

Abstract
The Oxford Nanopore MinION sequences individual DNA molecules using an array of pores that read nucleotide identities based on ionic current steps. We evaluated and optimized MinION performance using M13 genomic dsDNA. Using expectation-maximization (EM) we obtained robust maximum likelihood (ML) estimates for read insertion, deletion and substitution error rates (4.9%, 7.8%, and 5.1% respectively). We found that 99% of high-quality ‘2D’ MinION reads mapped to reference at a mean identity of 85%. We present a MinION-tailored tool for single nucleotide variant (SNV) detection that uses ML parameter estimates and marginalization over many possible read alignments to achieve precision and recall of up to 99%. By pairing our high-confidence alignment strategy with long MinION reads, we resolved the copy number for a cancer/testis gene family (CT47) within an unresolved region of human chromosome Xq24.
Nanopore sequencing with its speed, single base sensitivity, and long read lengths is a promising next generation method for sequencing DNA and RNA. Earlier in 2014 Oxford Nanopore Technologies (ONT) enlisted several hundred laboratories to beta test their pocket-sized MinION DNA sequencing device. We set out to characterize the performance and characteristics of the MinION sequencing platform, developing the platform for single nucleotide variant calling and repeat structure resolution of highly repetitive regions of the human genome.
The MinION reads the nucleotide sequence of individual DNA strands as they are driven through biological nanopores by an applied electric field. The rate at which each DNA strand moves through the nanopore is controlled by a processive enzyme bound to the DNA at the pore orifice. Up to 512 DNA molecules can be read simultaneously using amplifiers that independently address each nanopore. Ionic current changes, each associated with a 5-nucleotide DNA k-mer, are detected as DNA molecules translocate through the nanopores at 1 nucleotide precision. DNA base calls are performed using cloud-based (Metrichor) software provided by ONT that employs Hidden Markov Models (HMMs) to infer sequences from these ionic current changes.
As part of the MinION Access Program (MAP), we determined MinION sequence read quality and errors by analyzing the M13mp18 genome (a phage from E. coli host strain ER2738 with 42% average GC content and 7.2 kb genome size; see Methods). Using expectation-maximization (EM) we inferred maximum likelihood estimates (MLE) for the rates of insertions, deletions, and substitutions in MinION reads. We then re-aligned the reads to generate high-confidence alignments and used the MLE models to demonstrate that MinION reads can be used for accurate single nucleotide variant (SNV) calling. We coupled this highconfidence alignment strategy with long MinION reads to resolve tandem repeat organization of a CT47 cancer-testis gene family on an unfinished segment of human chromosome Xq24. Our results document the substantial improvements in MinION performance achieved during MAP.
RESULTS
The MinION reads both strands of duplex DNA
Library preparation was performed as recommended by ONT, with modifications to ensure the integrity of high-molecular weight DNA (see METHODS). A DNA construct analyzed on the MinION (Fig. 1) is composed of: a lead adaptor that loads the processive enzyme and facilitates DNA capture in the applied electric field across the pore; the DNA insert of interest (M13mp18 dsDNA in the example shown); a hairpin adaptor that permits consecutive reading of the template and complement strands by the nanopore; and a tethering adaptor that concentrates DNA at the membrane surface.
Fig. 1.
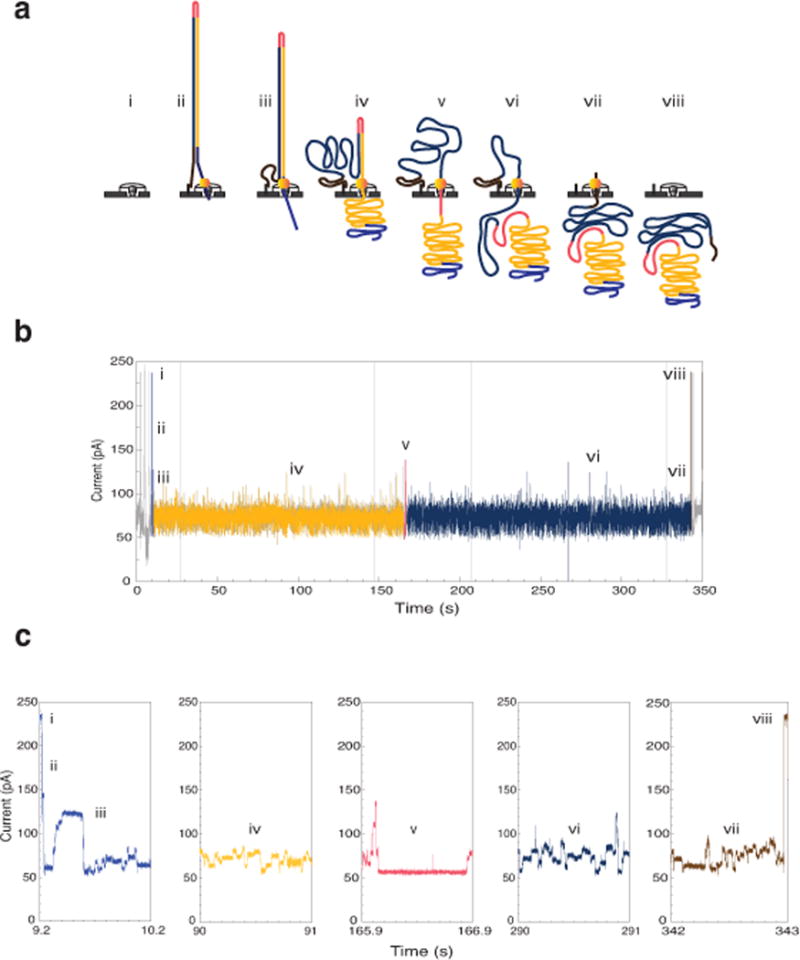
