

Abstract
Motivation: The Optical Mapping System discovers structural variants and potentiates sequence assembly of genomes via scaffolding and comparisons that globally validate or correct sequence assemblies. Despite its utility, there are few publicly available tools for aligning optical mapping datasets.
Results: Here we present software, named ‘Maligner’, for the alignment of both single molecule restriction maps (Rmaps) and in silico restriction maps of sequence contigs to a reference. Maligner provides two modes of alignment: an efficient, sensitive dynamic programming implementation that scales to large eukaryotic genomes, and a faster indexed based implementation for finding alignments with unmatched sites in the reference but not the query. We compare our software to other publicly available tools on Rmap datasets and show that Maligner finds more correct alignments in comparable runtime. Lastly, we introduce the M-Score statistic for normalizing alignment scores across restriction maps and demonstrate its utility for selecting high quality alignments.
Availability and implementation: The Maligner software is written in C ++ and is available at https://github.com/LeeMendelowitz/maligner under the GNU General Public License.
Contact: mpop@umiacs.umd.edu
1 Introduction
Optical Mapping, a single molecule system (Aston et al., 1998; Dimalanta et al., 2004; Jo et al., 2007; Lin et al., 1999; Valouev et al., 2006,a,b), constructs genome-wide physical maps through the acquisition and analysis of large datasets comprising restriction maps created from very long genomic DNA molecules (≈400–500 kb). Such genome-wide restriction maps, through analysis, elucidate genomic structure with a resolution of about 2 kb, thereby complementing DNA sequencing technologies which provide relatively short range information at base pair resolution. Optical Mapping analysis has been used as part of large-scale sequencing efforts (Armbrust et al., 2004; Church et al., 2009; Ivens et al., 2005; Schnable et al., 2009; Wei et al., 2009; Young et al., 2011; Zhou et al., 2004, 2007, 2009) and as a means for discovery of structural variants in normal human (Antonacci et al., 2010; Kidd et al., 2008; Teague et al., 2010) and cancer (Ray et al., 2013; Gupta et al., 2015) genomes.
Despite such promise, very few software tools are freely available for working with genomic mapping data for large genomes. SOMA (Nagarajan et al., 2008) is an open-source software tool for aligning assembled sequence contigs to an optical map but does not scale to large genomes and often gives incorrect contig placements due to the greedy nature of its alignment algorithm. TWIN (Muggli et al., 2014) is a recently developed tool for efficiently aligning sequence contigs to an optical map, but as we show, it does not allow for alignments which have unmatched sites, limiting its applicability in noisy experimental datasets.
Here we present Maligner, an open-source software package for aligning single molecule restriction maps (Rmaps) and in silico maps of contigs from a sequence assembly to a reference restriction map at speeds that are comparable or faster than currently available tools. Maligner has two modes of alignment: one which uses traditional dynamic programming (malignerDP) and a second index-based mode of alignment that runs orders of magnitude faster but is more stringent in the alignments that it accepts (malignerIX). In addition, we present a novel method for normalizing the alignment scores across queries based on computing the median absolute deviation (MAD) across the best random alignments, which allows for the selection of an alignment score cutoff that is applicable across queries, thereby obviating the need for a computationally expensive permutation test for determining alignment significance.
2 Background
In an optical mapping experiment, genomic DNA is randomly sheared into molecules of length 200–1000 kb and then stretched onto positively charged glass surfaces for immobilization using microfluidics and electrostatic interactions between the negatively charged molecules and surface. The molecules are then digested by a restriction endonuclease, which cleaves DNA molecules at their cognate sites. Resulting DNA fragments relax at the cut sites, creating visible gaps. The molecules are then stained with fluorescent dye and imaged using automated epifluorescence microscopy. Machine vision is used to identify the cut sites and then estimate fragment size by integrated fluorescence intensity. Experimental errors include sizing error due to variability in the molecular stretch or incorporation of fluorescent dye, missed cut sites in the reference due to partial digestion, false cut sites in the Rmap due to random breakage of the molecule, or missing small fragments due to desorption.
