

Down-regulation of the HIPK interactor CNOT2 leads to reduced HIPK2 protein levels, identifying the CCR4-NOT complex as a new regulator of HIPK2 abundance. Functional assays reveal that HIPK2 and HIPK1 restrict CNOT2-dependent mRNA decay, thus extending the regulatory potential of these kinases to the level of posttranscriptional gene regulation.
Abstract
The serine/threonine kinase HIPK2 functions as a regulator of developmental processes and as a signal integrator of a wide variety of stress signals, such as DNA damage, hypoxia, and reactive oxygen intermediates. Because the kinase is generated in a constitutively active form, its expression levels are restricted by a variety of different mechanisms. Here we identify the CCR4-NOT complex as a new regulator of HIPK2 abundance. Down-regulation or knockout of the CCR4-NOT complex member CNOT2 leads to reduced HIPK2 protein levels without affecting the expression level of HIPK1 or HIPK3. A fraction of all HIPK family members associates with the CCR4-NOT components CNOT2 and CNOT3. HIPKs also phosphorylate the CCR4-NOT complex, a feature that is shared with their yeast progenitor kinase, YAK1. Functional assays reveal that HIPK2 and HIPK1 restrict CNOT2-dependent mRNA decay. HIPKs are well known regulators of transcription, but the mutual regulation between CCR4-NOT and HIPKs extends the regulatory potential of these kinases by enabling posttranscriptional gene regulation.
INTRODUCTION
The evolutionary conserved family of homeodomain-interacting protein kinases (HIPKs) consists of four related kinases, HIPK1–4. HIPK1–3 share a similar basic architecture and contain an N-terminal kinase region followed by various other domains mediating binding to further proteins. HIPKs shape signaling pathways mediating the response to various stress signals, including DNA damage, reactive oxygen species, and hypoxia (Saul and Schmitz, 2013). The kinases typically mediate proapoptotic functions and contribute to cell killing after exposure of cells to endangering signals such as DNA damage (D’Orazi et al., 2002; Hofmann et al., 2002). Thus down-regulation of HIPK2 and its absent proapoptotic function has been linked to chemotherapy resistance (Lin et al., 2014). Biochemical experiments revealed the relevance of HIPK2 cis-autophosphorylation at Tyr-354 and Ser-357 in the activation loop for its kinase function (Saul et al., 2013; Siepi et al., 2013). This mechanism ensures that newly translated HIPK2 is already constitutively active even in the absence of triggering signals. The constitutive HIPK2 activity can be further augmented in response to external cues by kinases such as transforming growth factor-β–activated kinase 1 (TAK1) or c-Abl (Shang et al., 2013; Reuven et al., 2015). Because HIPK2 is constitutively active after being synthesized at the ribosome, its protein amounts are strictly controlled by various mechanisms in order to prevent exaggerated expression that could be detrimental for the cell. This is achieved by several layers of regulation taking place at several levels, including the control of HIPK2 translation and protein stability. The RNA-binding protein programmed cell death protein 4 (PDCD4) suppresses the translation of Hipk2 mRNA and thus limits its de novo protein synthesis (Ohnheiser et al., 2015). In addition, the protein levels of HIPK2 are tightly controlled by degradative ubiquitination using at least four different ubiquitin E3 ligases and also caspase-mediated cleavage of the C-terminal autoinhibitory domain (Saul and Schmitz, 2013). The HIPKs are mainly found in nuclear bodies, but a fraction also occurs in cytosolic speckles (de la Vega et al., 2011; van der Laden et al., 2015). In the nucleus, HIPKs function as regulators of gene expression upon phosphorylation of transcription factors and transcription regulatory proteins (D’Orazi et al., 2012; Schmitz et al., 2014). Various HIPK family members show functional redundancy at the biochemical level, as seen by partially overlapping interaction partners (Kim et al., 1998). The partial redundancy is also observed at the genetic level, where the individual knockout of HIPK1 or HIPK2 results in viable mice, whereas the simultaneous knockout of both kinases leads to embryonic lethality (Isono et al., 2006).
Synthesis and decay of mRNAs must be strictly controlled and coordinated to allow for mRNA and protein homeostasis. Whereas various protein complexes mediate capping and polyadenylation of mRNAs, other protein complexes counteract these processes. Stress conditions such as virus infections lead to sequestering of mRNAs in ribonucleoprotein complexes known as stress granules (SGs) or processing bodies (PBs) to prevent translation (Aulas et al., 2015). One important multiprotein complex affecting all major steps of mRNA metabolism is the carbon catabolite repressor 4 (CCR4)–negative on TATA (NOT) complex. This well-conserved multisubunit complex consists of scaffolding proteins and enzymatically active subunits. In the human CCR4-NOT complex, the proteins CNOT2, CNOT7, CNOT8, and CNOT9 interact with the scaffold protein CNOT1 (Lau et al., 2009). The multiprotein assembly contains four deadenylases (CNOT6, CNOT6L, CNOT7, and CNOT8), but these are not bound simultaneously. CNOT7 and CNOT8 associate with the complex in a mutually exclusive manner, and either CNOT6 or CNOT6L associates only with complexes containing CNOT7 (Miller and Reese, 2012). The CNOT4 subunit has ubiquitin E3 ligase activity but associates only loosely with the entire CCR4-NOT complex (Lau et al., 2009; Bartlam and Yamamoto, 2010). The catalytically inactive subunits are important for the integrity of the entire multiprotein complex (Russell et al., 2002; Ito et al., 2011). The CCR4-NOT complex is involved in virtually the entire spectrum of functions spanning the sequential process of gene expression from transcription to translation and even protein degradation. The CCR4-NOT complex regulates all steps of transcription, ranging from transcription initiation to elongation and rescue of arrested RNA polymerase II (Lemaire and Collart, 2000; Babbarwal et al., 2014; Dutta et al., 2015). Among the interactors of the CCR4-NOT complex are also several components of the inner nuclear basket of the nuclear pore complex, suggesting an interaction that takes place on the nuclear face of the nuclear pore complex, where CCR4-NOT might escort mRNAs for nuclear export (Kerr et al., 2011). The CCR4-NOT subunits with ribonuclease activities mediate deadenylation of mRNAs, which is the initial and often rate-limiting step in mRNA degradation, which then proceeds via decapping and exonucleolytic degradation from the 5′ and 3′ ends (Tucker et al., 2001; Maryati et al., 2015). CNOT4 also plays a crucial role in cotranslational quality control and is associated with polysomes. Gene deletion experiments showed the requirement of CNOT4 for global translational repression under conditions of nutritional limitation (Preissler et al., 2015). The ribosome-associated NOT4 protein also participates in ubiquitination and degradation of aberrant peptides and contributes to the assembly of the 26S proteasome (Laribee et al., 2007; Panasenko and Collart, 2011).
Here we identify CNOT2 and CNOT3 as new interaction partners for HIPKs to allow association of these kinases with the CCR4-NOT complex. Down-regulation of CNOT2 by short hairpin RNAs (shRNAs) or clustered regularly interspaced short palindromic repeats (CRISPR)-Cas9–mediated gene editing leads to reduced levels of the HIPK2 protein. Simultaneous down-regulation of HIPK2 and the highly related kinase HIPK1 enhances CNOT2-dependent mRNA decay, allowing the coupling of HIPKs to posttranscriptional gene regulation.
RESULTS
HIPKs directly bind to the CCR4-NOT complex
To identify new HIPK2 interaction partners, we performed a yeast two-hybrid screen using a kinase-inactive version of HIPK2 as a bait. Among the interactors, we found several copies of CNOT2 (Supplemental Figure S1). To confirm this finding by an independent experimental approach, we transfected cells to express epitope-tagged versions of CNOT2 and HIPK2. Coimmunoprecipitations with specific antibodies allowed detection of CNOT2 in immunoprecipitates of HIPK2 (Figure 1A), indicating an interaction between the two proteins. To test whether this interaction also occurs in intact cells before their lysis, we treated cells with the membrane-permeable cross-linking agent dimethyl dithiobispropionimidate (DTBP), which allows covalent cross-linking of interacting proteins located in very close proximity. Coimmunoprecipitation experiments detected a specific interaction between CNOT2 and HIPK2 (Figure 1B), showing an association between the endogenous proteins. The intracellular localization of HIPK2 and CNOT2 was compared by immunofluorescence experiments. Whereas CNOT2 localized to cytosolic speckles, the HIPK2 protein was mainly found in the nucleus and to a certain extent also in cytosolic structures (Supplemental Figure S2). This localization was not changed upon coexpression of both proteins together, but a minor fraction of the kinase was found in cytoplasmic foci, where it colocalized with CNOT2 (Figure 1C). We also tested whether the association between HIPK2 and CNOT2 might be changed upon stressors leading to the accumulation of untranslated mRNAs (Buchan and Parker, 2009; Houseley and Tollervey, 2009). HeLa cells expressing green fluorescent protein (GFP)–tagged HIPK2 and FLAG-tagged CNOT2 were treated with the oxidative stress inducer sodium arsenite or the translation inhibitors anisomycin and puromycin. Immunofluorescence analysis showed that all three agents did not change the degree of colocalization between both interactors. Puromycin and sodium arsenite caused a strong relocalization of the interaction partners to cytosolic aggregates and to the perinuclear region, respectively (Figure 1D). Because HIPK2 and CNOT2 colocalize in discrete cytoplasmic foci, it was interesting to test whether those dots match with known cytoplasmic structures such as SGs and PBs. Costaining between HIPK2 and marker proteins for SGs (TIA1) and PBs (PAN3) showed no significant colocalization under normal conditions (Supplemental Figure S3) or after treatment with anisomycin, puromycin, or sodium arsenite (Supplemental Figures S4 and S5). The CNOT2 protein showed no colocalization with TIA1 and only a partial colocalization with PAN3 (Supplemental Figure S6). Collectively these data show colocalization of HIPK2 with CNOT2-containing structures that do not overlap with SGs or PBs.