Molecular events and ionic current trace for a 2D read of a 7.25 kb M13 phage dsDNA molecule. (a) Schematic for the steps in DNA translocation through the nanopore. (i) Open channel; (ii) dsDNA with a ligated lead adaptor (blue), with a molecular motor bound to it (orange), and a hairpin adaptor (red), is captured by the nanopore. DNA translocation through the nanopore begins through the effect of an applied voltage across the membrane and the action of a molecular motor; (iii) Translocation of the lead adaptor (blue); (iv) Translocation of the template strand (gold); (v) Translocation of the hairpin adaptor (red); (vi) Translocation of the complement strand (dark blue); (vii) Translocation of the trailing adaptor (brown); (viii) Return to open channel. (b) Raw current trace for the passage of the M13 dsDNA construct through the nanopore. Regions of the ionic current trace corresponding to steps i-viii are labeled. (c) Expanded time and current scale for raw current traces corresponding to steps i–viii. Each adaptor generates a unique current signal used to aid base calling.
The translocation of a single M13 genomic dsDNA copy through a MinION pore consists of a series of steps, each associated with an identifiable ionic current pattern (Fig. 1). These steps include: i) the open pore; ii–iii) capture and translocation of the lead adaptor; iv) translocation of the template strand; v) translocation of the hairpin adaptor; vi) translocation of the complement strand; vii) translocation of the tethering adaptor; and viii) release of the DNA strand into the trans compartment with return to the open channel ionic current. At this point another DNA molecule can be captured and analyzed by the pore.
Over the six-month period of MAP to date, there have been three MinION chemistry versions and numerous base-calling algorithm updates that have resulted in improvements in device performance (Supplemental Note Fig. 1). For example, at UCSC the average % identity (the proportion of bases in a read aligned to a matching base in a reference sequence) observed for 2D reads was 66% in June 2014 (R6.0 chemistry release), 70% in July 2014 (R7.0 chemistry release), 78% in October 2014 (R7.3 chemistry release) and 85% in November 2014 (Metrichor R.7X 2D Version 1.9 update). The present study was based on MinION R7.3 chemistry and R7.X version 1.9 base-calling algorithms.
MinION throughput
We sequenced intact replicative phase M13 phage dsDNA using three MinION flow cells that contained 337-to-473 functional channels. Covalent attachment of the linker and hairpin adaptors enabled consecutive reads of both strands of each DNA duplex molecule. These reads were characterized as: template, representing the forward strand; complement, representing the reverse strand; or ‘2D’, representing reads obtained by computationally merging template and complement data. Each replicate run was 48-hours long, and read between 184 and 450 million bases (Supplementary Note Table 1). In our experiments using R7.3 chemistry, we observed ~63% template, ~24% complement, and ~13% 2D reads (Supplemental Note Table 1). Unless otherwise noted, all results presented in this study were based on reads classified by Metrichor as high-quality. These reads totaled between 60 and 189 million bases per M13 sequencing run.
Establishing a mapping pipeline for MinION reads
To evaluate the quality of these MinION reads, we experimented with four different alignment programs1,2,3,4 (Supplemental Note 3). Each was run with its default parameters and with tuned parameters that were selected either by experimentation or by expert advice from other MAP participants.
Among these programs, we found variation in the proportion of reads that mapped to reference sequences (M13 or ONT lambda DNA control; Supplemental Note Fig. 3). LAST3 was the most inclusive alignment program when using tuned parameters. Stringency analysis indicated that few of the LAST alignments were false positives (Supplemental Note Fig. 4). For data pooled from the three M13 experiments, tuned LAST mapped 95.26% of template, 98.31% of complement and 98.96% of 2D reads. To test if the unmapped reads resulted from DNA contamination, we compared them against the NCBI NT database5 using BLAST 2.296 and found most of the unmapped reads were homologous to E. coli, indicating some minor contamination (Fig. 2a–c, Supplemental Note 4).
Fig. 2.
