

Excess centrosomes preclude proper interphase MTOC reorientation during sprouting morphogenesis. Normal centrosome numbers are required for dynamic repolarization and migration of sprouting ECs, which contribute to blood vessel formation.
Abstract
Blood vessel formation requires dynamic movements of endothelial cells (ECs) within sprouts. The cytoskeleton regulates migratory polarity, and centrosomes organize the microtubule cytoskeleton. However, it is not well understood how excess centrosomes, commonly found in tumor stromal cells, affect microtubule dynamics and interphase cell polarity. Here we find that ECs dynamically repolarize during sprouting angiogenesis, and excess centrosomes block repolarization and reduce migration and sprouting. ECs with excess centrosomes initially had more centrosome-derived microtubules but, paradoxically, fewer steady-state microtubules. ECs with excess centrosomes had elevated Rac1 activity, and repolarization was rescued by blockade of Rac1 or actomyosin blockers, consistent with Rac1 activity promoting cortical retrograde actin flow and actomyosin contractility, which precludes cortical microtubule engagement necessary for dynamic repolarization. Thus normal centrosome numbers are required for dynamic repolarization and migration of sprouting ECs that contribute to blood vessel formation.
INTRODUCTION
Blood vessel formation extends vessel networks that deliver oxygen and nutrients to cells, a process coopted in diseases such as cancer (Carmeliet and Jain, 2011). The topology of angiogenic sprouts is dynamic, with extensive cell rearrangements required for proper blood vessel formation (Jakobsson et al., 2010; Bentley et al., 2014), but the consequences of perturbing these behaviors are poorly understood. Centrosomes orient and sustain migratory polarity via nucleation of microtubules (MTs); centrosome reorientation relative to the nucleus is believed to be required for proper migration (Doxsey et al., 2005; Cheng et al., 2008; Tang and Marshall, 2012). In culture, endothelial cells (ECs) establish a centrosome-forward orientation, with the centrosome in front of the nucleus relative to migration direction (Luxton and Gundersen, 2011). As blood vessels extend sprouts, individual ECs migrate both toward and away from the sprout tip (Perryn et al., 2008; Arima et al., 2011), and EC migration away from the tip is linked to proper formation of arteries during zebrafish fin regeneration (Xu et al., 2014). In epithelial Madin–Darby canine kidney cysts, centrosomes reorient as cells begin sprouting during tube formation (Gierke and Wittmann, 2012); however, how centrosomes are oriented in ECs during angiogenic sprouting is not known.
Tumor blood vessels have an abnormal morphology and are leaky, and the ECs of these vessels have a high frequency of excess centrosomes (more than two) (Hida et al., 2004). We previously reported that excess centrosomes alter directional migration and polarized vesicle trafficking because centrosomes do not cluster properly in two dimensions (Kushner et al., 2014). Here we show that, in topologically constrained ECs of three-dimensional (3D) angiogenic sprouts or micropatterns, excess centrosomes do not scatter. However, the clustered excess centrosomes do not behave normally, as they cannot efficiently repolarize, leading to reduced EC migration and sprouting. These ECs have abnormal microtubule dynamics and supraphysiological activation of Rac1. These findings reveal an unexpected role for dynamic reorientation of EC migratory polarity in blood vessel sprouting and a requirement for proper centrosome numbers, even in the clustered conformation, for this behavior.
RESULTS
Endothelial cells with excess centrosomes fail to repolarize in sprouts
We previously manipulated centrosome duplication in ECs using lentiviruses that knocked down CDC14 or overexpressed Plk4; however, we were unable to temporally regulate expression changes in this system. To temporally regulate centrosome overduplication in ECs, we used a tetracycline (tet)-inducible lentivirus for Polo-like kinase 4 (Plk4) overexpression in human umbilical vein ECs (HUVECs). Plk4 regulates centriole duplication without affecting the cell cycle (Habedanck et al., 2005). Exposure of tet-Plk4 ECs to doxycycline (DOX) for 24 h before analysis showed elevated Plk4 transcript expression by quantitative PCR (Supplemental Figure S1A) accompanied by increased frequencies of ECs with excess centrosomes compared with controls (36 vs. 6%; Supplemental Figure S1, B and C). Most effects of centrosome overduplication in ECs were analyzed within 24 h of DOX treatment to avoid complications from aneuploidy downstream of cell division, or the EC were placed on beads in a fibrin matrix, where their division rate is reduced. As for other cell types (Godinho et al., 2014), short-term Plk4 overexpression did not significantly affect the growth curve of ECs (Supplemental Figure S1D) or apoptosis, as assessed by activated caspase-3 expression (Supplemental Figure S1E).
To analyze effects of excess centrosomes on EC behaviors during sprouting angiogenesis, we used a 3D sprouting assay in which beads coated with HUVECs were embedded in a fibrin matrix (Nakatsu et al., 2007). Analysis of sprout morphology revealed that DOX-induced, Plk4-mediated overexpression significantly reduced the number of sprouts/bead and sprout length compared with controls (Figure 1, A and B, and Supplemental Figure S1F). Although ECs with excess centrosomes exhibited centrosome scattering in two dimensions (Kushner et al., 2014), ECs with excess centrosomes (averaging three to six centrosomes) had a single centrosome cluster in angiogenic sprouts (Figure 1C). Although cell divisions were rare in sprouts, the few divisions we saw in sprouting ECs with excess centrosomes did not have obvious multipolar spindles, consistent with findings of others that clustered excess centrosomes usually form bipolar spindles (Kwon et al., 2008). We found that ECs with more than seven centrosomes had scattered centrosomes in sprouts (Supplemental Figure S1G), but these ECs were rare (Supplemental Figure S1C) and were not used for further analysis except where noted. We next imaged centrosome dynamics during sprout morphogenesis via centrin::green fluorescent protein (GFP) expression. Control ECs frequently alternated between migrating toward the proximal and distal ends of the sprout, with repolarization of the nucleus–centrosome axis paralleling the direction of migration (Figure 1, D–F, and Supplemental Movie S1). In contrast, in ECs with excess centrosomes, the nucleus–centrosome axis was often perpendicular to the sprout axis, and these ECs had reduced motility (0.08 ± 0.01 μm/h) compared with controls (0.11 ± 0.01 μm/h; (Figure 1, D–F, and Supplemental Movie S2).
FIGURE 1:

Excess centrosomes prevent centrosome repolarization. (A) Top, schematic of DOX treatment for Plk4 overexpression in sprouting HUVECs. Bottom, images of d6 sprouts stained for actin (phalloidin; green) and DNA (DRAQ; pink) with indicated treatments. Scale bar, 100 μm. (B) Scatter plots of sprouting parameters. –DOX, 22 beads; Plk4 + DOX, 29 beads. (C) Representative images of centrosomes in sprouting HUVECs infected with centrin::GFP– expressing virus (green) and stained for DNA (blue; 4′,6-diamidino-2-phenylindole). 1-2C, one or two centrosomes; >2C, more than two centrosomes. Right, area of higher magnification. Scale bar, 10 μm. (D) Left, diagram of nucleus (N)–centrosome (C) axis. Right, representative time-lapse images of HUVECs in sprouts expressing centrin::GFP, with N-C axis superimposed. D, distal (toward bead); P, proximal (toward sprout tip). Green fiduciary mark denotes starting position of the nucleus. Scale bar, 20 μm. (E) Histogram of N-C angles between groups (1-2C: 227 time points, 10 ECs; >2C: 186 time points, 10 ECs). (F) Scatter plot of mean velocity of individual HUVECs in sprouts. 1-2C: 33 ECs; >2C: 21 ECs. (G) Centrosome orientation classifications in angiogenic sprouts. D, distal; F, forward; N, nuclear; P, proximal; R, reverse. (H) Scatter plot of mean velocity of individual HUVECs in sprouts. F/R, forward/reverse. 1-2C: F/R, 19 ECs, and N, 14 ECs; >2C: F/R, 9 ECs, and N, 12 ECs. (I) Frequency of successful centrosome repolarization events per cell in indicated groups. For all experiments, black bars indicate comparison groups with indicated p values. Error bars are 95% confidence intervals. All p values are from two-tailed Student’s t test from at least three experiments. *p ≤ 0.05; **p ≤ 0.01; NS, not significant.