FIGURE 1:
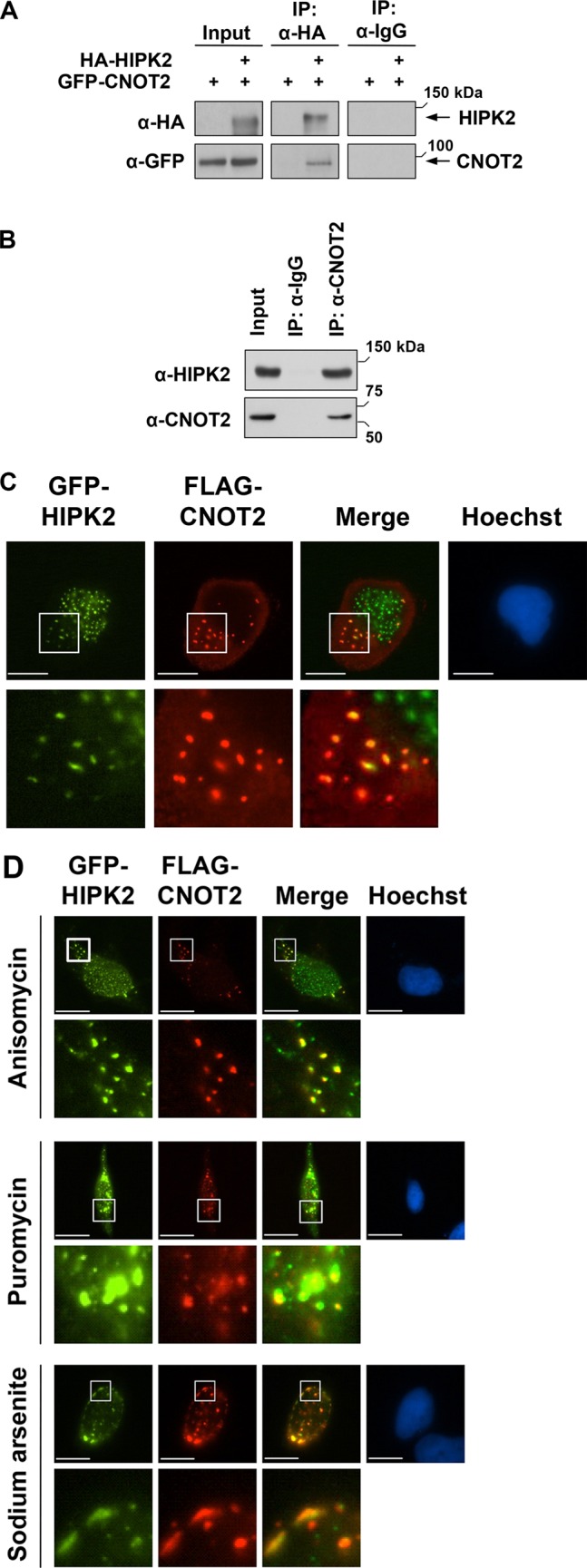
HIPK2 and CNOT2 interact and colocalize in cytosolic speckles. (A) 293T cells were transfected with GFP-CNOT2 either alone or in combination with HA-HIPK2. At 24 h after transfection, cells were lysed, and proteins were immunoprecipitated (IP) using an anti-HA antibody or a control rat IgG antibody. Precipitated samples were analyzed by Western blotting using appropriate antibodies, and input samples of the cell lysates were analyzed for correct protein expression. The positions of molecular weight markers are indicated. (B) Cells were treated for 30 min with a cell-permeable cross-linker to preserve protein–protein interactions in intact cells. After removal of the cross-linker and quenching, cells were lysed and subjected to immunoprecipitation using control or anti-CNOT2 antibodies. The precipitates and input controls were analyzed by immunoblotting. (C) U2OS cells coexpressing GFP-HIPK2 and FLAG-CNOT2 were stained and analyzed by indirect immunofluorescence to reveal the localization of HIPK2 and CNOT2; nuclear DNA was stained by Hoechst. Scale bar, 10 μm. Bottom, areas indicated by white boxes in 5× magnification. The merge shows areas of colocalization in yellow. (D) HeLa cells expressing GFP-HIPK2 and FLAG-CNOT2 were treated with anisomycin (10 μg/ml for 30 min), puromycin (100 μg/ml for 60 min), or sodium arsenite (0.5 mM for 60 min). The cells were fixed and analyzed by immunofluorescence as shown. Yellow in the merged images indicates colocalization of the two proteins. Scale bar, 10 μm. Bottom, the boxed areas in 5× magnification.
To determine the HIPK2 region mediating the interaction with CNOT2, we examined deletion mutants of HIPK2 lacking either the N- or the C-terminus for their ability to bind to CNOT2. Coimmunoprecipitation experiments showed that deletion of the C-terminal part still allowed for the protein–protein interaction (Figure 2A), thus identifying the N-terminal part as the interacting region. Because the binding region contains the evolutionary conserved kinase domain shared with the other HIPK family members, it was of interest to test whether HIPK1 and HIPK3 also have the ability to bind CNOT2. Coimmunoprecipitation experiments showed binding of the other HIPK family members to CNOT2 (Figure 2, B and C). Accordingly, HIPK1 and HIPK3 also colocalize with CNOT2 in cytoplasmic foci (Figure 2D), thus defining CNOT2 as a shared substrate for the HIPKs.
FIGURE 2:
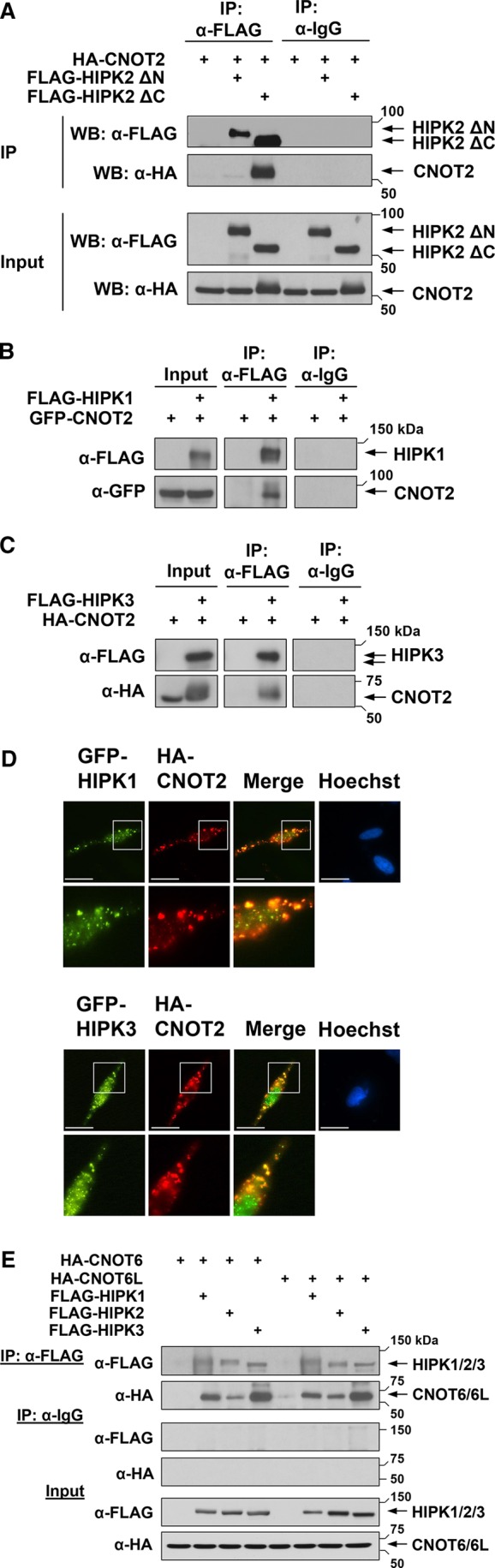
CNOT2 binds to HIPKs. (A) 293T cells were transfected to express HA-CNOT2 alone or in combination with either FLAG-HIPK2 ΔN or FLAG-HIPK2 ΔC. An aliquot of the lysates was analyzed for correct protein expression (Input). Another aliquot was used for coimmunoprecipitation using anti-FLAG antibody or an isotype-matched control mouse IgG antibody. Samples were analyzed by Western blotting as shown. (B) The indicated plasmids encoding FLAG-HIPK1 or GFP-CNOT2 were transfected into 239T cells, and lysates were used either for input controls or coimmunoprecipitation with the indicated antibodies as displayed. (C) The experiment was done as in B, with the difference that HA-CNOT2 and FLAG-HIPK3 were coexpressed. (D) HeLa cells coexpressing HA-CNOT2 and GFP-tagged HIPKs were stained and analyzed by indirect immunofluorescence to reveal the localization of HIPK1/3 and CNOT2. Nuclear DNA was stained by Hoechst. Scale bar, 10 μm. Bottom, areas indicated by white boxes in 5× magnification. The merge shows areas of colocalization in yellow. (E) 293T cells were transfected with the indicated plasmids coding for epitope-tagged CNOT6/6L, HIPK1, HIPK2, and HIPK3. Cells were lysed, and lysates were analyzed for correct protein expression (Input). Immunoprecipitation was performed using anti-FLAG antibody or a control mouse IgG antibody. Precipitated proteins and input samples were separated by SDS–PAGE and analyzed by Western blotting using appropriate antibodies.
Are the HIPKs associated with only the CNOT2 subunit or the complete CCR4-NOT complex? To address this question, we tested binding of HIPK to other subunits of the CCR4-NOT complex by coimmunoprecipitation experiments. Because the deadenylases CNOT6 and CNOT6L are integral parts of the complex, they were expressed either alone or together with the three kinases in 293T cells. All immunoprecipitated HIPKs were found to interact with CNOT6 and CNOT6L (Figure 2E), showing that the binding of HIPK family members occurs with the entire CCR4/NOT complex.