Consider a DNA molecular of length L bp which is digested with a restriction enzyme which cuts (or nicks) the DNA at n integral positions p0, p1, where pi represents the zero-based base pair location of the ith cut site. We can represent this restriction pattern by an ordered listing of n + 1 fragment lengths: where f0 = p0, for and . Since the DNA molecule is produced by random shearing of chromosomal DNA, the fragments f0 and fn which appear at the start and end of the molecule are not bounded by restriction sites at both ends. We refer to these fragments as boundary fragments. On the other hand, are interior fragments, as they are bounded on both ends by restriction sites.
A ordered restriction map is given by its ordered listing of fragment sizes . The number of restriction fragments in the map is given by . In this case, .
A chunk is an ordered list of consecutive restriction fragments from a single map . For example: is a chunk of three consecutive fragments from restriction map . We represent a chunk more concisely by the triple which corresponds to the consecutive fragments mi from where . For example, corresponds to the consecutive fragments . A chunk is a boundary chunk if s = 0 or .
Two chunks and are adjacent if , and e1 = s2 or e2 = s1. The number of fragments in a chunk is denoted as , and the number of interior sites in a chunk is . The length of a chunk is given by the sum of the lengths of the restriction fragments in the chunk: . A chunk is empty if e = s, meaning the chunk contains zero fragments and has zero length.
A matched chunk is an ordered pair of chunks. We refer to the first chunk as the query chunk and the second chunk as the reference chunk. Given query chunk and reference chunk , the matched chunk is the ordered pair . A matched chunk is a boundary chunk if either cq or cr are boundary chunks. Two matched chunks and are adjacent if both and are adjacent and and are adjacent.
An alignment of a query map to a reference map is given by an ordered listing of matched chunks where:
All of the query chunks are from map Q and the reference chunks are from map R.
The matched chunks are adjacent
The starting indices of the reference chunks are monotonically increasing.
The starting indices of the query chunks are monotonically increasing (for forward alignments) or monotonically decreasing (for reverse alignments).
We say that restriction site i in the query is aligned to restriction site j in the reference if the alignment has a non-boundary matched chunk (Cq, Cr) with and where: (i) sq = i and or eq = i and er = j for forward alignments or (ii) sq = i and er = j or and sr = j for reverse alignments. In the context of an alignment, we refer to the interior sites of a query chunk or reference chunk as unmatched sites. An example of an alignment using this notation is shown in Figure 1.
Fig. 1.

Alignment notation. The forward alignment of query map against a reference illustrated above can be represented as the following ordered listing of matched chunks: . Since the starting indices of the query chunks are monotonically increasing, the query is aligned in the forward direction
3 Methods
The maligner software has two modes of alignment: (i) a dynamic programming implementation malignerDP that allows for unmatched sites in both the query and reference and a faster mode malignerIX which builds an index on the reference which is queried through binary search but does not allow unmatched sites in the query.
3.1 Dynamic programming based alignment
We extend the work of Nagarajan et al. (2008) to build a map aligner which can scale to large eukaryotic genomes. Given a matched chunk with query length q bp, reference length r bp, m interior unmatched query sites, and n interior unmatched reference sites, we use scoring function which represents an edit distance between a query chunk and a reference chunk:
| (1) |
| (2) |
| (3) |
where Cq is the fixed cost for an unmatched query site, Cr is the fixed cost for an unmatched site in the reference, and S(q, r) is the cost of the sizing difference between the query chunk and the reference chunk. Note that we make an adjustment for small reference fragments, which can exhibit higher relative error rates (Valouev et al., 2006c), by putting a lower bound σmin on the sizing error scaling parameter.
Given a query map and a reference map , we seek an alignment which minimizes the sum of the cost of the matched chunks. Since each chunk is scored independently, this permits a dynamic programming solution given by Algorithm 1. We bound the number of consecutive unmatched sites in the query by δq and reference by δr, giving an algorithm that is
Algorithm 1 The dynamic programming algorithm for malignerDP for the global alignment of query map with m fragments to the reference map with n fragments. The alignment allows at most δq and δr unmatched sites in the query and reference, respectively.
1: procedure malignDP()
2: ▹ Initialize score matrix.
3: for do
4:
5: end for
6: for do
7:
8: end for
9: ▹ Fill the score matrix.