To determine whether the reduced EC velocity in angiogenic sprouts was due to defects in centrosome repolarization, we binned centrosome position into three categories based on relationship to the nucleus: forward or reverse (relative to nucleus and migration direction) or nuclear (laterally positioned or centered over the nucleus; Figure 1G). Evaluation of migration velocities revealed that sprouting ECs with centrosomes in the nuclear zone (N), regardless of number, had reduced velocity compared with ECs with centrosomes in the forward or reverse position (F/R; Figure 1H). ECs in F/R positions with excess centrosomes were also slower than their counterparts with one or two centrosomes, suggesting that EC processes in addition to polarization are affected by excess centrosomes (Figure 1H). Consistent with the idea that excess centrosomes affect migration via effects on polarization, we observed that ECs with excess centrosomes in sprouts had significantly fewer centrosome repolarization events over time (41% for one or two centrosomes vs. 5% for more than two centrosomes over 26 h; Figure 1I, Supplemental Figure S1, H–J, and Supplemental Movie S3), suggesting that excess centrosomes hindered centrosome repolarization. Thus, although excess centrosomes cluster in sprouting ECs, our results demonstrate that centrosomes do not efficiently repolarize during sprouting; this behavior is associated with impaired migration in angiogenic sprouts and dysfunctional sprouting morphogenesis.
Cell-autonomous polarization defects in endothelial cells with excess centrosomes
We asked whether excess centrosome-mediated orientation defects in ECs are cell autonomous. Centrosome orientation was assessed on two-dimensional crossbow micropatterns that enable stereotyped cell polarization with reproducible centrosome polarization in solitary cells (Thery et al., 2006). Consistent with the findings in sprouts, ECs with excess centrosomes on micropatterns exhibited centrosome clustering (Figure 2A). Fixed image analysis showed that uninduced tet-Plk4 ECs with normal centrosome numbers were able to position their centrosomes in a forward orientation on micropatterns, with 47% in the forward position after 6.5 h, whereas ECs with excess centrosomes had a lower frequency of centrosome-forward orientation (13%; Figure 2B). Of note, ECs on the crossbow micropatterns do not divide during the reorientation period, eliminating the possibility of effects downstream of cell division. We next overexpressed a truncated Plk4 (Plk41-608) protein that has intact kinase activity but does not promote centrosome overamplification (Guderian et al., 2010). Overexpression of Plk41-608 did not affect centrosome number (Supplemental Figure S2A) or micropattern-induced centrosome orientation (Supplemental Figure S2B), suggesting that excess centrosomes rather than off-target Plk4 kinase activity are responsible for orientation defects. Live imaging confirmed that ECs with excess centrosomes had a 60% reduction in a forward orientation and “stalled” in the nuclear zone twice as often as controls (Figure 2C, Supplemental Figure S2C, and Supplemental Movies S4 and S5). These results indicate that ECs with excess centrosomes have cell-autonomous centrosome orientation defects, and these defects likely mirror the repolarization defect of ECs with excess centrosomes during angiogenic sprouting.
FIGURE 2:
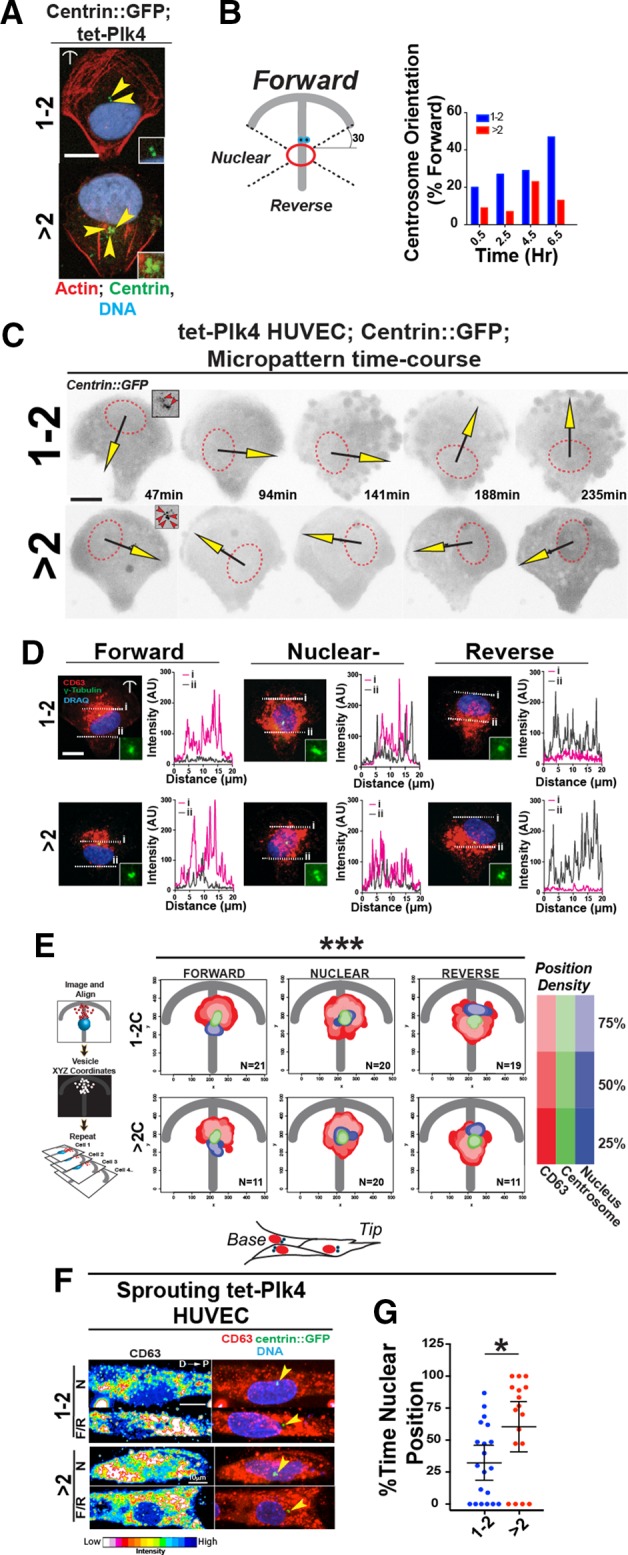
ECs with excess centrosomes have perturbed centrosome orientation and polarization. (A) Representative images of ECs with 1-2 (top) or >2 centrosomes (bottom) on crossbow micropatterns stained for actin (phalloidin; red), centrosomes (centrin::GFP; green), and DNA (DRAQ; blue). Yellow arrowheads, individual centrosomes. Scale bar, 10 μm. Insets, higher magnification of centrosomes. (B) Diagram (left) of centrosome orientation scoring. Right, percentage of ECs in forward position over time between ECs containing 1-2 or >2 centrosomes. (C) Time-lapse images of ECs expressing centrin::GFP with 1-2 or >2 centrosomes on micropatterns; black/yellow arrows, N-C angles. Dotted red line, nucleus outline. Insets, images of centrosomes. Scale bar, 10 μm. (D) Image and line scans of CD63 staining in ECs on micropatterns with indicated centrosome orientation and numbers. Dotted white lines, line scans. Scale bar, 10 μm. (E) Left, diagram of how probabilistic density maps were generated. Right, probabilistic density maps of CD63, centrosome, and nuclear staining grouped by centrosome position (F, N, or R) and centrosome number. Key is on the right. Statistical comparisons (see Materials and Methods) of F vs. N and R vs. N maps, ***p ≥ 0.001. (F) Images of tet-Plk4 HUVECs expressing centrin::GFP (green) in sprouts stained for CD63 (left, pseudocolored; right, red) and DNA (DRAQ; blue) and binned by centrosome orientation and centrosome number. Yellow arrowheads, centrosomes. Scale bar, 10 μm. (G) Quantification of percentage of time individual sprouting ECs with one or two or more than two centrosomes spent in N position. 1-2C: 20 ECs; >2C: 17 ECs. For all experiments, black bars indicate comparison with indicated p values. Error bars are 95% confidence intervals. All p values are from two-tailed Student’s t test from at least three experiments, unless indicated otherwise. *p ≤ 0.05; ***p ≤ 0.001; NS, not significant.