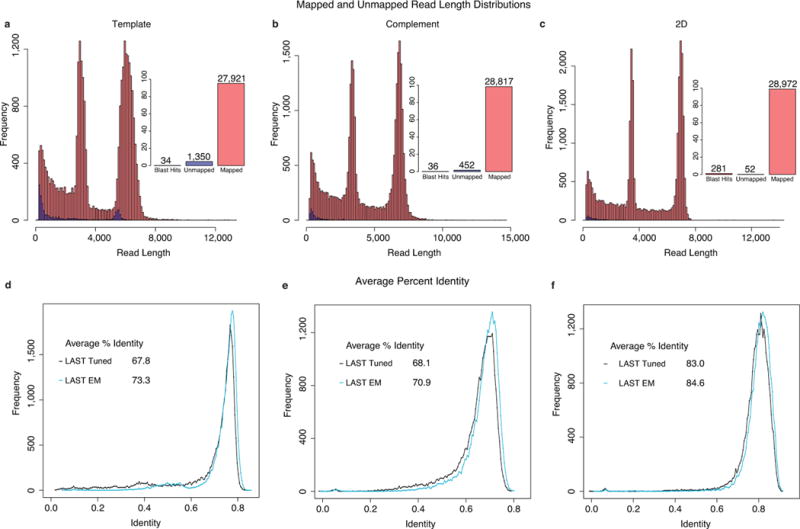
Read length distributions and identity plots for M13. Read length histograms for mapped vs. unmapped reads across three replicate M13 experiments for (a) template; (b) complement; and (c) 2D reads. Most of the reads mapped to a known reference, with two distinct peaks at about 7.2 kb, corresponding to full-length M13, and 3.8 kb, corresponding to the phage lambda DNA (control fragment). Insets show the proportion of mappable vs. unmappable reads and the proportion of unmappable reads that found hits when compared against the NCBI NT database using BLAST (to check for contamination or missed homology). Read alignment identities for mappable reads using tuned LAST, realigned LAST, and EM trained LAST for (d) template; (e) complement; and (f) 2D reads.
We observed two distinct peaks for reads, one at about 7.2 kb, corresponding to full-length M13 DNA, and one at 3.8 kb, corresponding to the ONT lambda phage DNA control (Fig. 2a–c). A large number of reads spanned the full length of the M13 genome (Fig. 2). The proportion of unmappable reads was small, and generally shorter than the mappable reads (Supplemental Note 5).
EM generates high-confidence alignments for MinION reads
We found substantial disagreement among rates of substitution, insertion, and deletion for alignments generated by different mapping programs (Fig. 3a–b). A more principled way to estimate the true rates of these errors is to propose a reasonable model of the error process and calculate MLEs of the parameters (Supplemental Note 6)7. Using EM to train an HMM model (Supplemental Note Fig. 5, and using alignment banding heuristics necessary for efficiency8, we obtained robust convergence to effectively the same parameter MLEs across all replicate experiments, guide alignments, and random starting parameterizations (Fig 3a–b, Supplemental Note Fig. 6). This showed that insertions were less frequent than deletions by about two-fold in 2D reads and about three-fold in template and complement reads. The combined insertion/deletion (indel) rate was between 0.13 (2D reads) and 0.2 (template/complement reads) events per aligned base. For all read types, indels were predominantly single bases (Supplemental Note Fig. 7; Supplemental Note 6). Substitutions varied from 0.21 (for template reads) to 0.05 (for 2D reads) events per aligned base (Fig. 3c, Supplemental Note Fig. 8–9). Substitutions errors were not uniform, in particular A-to-T and T-to-A errors were estimated to be very low at 0.04% and 0.1% respectively (Supplemental Note 6.1).
Fig. 3.
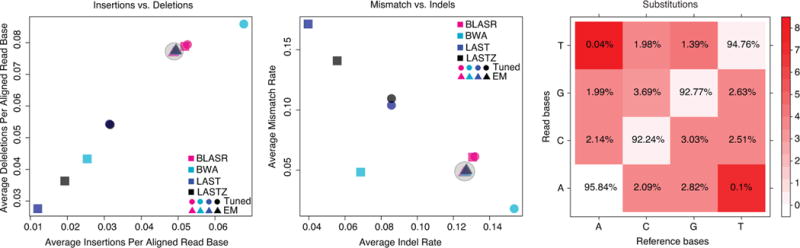
Maximum-likelihood (ML) alignment parameters derived using expectation-maximization (EM). The process starts from four guide alignments each generated with a different mapper using tuned parameters. (a) Insertion vs. deletion rates, expressed as events per aligned base. (b) Insertion or deletion (indel) events per aligned base vs. rate of mismatches per aligned base (see Supplement). Rates vary strongly between different guide alignments, however, EM training and realignment results in very similar rates (grey circles), regardless of the initial guide alignment. (c) Matrix for substitution emissions determined using EM reveals very low rates of A-to-T and T-to-A substitutions.
Re-alignment of the reads using the MLE parameters and the AMAP objective function9 substantially improved the identity of the alignments over the starting tuned alignments for every mapping program (Fig. 2d–f, Supplemental Note 7). Looking at our high-confidence alignments, there were no clear correlations between read length and errors (Supplemental Note 8). However, there was a positive correlation between the rate of insertions, deletions and substitutions in 2D reads (Supplemental Note 9).