10: for do
11: for do
12:
13:
14: for do
15: for do
16: if then
17: ▹ query chunk size
18: ▹ ref chunk size
19: ▹ Compute score of this alignment extension
20:
21: if then
22:
23:
24: end if
25: end if
26: end for
27: end for
28: end for
29: end for
30: return (SM, BackPointer)
31: end procedure
3.1.1 Alignment significance
The dynamic programming method used by malignerDP will find one alignment for each position in the reference. However, it is not clear whether the best scoring alignment is significantly better than random. One method for determining whether an alignment is significant is to perform a permutation test whereby a given query is aligned to a population of permuted references. Each alignment is assigned a p-value under the null hypothesis H0 that the query is not related to the reference by determining the fraction of permuted references that have a better scoring alignment for that query. We do not consider the permutation test for this problem because it is computationally expensive and impractical for alignment to large genomes.
Since our scoring function is a measure of edit distance, we cannot simply choose a single score cutoff for accepting or rejecting alignments that will apply to all queries, as the score for a quality alignment varies with the number of fragments and the size of the fragments in each query (Sarkar et al., 2012). Instead, we propose computing a cutoff score for each query map based upon the distribution of alignment scores of the best non-overlapping alignments. If a query has a single acceptable alignment to the reference, we expect that the best scoring alignment will be correct and the rest of the alignments to be random. Using this intuition, we formulate the M-Score as follows:
| (4) |
| (5) |
| (6) |
where we take to be the top 100 non-overlapping alignments for the given query against the reference. The M-Score normalizes all alignment scores across queries by shifting each query’s scores to the median and scaling by the median absolute deviation (MAD) of the top ranking alignments for that query. This allows us to select an M-Score cutoff for selecting significant alignments that can be applied across queries. We note that using dynamic programming as the method of alignment (as compared to index-based methods) gives us the distribution of alignment scores at no additional cost since the best alignment score at each position of the reference is computed when the score matrix is populated in Algorithm 1. The M-score method uses this distribution of alignment scores in place of the null distribution one could obtain through a more expensive permutation test.
We typically select an M-Score cutoff which maximizes the fraction of queries with a unique alignment below the cutoff. The M-Score cutoff can vary dataset from dataset based on enzyme digestion rate, uniformity of molecular stretch, and the number of restriction fragments per Rmap. Higher quality datasets will have higher quality alignments and therefore allow for the selection of stricter M-Score cutoff. Note that more negative M-Scores correspond to better quality alignments (since lower alignment scores are better).
3.2 Index-based alignment
If we do not allow for unmatched sites in the query map, we can leverage an indexed based method of alignment that avoids dynamic programming and instead uses an index built on the reference map. The reference can be indexed by extracting all possible chunks with at most δr interior missed restriction sites. Specifically, we consider the set of all chunks from the reference .
The adjacencies of the reference chunks can be represented as a directed acyclic graph (DAG), where we include an edge between chunks and if sb = ea. We build an index on the graph by storing all nodes (i.e. chunks) in an array sorted by chunk length. For a given query chunk cq and a lower bound function L(C) and upper bound function U(C) we can find all reference chunks that are compatible with cq in length from the index in time by binary search. Our implementation uses lower and upper bound functions and where specifies the relative error and specifies the minimum absolute error tolerance.
We can leverage the reference chunk index and the DAG to find alignments for a query map with k fragments if we restrict our search space to the set of alignments with no unmatched sites in Q. Specifically, we search for one or more paths of adjacent reference chunks where: (i) each chunk ci is compatible with the ith fragment in for and (ii) the rate of unmatched sites in the reference is less than a user selected threshold.
We find such alignments by first seeding on the largest interior restriction fragment in the query. Since genomic restriction fragment lengths are approximately exponentially distributed, the largest fragment in the query will have the fewest number of compatible fragments. For each reference chunk compatible with the seed fragments we perform bounded DFS to find all possible compatible right extensions and left extensions with respect to the reference which compatibly align all remaining fragments in the query. With each step in the DFS, we only consider taking an edge to a reference chunk if that reference chunk is compatible with the next fragment in the query. Of all extensions found (if any), we take the left and right extension which has the smallest number of unmatched sites in the reference. We concatenate the best left extension in the reference with the seed chunk and the best right extension to produce the best forward alignment for each seed hit. We align in the reverse direction by considering aligning to the DAG corresponding to the reverse of the reference, reusing the seed hits found for forward alignment. In a final post-processing step, we apply the same score function given by Eq. 1 to rank all alignments found and output a set of non-overlapping alignments selected in order of alignment score.