We previously showed that in unconstrained ECs, excess centrosomes scattered and caused disruption and fragmentation of the Golgi (Kushner et al., 2014). Here we determined the effects of excess centrosomes in topologically constrained ECs that more faithfully mimic the constraints on ECs found in sprouts. In contrast to unconstrained ECs, HUVECs with excess centrosomes on micropatterns did not exhibit obvious Golgi fragmentation in any of the orientations (Supplemental Figure S2D). We next determined the relative position of CD63, a vesicle-associated protein that is trafficked from the Golgi complex to the leading edge in polarized cells (Verweij et al., 2011) (Figure 2, D and E). Line scans flanking either side of the nucleus showed an expected accumulation of CD63 that paralleled centrosome orientation. In contrast, when centrosomes were oriented in the nuclear zone, regardless of centrosome number, CD63 intensity was equal on both sides of the nucleus (Figure 2D). Probability density mapping of CD63 vesicles in multiple ECs showed similar patterns, and distributions in centrosome-forward, nuclear, and reverse orientations were significantly different (Figure 2E). We stained for CD63 in angiogenic sprouts and confirmed localization toward the appropriate direction (forward or reverse) when centrosomes were polarized but lack of localization when centrosomes were in the nuclear region (Figure 2F). We next determined how much time ECs with excess centrosomes spent in the different orientations in 3D sprouts. Live imaging over 24–36 h showed that sprouting ECs with excess centrosomes spent significantly more time in the nuclear–centrosome orientation (70%) than did controls (30%; Figure 2G). Thus mispolarization as determined by CD63 vesicle distribution, a consequence of centrosome stalling in the nuclear region, is significantly more likely in ECs with excess centrosomes and occurs despite the fact that excess centrosomes remain clustered and do not cause obvious Golgi fragmentation. These findings suggest that, although excess centrosomes are clustered rather than scattered in sprouting ECs, they contribute to a loss of polarization that likely hinders angiogenic sprout formation.
Endothelial cells with excess centrosomes have aberrant microtubule dynamics
Because centrosomes function as a MT organizing center, we asked whether MT dynamics influenced dynamic centrosome repolarization on micropatterns. Low doses of either nocodazole or Taxol suppress MT dynamic instability (Vasquez et al., 1997) but do not completely depolymerize or overstabilize MTs, respectively (Figure 3A, top). Both drugs significantly inhibited the ability of control ECs to dynamically repolarize their centrosomes to a forward orientation (Figure 3A, bottom), indicating that centrosome orientation is sensitive to small perturbations in MT dynamics. In contrast, neither drug altered the centrosome orientation frequencies of ECs with excess centrosomes, suggesting that in these cells, centrosome orientation is randomized and cannot be further perturbed by altered MT dynamics.
FIGURE 3:

Excess centrosomes perturb microtubule regulation. (A) Top, images of tet-Plk4 HUVECs treated as indicated (100 nM concentration of drugs) and stained for MTs (α-tubulin). Bottom, left, diagram of centrosome orientation scoring; right, percentage of ECs with indicated orientation and treatments. 1-2C, one or two centrosomes; >2C, more than two centrosomes. Comparisons by c2 analysis; *p ≤ 0.05; NS, not significant. Scale bar, 10 μm. (B) Left, SIM images of ECs on micropatterns stained for MTs (α-tubulin; red), centrosomes (γ-tubulin; green), and DNA (DRAQ; blue). Yellow arrowheads, centrosomes; insets, higher magnification of centrosomes. Middle, inverted α-tubulin staining images. Right, magnified front cortex areas. Scale bars, 10 μm (left), 5 μm (right). (C) Whole-cell and front cortex MTs (α-tubulin intensity) between indicated groups. 1-2C: 46 ECs; >2C: 32 ECs. (D) Representative images of ECs on micropatterns treated with SiR-tubulin to visualize MT. (E) Indicated quantifications of MT parameters in ECs on micropatterns treated with SiR-tubulin. Each point was averaged from multiple MTs in a given cell. Shrink rate: 1-2C, 16 ECs; >2C, 18 ECs. Stall frequency: 1-2C, 17 ECs; >2C, 22 ECs. Growth rate: 1-2C, 16 ECs; >2C, 21 ECs. Catastrophes: 1-2C, 16 ECs; >2C, 19 ECs. (F) Left, SIM images of EC on micropatterns 15 s after cold washout stained for MTs (α-tubulin; red), centrosomes (γ-tubulin; green), and DNA (DRAQ; blue). Insets, higher magnification of centrosomes. Middle, inverted α-tubulin staining images. Right, higher magnification of MT-nucleating center. Scale bars, 10 μm (left), 5 μm (right). (G) Quantification of whole-cell MTs (α-tubulin intensity) between indicated groups; 22 ECs for each group. (H) Quantification of MTs (α-tubulin intensity) over time, normalized to one- or two-centrosome controls at the same time point. (I) Representative images of ECs with different numbers of centrosomes after rewarming to initiate MT growth and stained for MTs (α-tubulin; red), centrosomes (γ-tubulin; green), and DNA (DRAQ; blue). (J) Quantification of α-tubulin intensity by centrosome count; 49 ECs in two independent experiments. Error bars are SEM. For all experiments, black bars indicate comparison with indicated p values. Error bars are 95% confidence intervals unless indicated otherwise. All p values are from two-tailed Student’s t test from at least three experiments, unless indicated otherwise. *p ≤ 0.05; **p ≤ 0.01; NS, not significant.