Most recently, we analyzed our data using a newly available BWA mode (ont2d) optimized for nanopore reads. The average % identity for BWA ont2d mode declined slightly (Supplemental Note Table 6). However, the rates of insertions, deletions and substitutions were substantially closer to the MLE parameters estimated by EM, suggesting that it is an improvement over the pacbio mode that we used originally.
To see if our analysis pipeline produced similar results on larger, more complex genomes, we analyzed the E. coli dataset released by Quick et al.10 which used R7.3 chemistry and Metrichor R7.3 2D Version 1.5. The most recent Metrichor update was not available to Quick et al.10 at the time of their data release. We observed an improvement in average % identity from 80% with tuned LAST to 82% after realignment using the AMAP objective function with MLE parameters (Supplemental Note 10, Supplemental Note Table 7). In addition, the MLEs for the rates of insertions, deletions and substitutions were very similar to those found for the M13 data.
M13 sequencing depth and k-mer analysis
MinION sequencing depth was generally consistent across the 7.2 kb M13 genome (Fig. 4, Supplemental Note Fig. 13). However, 192 positions (2.6%) were underrepresented (Supplemental Note 11). Approximately 50% of these positions appear at the beginning and end of the reference, and are likely the result of adaptor trimming by Metrichor. A majority of the remaining underrepresented positions were associated with 5-mers rich in polymeric nucleotide runs (Supplemental Note Table 8). To determine if the MinION has an inherent bias towards certain k-mers, we compared counts of 5-mers for all three read types (template, complement, 2D) versus the M13 reference sequence. The most underrepresented 5-mers were homopolymers of poly-dA or poly-dT, while the most overrepresented 5-mers were G/C rich and absent homopolymer repeats (Supplemental Note 11.1, Supplemental Note Table 9). These findings are consistent with observations from Ashton et al.11.
Fig. 4.
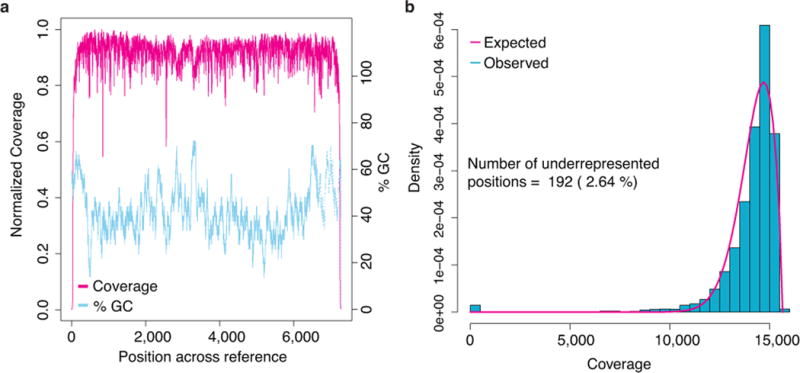
M13 sequencing depth. (a) The magenta line denotes coverage by position in the genome, and the dotted blue line depicts local G/C% for that position. Ignoring sites close to the ends of the reference, which appear to be affected by adaptor trimming, the coverage drops at polymeric nucleotide runs. Coverage was calculated by binning over a sliding 5 bp window. G/C content was calculated by binning over a 50 bp sliding window. (b) Coverage depth distribution fitted with a generalized extreme value distribution.
MinION reads can call SNVs with high recall and precision
SNV detection is important for metagenomics and microbial strain detection12,13,14. To determine if MinION reads could be used for SNV discovery in monoploid genomes, we computationally introduced random substitutions into the M13 reference sequence at 1-to-20% frequency. Using this altered sequence as an alignment reference we attempted to recover these substitutions using a Bayesian transducer framework15 (Supplemental Note 12.2). We assessed performance using standard information retrieval metrics (precision, recall and F-score). As well as assessing SNV detection ability, these experiments also served as an indicator of the accuracy of our alignments and models while avoiding issues of reference allele bias to which simple metrics, like alignment identity, are prone.
Using all the 2D read data and a posterior base calling threshold that gave the optimal F-score, we achieved a recall of 99% and precision of 99% at 1% substitution frequency (Fig. 5a). Reducing the sequencing depth down to a more reasonable 60X by sampling we achieved a recall and precision of 97%. Increasing the mutation frequency decreases the F-score progressively, presumably because the alignment challenge between the reads and the mutated reference becomes harder (Fig. 5b).
Fig. 5.