While this index-based method of alignment is not as flexible as full dynamic programming because it does not allow for unmatched sites in the query, it runs orders of magnitude faster.
4 Results
In this section we present results demonstrating the utility of the M-Score statistic in discerning correct alignments from spurious alignments. Next, we compare the Maligner software to other available software tools by aligning in silico digested contigs from an E.coli K12 sequence assembly to both an in silico digest of the reference sequence and an optical map. We also align E.coli K12 Rmaps to the in silico reference map. Lastly, we demonstrate our methods on a large genome by aligning both sequence contigs and Rmaps to an optical map of the budgerigar (Melopsittacus undulatus), an Australian parakeet.
4.1 M-score for alignment significance
Maligner scores alignments using an additive scoring function on each matched chunk given by Eq. 1, with fixed costs for interior unmatched sites in the query chunk and reference chunk and cost for sizing difference between the query and reference chunk. The score for an alignment is given by the sum of the scores of the matched chunks of the alignment. This scoring function represents an ‘edit distance’ between the query and the reference, with lower scores corresponding to alignments with greater similarity between the query and reference.
Since the scoring function is additive, the score of a query’s best alignment depends on the number of fragments, its length, and quality of the query (i.e. the sizing error, site cut rate, and desorption of small fragments). Therefore, one cannot select a simple cutoff that applies to all queries for selecting significant alignments. The M-Score, discussed in more detail in Section 3.1.1, seeks to normalize the scores for each query based on the distribution of alignment scores for its best non-overlapping alignments. This allows for the selection of a common cutoff that applies across queries.
We assessed the performance of the M-Score for selecting correct alignments on a simulated dataset under three different error settings. We simulated Rmaps from the human reference using enzyme BamHI by selecting map length uniformly at random from [100, 500] kb, selecting a genomic location and orientation at random, simulating a cutting pattern as a Bernoulli process with enzyme efficiency p, applying an Rmap scaling factor to model variability in molecular stretch, and finally adding fragment sizing measurement error with parameters p = 0.85, under the low error setting, p = 0.75, under the medium error setting, and p = 0.65, under the high error setting. We simulated 1000 Rmaps under each error setting from both the human reference and a permuted version of the human reference, requiring a minimum of 10 restriction fragments.
We aligned these simulated Rmaps with malignerDP and selected the best scoring alignment for each query against the human reference. From this set of alignments we consider an alignment of a query map sampled from the human reference aligned to its true location to be a true alignment, and all other alignments to be false alignments. We assessed how well the alignment score, alignment score per number of matched chunks, and M-score statistics performed at discriminating true alignments from false alignments. The receiver operator characteristic (ROC) curves are shown in Figure 2 and AUC statistics in Table 1, indicating that the M-Score statistic performs comparable to the alignment score and score per matched chunk statistics under low and medium error settings and superiorly under the high error setting. The score per matched chunk statistic attempts to normalize an alignment score by the number of fragments in each query but this does not perform as well as the M-Score, which indirectly takes into account the ‘alignability’ of a query based on the distribution of its best alignment scores.
Fig. 2.
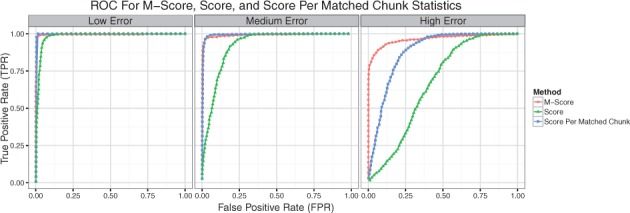
ROC for alignment significance. True Positive Rate vs. False Positive Rate for discriminating correct alignments from random alignments on a set of simulated BamHI Rmaps from the human reference using the M-Score, alignment score, and alignment score per matched chunk statistics under low, medium, and high error conditions (Color version of this figure is available at Bioinformatics online.)
Table 1.
AUC for alignment score, alignment score per matched chunk, and M-Score
| Error setting | Score | Score per Matched Chunk | M-score |
|---|---|---|---|
| Low | 0.982 | 0.997 | 0.995 |
| Medium | 0.918 | 0.994 | 0.991 |
| High | 0.664 | 0.881 | 0.965 |
The AUC for discriminating true alignments from false alignments using the alignment score, alignment score per matched chunk, and M-score statistics for simulated Rmaps from the human reference under low, medium and high error settings. M-Score is comparable to the other methods under the low and medium error and performs superiorly under high error.