The evolutionarily conserved C-terminal region of CNOT2 contains the SH3-like NOT-box, which is responsible for the direct association with other proteins, such as cyclin-dependent kinase 11 (Shi and Nelson, 2005). To test a possible contribution of the NOT-box for the binding to HIPK2, we created a deletion mutant of CNOT2 (CNOT2 ΔNOT-box) and tested it for interaction with HIPK2. Deletion of the NOT-box precluded interaction with HIPK2 (Figure 3A) and also HIPK3 (Figure 3B), indicating that this region is necessary for efficient binding to HIPKs. In addition, the CNOT3 protein contains a NOT-box, and therefore it was of interest to test its ability to bind to HIPK2. Glutathione S-transferase (GST) pull-down experiments showed that binding of HIPK2 to CNOT3 was even stronger than binding of the kinase to CNOT2 (Figure 3C).
FIGURE 3:

The NOT-box in CNOT2 is important for HIPK binding. (A) Top, schematic representation of CNOT2 and the CNOT2 deletion mutant ΔNOT-box. Bottom, HA-tagged CNOT2 and CNOT2 ΔNOT-box proteins were coexpressed with FLAG-HIPK2 as shown, followed by cell extraction and analysis of proteins for expression (Input) and mutual binding by coimmunoprecipitation. (B) The experiment was done as in A, with the exception that binding to HIPK3 was measured. (C) Cells transfected to express GFP-HIPK2 were lysed, and extracts were tested for interaction with bacterially produced GST, GST-CNOT2, and GST-CNOT3 proteins by pull-down experiments as shown. Top, Western blot displaying the input material and the eluates. Bottom, CBB-stained input material and GST fusion proteins used for this experiment.
CNOT2 controls HIPK2 levels
In further experiments, we noticed that shRNA-mediated depletion of CNOT2 resulted in markedly decreased HIPK2 protein levels (Figure 4A). In contrast, the levels of HIPK1 and HIPK3 were unchanged, suggesting that CNOT2-mediated regulation occurs specifically for the HIPK2 protein. To confirm this finding by an independent experimental approach, we eliminated CNOT2 protein expression using the CRISPR-Cas9 system. Further, these cells showed significantly reduced HIPK2 protein levels (Figure 4A), corroborating the finding that impaired CNOT2 expression results in reduced HIPK2 protein levels. The analysis of relative mRNA levels by quantitative PCR (qPCR) showed that CNOT2 down-regulation caused only a slight reduction of HIPK2 mRNA levels (Figure 4B). This reduction was seen only for some regions of the Hipk2 mRNA, suggesting that further mechanisms account for reduced HIPK2 protein levels. To test the effect of CNOT2 depletion on HIPK2 protein stability, we blocked protein synthesis with anisomycin in control cells or cells treated to down-regulate CNOT2. The degradation rates of HIPK2 were comparable in control and CNOT2-knockdown cells (Figure 4C), suggesting that reduced steady-state HIPK2 protein levels are also due to impaired translation.
FIGURE 4:
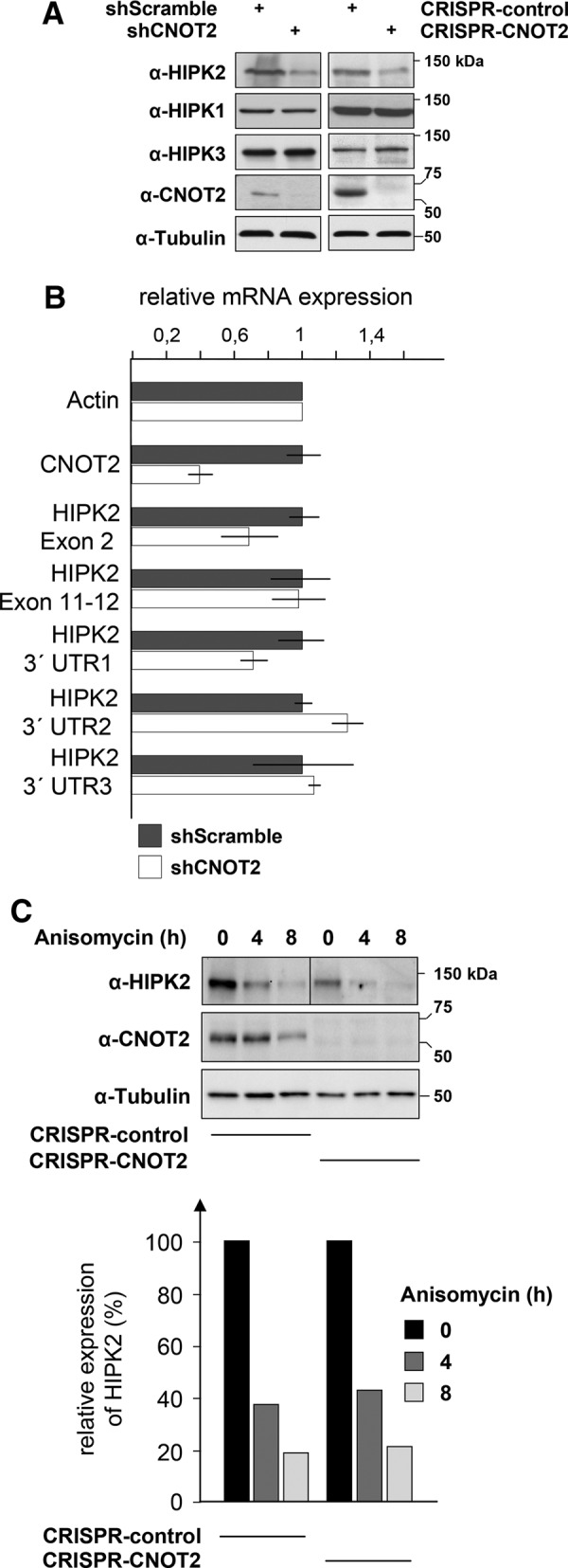
Regulation of HIPK2 protein amount by CNOT2. (A) Expression of the CNOT2 protein was either partially reduced by the expression of a specific shRNA or completely prohibited by CRISPR-Cas9–mediated genome engineering. Cell extracts were prepared and equal amounts of proteins analyzed for the expression of all HIPKs and the indicated controls as shown. (B) 293T cells transfected to express a CNOT2-specific shRNA or a scrambled control were analyzed for HIPK2 mRNA levels by qPCR. Five different primer pairs were used to quantify HIPK2 mRNA at different exons, and the 3′ untranslated region (3′ UTR) as shown. Values were normalized to the housekeeping gene β-actin, and relative expression levels were determined using the ΔΔCT method. Error bars show SDs from two independent experiments performed in triplicate. (C) Top, the protein stability of HIPK2 was compared between control 293T cells and a cell line in which CNOT2 expression was eliminated by CRISPR-Cas9–mediated gene editing. The cells were treated with anisomycin or vehicle for the indicated periods as shown. Equal numbers of cells were lysed, and lysates were analyzed for protein expression by immunoblotting. Bottom, the HIPK2 protein expression levels were quantified using the Chemidoc touch imaging system (Bio-Rad). To facilitate comparison, the protein levels in the respective vehicle controls were arbitrarily set as 100%.
Camptothecin-triggered cell death is regulated by HIPK2 and CNOT2
In a search for physiologically relevant modulators of CNOT2 expression, we noticed in a database search that camptothecin (CPT), a topoisomerase I inhibitor that induces DNA damage, leads to impaired CNOT2 mRNA expression (GEO DataSet gds1453). To validate this finding, we exposed 293T cells to various concentrations of this chemotherapeutic agent and quantified CNOT2 mRNA by qPCR. In agreement with the microarray data set, the treatment of cells with CPT led to markedly reduced CNOT2 levels. Further, the mRNA levels of HIPK2 decayed in the presence of CPT, whereas the transcript levels of HIPK1 and HIPK3 were unaffected (Figure 5A). CPT also resulted in significantly reduced protein amounts of CNOT2 and of all three HIPK family members (Figure 5B). Because CNOT2 does not regulate HIPK1 and HIPK3 mRNA and protein levels, this CPT-mediated effect seems to be independent of CNOT2. It was then of interest to test the role of CNOT2 and HIPK2 in CPT-triggered cell death. Cells were transfected to express shRNAs specific for CNOT2 or HIPK2 or a scrambled control, followed by induction of cell death by CPT or the anticancer drug etoposide. HIPK2-depleted cells were protected from etoposide-induced death (Figure 6A), as expected (Sakamoto et al., 2010). In contrast, knockdown of HIPK2 sensitized cells to CPT-induced cell death, indicating an antiapoptotic function of HIPK2 in response to this stimulus. Knockdown of CNOT2 increased CPT- and etoposide-triggered cell death (Figure 6A), indicating an antiapoptotic function of this protein. These proapoptotic and antiapoptotic functions of HIPK2 and CNOT2 were also reflected by an independent experimental approach at the level of caspase activities (Supplemental Figure S7). To test whether the antiapoptotic function of CNOT2 involves the depletion of HIPK2, we transfected CNOT2-knockdown cells to express GFP-tagged active and inactive versions of this kinase. CNOT2-dependent sensitization to CPT-induced cell death was not affected by HIPK2 (Supplemental Figure S8), indicating that CNOT2- and HIPK2-regulated pathways are not connected at the level of apoptosis induction. Similar results were obtained when HIPK2-deficient mouse embryonic fibroblasts (MEFs) and wild-type controls were compared for CPT- or H2O2-induced cell death (Figure 6, B and C), showing that the nature of the apoptotic stimulus dictates the functional role of HIPK2 in apoptosis.