To determine whether ECs with excess centrosomes have changes in steady-state MTs, we used structured illumination microscopy (SIM) to closely examine MT architecture at the cell cortex (within 7.5 μm of the leading edge). In control ECs, the leading edge had numerous MTs that contacted the front cortex, bending back into the cell interior in a stereotypical manner (Figure 3B, top right). In contrast, ECs with excess centrosomes had fewer MTs in this region, and these MTs were truncated and less organized (Figure 3B; bottom right). Quantification of whole-cell and cortex-restricted MT intensities showed reduced α-tubulin polymer levels in ECs with excess centrosomes (13.4% whole cell and 19.1% cortical reduction vs. control, respectively; Figure 3C). We next live-imaged MTs of EC on micropatterns using an MT-specific dye (Lukinavicius et al., 2014). ECs with excess centrosomes had normal shrink, stall, and growth rates of MTs compared with controls but they had significantly elevated MT catastrophes (Figure 3, D and E). These data show that topologically constrained ECs with excess centrosomes have reduced polymerized MTs at steady state, and this reduction is associated with increased MT catastrophes.
To test whether defective centrosome-derived MT nucleation contributed the reduced MTs, we examined MT nucleation capacity after cold depolymerization and washout. MT nucleation from centrosome clusters was significantly enhanced (16.5% higher α-tubulin intensity at 15 s after renucleation) in ECs with excess centrosomes compared with controls (Figure 3, F and G). Increased MT nucleation with excess centrosomes has been reported in other cell types (Lingle et al., 1998; Godinho et al., 2014) but was inconsistent with our steady-state MT results. To resolve this paradox, we assessed MT intensities at different time points after recovery from MT depolymerization. ECs with excess centrosomes had elevated polymerized tubulin compared with controls up to 2 h after rewarming. However, at 8 h, polymerized tubulin intensities in ECs with excess centrosomes dropped below control levels (Figure 3H). Our finding that MT density diminishes over time in ECs with excess centrosomes is unexpected and indicative of a possible secondary pathway involved in MT defects.
To determine whether centrosome number affected MT renucleation capacity, we quantified MT nucleation capacity after cold depolymerization and washout relative to centrosome number (Figure 3, I and J). Of interest, there was a stepwise increase (20–30% per centrosome) in centrosome MT nucleation capacity up to four centrosomes. Thereafter, increasing centrosome number did not result in increased MT nucleations. These results indicate that MT nucleation depends on centrosome number when clustered (i.e., topologically constrained) but that this response has an upper limit.
Abnormal microtubule dynamics is associated with elevated Rac1 activation and reorientation defects in endothelial cells
Initially, there were more MTs after renucleation in ECs, but subsequently MTs were reduced in ECs with excess centrosomes. MT nucleations stimulate Rac1 activation and actin remodeling (Waterman-Storer et al., 1999). Thus we hypothesized that excess centrosomes initially generate a robust MT array from increased nucleations that stimulates elevated and sustained Rac1 activity, which in turn drives the increased actin remodeling at the cell periphery that subsequently suppresses MT density downstream of MT nucleations. To determine Rac1 activity in tet-Plk4 HUVECs with excess centrosomes on micropatterns, we used a Rac1 fluorescence resonance energy transfer (FRET) biosensor (Kraynov et al., 2000). FRET/cyan fluorescent protein (CFP) ratios revealed that ECs with excess centrosomes had 50% more Rac1 activity than controls (Figure 4, A and B). Nocodazole-induced MT depolymerization, which also prevents MT nucleation, significantly reduced Rac1 activation and abolished the differences (Figure 4B), suggesting that the increased Rac activity with excess centrosomes depends on MT nucleations. We next overexpressed either constitutively active (CA; Q61L mutation) or dominant-negative (DN; T17N mutation) enhanced GFP (eGFP)::Rac1 (Kraynov et al., 2000) and assessed centrosome-forward orientation. Expression of CA eGFP::Rac1 did not affect centrosome orientation in ECs with excess centrosomes, whereas expression of DN eGFP::Rac1 partially rescued forward-centrosome orientation (Figure 4C). Next we pharmacologically blocked Rac1 or one of its downstream effectors that affects actomyosin remodeling, Arp2/3. Strikingly, blockade of either Rac1 or Arp2/3 increased forward centrosome orientation on micropatterns in ECs with excess centrosomes (Figure 4D). Previous studies showed that Rac and/or Arp2/3 inhibition increased cortical MTs (Wittmann et al., 2003). The leading edge of ECs with excess centrosomes and Rac1 or Arp2/3 inhibition had increased cortical MT staining, and both treatments increased polymerized tubulin levels relative to untreated ECs with excess centrosomes (Figure 4, E and F). These results suggest that repolarization in ECs with excess centrosomes can be rescued by reducing Rac and Arp2/3 activity.
FIGURE 4:

ECs with excess centrosomes have ectopic Rac1 activation and centrosome orientation defects. (A) Images of EC with one or two (1-2C) or more than two (>2C) centrosomes expressing the Rac1 biosensor and pseudocolored to indicate Rac1 activity. Scale bar, 10 μm. (B) Scatter plot of average FRET/CFP ratios between EC with 1-2 or >2 centrosomes and with or without nocodazole treatment (NOC). –NOC: 1-2C, 60 ECs; >2C, 58 ECs. +NOC: 1-2C, 19 ECs; >2C, 19 ECs. (C) Percentage of forward-oriented ECs with indicated treatments between ECs with 1-2 or >2 centrosomes, normalized to controls. CA, constitutively active; DN, dominant negative. Comparisons are to 1-2C group by c2 analysis. (D) Percentage of forward-oriented ECs with indicated treatments between ECs with one or two or more than two centrosomes, normalized to controls. Rac inhibitor (NSC23766); Arp 2/3 inhibitor (Ck-666). Comparisons are to 1-2C group compared by c2 analysis. (E) Images of front cortex of HUVECs on micropatterns stained for α-tubulin; treatments as indicated. Scale bar, 10 μm. Right, higher magnification of leading edge. (F) Quantification of whole-cell and cortex MTs (α-tubulin intensity) with indicated treatments, normalized to 1-2C EC controls. >2C, 20 ECs; >2C + Rac Inh, 20 ECs; >2C + Arp 2/3 Inh, 18 ECs. Error bars, 95% confidence intervals. (G) Time-lapse images of a representative EC on micropattern expressing mCherry-PA-Rac1 without 488-nm light exposure (dark; left) and subsequent to light exposure (light; right). Red boxes, higher-magnification areas to left. Time is indicated in seconds. (H) Time-lapse images of a representative EC on a micropattern expressing Venus-PA-Rac1 (C450A) light-insensitive mutant in conditions similar to those in G. Red boxes, higher-magnification areas to left. Time is indicated in seconds. (I) Time-lapse images of EC in sprouts expressing centrin::tdTomato to mark centrosomes, GFP::Rac1 (DN), and N-C axis superimposed. Scale bar, 10 μm. (J) Percentage of sprouting ECs that repolarize with expression of DN Rac::GFP. Control: 1-2C, 22 ECs; >2C, 17 ECs. DNRac: 1-2C, 10 ECs; >2C, 12 ECs. All p values are from two-tailed Student’s t test from at least three experiments, unless indicated otherwise. *p < 0.05; **p ≤ 0.01; ***p ≤ 0.001; NS, not significant; N, number of ECs analyzed.