4.2 Escherichia coli K-12
4.2.1 Contig alignment
One practical use of optical mapping is to aid in the scaffolding and finishing of sequence assemblies. By placing contigs on an optical map, one is able to arrange and orient assembled sequence contigs onto a chromosome wide scaffold. To test our software for this purpose we produced a sequence assembly of E.coli K-12 using short reads (SRA accession SRX298884) trimmed with PRINSEQ (Schmieder et al., 2011) and assembled with SPAdes (Bankevich et al., 2012) using default parameters, giving an assembly of 149 contigs 4.58 Mb in length (N50 112 kb).
We ran SOMA, TWIN (Muggli et al., 2014), malignerIX and malignerDP and evaluated the alignment accuracy and runtime performance on a set of 31 contigs (2.81 Mb) that had five or more restriction fragments (including boundary fragments) and a unique placement as determined by nucmer. For our evaluation, we ignored contigs with 4 or fewer restriction fragments as these are difficult to uniquely align to the optical map.
We aligned the contigs to an experimentally produced optical consensus map (425 restriction fragments, 4.17 Mb), assembled from individual molecular maps (Rmaps) produced using enzyme BamHI (with recognition sequence GGATCC). For comparison, we also produced an error free optical map from the E.coli K-12 MG1655 reference sequence by performing an in silico digest (448 restriction fragments, 4.64 Mb). We note that while the restriction pattern of the experimental optical map is faithful to the reference, it exhibits an overall fragment undersizing bias and is missing small fragments and sites due to desorption (Fig. 3). Before alignment we smoothed out small restriction fragments less than 1 kb by merging them with neighboring restriction fragments.
Fig. 3.

Comparison of E.coli K12 experimental optical map with reference. (A) Overall, the consensus optical map (gold) assembled from experimental molecular maps shows great concordance with the in silico digest of the reference sequence (blue). (B) A close view of the left end of the same alignment shown in A. (C) An example of a matched chunk containing two fragments in reference (light blue) aligned to one fragment in the optical map (green). This indicates that a small restriction fragment is missing from the optical map. (D) Illustration of the overall undersizing bias of the optical map. Fragment sizes are given in kilobases (Color version of this figure is available at Bioinformatics online.)
We used nucmer (Kurtz et al., 2004) to determine the true placement of the contigs on the reference sequence. We took a contig placement to be correct if 90% or more of the contig aligned to a unique location with 95% identity or better.
Alignment to error-free optical map
Results for alignment to the error free optical map are shown in Table 2. We consider a contig to be placed correctly if its starting location is within 50 kb of the location reported by nucmer. TWIN, malignerIX, and malignerDP all perform similarly, finding correct alignments for all 31 contigs. SOMA does not perform as well, only finding correct alignments for 11 of the 28 contigs. TWIN has a bug that results in reported alignment locations that have several kb in error, as shown in Figure 4. This issue prevented us from using a more strict criterion for alignment correctness for this comparison. We have e-mailed the authors of TWIN to inform them of this issue.
Table 2.
Alignment results against error free E.coli optical map
| Software | Total alignments | Contigs with alignment | Contigs with correct alignment | Contigs with unique alignment | Contigs with unique and correct alignment | Runtime |
|---|---|---|---|---|---|---|
| TWIN | 37 | 31 | 31 | 28 | 28 | 0.47s |
| (2.99 Mb) | (2.81 Mb) | (2.81 Mb) | (2.71 Mb) | (2.71 Mb) | ||
| SOMA | 28 | 28 | 11 | 28 | 11 | 14.22s |
| (2.66 Mb) | (2.66 Mb) | (1.48 Mb) | (2.66 Mb) | (1.48 Mb) | ||
| malignerIX | 36 | 31 | 31 | 29 | 29 | 0.03s |
| (2.93 Mb) | (2.81 Mb) | (2.81 Mb) | (2.75Mb) | (2.75 Mb) | ||
| malignerDP | 39 | 31 | 31 | 27 | 27 | 0.40s |
| (3.01 Mb) | (2.81 Mb) | (2.81 Mb) | (2.68 Mb) | (2.68 Mb) |
Number of contigs (and bp) aligned to the error free E.coli optical map. A contig is considered to be placed correctly if its location is within 50kb of the location reported by nucmer.