FIGURE 5:
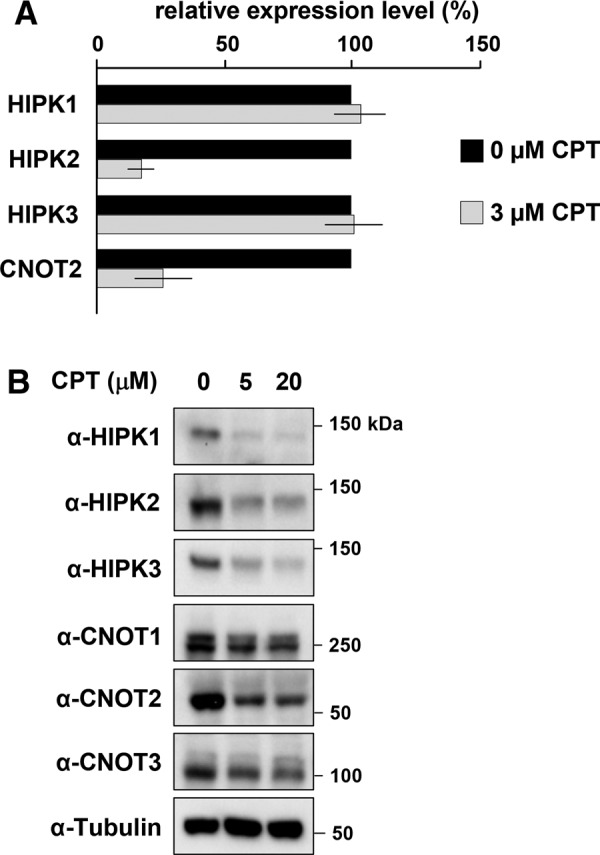
CPT-mediated down-regulation of CNOT2 and HIPK2. (A) 293T cells were treated with indicated concentrations of CPT for 16 h. Total RNA was isolated, and mRNA levels of the indicated genes were analyzed with real-time qPCR using gene-specific primers. Error bars display the SDs from two independent experiments. (B) Cells were treated for 24 h with different concentrations of CPT, and the levels of the indicated proteins were detected by Western blot analysis using specific antibodies.
FIGURE 6:

HIPK2 depletion sensitizes cells toward CPT-induced apoptosis. (A) 293T cells were transfected with vectors directing the expression of HIPK2- or CNOT2-specific shRNAs or an unspecific control shRNA. Transfected cells were selected for 2 d in puromycin to eliminate untransfected cells. Subsequently, 5 μM CPT or 60 μM etoposide was added for 24 h. Most of the cells were used to measure apoptosis in the flow cytometer using the Annexin V FITC Apoptosis Detection Kit. Error bars show SDs from two independent experiments performed in triplicate. An aliquot of the cells was used to control the knockdown efficiencies by Western blotting. (B) HIPK2-knockout MEFs reconstituted either with an empty vector (HIPK2−/−) or with HIPK2 wild-type (HIPK2 wt) were treated with 20 μM CPT or 2 mM hydrogen peroxide for 24 h. Apoptosis was measured in the flow cytometer. Results show a quantification of cell death. Error bars indicate SDs from two independent experiments performed in triplicate. An aliquot of cells was used as a control to ensure HIPK2 expression (right). (C) Equal numbers of the HIPK2-knockout and reconstituted MEFs used in B were seeded in 10-cm dishes. After addition of 2 μM CPT for 24 h, cells were washed twice, and complete DMEM was added for 2 d. Cells were fixed and subsequently stained with crystal violet. A representative experiment is shown. Right, control Western blot ensuring correct HIPK2 expression.
HIPKs phosphorylate and regulate the CCR4-NOT complex
The coexpression of HIPKs together with CNOT2 frequently resulted in the occurrence of a slower-migrating CNOT2 form (compare Figure 2A), suggesting a possible posttranslational modification of CNOT2 by the kinases. To test whether HIPK2 can phosphorylate CNOT2, we coexpressed both proteins and left cell lysates untreated or incubated them with λ phosphatase. Analysis of the gel electrophoretic behavior of CNOT2 showed that the HIPK2-induced occurrence of a slower-migrating CNOT2 form was not seen after phosphatase treatment (Figure 7A), revealing a HIPK2-mediated phosphorylation event. We also observed that phosphorylation was strongly induced by HIPK3 and various deletion mutants thereof in a kinase-dependent manner (Figure 7B). To identify HIPK-mediated CNOT2 phosphorylation by an unbiased approach, we performed a mass spectrometric analysis. We transfected 293T cells to express HA-CNOT2 alone or together with FLAG-HIPK3 or FLAG-HIPK2 ΔC, which efficiently phosphorylates CNOT2, followed by purification of the CNOT2 protein by immunoprecipitation. After SDS–PAGE and Coomassie brilliant blue (CBB) staining of the precipitated protein (Supplemental Figure S9), the CNOT2 bands were excised and subjected to tryptic in-gel digestion and high-resolution mass spectrometric analysis. These experiments showed HIPK-mediated phosphorylation of five serines within the CNOT2 protein by both HIPK2 and HIPK3 (Supplemental Table S1 and the Andromeda score in Supplemental Table S2). As schematically displayed in Figure 7C, these sites cluster in the N-terminal portion of CNOT2. To test a possible function of these CNOT2 phosphorylations we performed tethering assays in which CNOT2 was fused to the RNA-binding domain of the λN protein, which allows binding to a luciferase-encoding mRNA containing several copies of λN-binding B boxes (Gehring et al., 2008). Expression of λN-CNOT2 resulted in a dose-dependent reduction of luciferase activity, indicating mRNA decay (Supplemental Figure S10). This reduction was also mirrored at the level of mRNA, as revealed by qPCR (unpublished data). To test a possible function of CNOT2 phosphorylation for this activity, we generated phosphorylation-deficient (serine-to-alanine) or phosphorylation mimicking (serine-to–glutamic acid) CNOT2 mutants. Both mutants had largely unchanged activities in mRNA decay assays (Supplemental Figure S10), showing that at least CNOT2 activity is not affected by these phosphorylation events. Because several HIPKs bind to the entire CCR4-NOT complex, we wanted to test the effect of these kinases on mRNA decay in a more general approach. To circumvent the described functional redundancy between the kinases (Isono et al., 2006), we created a cell line in which HIPK2 was knocked out by CRISPR-Cas9 and HIPK1 was strongly down-regulated by a doxycycline (dox)-inducible shRNA (Figure 7D). Tethering assays showed that elimination of HIPK2 alone did not affect CNOT2-mediated mRNA degradation, whereas simultaneous interference with HIPK1 and HIPK2 expression resulted in increased mRNA decay (Figure 7E). These data suggest that HIPK1 and HIPK2 serve to restrict CCR4-NOT mRNA decay activity.
FIGURE 7:
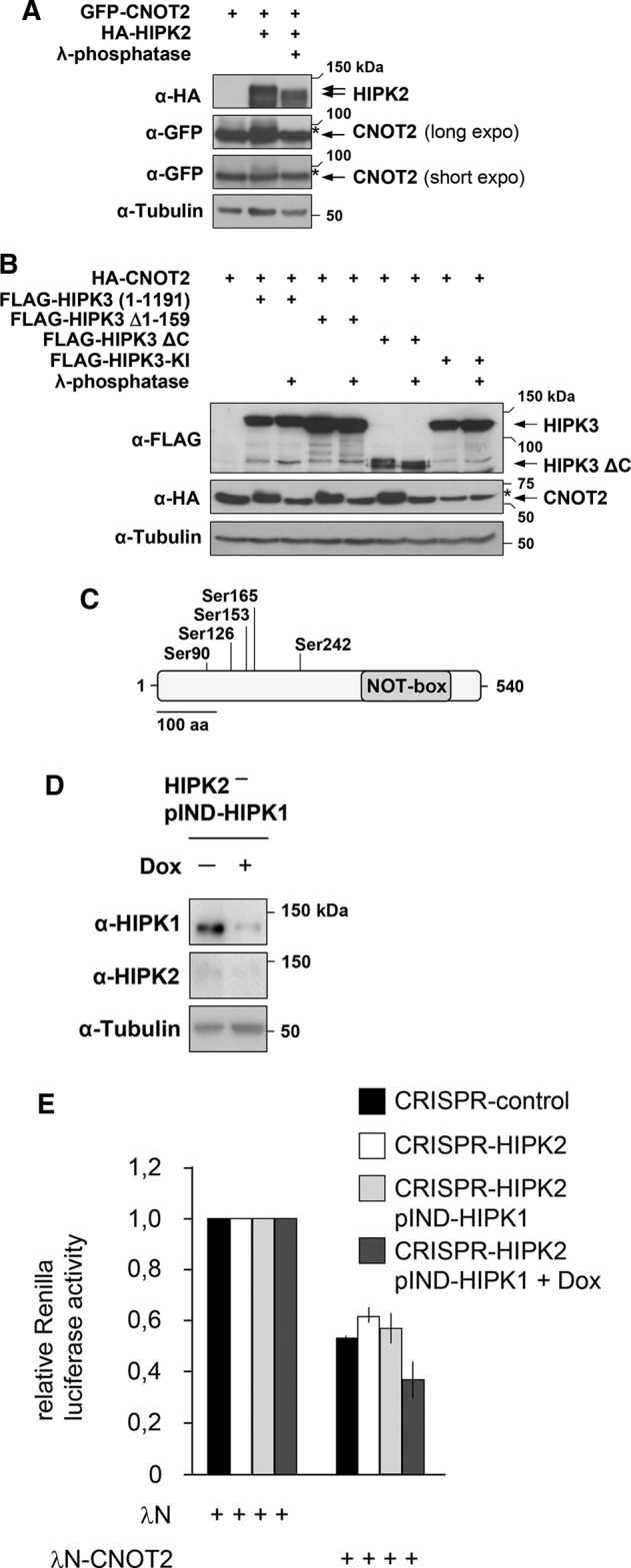
Phosphorylation and regulation of CNOT2. (A) 293T cells were transfected to express epitope-tagged CNOT2 together with full-length HIPK2. Cell lysates were treated with λ-phosphatase for 30 min as indicated and analyzed by Western blotting using appropriate antibodies. Two different exposure times for the detection of CNOT2 are shown. The phosphorylated form is marked by a star. (B) HA-CNOT2 was coexpressed along with HIPK3 or the indicated variants thereof, including a kinase-inactive point mutant (FLAG-HIPK3 KI). Cell lysates were treated with λ-phosphatase as indicated, and proteins were analyzed by Western blot with appropriate antibodies. The phosphorylated forms are marked by a star. (C) Schematic representation of CNOT2, indicating the positions of the identified HIPK phosphorylation sites. (D) HIPK2 CRISPR-Cas9–knockout cells characterized in Supplemental Figure S7A were stably transfected with pIND-HIPK1, and cells were treated for 3 d with doxycycline (1 μg/ml) to allow down-regulation of HIPK1. Cells were characterized by Western blotting as shown. (E) The indicated cells were left untreated or treated with doxycycline (1 μg/ml) for 2 d. Cells were then transfected with the Renilla luciferase reporter plasmid containing five B-boxes in its 3′ untranslated region along with the λN expression vectors (50 ng) and the firefly luciferase used for normalization. Cells were grown for further 24 h in the absence or presence of doxycycline, and luciferase activities were determined. To facilitate comparison, the luciferase activity in the presence of λN was set as 1. Error bars indicate SDs obtained from two independent experiments performed in triplicate.