We reasoned that if Rac1 blockade rescued MT levels in ECs with excess centrosomes, then increasing Rac1 in normal ECs would reduce cortical MTs, similar to the effects of excess centrosomes. To achieve this, we used a blue-light photoactivatable (PA)-Rac1 to conditionally activate Rac1 in EC with a normal centrosome complement (Kraynov et al., 2000). ECs on micropatterns maintained in the dark state had little membrane ruffling (unpublished data), and MTs had a typical conformation at the cortex, smoothly bending backward toward the cell interior (Figure 4G, left). On light exposure to activate Rac1, membrane ruffling rapidly increased (unpublished data), and MTs at the front cortex gradually retreated away from the cortex, decreasing overall MT density (Figure 4G, right). In contrast, light exposure in ECs expressing the light-insensitive mutant (PA-Rac1 C450A) did not elicit obvious effects on cortical MT density (Figure 4H). These results are consistent with the biosensor and inhibitor experiments and suggest that Rac1 activation downstream of excess centrosomes affects cortical MT architecture.
To investigate further the effects of reducing Rac1 activity on EC repolarization in angiogenic sprouts, we overexpressed DN Rac1 in sprouting ECs. Strikingly, expression of DN Rac1 rescued the repolarization defects seen in sprouting EC with excess centrosomes to levels indistinguishable from those for unmanipulated ECs with normal centrosome numbers (Figure 4, I and J). Collectively these results show that ectopic Rac1 activation promotes defects in reorientation of topologically constrained EC on micropatterns and in sprouts, likely downstream of robust MT nucleation by excess centrosomes.
We next sought to determine whether elevated centrosome-derived MT nucleations, independent of centrosome overduplication, mimicked the effects of excess centrosomes on the MT cytoskeleton and cell polarization. We used Plk1, a well-characterized positive regulator of centrosome-derived MT nucleation that we previously reported increased γ-tubulin levels and MT nucleation in ECs (Kushner et al., 2014). As predicted, introduction of tet-inducible Plk1 (tet-Plk1 DBN; Lindon and Pines, 2004) into ECs increased polymerized tubulin levels during recovery from cold depolymerization (Figure 5, A and B). Of note, Plk1 overexpression also resulted in significantly elevated MT-dependent Rac1 activity, as assessed by the Rac1 biosensor, and impeded micropattern-induced centrosome-forward orientation, whereas blockade of Rac1 activity rescued centrosome orientation in Plk1-overexpressing ECs (Figure 5, C–E). These results suggest that increased centrosome-derived MT nucleation leads to deregulated Rac1 activation and reduced ability of centrosomes to dynamically repolarize.
FIGURE 5.

Plk1 overexpression phenocopies excess centrosome defects. (A) Images of ECs expressing stabilized tet-inducible Plk1 without or with DOX treatment and stained for MTs (α-tubulin). Top, 60 s after cold washout, showing renucleated centrosome-derived MTs; bottom, steady-state MTs. Arrows denote differences in MT density between groups. (B) Scatter plot comparing α-tubulin intensity between control ECs (–DOX) and ECs expressing Plk1 (+DOX) 60 s after cold washout (two experiments). Scale bar, 10 μm. (C) Representative pseudocolored (to indicate Rac activity) images of ECs expressing DOX-inducible Plk1 and Rac1 biosensor. Scale bar, 10 μm. (D) Plot of FRET/CFP ratio between controls (–DOX) and ECs expressing Plk1 (+DOX) with or without nocodazole treatment (NOC). –DOX, 56 ECs; +DOX, 55 ECs. (E) Percentage of forward-oriented ECs with indicated treatments. Rac inhibitor (NSC23766). Comparisons are to –DOX control by c2 . N, number of observations. For all experiments, black bars indicate comparison with indicated p values. Error bars are 95% confidence intervals. All p values are from two-tailed Student’s t test from three experiments, unless indicated otherwise. *p ≤ 0.05; **p ≤ 0.01; NS, not significant.
Rac1-mediated actomyosin contractility leads to MT bending, buckling, and breakage. Thus we blocked myosin light chain kinase activity, which is a major regulator of actomyosin contractility, in induced tet-Plk4 ECs. This inhibition rescued micropattern-induced forward-centrosome orientation in ECs with excess centrosomes (Supplemental Figure S3, A and B). These results suggest that aberrant remodeling of the actin cytoskeleton downstream of Rac1 activation contributes to reduced MT levels and centrosome orientation defects in ECs with excess centrosomes.
DISCUSSION
Although excess centrosomes are implicated in generating chromosome instability and aneuploidy, only recently have excess centrosomes been implicated in interphase cell phenotypes linked to cancer progression (Godinho et al., 2014; Kushner et al., 2014). We sought a better understanding of the effects of excess centrosomes during sprouting angiogenesis, given that tumor ECs have high frequencies of centrosome overamplification. Here we show that excess centrosomes block the dynamic repolarization of ECs, which is part of normal sprout extension, and this defect is recapitulated on cell polarization–inducing micropatterns. Effects of excess centrosomes occur via effects on MT nucleations and dynamics, Rac1 activation, and remodeling of the actin cytoskeleton and, given the timing, likely do not involve aneuploidy.
Our previous work showed that, in ECs with excess centrosomes that were not topologically constrained, the centrosomes were more dynamic and scattered, and as a result, ECs had aberrant migration and vesicle trafficking in two dimensions (Kushner et al., 2014). Here we extended this analysis to ECs that are topologically constrained, either in angiogenic sprouts or by placement onto micropatterns that confer a leading/trailing-edge polarization. To our surprise, under these more physiological conditions, the excess centrosomes remained clustered, but nevertheless the ECs exhibited defects in repolarization. Normal ECs dynamically repolarize and migrate both proximally and distally in extending sprouts (Xu et al., 2014; Sugihara et al., 2015; this study); however, ECs with excess centrosomes did not repolarize efficiently, but instead remained “stalled” with the centrosomes more central over the nucleus, a configuration that was recapitulated by ECs with excess centrosomes on micropatterns. The repolarization defect correlates with loss of polarity—for example, that seen in localization of the vesicle protein CD63. The reduced spouting seen when sprouts contain significant numbers of ECs with excess centrosomes implies that dynamic repolarization is an essential component of vascular sprouting. This extends recent studies showing that ECs dynamically compete during sprout extension, with ECs at the tip exchanging with ECs in the stalk (Jakobsson et al., 2010; Bentley et al., 2014).
We found that repolarization defects downstream of excess centrosomes affected the MT cytoskeleton in paradoxical ways. ECs with clustered excess centrosomes had more MT nucleations, consistent with other reports (Lingle et al., 1998; Godinho et al., 2014), but more catastrophes and fewer MTs at steady state. The increased MT nucleations correlated with higher Rac1 activity, and conditionally activated Rac1 reduced cortical MTs, whereas Rac inhibition rescued the repolarization defects of ECs. Because manipulations of actomyosin contractility also rescued repolarization, we hypothesize that MT nucleation–induced elevated Rac activity promotes increased actomyosin remodeling near the cortex, which leads to more MT catastrophes, or breakage, and this process eventually reduces MT levels and affects the ability of ECs to repolarize. In this model, Rac activation is sustained because elevated MT nucleations at the centrosome and from the broken MT ends promote elevated Rac1 activity. Our finding that a delicate balance of MT dynamics and actomyosin contractility is perhaps required for EC repolarization is in line with reports linking centrosome positioning and peripheral myosin (Burakov et al., 2003; Gomes et al., 2005; Rodriguez-Fraticelli et al., 2012). We show, however, that excess centrosomes might be capable of tipping this important balance. In sprouting ECs, clusters of excess centrosomes become “trapped” in a nonpolarized state, and these cells have reduced migration and sprouting defects. Thus vessels with high frequencies of excess centrosomes, such as those in tumors, are predicted to have reduced migration within the tumor and aberrant network formation, limiting chemotherapeutic drug penetration.