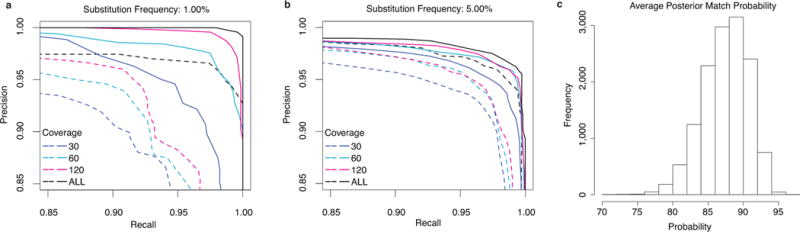
Exploring single nucleotide variant (SNV) calling with MinION reads. (a) Variant calling with substitution frequency of 1%. (b) Variant calling with substitution frequency of 5%. Dotted lines in both (a) and (b) represent variant calling using a simple transducer model, using a tuned LAST alignment and giving all substitutions equal probability. Different sampled read coverages are shown. Each curve is produced by varying the posterior base calling threshold to trade off precision for recall. Solid lines in both (a) and (b) represent variant calling using the same simple transducer model as in the dotted lines, but trained by EM and incorporating marginalization over the read to reference alignments. Results shown are averaged over three replicate M13 experiments, and for each coverage level, three samplings of the reads. ALL curve reflects all the available data for each experiment. (c) The distribution of posterior match probabilities show that there is substantial uncertainty in most matches and explain why marginalizing over the read alignments is a powerful approach.
One particularly powerful strategy of the method we employed was the marginalization over many possible alignments for each read, which helped factor out the considerable alignment uncertainty (Fig. 5c). In contrast, using fixed LAST alignments but otherwise keeping the method the same resulted in substantially higher rates of false positives for a given recall value (Fig. 5a–b).
Resolving organization of a cancer-testis gene family
One of the strengths of the MinION device is long, single molecule reads. For example, 7.2 kb full length 2D reads of M13 genomic DNA (that constituted the core of this study) were routinely observed (Fig. 2). Substantially longer reads were also observed, but at lower frequency, when intact very large DNA fragments were delivered to the MinION (e.g., a full length 48 kb 2D read of intact phage lambda DNA that mapped back to reference with 87% identity (Supplemental Note Fig. 2)). We reasoned that long MinION reads, coupled with our high-confidence alignment strategy, could be used to resolve complex and often unfinished regions of genomes.
As a test, we examined the organization of a human-specific tandem repeat cluster spanning a putative 50 kb assembly gap on human Xq24 (hg38 chrX:120,814,747–121,061,920) (Fig. 6a). Each 4,861 bp tandem repeat in this region contains a single annotated testis/cancer gene (CT47), with observed expression in testes, and in lung and esophageal cancer cells16. The high level of sequence homology between adjacent copies (ranging from 95–100% sequence identity) is likely to result in recombination or replication errors, leading to alleles with different numbers of repeats that are often difficult to represent accurately by standard short read assembly17. Furthermore, copy number expansion and contraction involving genes contribute to variability in gene expression, epigenetic regulation, and association with human disease18,19.
Fig. 6.
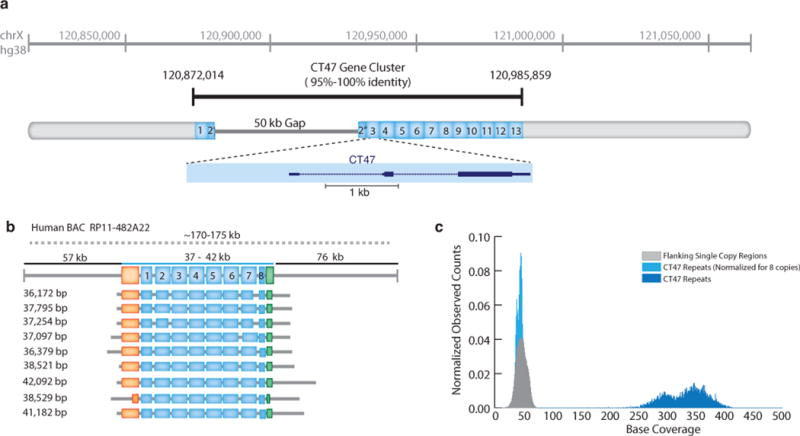
Resolution of CT47 repeat copy number estimate on human chromosome Xq24. (a) BAC end sequence alignments (RP11-482A22: AQ630638 and AZ517599) span a 247 kb region, including thirteen annotated CT47 genes (each defined within a 4.8 kb tandem repeat) and a 50 kb scaffold gap in the GRCh38/hg38 reference assembly. (b) Utilizing MinION long-reads obtained from RP11-482A22 high-molecular weight BAC DNA, nine reads span the length of the CT47-repeat region providing evidence for eight tandem copies of the CT47-repeat. Insert size estimate (170–175 kb, as determined by pulse-field gel electrophoresis) is noted as a dotted line, with flanking regions (upstream: 57 kb and downstream region: 73 kb, black line) and repeat region (37-to-42 kb, or 7.5-to-8.75 copies of the repeat, blue line). Single copy regions directly before the CT47 repeats are shown in orange (6.6 kb) and green (2.6 kb), repeat copies are labeled in blue, and grey lines describe read alignment into flanking region. The size of the repeat region are provided on the left (range 36 kb ’ 42 kb). (c) Shearing the BAC DNA to increase sequence coverage provided copy number estimates by read depth. All bases not included in the CT47 repeat unit are labeled as flanking region (grey distribution, mean: 46.2 base coverage). Base coverage across the CT47 repeats are summarized over one copy of the repeat to provide an estimate of the combined number (dark blue distribution, mean: 329.3 base coverage), and are similar to single copy estimates when normalized for eight copies (light blue distribution, mean: 41.15 base coverage).