DISCUSSION
Regulation of HIPK2 by the CCR4-NOT complex
This study shows a new mechanism allowing the regulation of HIPK2 amount via the CCR4-NOT complex. It is not clear whether impaired HIPK2 expression can really be attributed to the CNOT2 subunit, as interference with CNOT2 expression also affected expression of CNOT1 (Supplemental Figure S11), CNOT3 (unpublished data), and probably further subunits as well. Interference with CNOT2 expression affects HIPK2 mRNA levels only to a minor extent and leaves HIPK2 degradation unaffected. Thus the reduced HIPK2 protein amounts in the absence of CNOT2 are likely due to a mixture of impaired transcription and reduced de novo synthesis of HIPK2 at the ribosome. A further complication comes from the fact that the vast majority of HIPK2 mRNA occurs in the recently discovered species of circular RNA (Jeck et al., 2013), and it is unclear whether this circular RNA is translated into a protein. The molecular mechanism allowing decreased HIPK2 synthesis is not known and might involve further regulatory proteins, such as PDCD4, which was recently identified as an antagonist of HIPK2 translation (Ohnheiser et al., 2015). Although previous data also showed a repressive function of the CCR4-NOT complex on the translation of several proteins (Cooke et al., 2010; Zekri et al., 2013), the data described here are compatible with a situation in which HIPK2 protein translation is supported by a functional CCR4-NOT complex. Our immunofluorescence data suggest that only a minor fraction of HIPK proteins is found in association with the CCR4-NOT complex. HIPK2 is a mainly nuclear protein, but the fraction of cytosolic HIPK2 proteins can be increased by removal of covalently bound small ubiquitin-related modifier (SUMO) or acetylation of lysines contained in nuclear localization signal 1 (de la Vega et al., 2011, 2012). Similarly, HIPK1 also can be transported to the cytosol after removal of SUMO by sentrin-specific protease (Li et al., 2008). HIPK2 and also the other family members HIPK1 and HIPK3 contact the CCR4-NOT complex by their highly homologous kinase domain. Direct binding was observed to the NOT-box containing proteins CNOT2 and CNOT3, which heterodimerize through their NOT-boxes to form an exposed surface that might provide a binding platform for interaction partners (Boland et al., 2013), as schematically summarized in Figure 8A.
FIGURE 8:
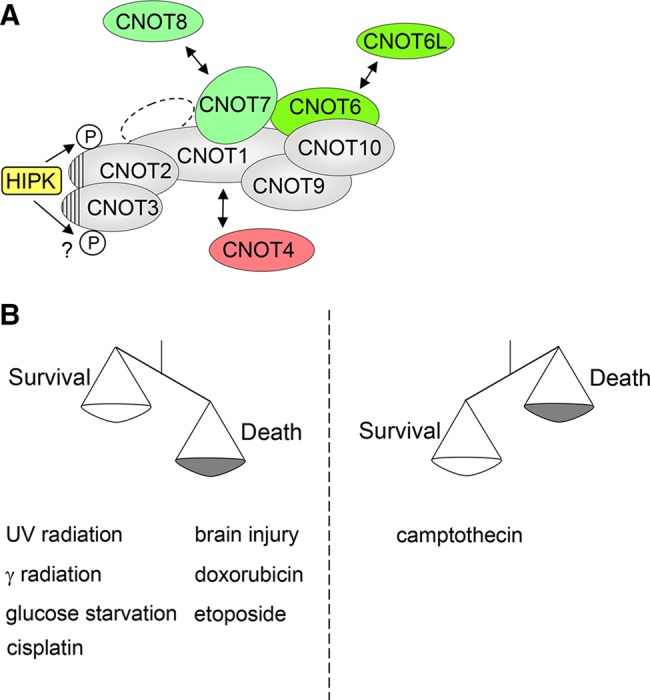
Schematic models summarizing the results. (A) Association with and phosphorylation by the HIPKs. The NOT-boxes contained in CNOT2 and CNOT3 are indicated by striated areas. The subunits with deadenylation activity are shown in green, and the loosely associated subunit with ubiquitin E3 ligase activity is shown in red. (B) Schematic model showing the proapoptotic and antiapoptotic functions of HIPK2.
An antiapoptotic function of HIPK2: implications for tumor therapy
Many studies describe a proapoptotic function of HIPK2 in response to many different agents, such as ultraviolet radiation, doxorubicin, and etoposide (D’Orazi et al., 2012; Hofmann et al., 2013; Schmitz et al., 2014). The ability of HIPK2 to induce cell death relies on p53-dependent and -independent pathways. HIPK2-mediated p53-dependent apoptosis critically involves p53 Ser-46 phosphorylation, which in turn allows recruitment of the acetylase CREB-binding protein to acetylate p53 at Lys-382 (D’Orazi et al., 2002; Hofmann et al., 2002). The p53 Ser-46 phosphorylation affects cell cycle–modulatory genes only to a minor extent, whereas it preferentially activates proapoptotic p53 target genes, as revealed by experiments using gene array and chromatin immunoprecipitation coupled to massively parallel sequencing (Smeenk et al., 2011). However, HIPK2 can also trigger cell death via p53-independent cell death pathways. These processes involve, for example, HIPK2-mediated phosphorylation of the antiapoptotic transcriptional corepressor C-terminal binding protein (Zhang et al., 2005) or phosphorylation of ΔNp63α, a dominant-negative isoform of the p53 family member p63 (Lazzari et al., 2011).
Here we show for the first time an antiapoptotic function of HIPK2, implying that HIPK2 plays a dual role in the regulation of cell death, as shown schematically in Figure 8B. Other apoptosis regulators also can have such a dual role, as seen in the case of p53, which is typically proapoptotic but in some cases functions to prevent cell death (Lassus et al., 1996; Vousden, 2006). Another example is provided by the largely antiapoptotic transcription factor NF-κB, which can also promote apoptosis, depending on the cell type and apoptotic stimulus (Dumont et al., 1999; Radhakrishnan and Kamalakaran, 2006). CPT is a quinoline alkaloid that binds to DNA, and the water-soluble CPT derivatives topotecan and irinotecan are clinically used for the treatment of cancer (Houghton et al., 1995). Thus it will be very relevant to investigate in future studies whether the efficacy of these drugs or the clinically used topoisomerase I inhibitors are also limited by the antiapoptotic function of HIPK2. In such a case, the reduced HIPK2 amounts would still be sufficient to protect from the early steps of CPT-induced cell death, and kinase inhibitors would be beneficial to increase the efficacy of chemotherapeutic treatment.
Regulation of CCR4-NOT by HIPKs?
Because the CCR4-NOT complex has been identified as a central integration point coordinating virtually all aspects of gene expression, it is important to control its functions and activities (Miller and Reese, 2012). Thus the composition of the CCR4-NOT complex is dynamic, and individual subunits might function both within and outside of the complex in different cellular compartments (Collart and Panasenko, 2012). In addition, the relative expression levels of subunits can change, as exemplified by the down-regulation of yeast CNOT1 in the absence of glucose (Norbeck, 2008) and the reduced expression of CNOT2 in the presence of CPT (this study). The complex is also regulated by different posttranslational modifications, such as phosphorylation (Lau et al., 2010). Here we identify five HIPK-mediated phosphorylation sites in CNOT2, but it is highly probable that these kinases also modify other subunits of the complex. It remains to be seen whether the restriction of CNOT2 activity by HIPK1/2 is due to kinase-independent events or regulatory phosphorylations. Of interest, the YAK1 kinase from yeast, a phylogenetic ancestor of the HIPK family, also was shown to phosphorylate POP2/CAF1, a homologue of the human CNOT7 protein, upon glucose limitation (Moriya et al., 2001). YAK1 also associates with other components of this complex, such as cell division cycle (CDC) 39/NOT1 (www.yeastgenome.org/). It will thus be very interesting to study the functional consequences of CCR4-NOT phosphorylation after comprehensively mapping basal and stimulus-regulated HIPK-dependent phosphorylation sites in this complex.
MATERIALS AND METHODS
Antibodies, plasmids, and reagents
See Supplemental Table S3 for the antibodies, plasmids, and reagents used in this work.
Cell culture and transfections
Human embryonic kidney HEK293T cells, HeLa, HIPK2−/−, and control MEFs were grown in DMEM containing 10% fetal calf serum (FCS) and 1% (wt/vol) penicillin/streptomycin at 37°C and 5% CO2. Cells were seeded in dishes and transfected using the transfection reagent Rotifect (Roth) or linear polyethylenimine. After pipetting up and down several times, complex formation occurred in serum- and antibiotic-free DMEM for 20 min at room temperature. After addition of the transfection mixture to antibiotic-free DMEM containing FCS, the cells were incubated for 3–5 h before the medium was changed and the cells were further grown. Stable cell clones containing the pINDUCER plasmids were produced by selection for 10 d in puromycin, followed by single clone picking and analysis of doxycycline-dependent protein and mRNA expression.