Fig. 4.

TWIN and malignerDP Contig Location Errors. Errors in the placement of the contig by malignerDP and TWIN, as compared to the true placement given by nucmer. TWIN and malignerDP place all 31 contigs correctly, but TWIN reports contig alignment locations with several kb of error that cannot be explained by the sizes of the contig boundary restriction fragments (Color version of this figure is available at Bioinformatics online.)
Alignment to experimental optical map
We aligned the set of contigs to an optical consensus map assembled from experimental molecular restriction maps (Rmaps) using Gentig (Anantharaman et al., 1999). The optical map has more noise compared to the error free map, as there is an undersizing bias in the size of the fragments and some cut sites and small fragments are missing. We see from Table 3 that malignerDP is able to find correct alignments for 26 of the 31 contigs, outperforming the index-based methods malignerIX (13 contigs) and TWIN (7 contigs) as well as SOMA (0 contigs).
Table 3.
Alignment results against experimental E.coli optical map
| Software | Total alignments | Contigs with alignment | Contigs with correct alignment | Contigs with unique alignment | Contigs with unique and correct alignment | Runtime |
|---|---|---|---|---|---|---|
| TWIN | 101 | 11 | 7 | 4 | 0 | 0.76s |
| (3.44 Mb) | (0.49 Mb) | (0.26 Mb) | (0.23 Mb) | (0.00 Mb) | ||
| SOMA | 6 | 6 | 0 | 6 | 0 | 14.45s |
| (0.25 Mb) | (0.25 Mb) | (0.00 Mb) | (0.25 Mb) | (0.00 Mb) | ||
| malignerIX | 81 | 15 | 13 | 7 | 5 | 0.15s |
| (3.23 Mb) | (0.99 Mb) | (0.87 Mb) | (0.65 Mb) | (0.53 Mb) | ||
| malignerDP | 208 | 28 | 26 | 13 | 13 | 0.31s |
| (8.35 Mb) | (2.37 Mb) | (2.24 Mb) | (1.60 Mb) | (1.60 Mb) |
Number of contigs (and bp) aligned to the experimental E.coli consensus optical map. A contig is considered to be placed correctly if its location given as percentage of optical map length is within 5% of the location reported by nucmer. Among these software, the only alignment method which is able to handle the experimental error characteristics of real data is malignerDP. Index-based methods, being less flexible, are not able to find the same number of correct placements.
4.2.2 RMap alignment
We used malignerDP, malignerIX, and TWIN to align a high coverage set (1159×) of 14 734 Rmaps (average 364 kb, 20 fragments) to the error-free in silico digest of the E.coli K12 reference sequence.
malignerDP. We ran malignerDP on the RMap set and selected the subset of alignments with at most 40% unmatched sites in the reference, 15% unmatched sites in the Rmap, and an M-Score of 5 or better. 2831 (19.2%) of the Rmaps had a unique alignment with this criteria, resulting in an overall alignment coverage of 245×. No Rmaps had duplicate alignments matching this criteria. The low mapping rate is due to the fact that we used raw instrument output rather than carefully filtered Rmap datasets, for which alignment rates are much higher (Schwartz, personal communication). We chose to use raw data to demonstrate the potential of using Maligner as a component in a Q/C pipeline for optical or Nanocoding (Jo et al., 2007) mapping data. We visualized the alignments in GnomSpace (Fig. 5), showing that the Rmaps align to the reference with good fidelity. malignerDP completed in 169.86 s (11.5 ms/RMap).
Fig. 5.

Alignment of E.coli K12 Rmaps to reference sequence. A pileup of Rmap alignments (gold) to the in silico digest of the E.coli K12 MG1655 reference (blue) produced using the malignerDP software (Color version of this figure is available at Bioinformatics online.)
malignerIX. We ran malignerIX with a maximum allowed relative fragment sizing error of 15% and absolute error of 5 kb per fragment (whichever is greater), at most 40% unmatched sites in the reference, and at most an edit score per interior chunk of 5.0. 553 (3.8%) of the Rmaps had a unique alignment with this criteria, resulting in an overall alignment coverage of 39.7×. malignerIX completed in 5.72 s (0.39 ms/RMap).