We used the MinION to acquire very long reads from a human BAC (RP11-482A22) that contained the CT47 repeats within the unresolved Xq24 segment. Our intent was to acquire reads that spanned the entirety of the tandem repeat array, thereby resolving the sequence organization by consensus. We identified nine 2D reads, ranging in length from 36 kb to 42 kb, which together indicated eight tandem repeat copies within the gap (Fig. 6b, Supplemental Note 13, Supplemental Data 1–3). This copy number prediction was supported by pulse-field gel electrophoresis, which revealed a repeat array of 37-to-42 kb, or 7.5-to-8.6 copies of the 4.8 kb repeat (Supplemental Note Fig. 22). As an additional test, we obtained 40-to-60X sequence coverage of the unresolved Xq24 segment using short (~10 kb) MinION reads from sheared BAC DNA. A copy number estimate, based on these reads, also indicated 8 CT47 repeats within the unresolved region (Fig. 6c).
DISCUSSION
We began this study by documenting MinION performance using M13. We found that consecutive reads of adaptor-linked template and complement DNA strands (circa 14.4 kb total bases) were routinely achieved. Approximately 99% of 2D reads mapped to a reference (M13 or phage lambda DNA control) and yielded 85% average identity. Using EM training of an HMM model we were able to robustly parse the error sources into mismatches, insertions, and deletions. This information was used to generate high-confidence alignments that both allowed us to call SNVs accurately and to characterize an unresolved region of human Xq24 enriched in repetitive DNA. A dual MinION sequencing strategy that employed both long-read scaffolds and higher coverage shorter reads was essential for copy number estimates in that region.
Our results differ markedly from an early MinION study by Mikheyev and colleagues20. Their results, produced during the first weeks of MAP using phage lambda DNA, gave only 8% of 2D reads that mapped to reference, and very low read identities. This was in part due to their use of an early MinION release (R6.0). In addition, they used standard alignment tools designed either for short reads (BLASTN) or for the PacBio sequencing platform (BLASR), and did not employ tuning or realignment to optimize performance.
Given the pace of read quality improvements during MAP, we anticipate that correct base calls will continue to increase beyond the average 85% identity we observed in this study. We also anticipate that the MinION will be used to report features of genomic DNA that are observable because the nanopore sensor directly touches each base on native DNA strands. These features include epigenetic modifications21,22,23, abasic residues24,25, DNA adducts26, thymine-thymine dimers, and strand breaks.
In summary, the MinION is a portable device that sequences individual, long native DNA strands. Its accuracy can resolve important biological questions, and is improving rapidly. It is a new paradigm for DNA sequencing.
ONLINE METHODS
M13 MinION Experiments
We generated three replicate experiments of M13mp18 phage double stranded DNA to establish the reproducibility and performance characteristics of the MinION. Below we describe the M13 sequencing standard preparation and MinION sequencing protocols.
M13mp18 DNA sequencing standard
M13mp18 dsDNA was obtained from New England Biolabs (Cat No. N4018S). The host for this phage is E. coli strain ER2738, and the genome is 7.2 kb in size with 42% average GC content. Thirty micrograms of M13mp18 was linearized by overnight double digestion with High-Fidelity HindIII (NEB, Cat No. R3104S), and High-Fidelity BamHI (NEB, Cat No. R3136S). Digests were performed according to NEB recommendations using Cut Smart Buffer supplied with restriction enzymes. Two hundred nanograms of M13mp18 double digest were run on a 1% TBE agarose gel to confirm complete linearization of the circular RF genome. The restriction digest was then extracted once with equal volume of TE (10 mM Tris, 1 mM EDTA pH 8) equilibrated Phenol:Chloroform (OmniPur, Cat No. 6805), twice with TE equilibrated Chloroform pH 8, and then ethanol precipitated by addition of 1/10 volume of 5 M sodium acetate pH 5.2 (Teknova, Cat No. S0296) and 2 volumes of ice cold 100% ethanol. Samples were centrifuged to pellet DNA and M13mp18 pellet washed twice with 70% ethanol, allowed to dry to remove ethanol and then resuspended in MilliQ water and quantitated using a Nanodrop. M13 sequence was confirmed using Sanger sequencing (UC Berkeley DNA Sequencing Facility, with ABI Model 3730 XL DNA Sequencer (Applied Biosystems, Life Technologies, Thermo Fisher Scientific Inc.)). Sequencing primers TAAGGTAATTCACAATGATTAAAGTTG; CTGTGGAATGCTACAGGC; CACCTTTAATGAATAATTTCCGTC; CATGCTCGTAAATTAGGATGG; GTTTTACGTGCTAATAATTTTGATATG; CAAGGCCGATAGTTTGAGT; CACTGGCCGTCGTTTTA; GAGGCTTTATTGCTTAATTTTGC; AGGTCTTTACCCTGACTATTATAG; AGGCTTTGAGGACTAAAGAC; AATGGATCTTCATTAAAGCCAG; CAGCCTTTACAGAGAGAATAAC; TCCGGCTTAGGTTGGG; GTGAGGCGGTCAGTATTAAC; GAGATAGGGTTGAGTGTTGT; TTCTCCGTGGGAACAAAC; were obtained from Integrated DNA Technologies (http://www.idt.com/).