Cell lysis
To prepare cell extracts under native conditions, the cells were washed once with 1× phosphate-buffered saline (PBS), harvested by scraping, and collected by centrifugation for 4 min at 350 × g. The pellet was resuspended in an appropriate amount of NP-40 buffer (20 mM Tris/HCl, pH 7.5, 150 mM NaCl, 1 mM phenylmethylsulfonyl fluoride, 10 mM NaF, 0.5 mM sodium orthovanadate, leupeptin [10 μg/ml], aprotinin [10 μg/ml], 1% NP-40, and 10% glycerol) and incubated on ice for 20 min. The lysate was cleared by centrifugation for 10 min at 16,000 × g. The supernatant was then used for immunoprecipitation experiments or mixed with 5× SDS sample buffer, boiled at 95°C for 5 min. and analyzed by Western blotting. Alternatively, cells were directly lysed in 1× SDS sample buffer, sonified to shear the genomic DNA, and used for Western blotting.
Immunoprecipitation and Western blotting
Coimmunoprecipitation was done either after cross-linking of proteins with the membrane-permeable cross-linker DTBP as described (Renner et al., 2011) or after lysis of cells under native conditions using NP-40 buffer. The lysate was cleared by centrifugation at 12,000 × g for 10 min. The supernatant was transferred to a fresh tube, and 10% of the volume was removed as input sample, mixed with 5× SDS sample buffer, and heated at 95°C for 5 min. The remaining lysate was precleared by the addition of 20 μl of A/G-agarose bead slurry and incubation for 1 h at 4°C. After centrifugation, the precleared lysate was transferred to a new tube, and 1 μg of the precipitating antibody or control immunoglobulin G (IgG) was added. After incubation at 4°C for at least 2 h, 30 μl of A/G-agarose bead slurry was added, and the lysates were incubated for another 1 h at 4°C. To remove all proteins that were not precipitated, the beads were washed five times for 10 min with NP-40 buffer. After elution in 1.5× SDS sample buffer, the samples were analyzed by Western blotting. This was done by separation of proteins via SDS–PAGE, followed by semidry blotting to a polyvinylidene difluoride membrane (Millipore) as previously described (Milanovic et al., 2014).
Immunofluorescence
Cells were grown in 12-well plates on coverslips, washed once with cold 1× PBS, and fixed for 1 min with an ice-cold methanol:acetone (1:1) solution. The fixing solution was aspirated off, and the cells were rehydrated with 1× PBS for 10 min and then blocked for 60 min with 1× PBS containing 10% (vol/vol) goat serum. Afterward, the cells were incubated with the primary antibody diluted in 1× PBS containing 1% (vol/vol) goat serum either overnight at 4°C or for 2 h at room temperature. The cells were then washed three times for 5 min with 1× PBS and incubated with the appropriate secondary dye-coupled antibody diluted in 1× PBS containing 1% (vol/vol) goat serum for 2 h in the dark. The incubation was followed by three washing steps for 5 min with 1× PBS. Nuclear DNA was stained by incubating the cells with Hoechst 33324 for 5 min. Cells were again washed three times for 5 min and then mounted on microscope slides with Kaiser’s glycerol gelatin. The stained proteins were analyzed using an inverted Nikon Eclipse 2000E microscope.
Tethering assays
A Renilla luciferase reporter construct harboring five B-box elements in its 3′ untranslated region was coexpressed with the B-box RNA-binding bacteriophage λN protein to tether the CNOT2 protein to the reporter mRNA. The reporter gene was coexpressed together with the λN fusion protein and the firefly luciferase lacking B-box motifs as an internal control for normalization. Transfections were done in six-well plates, and 24 h later cell lysates were prepared. The emitted bioluminescence was detected with a Berthold DuoLumat LB 9501 luminometer. The relative activities were calculated after the normalization of the Renilla luciferase activities to the activities of the firefly luciferase.
GST pull-down experiments
GST fusion proteins were produced and purified from Escherichia coli. The GSH Sepharose beads containing the attached GST fusion proteins were washed with PBS and then incubated with 100 μl of NP-40 extracts containing the GFP-HIPK2 protein. Another 500 μl of NP-40 buffer was added and incubated on a rotating device for at least 4 h at 4°C. The beads were washed five times in NP-40 lysis buffer. Bound proteins were eluted by the addition of 40 μl 1.5× SDS sample buffer and subsequent boiling for 5 min. Proteins associated with GST fusion proteins were analyzed by Western blotting, and the recombinant GST proteins were controlled by SDS–PAGE and CBB staining.
Identification of CNOT2 phosphorylation sites by mass spectrometry
We transfected 293T cells to express HA-CNOT2 alone or together with FLAG-HIPK2 ΔC or FLAG-HIPK3 and purified the CNOT2 protein by immunoprecipitation using anti-hemagglutinin (HA) antibodies. The CNOT2 bands were excised from the Coomassie-stained gel and subjected to tryptic in-gel digestion. Extracted peptides were desalted using C18-based Stop and Go Extraction Tips, and mass spectrometric measurements were performed with a nano liquid chromatography system coupled to an LTQ-Orbitrap Velos mass spectrometer (Thermo Fisher Scientific, Waltham, MA) via a nanoelectrospray source (Proxeon) as described in Hölper et al. (2015). For mass spectrometric (MS) measurements, full MS scan spectra (m/z = 300–1650) were acquired in the Orbitrap with a resolution of 60,000 after accumulation of 1,000,000 ions. The 15 most intense peaks from full MS scan were isolated and fragmented in the linear ion trap after accumulation of 5000 ions. Fragmentation of precursor ions was performed using collision-induced dissociation (35% normalized collision energy) before acquisition of MS/MS scan spectra. The raw data were processed and analyzed using MaxQuant (Cox and Mann, 2008) software (version 1.2.2.9), and peptides were searched against a human FASTA database (version 3.68). Enzyme specificity was set to trypsin with an additional allowance of cleavage N-terminal to proline. A maximum of two missed cleavages was allowed. Cysteine carbamidomethylation was set as fixed modification, and oxidation of methionine, acetylation of protein N-terminus, and phosphorylation of STY (serine, threonine, and tyrosine) were set as variable modifications. The initial precursor ion mass deviation was set to 7 ppm, and the maximum allowed mass deviation was set to 20 ppm. MS/MS tolerance was set to 0.5 Da. A false discovery rate of 0.01 and minimum peptide length of seven amino acids were used for peptide identifications.
CRISPR-Cas9–mediated gene targeting
Oligos targeting the first exon of the CNOT2 or Hipk2 genes were cloned into pX459 (Addgene) to obtain the pX459-CNOT2 and pX459-HIPK2 plasmids. 293T cells were transfected with either pX459 targeting the luciferase gene or pX459-CNOT2, and HeLa cells were transfected with the plasmid pX459-HIPK2. The next day, puromycin (1 μg/ml) was added for 3 d to kill the untransfected cells. Single-cell clones were isolated and tested for expression of the CNOT2 or HIPK2 proteins and Cas9 by Western blotting. Cells expressing neither CNOT2 nor HIPK2 and also lacking Cas9 expression were used for the subsequent experiments. The Indel mutations in the knockout clones were characterized by sequencing of the genomic DNA.
Quantitative real-time PCR
The RNeasy mini kit (Qiagen) was used to extract total RNA from cells, and RNA quality was tested on ethidium bromide–stained agarose gels. cDNA was synthesized using Oligo (dT) 20 primers and the Superscript II first-strand synthesis system (Invitrogen). Real-time PCR was performed using Absolute SYBR Green ROX Mix (Thermo Scientific) with specific primers (Supplemental Table S3). Gene expression was determined using an Applied Biosystems 7300 real-time PCR system. All experiments were performed in triplicate, and quantification was done using the comparative ΔΔCT method. Data were normalized to the housekeeping gene β-actin, and the resulting ΔCT values were compared with a sample that was chosen as a calibrator. The relative expression level was then calculated according to the formula R = 2−ΔΔCT.
Measurement of apoptosis
Apoptosis was measured by double staining with fluorescein isothiocyanate (FITC)–annexin V and propidium iodide (PI) to allow distinguishing between cells in early apoptosis (FITC-annexin V positive/PI negative) or late apoptotic stages (FITC-annexin V positive/PI positive). The staining was performed using the Annexin V FITC Apoptosis Detection Kit (eBioscience), following the manufacturer’s protocol. In brief, cells were detached from the plate using TrypLETM Express and washed once in PBS and once in 1× binding buffer. Then the cells were resuspended in 1× binding buffer, (1–5) × 106 cells/ml, and 5 μl of FITC-annexin V was added to 100 μl of this cell suspension. After incubation for 10–15 min at room temperature the cells were washed once again with 1× binding buffer and subsequently resuspended in 200 μl of 1× binding buffer. After the addition of 5 μl of PI staining solution, the cells were analyzed by flow cytometry within 4 h.
Colony formation assays
Cells in the amount of 5 × 105 were seeded on a 10-cm plate, followed by treatment with CPT as specified in the figure legends. Afterward, the medium was removed, and cells were washed with PBS and grown in complete DMEM for a further 2 d. Then the cells were washed with PBS, fixed with ice-cold methanol for 10 min, and stained with 0.5% (wt/vol) crystal violet (in 25% ethanol) for 10 min. The excess dye was removed by rinsing the cells twice with distilled water, and the plates were photographed.