TWIN. We ran TWIN on the RMap dataset. Note that this comparison is unfair, as TWIN was designed as an aligner for in silico restriction maps and as such it does not handle unmatched sites in the query or the reference. Running TWIN with lenient alignment settings (search radius of 5 kb, fval 1000) only produced alignments for 14 Rmaps. We could not determine the alignment locations or coverage due to runtime errors encountered in TWIN’s post processing scripts. TWIN completed in 20.35 s.
4.3 Budgerigar
To show that our methods scale to larger genomes, we aligned both contigs and SwaI Rmaps to an assembled budgerigar parakeet optical map comprising 93 contigs (889 Mb).
4.3.1 Contig alignment
We aligned the budgerigar v6.3 assembly contigs to the budgerigar optical map (Ganapathy et al., 2014). Before aligning, we filtered the total assembly (70 863 contigs, 1.09 Gb, 55.6 kb N50) down to those contigs with 5 or more restriction fragments (6008 contigs, 406 Mb). We aligned these contigs using TWIN, malignerDP, and malignerIX to evaluate the number of alignments and run time performance. We chose not to run SOMA since SOMA does not scale to genomes of this size, as documented in (Muggli et al., 2014). The alignment results are summarized in Table 4. We find that malignerIX runs in comparable time to TWIN, but aligns more contigs and places more contigs uniquely, as it is able to handle more unmatched sites in the reference. MalignerDP runs much slower than the index-based methods, but finds more unique alignments.
Table 4.
Contig alignment to budgerigar optical map
| Software | Contigs aligned | Contigs aligned uniquely | Runtime |
|---|---|---|---|
| TWIN | 3889 (267.0 Mb) | 1340 (130.8 Mb) | 51.01 s |
| malignerDP | 5093 (427.7 Mb) | 2635 (299.7 Mb) | 46m 16.0s |
| malignerIX | 5142 (422.8 Mb) | 2148 (249.7 Mb) | 51.53 s |
Number of contigs (and bp) aligned to the budgerigar optical map.
4.3.2 RMap alignment
We aligned a set of 671 896 Rmaps (352 kb avg, 18.3 fragments avg, 236.6 Gb total) to the budgerigar optical map using malignerDP, filtering our alignment set to those with a query unmatched site rate ≤15%, a reference unmatched site rate ≤40%, and an M-Score ≤20. 69 537 (10.3%, 30.9 Gb) of the Rmaps had at least one alignment fitting this criteria, 69 130 (30.5 Gb) of which were aligned uniquely. Total alignment took 334 h 33 m of CPU time (1.8 s/Rmap).
5 Discussion and conclusions
Alignment is an important first step in the analysis of optical mapping data. We have presented the malignerDP and malignerIX software for restriction map alignment and evaluated their performance on experimental datasets for E.coli and budgerigar parakeet. We have shown that on E.coli, malignerDP finds more correct alignments than other available methods. We have also demonstrated that malignerDP can align a high coverage Rmap set for a large genome within a couple hours on a moderately sized computing cluster.
We have also introduced the M-Score, which provides a method for normalizing alignment scores found through dynamic programming by adjusting the scores for each query based on the distribution of the best scoring but random alignments for that query. The normalization allows one to apply a consistent score threshold across queries for accepting or rejecting alignments and thereby avoid a permutation test for determining alignment significance.
Finally, we have shown that while the index-based methods malignerIX and TWIN run significantly faster than malignerDP, these methods are less sensitive than full dynamic programming, finding fewer alignments (for Rmap alignment) and fewer correct alignments (for contig alignment) against experimental datasets.
Acknowledgement
LMM would like to acknowledge Martin Muggli for his assistance in running the TWIN software and Shiguo Zhou for providing the Rmaps and optical map for E.coli K-12.
Funding
This work has been funded by NSF IIS-1117247 to MP and NIH R01-HG-000225 to DCS.
Conflict of Interest: none declared.
