms Yi Lasanajak and Ms Hong Ju, and Dr Jamie Heimburg-Molinaro for helpful discussion and editing the article.
Funding
This work has been funded by the National Institutes of Health (GM0987912 and P41GM103694) (RDC), which support the National Center for Functional Glycomics. This work was also supported in part by the Emory Comprehensive Glycomics Core (ECGC), which is subsidized by the Emory University School of Medicine as one of the Emory Integrated Core Facilities.
Conflict of Interest: none declared.
References
- Aoki-Kinoshita K. et al. (2015) GlyTouCan 1.0 - The international glycan structure repository. Nucleic Acids Res., 44, D1237–D1242. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ashline D.J. et al. (2014a) Structural documentation of glycan epitopes: sequential mass spectrometry and spectral matching. J. Am. Soc. Mass. Spectrom., 25, 444–453. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ashline D.J. et al. (2014b) Structural characterization by multistage mass spectrometry (MSn) of human milk glycans recognized by human rotaviruses. Mol. Cell Proteomics, 13, 2961–2974. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Banin E. et al. , (2002) A novel linear code nomenclature for complex carbohydrates. Trends Glycosci. Glycotechnol., 14, 127–137. [Google Scholar]
- Bertozzi C.R., Rabuka D. (2009). Structural basis of glycan diversity In Varki A., Cummings R.D., Esko J.D., Freeze H.H., Stanley P., Bertozzi C.R., Hart G.W., Etzler M.E. (eds.) Essentials of Glycobiology, 2nd edn. Cold Spring Harbor, NY. [PubMed] [Google Scholar]
- Bienenstock J. et al. (2013) Fucosylated but not sialylated milk oligosaccharides diminish colon motor contractions. Plos One, 8, e76236. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bode L. (2012) Human milk oligosaccharides: Every baby needs a sugar mama. Glycobiology, 22, 1147–1162. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Campbell M.P. et al. (2014) UniCarbKB: building a knowledge platform for glycoproteomics. Nucleic Acids Res., 42, D215–D221. (Database issue), [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ceroni A. et al. (2008) GlycoWorkbench: a tool for the computer-assisted annotation of mass spectra of glycans. J. Proteome Res., 7, 1650–1659. [DOI] [PubMed] [Google Scholar]
- Cummings R.D. (2009) The repertoire of glycan determinants in the human glycome. Mol. Biosyst., 5, 1087–1104. [DOI] [PubMed] [Google Scholar]
- Duska-McEwen G. et al. (2014) Oligosaccharides enhance innate immunity to respiratory syncytial virus and influenza in vitro. Food Nutr. Sci., 5, 1387–1398. [Google Scholar]
- Gaucher S.P. et al. (2000) STAT: a saccharide topology analysis tool used in combination with tandem mass spectrometry. Anal. Chem., 72, 2331–2336. [DOI] [PubMed] [Google Scholar]
- Goehring K.C. et al. (2014) Direct evidence for the presence of human milk oligosaccharides in the circulation of breastfed infants. PLOS One, 9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Goldberg D. et al. (2005) Automatic annotation of matrix-assisted laser desorption/ionization N-glycan spectra. Proteomics, 5, 865–875. [DOI] [PubMed] [Google Scholar]
- Holscher H.D. et al. (2014) Human milk oligosaccharides influence maturation of human intestinal Caco-2Bbe and HT-29 cell lines. J. Nutr., 144, 586–591. [DOI] [PubMed] [Google Scholar]
- Hu H. et al. (2014) A computational framework for heparan sulfate sequencing using high-resolution tandem mass spectra. Mol. Cell. Proteomics, 13, 2490–2502. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jantscher-Krenn E. et al. (2012) Human milk oligosaccharides reduce Entamoeba histolytica attachment and cytotoxicity in vitro. Br. J. Nutr., 108, 1839–1846. [DOI] [PubMed] [Google Scholar]
- Joshi H.J. et al. (2004) Development of a mass fingerprinting tool for automated interpretation of oligosaccharide fragmentation data. Proteomics, 4, 1650–1664. [DOI] [PubMed] [Google Scholar]
- Kameyama A. et al. (2005) A strategy for identification of oligosaccharide structures using observational multistage mass spectral library. Anal. Chem., 77, 4719–4725. [DOI] [PubMed] [Google Scholar]
- Kuhn L. et al. (2008) Effects of early, abrupt weaning on HIV-free survival of children in Zambia. N. Engl. J. Med., 359, 1859. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lapadula A.J. et al. (2005) Congruent strategies for carbohydrate sequencing. 3. OSCAR: an algorithm for assigning oligosaccharide topology from MSn data. Anal. Chem., 77, 6271–6279. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Maass K. et al. (2007) “Glyco-peakfinder”–de novo composition analysis of glycoconjugates. Proteomics, 7, 4435–4444. [DOI] [PubMed] [Google Scholar]
- Mulloy B. et al. (2009). Structural Analysis of Glycans In Varki A., Cummings R.D., Esko J.D., Freeze H.H., Stanley P., Bertozzi C.R., Hart G.W., Etzler M.E. (eds.) Essentials of Glycobiology, 2nd edn. Cold Spring Harbor, NY. [PubMed] [Google Scholar]
- Ruiz-Palacios G.M. et al. (2003) Campylobacter jejuni binds intestinal H(O) antigen (Fuc alpha 1, 2Gal beta 1, 4GlcNAc), and fucosyloligosaccharides of human milk inhibit its binding and infection. J. Biol. Chem., 278, 14112–14120. [DOI] [PubMed] [Google Scholar]
- Smith D.F., Cummings R.D. (2013) Application of microarrays for deciphering the structure and function of the human glycome. Mol. Cell. Proteomics, 12, 902–912. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Smith D.F. et al. (2010) Use of glycan microarrays to explore specificity of glycan-binding proteins. Methods Enzymol., 480, 417–444. [DOI] [PubMed] [Google Scholar]
- Song X. et al. (2011) Shotgun glycomics: a microarray strategy for functional glycomics. Nat. Methods, 8, 85–90. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Varki A., Lowe J.B. (2009). Biological Roles of Glycans In Varki A., Cummings R.D., Esko J.D., Freeze H.H., Stanley P., Bertozzi C.R., Hart G.W., Etzler M.E. (eds.) Essentials of Glycobiology, 2nd edn. Cold Spring Harbor, NY. [PubMed] [Google Scholar]
- von der Lieth C.W. et al. (2006) The role of informatics in glycobiology research with special emphasis on automatic interpretation of MS spectra. Biochim. Biophys. Acta, 1760, 568–577. [DOI] [PubMed] [Google Scholar]
- Yu Y. et al. (2014) Human milk contains novel glycans that are potential decoy receptors for neonatal rotaviruses. Mol. Cell. Proteomics, 13, 2944–2960. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yu Y. et al. (2012) Functional glycomic analysis of human milk glycans reveals the presence of virus receptors and embryonic stem cell biomarkers. J. Biol. Chem., 287, 44784–44799. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.