Monoclonal anti-HIPK2 and anti-HIPK3 antibodies
The development of HIPK2 monoclonal antibodies was done as described (de la Vega et al., 2013), and one of the clones recognizing HIPK2 with a high affinity (C-5C6, rat IgG2a) was used in this study. To develop monoclonal antibodies recognizing HIPK3, the region encompassing sequences between amino acids 881 and 1050 of human HIPK3 was expressed as a histidine-tagged protein in E. coli. The HIPK3 fragment was purified under denaturing conditions and dialyzed against PBS. A 50-μg amount of the purified protein was injected intraperitoneally and subcutaneously into LOU/C rats using incomplete Freund’s adjuvant supplemented with 5 nmol of CpG 2006 (TIB MOLBIOL, Berlin, Germany). After a 6-wk interval, a final boost with 50 μg of HIPK3 protein was given intraperitoneally and subcutaneously 3 d before fusion. Fusions of the myeloma cell line P3×63-Ag8.653 with the rat immune spleen cells were performed according to standard procedures. Hybridoma supernatants were tested in a solid-phase immunoassay with HIPK3 coated to enzyme-linked immunosorbent assay plates. Antibodies from tissue culture supernatant bound to HIPK3 were detected with horseradish peroxidase (HRP)–conjugated monoclonal antibodies (mAbs) against the rat IgG isotypes (TIB173 IgG2a, TIB174 IgG2b, TIB170 IgG1 [all from the American Type Culture Collection], and R-2c IgG2c [home made]), thus avoiding mAbs of IgM class. HRP was visualized with ready-to-use TMB (1-StepTM Ultra TMB-ELISA; Thermo Fisher). The mAbs that reacted specifically with HIPK3 were further analyzed in Western blotting. The clones 5C1 (R-G1) and 19A8 (R-G1) were selected and subsequently used for Western blotting.
Supplementary Material
Acknowledgments
We are grateful to Elisa Izaurralde (Max Planck-Institute for Developmental Biology, Tübingen, Germany) for helpful discussions, the plasmids for tethering assays, and antibodies recognizing CNOT1. We thank all colleagues who generously provided plasmids, especially O. S. Gabrielsen (University of Oslo, Norway), J. J. Palvimo (University of Helsinki, Finland), M. Timmers (University of Utrecht, Netherlands), S. Winkler (University of Nottingham, United Kingdom), F. Zhang (Massachusetts Institute of Technology, Boston, MA), R. Singh (Iowa State University, Ames, IA), and S. Hoshino (University of Nagoya, Japan). We are also indebted to Issay Kitabayashi (National Cancer Center Research Institute, Tokyo, Japan) for sharing HIPK2-deficient MEFs. This work was supported by grants from the Deutsche Forschungsgemeinschaft (SCHM 1417/8-3, SCHM 1417/9-1), the Deutsche Krebshilfe (111447), SFB/TRR81, SFB1021, and the Excellence Cluster Cardio-Pulmonary System.
Abbreviations used:
- CBB
Coomassie brilliant blue
- CCR4
carbon catabolite repressor 4
- CPT
camptothecin
- CRISPR
clustered regularly interspaced short palindromic repeats
- DTBP
dimethyl dithiobispropionimidate
- HIPK
homeodomain-interacting protein kinase
- MEF
mouse embryonic fibroblast
- NOT
negative on TATA
- PB
processing body
- PDCD4
programmed cell death protein 4
- PI
propidium iodide
- SUMO
small ubiquitin-related modifier
- SG
stress granule.
Footnotes
This article was published online ahead of print in MBoC in Press (http://www.molbiolcell.org/cgi/doi/10.1091/mbc.E15-09-0629) on April 27, 2016.
REFERENCES
- Aulas A, Caron G, Gkogkas CG, Mohamed NV, Destroismaisons L, Sonenberg N, Leclerc N, Parker JA, Vande Velde C. G3BP1 promotes stress-induced RNA granule interactions to preserve polyadenylated mRNA. J Cell Biol. 2015;209:73–84. doi: 10.1083/jcb.201408092. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Babbarwal V, Fu J, Reese JC. The Rpb4/7 module of RNA polymerase II is required for carbon catabolite repressor protein 4-negative on TATA (Ccr4-not) complex to promote elongation. J Biol Chem. 2014;289:33125–33130. doi: 10.1074/jbc.C114.601088. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bartlam M, Yamamoto T. The structural basis for deadenylation by the CCR4-NOT complex. Protein Cell. 2010;1:443–452. doi: 10.1007/s13238-010-0060-8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Boland A, Chen Y, Raisch T, Jonas S, Kuzuoglu-Ozturk D, Wohlbold L, Weichenrieder O, Izaurralde E. Structure and assembly of the NOT module of the human CCR4-NOT complex. Nat Struct Mol Biol. 2013;20:1289–1297. doi: 10.1038/nsmb.2681. [DOI] [PubMed] [Google Scholar]
- Buchan JR, Parker R. Eukaryotic stress granules: the ins and outs of translation. Mol Cell. 2009;36:932–941. doi: 10.1016/j.molcel.2009.11.020. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Collart MA, Panasenko OO. The Ccr4–not complex. Gene. 2012;492:42–53. doi: 10.1016/j.gene.2011.09.033. [DOI] [PubMed] [Google Scholar]
- Cooke A, Prigge A, Wickens M. Translational repression by deadenylases. J Biol Chem. 2010;285:28506–28513. doi: 10.1074/jbc.M110.150763. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cox J, Mann M. MaxQuant enables high peptide identification rates, individualized p.p.b.-range mass accuracies and proteome-wide protein quantification. Nat Biotechnol. 2008;26:1367–1372. doi: 10.1038/nbt.1511. [DOI] [PubMed] [Google Scholar]
- de la Vega L, Frobius K, Moreno R, Calzado MA, Geng H, Schmitz ML. Control of nuclear HIPK2 localization and function by a SUMO interaction motif. Biochim Biophys Acta. 2011;1813:283–297. doi: 10.1016/j.bbamcr.2010.11.022. [DOI] [PubMed] [Google Scholar]
- de la Vega L, Grishina I, Moreno R, Kruger M, Braun T, Schmitz ML. A redox-regulated SUMO/acetylation switch of HIPK2 controls the survival threshold to oxidative stress. Mol Cell. 2012;46:472–483. doi: 10.1016/j.molcel.2012.03.003. [DOI] [PubMed] [Google Scholar]
- de la Vega L, Hornung J, Kremmer E, Milanovic M, Schmitz ML. Homeodomain-interacting protein kinase 2-dependent repression of myogenic differentiation is relieved by its caspase-mediated cleavage. Nucleic Acids Res. 2013;41:5731–5745. doi: 10.1093/nar/gkt262. [DOI] [PMC free article] [PubMed] [Google Scholar]
- D’Orazi G, Cecchinelli B, Bruno T, Manni I, Higashimoto Y, Saito S, Gostissa M, Coen S, Marchetti A, Del Sal G, et al. Homeodomain-interacting protein kinase-2 phosphorylates p53 at Ser 46 and mediates apoptosis. Nat Cell Biol. 2002;4:11–19. doi: 10.1038/ncb714. [DOI] [PubMed] [Google Scholar]
- D’Orazi G, Rinaldo C, Soddu S. Updates on HIPK2: a resourceful oncosuppressor for clearing cancer. J Exp Clin Cancer Res. 2012;31:63. doi: 10.1186/1756-9966-31-63. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dumont A, Hehner SP, Hofmann TG, Ueffing M, Droge W, Schmitz ML. Hydrogen peroxide-induced apoptosis is CD95-independent, requires the release of mitochondria-derived reactive oxygen species and the activation of NF-kappaB. Oncogene. 1999;18:747–757. doi: 10.1038/sj.onc.1202325. [DOI] [PubMed] [Google Scholar]
- Dutta A, Babbarwal V, Fu J, Brunke-Reese D, Libert DM, Willis J, Reese JC. Ccr4-Not and TFIIS function cooperatively to rescue arrested RNA polymerase II. Mol Cell Biol. 2015;35:1915–1925. doi: 10.1128/MCB.00044-15. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gehring NH, Hentze MW, Kulozik AE. Tethering assays to investigate nonsense-mediated mRNA decay activating proteins. Methods Enzymol. 2008;448:467–482. doi: 10.1016/S0076-6879(08)02623-2. [DOI] [PubMed] [Google Scholar]
- Hofmann TG, Glas C, Bitomsky N. HIPK2: A tumour suppressor that controls DNA damage-induced cell fate and cytokinesis. Bioessays. 2013;35:55–64. doi: 10.1002/bies.201200060. [DOI] [PubMed] [Google Scholar]
- Hofmann TG, Moller A, Sirma H, Zentgraf H, Taya Y, Droge W, Will H, Schmitz ML. Regulation of p53 activity by its interaction with homeodomain-interacting protein kinase-2. Nat Cell Biol. 2002;4:1–10. doi: 10.1038/ncb715. [DOI] [PubMed] [Google Scholar]
- Hölper S, Nolte H, Bober E, Braun T, Kruger M. Dissection of metabolic pathways in the Db/Db mouse model by integrative proteome and acetylome analysis. Mol Biosystems. 2015;11:908–922. doi: 10.1039/c4mb00490f. [DOI] [PubMed] [Google Scholar]
- Houghton PJ, Cheshire PJ, Hallman JD, 2nd, Lutz L, Friedman HS, Danks MK, Houghton JA. Efficacy of topoisomerase I inhibitors, topotecan and irinotecan, administered at low dose levels in protracted schedules to mice bearing xenografts of human tumors. Cancer Chemother Pharmacol. 1995;36:393–403. doi: 10.1007/BF00686188. [DOI] [PubMed] [Google Scholar]
- Houseley J, Tollervey D. The many pathways of RNA degradation. Cell. 2009;136:763–776. doi: 10.1016/j.cell.2009.01.019. [DOI] [PubMed] [Google Scholar]