References
- Anantharaman T. et al. (1999) Genomics via optical mapping. III: contiging genomic DNA. Seventh Int. Conf. Intell. Syst. Mol. Biol., 7, 18–27. [PubMed] [Google Scholar]
- Antonacci F. et al. (2010) A large and complex structural polymorphism at 16p12.1 underlies microdeletion disease risk. Nat. Genet., 42, 745–750. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Armbrust E.V. et al. (2004) The genome of the diatom Thalassiosira pseudonana: ecology, evolution, and metabolism. Science, 306, 79–86. [DOI] [PubMed] [Google Scholar]
- Aston C. et al. (1998) Optical mapping: an approach for fine mapping. Methods Enzymol., 303, 55–73. [DOI] [PubMed] [Google Scholar]
- Bankevich A. et al. (2012) SPAdes: a new genome assembly algorithm and its applications to single-cell sequencing. J. Comput. Biol., 19, 455–477. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Church D.M. et al. (2009) Lineage-specific biology revealed by a finished genome assembly of the mouse. PLoS Biol., 7, 1089. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dimalanta E.T. et al. (2004) A microfluidic system for large DNA molecule arrays equally large data sets. Anal. Chem., 76, 5293–5301. [DOI] [PubMed] [Google Scholar]
- Ganapathy G. et al. (2014) High-coverage sequencing and annotated assemblies of the budgerigar genome. Gigascience, 3, 19. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gupta A. et al. (2015) Single-molecule analysis reveals widespread structural variation in multiple myeloma. PNAS, 112, 7689–7694. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ivens A.C. et al. (2005) The genome of the kinetoplastid parasite, Leishmania major. Science, 309, 436–442. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jo K. et al. (2007) A single-molecule barcoding system using nanoslits for DNA analysis. PNAS, 104, 2673–2678. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kidd J.M. et al. (2008) Mapping and sequencing of structural variation from eight human genomes. Nature, 453, 56–64. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kounovsky-Shafer K.L. et al. (2013) Presentation of large DNA molecules for analysis as nanoconfined dumbbells. Macromolecules, 46, 8356–8368. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kurtz S. et al. (2004) Versatile and open software for comparing large genomes. Genome Biol., 5, R12. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lin J. et al. (1999) Whole-genome shotgun optical mapping of Deinococcus radiodurans. Science, 285, 1558–1562. [DOI] [PubMed] [Google Scholar]
- Muggli M.D. et al. (2014) Efficient indexed alignment of contigs to optical maps In: Brown D., Morgenstern B. (eds), Algorithms in Bioinformatics. Vol. 8701, Springer, Berlin Heidelberg, pp. 68–81. [Google Scholar]
- Nagarajan N. et al. (2008) Scaffolding and validation of bacterial genome assemblies using optical restriction maps. Bioinformatics, 24, 1229–1235. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ray M. et al. (2013) Discovery of structural alterations in solid tumor oligodendroglioma by single molecule analysis. BMC Genomics, 14, 505. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sarkar D. et al. (2012) Statistical significance of optical map alignments. J. Comput. Biol., 19, 478–492. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schmieder R., Edwards R. (2011) Quality control and preprocessing of metagenomic datasets. Bioinformatics, 27, 863–864. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schnable P.S. et al. (2009) The B73 maize genome: complexity, diversity, and dynamics. Science, 326, 1112–1115. [DOI] [PubMed] [Google Scholar]
- Teague B. et al. (2010) High-resolution human genome structure by single-molecule analysis. PNAS, 107, 10848–10853. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Valouev A. et al. (2006a) Alignment of optical maps. J. Comput. Biol., 13, 442–462. [DOI] [PubMed] [Google Scholar]
- Valouev A. et al. (2006b) An algorithm for assembly of ordered restriction maps from single DNA molecules. PNAS, 103, 15770–15775. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Valouev A. et al. (2006c) Refinement of optical map assemblies. Bioinformatics, 22, 1217–1224. [DOI] [PubMed] [Google Scholar]
- Wei F. et al. (2009) The physical and genetic framework of the maize B73 genome. PLoS Genet., 5, e1000715. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Young N.D. et al. (2011) The Medicago genome provides insight into the evolution of rhizobial symbioses. Nature, 480, 520–524. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhou S. et al. (2007) Validation of rice genome sequence by optical mapping. BMC Genomics, 8, 278. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhou S. et al. (2004) Shotgun optical mapping of the entire Leishmania major Friedlin genome. Mol. Biochem. Parasitol., 138, 97–106. [DOI] [PubMed] [Google Scholar]
- Zhou S. et al. (2009) A single molecule scaffold for the maize genome. PLoS Genet, 5, e1000711. [DOI] [PMC free article] [PubMed] [Google Scholar]