

Abstract
Summary: Precise regulatory control of genes, particularly in eukaryotes, frequently requires the joint action of multiple sequence-specific transcription factors. A cis-regulatory module (CRM) is a genomic locus that is responsible for gene regulation and that contains multiple transcription factor binding sites in close proximity. Given a collection of known transcription factor binding motifs, many bioinformatics methods have been proposed over the past 15 years for identifying within a genomic sequence candidate CRMs consisting of clusters of those motifs.
Results: The MCAST algorithm uses a hidden Markov model with a P-value-based scoring scheme to identify candidate CRMs. Here, we introduce a new version of MCAST that offers improved graphical output, a dynamic background model, statistical confidence estimates based on false discovery rate estimation and, most significantly, the ability to predict CRMs while taking into account epigenomic data such as DNase I sensitivity or histone modification data. We demonstrate the validity of MCAST’s statistical confidence estimates and the utility of epigenomic priors in identifying CRMs.
Availability and implementation: MCAST is part of the MEME Suite software toolkit. A web server and source code are available at http://meme-suite.org and http://alternate.meme-suite.org.
Contact: t.bailey@imb.uq.edu.au or william-noble@uw.edu
Supplementary information: Supplementary data are available at Bioinformatics online.
1 Introduction
Eukaryotic gene regulation is carried out by a variety of mechanisms, involving transcription factors, microRNAs, DNA methylation, nucleosome occupancy and local as well as large-scale chromatin structure. The relative importance of these various mechanisms is subject to debate; however, the joint influence of multiple transcription factors on gene regulation is clearly an important component of the eukaryotic gene regulatory system. In practice, a single transcription factor is neither specific enough nor binds strongly enough to achieve and maintain access to the genomic DNA; consequently, most regulatory loci contain binding sites for multiple factors that act in concert to upregulate or downregulate one or more target genes. Such sites are known as cis-regulatory modules (CRMs).
An important and well-studied problem in computational biology is the CRM scanning problem. The input to this task is a collection of transcription factor binding motifs plus a set of DNA sequences. The latter may comprise a complete genome or a set of regions of interest, such as upstream regions of annotated genes. The CRM scanning algorithm searches for clusters of occurrences of the given motifs; hence, the output is a collection of candidate CRMs, along with annotations indicating which motifs occur within each one.
The first CRM scanning algorithm was LRA (Wasserman and Fickett, 1998). Since its introduction, numerous other algorithms have been described in the literature, some with accompanying software and web servers (see Supplementary materials for a review of existing methods). Some of these methods employ a sliding window approach, whereas others use probabilistic models such as hidden Markov models.
Here, we introduce an improved implementation of the MCAST algorithm (Bailey and Noble, 2003). MCAST uses a motif-based hidden Markov model to scan for clusters of motifs. Its key features include a scoring scheme based on P-values and a method for calibrating the resulting scores to obtain statistical confidence estimates. We have recently improved MCAST in four ways. First, we have dramatically improved the graphical output produced by the tool (Supplementary Fig. 1). The new output provides a schematic view showing the relative position and statistical significance of the motif occurrences in the CRM. The schematic view can be expanded into a detailed view displaying any region of the CRM at the nucleotide level, and the region of the CRM displayed in the detailed view may be chosen interactively. Second, to reduce false positives due to unusual local base content, we have added a dynamic background model, selected based on the local GC content around the current position in the genome. Third, we have improved the multiple testing correction methodology, replacing the Bonferroni adjustment with a false discovery rate (FDR) estimation procedure. This change employs a reservoir sampling scheme to allow MCAST to accurately estimate the percentage of motif scores drawn according to the null hypothesis (Storey, 2002) without running out of memory when scanning a large genome with many motifs. Fourth, and most significantly, to improve accuracy we have incorporated into MCAST a method for converting epigenomic data—DNase I sensitivity or histone modification data from ChIP-seq experiments—into a probabilistic prior (Cuellar-Partida et al., 2011). We demonstrate that MCAST provides state-of-the-art discrimination power, and that the incorporation of a DNase I sensitivity prior significantly improves its performance.
2 Implementation
MCAST is implemented in C and is available as a command line tool or via the web interface. Motifs are accepted in the MEME or TRANSFAC formats, and utilities are provided for converting a variety of common motif formats to MEME format. The command line version of MCAST accepts FASTA-formatted DNA sequences, and the web server additionally offers a large variety of genome databases that are automatically downloaded from the NCBI. MCAST supports a variety of user-level parameters, the most important of which are the P-value threshold at which a motif is deemed significant (motif-pthresh), the maximum allowed distance between adjacent motif occurrences within a CRM (max-gap) and the FDR threshold for reporting CRMs (output-qthresh). MCAST provides HTML, tab-delimited text and XML output. The HTML output provides interactive graphical representations of the CRMs using embedded JavaScript. Key features of the MCAST output include summaries of the input sequences, motifs and analysis parameters. The coordinates, log-odds score, P-value, E-value and q-value are provided for each putative CRM.