M13 MinION sequencing
The libraries for MinION runs were prepared as recommended by ONT. Unsheared DNA was used for M13 sequencing library preparation. For BAC DNA, sequencing libraries were prepared using unshared DNA as well as DNA sheared to an average length of 10 kb using g-TUBE (Covaris, Cat. No. 520079). Briefly, the DNA sample was spiked with ONT lambda DNA control, end-repaired using NEBNext End Repair Module (NEB, Cat. No. E6050S), and cleaned up using Agencourt AMPure XP beads (Beckman Coulter; Cat. No. A63880). The purified end-repaired DNA then underwent dA tailing using the NEB dA-Tailing Module (NEB, Cat. No. E6053S). This was followed by ligation of ONT sequencing adaptors (adaptor Mix and HP adaptor) using Blunt/TA Ligase Master Mix (NEB, Cat. No. M0367S). Using Dynabeads His-Tag Isolation and Pulldown (Life Technologies, Cat. No. 10103D), the library was enriched for DNA molecules ligated to the ONT HP adaptor. The adapted and enriched DNA was eluted in ONT supplied elution buffer. This prepared library was then mixed with proprietary ONT EP Buffer and ONT Fuel Mix prior to being added to the MinION flowcell. Three 48-hour sequencing runs were performed, each using a new flowcell.
The MinION data were base called using ONT Metrichor software (workflow R7.X 2D rev1.9). The basecaller classifies reads as pass and fail. Unless otherwise noted, all the analyses reported in this study were performed using the pass reads from R7.3 chemistry (also see Supplemental Notes).
Code availability
The analysis software is open-source and available (nanopore pipeline at https://github.com/mitenjain/ nanopore; and marginAlign pipeline at https://github.com/benedictpaten/marginAlign; see Supplemental Note 3).
Supplementary Material
Acknowledgments
Research reported in this publication was supported by the National Human Genome Research Institute of the National Institutes of Health under Award Numbers HG006321 (MA), HG007827 (MA), and U54HG007990 (BP). The authors would also like to thank Oxford Nanopore Technologies for their gift to the UCSC Nanopore Group. The authors thank D. Deamer for support, reading the manuscript, and helpful discussion. The authors gratefully acknowledge D. Haussler and J. Kent for their support. The content is solely the responsibility of the authors and does not necessarily represent the official views of the National Institutes of Health or Oxford Nanopore Technologies.
Footnotes
ACCESSION CODES
All the sequence data for M13 and BAC sequencing runs in the study are available via ENA. The primary accession number is PRJEB8230, and the secondary accession number is ERP009289.
AUTHOR CONTRIBUTIONS
MA conceived experiments and directed research. BP conceived and directed bioinformatics analysis. BP, MJ, IF, and KHM were responsible for bioinformatics analysis and software development. MJ and HEO were responsible for completion of sequencing experiments and data processing. MJ and HEO were responsible for preparing DNA sequencing standards. HEO was responsible for Sanger sequencing of M13 dsDNA. BP and IF were responsible for k-mer and BLAST analysis. BP and MJ were responsible for SNV analysis. BP developed and implemented EM and realignment strategies. KHM conceived and directed BAC experiments and data analysis. All authors contributed to manuscript writing, editing, and completion.
COMPETING FINANCIAL INTERESTS
MA is a consultant to Oxford Nanopore Technologies.