MATERIALS AND METHODS
Cell culture
HUVECs (CC-2517; Lonza, Houston, TX) and human lung fibroblasts (CC-2512; Lonza) were maintained at 37°C with 5% CO2 and grown under standard conditions. Rac1-GTPase inhibitor (NCS-23766; SelleckChem, Boston, MA) was at 100 μM, and Arp2/3 inhibitor (CK-666; Sigma-Aldrich, St. Louis, MO) was at 10 μM. Nocodazole (M1404; Sigma-Aldrich) and Taxol (T7402; Sigma-Aldrich) were used at indicated concentrations. Stable HUVEC populations were generated by inserting relevant transgenes into pLIX_402 (41394; Addgene, Cambridge, MA), a Gateway-compatible Tet-On lentivirus. P2 HUVECs were transfected with the pLIX virus, and puromycin at 2 μg/ml (P9620; Sigma-Aldrich) was added 4 d later. After 2 d, puromycin was added at 4 μg/ml. When HUVECs reached confluency, they were split 1:10 and frozen in liquid N2. Transgene expression was induced by overnight incubation with DOX (D9891; Sigma-Aldrich). For all experiments except the sprouting angiogenesis assay (described later), cells were assessed within 24 h of DOX treatment, and ECs with nuclear sizes <50% of median or ≥200% of median size were excluded from analysis.
Three-dimensional sprouting assay
The sprouting angiogenesis assay was performed as described (Nakatsu and Hughes, 2008; Kushner et al., 2014). DOX was added at 500 ng/ml at indicated times. Live-cell imaging was performed on an Olympus FV1200 live-imaging confocal system with a 40× silicone-immersion objective. HUVECs were infected with centrin::GFP lentivirus the day before embedding to label centrioles. Nuclei were identified using phase contrast microscopy. A cell was considered to have “repolarized” if its centrosomes passed through the nuclear position from a forward position to a reverse position or vice versa. Nuclear velocity was measured with the ImageJ plug-in MtrackJ (National Institutes of Health, Bethesda, MD). Phalloidin labeled with a fluorescent marker was added at 1:100 postfixation to reveal sprout morphology.
Micropatterns and centrosome orientation
Micropatterns were fabricated as described (Thery et al., 2006; Thery and Piel, 2009) or purchased directly from Cytoo (Bethesda, MD; 10-005-00-18). Briefly, a silicon master with microfeatures of interest was used to cast polydimethylsiloxane (PDMS) stamps. The PDMS stamps were coated with fibronectin (F1141; Sigma-Aldrich) or fluorescent fibrinogen (F13192; Invitrogen, Waltham, MA) and used to print the polystyrene-coated glass coverslips with a patterned adhesive surface. Coverslips were then immersed in poly-l-lysine–polyethylene glycol. Micropattern production was at the Chapel Hill Analytical and Nanofabrication Laboratory at the University of North Carolina. The stamp patterns were designed using computer-aided design (CAD) software, and the optical mask was produced by Photo Sciences. ECs were treated, sparsely plated on micropatterns, and then incubated for indicated times before paraformaldehyde (PFA) fixation and scoring as indicated in the text.
Immunohistochemistry
The antibodies used were as follows: γ-tubulin (1:5000, T6557; Sigma-Aldrich), GM130 (1:1000, G7295; Sigma-Aldrich), α-tubulin-555 (1:250, 05-829X-555; Millipore, Darmstadt, Germany), α-tubulin-488 (1:250, 16-232; Millipore), CD63 (1:500, ab8219; Abcam), pericentrin (1:1000, ab4448; Abcam), phalloidin (1:100, A34055; Invitrogen), and DRAQ7 (1:1000, ab109202; Abcam). For γ-tubulin staining, ECs were fixed in ice-cold MeOH for 10 min. For CD63 and pericentrin staining, ECs were fixed in 4% PFA for 6 min. For phalloidin staining, ECs were first rinsed in 37°C 1× PEM (100 mM 1,4-piperazinediethanesulfonic acid [PIPES]-KOH, 1 mM MgCl2, 1 mM ethylene glycol tetraacetic acid [EGTA], pH 6.9) and then fixed in 4% PFA in 1× PEM for 10 min. For α-tubulin staining, ECs were dipped in PHEM/Triton-X (60 mM PIPES, 21 mM 4-(2-hydroxyethyl)-1-piperazineethanesulfonic acid, 10 mM EGTA, 685 mM NaCl, 2 mM MgCl2, pH 6.9, 0.2% Triton-X) at 37°C for 15 s before fixation in MeOH for 10 min. Slides were washed in phosphate-buffered saline (PBS) with 0.01% Tween 20 (PBST) three times and blocked in 10% newborn calf serum diluted in PBST (blocking buffer) for 2 h. After blocking, primary antibodies were added at the indicated concentrations in blocking buffer and incubated overnight at 4°C. After incubation, slides were extensively washed three times in blocking buffer at room temperature and then incubated with secondary antibody in blocking buffer for 2 h. Secondary antibodies were Alexa Fluor conjugates (1:250; Invitrogen). Slides were washed in PBST at room temperature and mounted in Vectashield mounting medium (Vector Labs) before imaging.
Cold MT renucleation
ECs were placed on ice for 20 min, and then growth medium at 37°C was added for the indicated times. For experiments requiring PHEM/Triton-X treatment, the PHEM was at 37°C and added after growth medium. ECs were then immediately fixed in cold MeOH.
MT analysis
HUVECs expressing transgenes of interest via lentivirus infection were seeded onto custom-made micropatterns. Coverslips were dipped in PHEM/Triton-X for 15 s and then fixed in cold MeOH for 10 min. High-resolution images were acquired using a 60× objective on either an Olympus FV1200 confocal microscope or a DeltaVision OMX superresolution microscope (Applied Precision, Marlborough, MA). Whole-cell tubulin polymer was measured using the Measure function in ImageJ. To determine tubulin polymer in the cell periphery, a custom macro was written that drew an oval with the same curvature as the “bow” section of the crossbow micropattern. The oval was aligned with the edge of the cell and then displaced ∼7.5 um toward the “tail” of the pattern. The body of the oval was then filled with pixels of zero fluorescence intensity, so that only peripheral MT fluorescent signal was measured. Fluorescence measurements were corrected for background. In some cases, deconvolution using Huygens software was used.
For analyzing live MTs, HUVECs were treated for 18 h (150 nM final concentration) with SiR-tubulin, a fluorogenic compound for live-cell imaging (kind gift of Kai Johnsson, Institute of Chemical Sciences and Engineering, École Polytechnique Fédérale de Lausanne, Lausanne, Switzerland) as previously described and then plated on micropatterns and imaged. MTs were manually tracked using the ImageJ plug-in MTrackJ. The x- and y-coordinates were placed in a custom algorithm coded in Visual Basic using Excel (a gift of Dan Buster, University of Arizona Cancer Center, University of Arizona, Tucson). Movies of MTs were taken at 6-s intervals using an Olympus FV1200 confocal microscope at 37°C and an Olympus 60×/numerical aperture (NA) 1.4 objective.