3 Results
We tested the validity of MCAST’s statistical confidence estimates by searching a set of five motifs against a shuffled version of the mouse genome (see Supplementary materials for details). The resulting P-values show good calibration, as evidenced by the close fit between the P-values and a uniform distribution (Fig. 1A). In particular, the P-values are accurate to within a factor of 2 over nearly five orders of magnitude. This result is robust to variations in the primary parameters, max-gap and motif-pthresh, although setting motif-pthresh to a smaller value leads to fewer predicted CRMs and slightly conservative calibration in the tail of the p-value distribution (Supplementary Fig. 3).
Fig. 1.
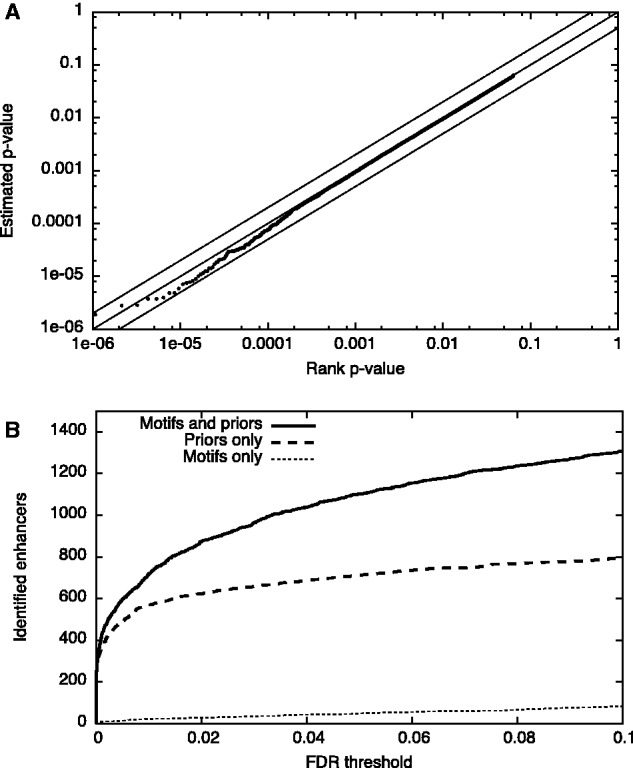
(A) Calibration of P-values. The panel plots, for a search using a shuffled genome, MCAST’s estimated P-value as a function of the rank P-value. The lines y = x, y = 2x and y = 0.5x are included for reference. The close fit of the observed data to the line y = x shows that the P-values are uniformly distributed. (B) Enhancer identification. MCAST predicts substantially more enhancers (‘Identified enhancers’) at all ‘FDR’ when using five motifs and a DNase I prior (‘Motifs and prior’ curve) compared with using just the five motifs (‘Motifs only’ curve). Prediction accuracy attributable to the prior alone was assessed by scanning a shuffled version of the genome using the same motifs and the DNase I prior (‘Priors only’ curve)
A recent study identified enhancers in several mouse embryonic stem cell lines, characterized by clusters of the motifs for Oct4, Sox2, Nanog, Klfr and Esrb and by interaction with the Mediator complex (Whyte et al., 2013). Even jointly, these five motifs do not have sufficient information content to enable detection of enhancers at a low FDR. However, the motifs are indeed valuable when combined with DNase I sensitivity data. We used MCAST to scan the mouse genome with these five motifs and a DNase I prior, and we compared the performance with searches in which (1) the DNA sequence is shuffled but the prior is left intact and (2) the prior is not used at all (see Supplementary materials for details). This experiment shows that, though the DNase I data alone are sufficient to give a reasonably good ranking of sites in conjunction with shuffled sequence data, the combination of true sequence data and DNase I data boosts performance considerably (Fig. 1B). At an FDR threshold of 1%, MCAST with a DNase I prior identifies 1307 out of 8794 enhancers, for a recall of 15%. Using only the DNase I prior or using only the motifs yields far fewer identifications (793 and 82 enhancers, respectively, at 1% FDR). The scans of the mouse genome, run on an Intel Xeon 5060 3.2 GHz processor, took 47 min. without the DNase I prior and 76 min with the DNase I prior.
Funding
This work was funded by National Institutes of Health award R01 GM103544. The MEME Suite server receives support from Google and Amazon Web Services.
Conflict of Interest: none declared.
Supplementary Material
References
- Bailey T.L., Noble W.S. (2003) Searching for statistically significant regulatory modules. Bioinformatics, 19, ii16–ii25. [DOI] [PubMed] [Google Scholar]
- Cuellar-Partida G. et al. (2011) Epigenetic priors for identifying active transcription factor binding sites. Bioinformatics, 28, 56–62. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Storey J.D. (2002) A direct approach to false discovery rates. J. R. Stat. Soc. Ser. B, 64, 479–498. [Google Scholar]
- Wasserman W.W., Fickett J.W. (1998) Identification of regulatory regions which confer muscle-specific gene expression. J. Mol. Biol., 278, 167–181. [DOI] [PubMed] [Google Scholar]
- Whyte W.A. et al. (2013) Master transcription factors and mediator establish super-enhancers at key cell identity genes. Cell, 153, 307–319. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.