References
- 1.Chaisson M, Tesler G. Mapping single molecule sequencing reads using basic local alignment with successive refinement (BLASR): application and theory. BMC bioinformatics. 2012;13:238. doi: 10.1186/1471-2105-13-238. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Li H. Aligning sequence reads, clone sequences and assembly contigs with BWA-MEM. 2013;00:3. [Google Scholar]
- 3.Frith MC, Wan R, Horton P. Incorporating sequence quality data into alignment improves DNA read mapping. Nucleic acids research. 2010;38:e100. doi: 10.1093/nar/gkq010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Harris RS. Ph.D. thesis. The Pennsylvania State University; 2007. Improved pairwise alignment of genomic DNA. [Google Scholar]
- 5.Benson DA, et al. GenBank. Nucleic acids research. 2013;41:D36–42. doi: 10.1093/nar/gks1195. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Altschup SF, Gish W, Miller W, Myers EW, Lipman DJ. Basic Local Alignment Search Tool. J Mol Biol. 1990;215:403–410. doi: 10.1016/S0022-2836(05)80360-2. [DOI] [PubMed] [Google Scholar]
- 7.Do CB, Batzoglou S. What is the expectation maximization algorithm? Nature biotechnology. 2008;26:897–9. doi: 10.1038/nbt1406. [DOI] [PubMed] [Google Scholar]
- 8.Paten B, Herrero J, Beal K, Fitzgerald S, Birney E. Enredo and Pecan: genome-wide mammalian consistency-based multiple alignment with paralogs. Genome research. 2008;18:1814–28. doi: 10.1101/gr.076554.108. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Schwartz AS, Pachter L. Multiple alignment by sequence annealing. Bioinformatics (Oxford, England) 2007;23:e24–9. doi: 10.1093/bioinformatics/btl311. [DOI] [PubMed] [Google Scholar]
- 10.Quick J, Quinlan A, Loman N. A reference bacterial genome dataset generated on the MinION portable single-molecule nanopore sequencer. GigaScience. 2014:1–6. doi: 10.1186/2047-217X-3-22. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Ashton PM, et al. MinION nanopore sequencing identifies the position and structure of a bacterial antibiotic resistance island. Nature Biotechnology. 2014 doi: 10.1038/nbt.3103. [DOI] [PubMed] [Google Scholar]
- 12.Davey JW, et al. Genome-wide genetic marker discovery and genotyping using next-generation sequencing. Nature reviews. Genetics. 2011;12:499–510. doi: 10.1038/nrg3012. [DOI] [PubMed] [Google Scholar]
- 13.Bourlat SJ, et al. Genomics in marine monitoring: new opportunities for assessing marine health status. Marine pollution bulletin. 2013;74:19–31. doi: 10.1016/j.marpolbul.2013.05.042. [DOI] [PubMed] [Google Scholar]
- 14.Stucki D, Gagneux S. Tuberculosis. Vol. 93. Edinburgh, Scotland: 2013. Single nucleotide polymorphisms in Mycobacterium tuberculosis and the need for a curated database; pp. 30–9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Holmes I, Bruno WJ. Evolutionary HMMs: a Bayesian approach to multiple alignment. Bioinformatics. 2001;17:803–820. doi: 10.1093/bioinformatics/17.9.803. [DOI] [PubMed] [Google Scholar]
- 16.Chen Y, Iseli C, Venditti C. Identification of a new cancer/testis gene family, CT47, among expressed multicopy genes on the human X chromosome. Genes, Chromosomes & Cancer. 2006;400:392–400. doi: 10.1002/gcc.20298. [DOI] [PubMed] [Google Scholar]
- 17.Treangen TJ, Salzberg SL. Repetitive DNA and next-generation sequencing: computational challenges and solutions. Nature reviews. Genetics. 2012;13:36–46. doi: 10.1038/nrg3117. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Tremblay DC, Alexander G, Moseley S, Chadwick BP. Expression, tandem repeat copy number variation and stability of four macrosatellite arrays in the human genome. BMC genomics. 2010;11:632. doi: 10.1186/1471-2164-11-632. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Brahmachary M, et al. Digital genotyping of macrosatellites and multicopy genes reveals novel biological functions associated with copy number variation of large tandem repeats. PLoS genetics. 2014;10:e1004418. doi: 10.1371/journal.pgen.1004418. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Mikheyev AS, Tin MM. A first look at the Oxford Nanopore MinION sequencer. Molecular Ecology Resources. 2014;14:1097–1102. doi: 10.1111/1755-0998.12324. [DOI] [PubMed] [Google Scholar]
- 21.Schreiber J, et al. Error rates for nanopore discrimination among cytosine, methylcytosine, and hydroxymethylcytosine along individual DNA strands. Proceedings of the National Academy of Sciences of the United States of America. 2013;110:18910–5. doi: 10.1073/pnas.1310615110. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Laszlo AH, et al. Detection and mapping of 5-methylcytosine and 5-hydroxymethylcytosine with nanopore MspA. Proceedings of the National Academy of Sciences of the United States of America. 2013;110:18904–9. doi: 10.1073/pnas.1310240110. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Wescoe ZL, Schreiber J, Akeson M. Nanopores discriminate among five C5-Cytosine variants in DNA. Journal of the American Chemical Society. 2014;136:16582–7. doi: 10.1021/ja508527b. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Cherf GM, et al. Automated forward and reverse ratcheting of DNA in a nanopore at 5-Å precision. Nature biotechnology. 2012;30:344–8. doi: 10.1038/nbt.2147. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Lieberman KR, Dahl JM, Mai AH, Akeson M, Wang H. Dynamics of the translocation step measured in individual DNA polymerase complexes. Journal of the American Chemical Society. 2012;134:18816–23. doi: 10.1021/ja3090302. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Schibel A, et al. Nanopore detection of 8-oxo-7,8-dihydro-2′-deoxyguanosine in immobilized singlestranded DNA via adduct formation to the DNA damage site. Journal of American Chemical Society. 2010;132:17992–17995. doi: 10.1021/ja109501x. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.