Rac1 activation assay
Tet-Plk4–expressing HUVECs were electroporated with a dual-chain Rac1 FRET biosensor (gift of Klaus Hahn, University of North Carolina at Chapel Hill; Kraynov et al., 2000) and centrin::tdTomato, using an Amaxa Nucleofector Kit (VPB-1492; Lonza) and then plated on fibronectin-coated (5 μg/ml) glass-bottom dishes. ECs were fixed (4% PFA) after overnight incubation with DOX to induce centrosome overamplification. For FRET imaging, a 453-nm laser was used for excitation, and both CFP (470/24 nm) and FRET/yellow fluorescent protein (535/40 nm) emission channels were collected with an Olympus 60×/NA 1.4 objective on an Olympus FV1200 confocal microscope. All images were acquired using the same parameters, optimized to reduce photobleaching. FRET images were processed as described previously (Kardash et al., 2011). Briefly, the acquired CFP/FRET ratios were corrected for background in ImageJ. Single-cell images were segmented in each channel in ImageJ using the minimum and mean threshold method, and regions of interest were analyzed. FRET ratio images were produced by dividing the processed FRET image by the CFP image. Individual cell mean FRET/CFP values corresponding to Rac1 activation were obtained by calculating the mean pixel value of the FRET ratio image for each single cell.
Photoactivation of Rac1
Photoactivatable Rac1 (mCherry-PA-Rac1) and light-insensitive mutants were used. Constructs were subcloned into lentiviral expression vectors (pLenti705, 17392; Addgene) and virus produced in the University of North Carolina Virus Core Facility. HUVECs were infected using standard techniques and plated onto micropatterns protected from light before image acquisition. Cells were first imaged using red fluorescent protein and far-red filter sets (568- and 633-nm lasers, respectively) to detect membrane dynamics and SiR-tubulin–labeled MTs. To activate Rac1, a 488-nm laser was used as previously described (Wu et al., 2009). Imaging was carried out on an Olympus FV1200 confocal microscope at 37°C, using an Olympus 60×/NA 1.4 objective.
Probabilistic density mapping
Density maps were computed as described previously (Schauer et al., 2010). Briefly, HUVECs expressing pLIX-PLK4 in the presence of DOX were seeded on either in-house or commercially available micropatterns (Cytoo) and allowed to polarize for 4 h. ECs were then stained for CD63 and imaged. Images were rotated and cropped around the micropattern. The centroids of the vesicle puncta or organelle centroids in each Z-slice were determined using the Analyze Particles function in ImageJ. Density maps were created using the ks library in R (http://cran.r-project.org), a free software package for statistical computing. Statistical analysis was carried out as reported by Schauer et al. (2010). Briefly, we took random points from each distribution and determined the probability that points from each group belong to the same distribution. In theory, lower probability indicates that the compared groups are less likely from the same distribution. The p values were determined using the kde.test function from the ks package.
Supplementary Material
Acknowledgments
We thank Joshua Boucher, Stephanie Gupton, Amy Maddox, Kevin Mouillesseaux, Steve Rogers, and Eleni Tzima for insightful comments on the manuscript. We thank Marie Rougie and Klaus Hahn for help with FRET and Bob Geil of the Chapel Hill Analytical and Nanofabrication Laboratory for help with micropattern fabrication. We are grateful to Applied Precision (GE) for the loan of their DeltaVision OMX microscope. This research was supported by grants from the National Institutes of Health to V.L.B. (R01 HL116719 and HL43174) and National Institutes of Health National Research Service Award Postdoctoral Fellowship F32 HL113296, American Heart Association Scientist Development Grant 5SDG24660001, and National Institutes of Health Grant 1K99HL124311-01A1 to E.J.K.
Abbreviations used:
- 1-2C
one or two centrosomes per cell
- >2C
greater than two centrosomes per cell
- 2D
two dimensional
- 3D
three dimensional
- CFP
cyan fluorescent protein
- EC
endothelial cells
- F/R
forward/reverse
- FRET
Forster resonance energy transfer
- GFP
green fluorescent protein
- HUVEC
human umbilical vein endothelial cells
- MT
microtubule
- N
nuclear zone
- Plk1
Polo-like kinase 1
- Plk4
Polo-like kinase 4
Footnotes
This article was published online ahead of print in MBoC in Press (http://www.molbiolcell.org/cgi/doi/10.1091/mbc.E15-09-0645) on April 20, 2016.
REFERENCES
- Arima S, Nishiyama K, Ko T, Arima Y, Hakozaki Y, Sugihara K, Koseki H, Uchijima Y, Kurihara Y, Kurihara H. Angiogenic morphogenesis driven by dynamic and heterogeneous collective endothelial cell movement. Development. 2011;138:4763–4776. doi: 10.1242/dev.068023. [DOI] [PubMed] [Google Scholar]
- Bentley K, Franco CA, Philippides A, Blanco R, Dierkes M, Gebala V, Stanchi F, Jones M, Aspalter IM, Cagna G, et al. The role of differential VE-cadherin dynamics in cell rearrangement during angiogenesis. Nat Cell Biol. 2014;16:309–321. doi: 10.1038/ncb2926. [DOI] [PubMed] [Google Scholar]
- Burakov A, Nadezhdina E, Slepchenko B, Rodionov V. Centrosome positioning in interphase cells. J Cell Biol. 2003;162:963–969. doi: 10.1083/jcb.200305082. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Carmeliet P, Jain RK. Principles and mechanisms of vessel normalization for cancer and other angiogenic diseases. Nat Rev Drug Discov. 2011;10:417–427. doi: 10.1038/nrd3455. [DOI] [PubMed] [Google Scholar]
- Cheng J, Turkel N, Hemati N, Fuller MT, Hunt AJ, Yamashita YM. Centrosome misorientation reduces stem cell division during ageing. Nature. 2008;456:599–604. doi: 10.1038/nature07386. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Doxsey S, McCollum D, Theurkauf W. Centrosomes in cellular regulation. Annu Rev Cell Dev Biol. 2005;21:411–434. doi: 10.1146/annurev.cellbio.21.122303.120418. [DOI] [PubMed] [Google Scholar]
- Gierke S, Wittmann T. EB1-recruited microtubule +TIP complexes coordinate protrusion dynamics during 3D epithelial remodeling. Curr Biol. 2012;22:753–762. doi: 10.1016/j.cub.2012.02.069. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Godinho SA, Picone R, Burute M, Dagher R, Su Y, Leung CT, Polyak K, Brugge JS, Thery M, Pellman D. Oncogene-like induction of cellular invasion from centrosome amplification. Nature. 2014;510:167–171. doi: 10.1038/nature13277. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gomes ER, Jani S, Gundersen GG. Nuclear movement regulated by Cdc42, MRCK, myosin, and actin flow establishes MTOC polarization in migrating cells. Cell. 2005;121:451–463. doi: 10.1016/j.cell.2005.02.022. [DOI] [PubMed] [Google Scholar]
- Guderian G, Westendorf J, Uldschmid A, Nigg EA. Plk4 trans-autophosphorylation regulates centriole number by controlling betaTrCP-mediated degradation. J Cell Sci. 2010;123:2163–2169. doi: 10.1242/jcs.068502. [DOI] [PubMed] [Google Scholar]
- Habedanck R, Stierhof YD, Wilkinson CJ, Nigg EA. The Polo kinase Plk4 functions in centriole duplication. Nat Cell Biol. 2005;7:1140–1146. doi: 10.1038/ncb1320. [DOI] [PubMed] [Google Scholar]
- Hida K, Hida Y, Amin DN, Flint AF, Panigrahy D, Morton CC, Klagsbrun M. Tumor-associated endothelial cells with cytogenetic abnormalities. Cancer Res. 2004;64:8249–8255. doi: 10.1158/0008-5472.CAN-04-1567. [DOI] [PubMed] [Google Scholar]
- Jakobsson L, Franco CA, Bentley K, Collins RT, Ponsioen B, Aspalter IM, Rosewell I, Busse M, Thurston G, Medvinsky A, et al. Endothelial cells dynamically compete for the tip cell position during angiogenic sprouting. Nat Cell Biol. 2010;12:943–953. doi: 10.1038/ncb2103. [DOI] [PubMed] [Google Scholar]
- Kardash E, Bandemer J, Raz E. Imaging protein activity in live embryos using fluorescence resonance energy transfer biosensors. Nat Protoc. 2011;6:1835–1846. doi: 10.1038/nprot.2011.395. [DOI] [PubMed] [Google Scholar]
- Kraynov VS, Chamberlain C, Bokoch GM, Schwartz MA, Slabaugh S, Hahn KM. Localized Rac activation dynamics visualized in living cells. Science. 2000;290:333–337. doi: 10.1126/science.290.5490.333. [DOI] [PubMed] [Google Scholar]
- Kushner EJ, Ferro LS, Liu JY, Durrant JR, Rogers SL, Dudley AC, Bautch VL. Excess centrosomes disrupt endothelial cell migration via centrosome scattering. J Cell Biol. 2014;206:257–272. doi: 10.1083/jcb.201311013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kwon M, Godinho SA, Chandhok NS, Ganem NJ, Azioune A, Thery M, Pellman D. Mechanisms to suppress multipolar divisions in cancer cells with extra centrosomes. Genes Dev. 2008;22:2189–2203. doi: 10.1101/gad.1700908. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lindon C, Pines J. Ordered proteolysis in anaphase inactivates Plk1 to contribute to proper mitotic exit in human cells. J Cell Biol. 2004;164:233–241. doi: 10.1083/jcb.200309035. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lingle WL, Lutz WH, Ingle JN, Maihle NJ, Salisbury JL. Centrosome hypertrophy in human breast tumors: implications for genomic stability and cell polarity. Proc Natl Acad Sci USA. 1998;95:2950–2955. doi: 10.1073/pnas.95.6.2950. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lukinavicius G, Reymond L, D’Este E, Masharina A, Gottfert F, Ta H, Guther A, Fournier M, Rizzo S, Waldmann H, et al. Fluorogenic probes for live-cell imaging of the cytoskeleton. Nat Methods. 2014;11:731–733. doi: 10.1038/nmeth.2972. [DOI] [PubMed] [Google Scholar]
- Luxton GW, Gundersen GG. Orientation and function of the nuclear-centrosomal axis during cell migration. Curr Opin Cell Biol. 2011;23:579–588. doi: 10.1016/j.ceb.2011.08.001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nakatsu MN, Davis J, Hughes CC. Optimized fibrin gel bead assay for the study of angiogenesis. J Vis Exp. 2007:186. doi: 10.3791/186. doi: 10.3791/186. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nakatsu MN, Hughes CC. An optimized three-dimensional in vitro model for the analysis of angiogenesis. Methods Enzymol. 2008;443:65–82. doi: 10.1016/S0076-6879(08)02004-1. [DOI] [PubMed] [Google Scholar]
- Perryn ED, Czirok A, Little CD. Vascular sprout formation entails tissue deformations and VE-cadherin-dependent cell-autonomous motility. Dev Biol. 2008;313:545–555. doi: 10.1016/j.ydbio.2007.10.036. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rodriguez-Fraticelli AE, Auzan M, Alonso MA, Bornens M, Martin-Belmonte F. Cell confinement controls centrosome positioning and lumen initiation during epithelial morphogenesis. J Cell Biol. 2012;198:1011–1023. doi: 10.1083/jcb.201203075. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schauer K, Duong T, Bleakley K, Bardin S, Bornens M, Goud B. Probabilistic density maps to study global endomembrane organization. Nat Methods. 2010;7:560–566. doi: 10.1038/nmeth.1462. [DOI] [PubMed] [Google Scholar]
- Sugihara K, Nishiyama K, Fukuhara S, Uemura A, Arima S, Kobayashi R, Kohn-Luque A, Mochizuki N, Suda T, Ogawa H, et al. Autonomy and non-autonomy of angiogenic cell movements revealed by experiment-driven mathematical modeling. Cell Rep. 2015;13:1814–1827. doi: 10.1016/j.celrep.2015.10.051. [DOI] [PubMed] [Google Scholar]
- Tang N, Marshall WF. Centrosome positioning in vertebrate development. J Cell Sci. 2012;125:4951–4961. doi: 10.1242/jcs.038083. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Thery M, Piel M. Adhesive micropatterns for cells: a microcontact printing protocol. Cold Spring Harb Protoc. 2009;2009 doi: 10.1101/pdb.prot5255. pdb.prot5255. [DOI] [PubMed] [Google Scholar]
- Thery M, Racine V, Piel M, Pepin A, Dimitrov A, Chen Y, Sibarita JB, Bornens M. Anisotropy of cell adhesive microenvironment governs cell internal organization and orientation of polarity. Proc Natl Acad Sci USA. 2006;103:19771–19776. doi: 10.1073/pnas.0609267103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Vasquez RJ, Howell B, Yvon AM, Wadsworth P, Cassimeris L. Nanomolar concentrations of nocodazole alter microtubule dynamic instability in vivo and in vitro. Mol Biol Cell. 1997;8:973–985. doi: 10.1091/mbc.8.6.973. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Verweij FJ, van Eijndhoven MA, Hopmans ES, Vendrig T, Wurdinger T, Cahir-McFarland E, Kieff E, Geerts D, van der Kant R, Neefjes J, et al. LMP1 association with CD63 in endosomes and secretion via exosomes limits constitutive NF-kappaB activation. EMBO J. 2011;30:2115–2129. doi: 10.1038/emboj.2011.123. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Waterman-Storer CM, Worthylake RA, Liu BP, Burridge K, Salmon ED. Microtubule growth activates Rac1 to promote lamellipodial protrusion in fibroblasts. Nat Cell Biol. 1999;1:45–50. doi: 10.1038/9018. [DOI] [PubMed] [Google Scholar]
- Wittmann T, Bokoch GM, Waterman-Storer CM. Regulation of leading edge microtubule and actin dynamics downstream of Rac1. J Cell Biol. 2003;161:845–851. doi: 10.1083/jcb.200303082. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wu YI, Frey D, Lungu OI, Jaehrig A, Schlichting I, Kuhlman B, Hahn KM. A genetically encoded photoactivatable Rac controls the motility of living cells. Nature. 2009;461:104–108. doi: 10.1038/nature08241. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Xu C, Hasan SS, Schmidt I, Rocha SF, Pitulescu ME, Bussmann J, Meyen D, Raz E, Adams RH, Siekmann AF. Arteries are formed by vein-derived endothelial tip cells. Nat Commun. 2014;5:5758. doi: 10.1038/ncomms6758. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.