- Isono K, Nemoto K, Li Y, Takada Y, Suzuki R, Katsuki M, Nakagawara A, Koseki H. Overlapping roles for homeodomain-interacting protein kinases hipk1 and hipk2 in the mediation of cell growth in response to morphogenetic and genotoxic signals. Mol Cell Biol. 2006;26:2758–2771. doi: 10.1128/MCB.26.7.2758-2771.2006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ito K, Inoue T, Yokoyama K, Morita M, Suzuki T, Yamamoto T. CNOT2 depletion disrupts and inhibits the CCR4-NOT deadenylase complex and induces apoptotic cell death. Genes Cells. 2011;16:368–379. doi: 10.1111/j.1365-2443.2011.01492.x. [DOI] [PubMed] [Google Scholar]
- Jeck WR, Sorrentino JA, Wang K, Slevin MK, Burd CE, Liu J, Marzluff WF, Sharpless NE. Circular RNAs are abundant, conserved, and associated with ALU repeats. RNA. 2013;19:141–157. doi: 10.1261/rna.035667.112. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kerr SC, Azzouz N, Fuchs SM, Collart MA, Strahl BD, Corbett AH, Laribee RN. The Ccr4-Not complex interacts with the mRNA export machinery. PLoS One. 2011;6:e18302. doi: 10.1371/journal.pone.0018302. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kim YH, Choi CY, Lee SJ, Conti MA, Kim Y. Homeodomain-interacting protein kinases, a novel family of co-repressors for homeodomain transcription factors. J Biol Chem. 1998;273:25875–25879. doi: 10.1074/jbc.273.40.25875. [DOI] [PubMed] [Google Scholar]
- Laribee RN, Shibata Y, Mersman DP, Collins SR, Kemmeren P, Roguev A, Weissman JS, Briggs SD, Krogan NJ, Strahl BD. CCR4/NOT complex associates with the proteasome and regulates histone methylation. Proc Natl Acad Sci USA. 2007;104:5836–5841. doi: 10.1073/pnas.0607996104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lassus P, Ferlin M, Piette J, Hibner U. Anti-apoptotic activity of low levels of wild-type p53. EMBO J. 1996;15:4566–4573. [PMC free article] [PubMed] [Google Scholar]
- Lau NC, Kolkman A, van Schaik FM, Mulder KW, Pijnappel WW, Heck AJ, Timmers HT. Human Ccr4-Not complexes contain variable deadenylase subunits. Biochem J. 2009;422:443–453. doi: 10.1042/BJ20090500. [DOI] [PubMed] [Google Scholar]
- Lau NC, Mulder KW, Brenkman AB, Mohammed S, van den Broek NJ, Heck AJ, Timmers HT. Phosphorylation of Not4p functions parallel to BUR2 to regulate resistance to cellular stresses in Saccharomyces cerevisiae. PLoS One. 2010;5:e9864. doi: 10.1371/journal.pone.0009864. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lazzari C, Prodosmo A, Siepi F, Rinaldo C, Galli F, Gentileschi M, Bartolazzi A, Costanzo A, Sacchi A, Guerrini L, et al. HIPK2 phosphorylates DeltaNp63alpha and promotes its degradation in response to DNA damage. Oncogene. 2011;30:4802–4813. doi: 10.1038/onc.2011.182. [DOI] [PubMed] [Google Scholar]
- Lemaire M, Collart MA. The TATA-binding protein-associated factor yTafII19p functionally interacts with components of the global transcriptional regulator Ccr4-Not complex and physically interacts with the Not5 subunit. J Biol Chem. 2000;275:26925–26934. doi: 10.1074/jbc.M002701200. [DOI] [PubMed] [Google Scholar]
- Li X, Luo Y, Yu L, Lin Y, Luo D, Zhang H, He Y, Kim YO, Kim Y, Tang S, et al. SENP1 mediates TNF-induced desumoylation and cytoplasmic translocation of HIPK1 to enhance ASK1-dependent apoptosis. Cell Death Differ. 2008;15:739–750. doi: 10.1038/sj.cdd.4402303. [DOI] [PubMed] [Google Scholar]
- Lin J, Zhang Q, Lu Y, Xue W, Xu Y, Zhu Y, Hu X. Downregulation of HIPK2 increases resistance of bladder cancer cell to cisplatin by regulating Wip1. PLoS One. 2014;9:e98418. doi: 10.1371/journal.pone.0098418. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Maryati M, Airhihen B, Winkler GS. The enzyme activities of Caf1 and Ccr4 are both required for deadenylation by the human Ccr4-Not nuclease module. Biochem J. 2015;469:169–176. doi: 10.1042/BJ20150304. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Milanovic M, Kracht M, Schmitz ML. The cytokine-induced conformational switch of nuclear factor kappaB p65 is mediated by p65 phosphorylation. Biochem J. 2014;457:401–413. doi: 10.1042/BJ20130780. [DOI] [PubMed] [Google Scholar]
- Miller JE, Reese JC. Ccr4-Not complex: the control freak of eukaryotic cells. Crit Rev Biochem Mol Biol. 2012;47:315–333. doi: 10.3109/10409238.2012.667214. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Moriya H, Shimizu-Yoshida Y, Omori A, Iwashita S, Katoh M, Sakai A. Yak1p, a DYRK family kinase, translocates to the nucleus and phosphorylates yeast Pop2p in response to a glucose signal. Genes Dev. 2001;15:1217–1228. doi: 10.1101/gad.884001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Norbeck J ( Carbon source dependent dynamics of the Ccr4-Not complex in Saccharomyces cerevisiae. J Microbiol. 2008;46:692–696. doi: 10.1007/s12275-008-0122-2. [DOI] [PubMed] [Google Scholar]
- Ohnheiser J, Ferlemann E, Haas A, Muller JP, Werwein E, Fehler O, Biyanee A, Klempnauer KH. Programmed cell death 4 protein (Pdcd4) and homeodomain-interacting protein kinase 2 (Hipk2) antagonistically control translation of Hipk2 mRNA. Biochim Biophys Acta. 2015;1853:1564–1573. doi: 10.1016/j.bbamcr.2015.03.008. [DOI] [PubMed] [Google Scholar]
- Panasenko OO, Collart MA. Not4 E3 ligase contributes to proteasome assembly and functional integrity in part through Ecm29. Mol Cell Biol. 2011;31:1610–1623. doi: 10.1128/MCB.01210-10. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Preissler S, Reuther J, Koch M, Scior A, Bruderek M, Frickey T, Deuerling E. Not4-dependent translational repression is important for cellular protein homeostasis in yeast. EMBO J. 2015;34:1905–1924. doi: 10.15252/embj.201490194. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Radhakrishnan SK, Kamalakaran S. Pro-apoptotic role of NF-kappaB: implications for cancer therapy. Biochim Biophys Acta. 2006;1766:53–62. doi: 10.1016/j.bbcan.2006.02.001. [DOI] [PubMed] [Google Scholar]
- Renner F, Saul VV, Pagenstecher A, Wittwer T, Schmitz ML. Inducible SUMO modification of TANK alleviates its repression of TLR7 signalling. EMBO Rep. 2011;12:129–135. doi: 10.1038/embor.2010.207. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Reuven N, Adler J, Porat Z, Polonio-Vallon T, Hofmann TG, Shaul Y. The tyrosine kinase c-Abl promotes homeodomain-interacting protein kinase 2 (HIPK2) accumulation and activation in response to DNA Damage. J Biol Chem. 2015;290:16478–16488. doi: 10.1074/jbc.M114.628982. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Russell P, Benson JD, Denis CL. Characterization of mutations in NOT2 indicates that it plays an important role in maintaining the integrity of the CCR4-NOT complex. J Mol Biol. 2002;322:27–39. doi: 10.1016/s0022-2836(02)00707-6. [DOI] [PubMed] [Google Scholar]
- Sakamoto K, Huang BW, Iwasaki K, Hailemariam K, Ninomiya-Tsuji J, Tsuji Y. Regulation of genotoxic stress response by homeodomain-interacting protein kinase 2 through phosphorylation of cyclic AMP response element-binding protein at serine 271. Mol Biol Cell. 2010;21:2966–2974. doi: 10.1091/mbc.E10-01-0015. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Saul VV, de la Vega L, Milanovic M, Krüger M, Braun T, Fritz-Wolf K, Becker K, Schmitz ML. HIPK2 kinase activity depends on cis-autophosphorylation of its activation loop. J Mol Cell Biol. 2013;5:27–38. doi: 10.1093/jmcb/mjs053. [DOI] [PubMed] [Google Scholar]
- Saul VV, Schmitz ML. Posttranslational modifications regulate HIPK2, a driver of proliferative diseases. J Mol Med (Berl) 2013;91:1051–1058. doi: 10.1007/s00109-013-1042-0. [DOI] [PubMed] [Google Scholar]
- Schmitz ML, Rodriguez-Gil A, Hornung J. Integration of stress signals by homeodomain interacting protein kinases. Biol Chem. 2014;395:375–386. doi: 10.1515/hsz-2013-0264. [DOI] [PubMed] [Google Scholar]
- Shang Y, Doan CN, Arnold TD, Lee S, Tang AA, Reichardt LF, Huang EJ. Transcriptional corepressors HIPK1 and HIPK2 control angiogenesis via TGF-beta-TAK1-dependent mechanism. PLoS Biol. 2013;11:e1001527. doi: 10.1371/journal.pbio.1001527. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shi J, Nelson MA. The cyclin-dependent kinase 11 interacts with NOT2. Biochem Biophys Res Commun. 2005;334:1310–1316. doi: 10.1016/j.bbrc.2005.07.026. [DOI] [PubMed] [Google Scholar]
- Siepi F, Gatti V, Camerini S, Crescenzi M, Soddu S. Homeodomain-interacting protein kinase 2 (HIPK2) catalytic activity and specificity are regulated by activation-loop Y354 autophosphorylation. Biochim Biophys Acta. 2013;1833:1443–1453. doi: 10.1016/j.bbamcr.2013.02.018. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Smeenk L, van Heeringen SJ, Koeppel M, Gilbert B, Janssen-Megens E, Stunnenberg HG, Lohrum M. Role of p53 serine 46 in p53 target gene regulation. PLoS One. 2011;6:e17574. doi: 10.1371/journal.pone.0017574. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tucker M, Valencia-Sanchez MA, Staples RR, Chen J, Denis CL, Parker R. The transcription factor associated Ccr4 and Caf1 proteins are components of the major cytoplasmic mRNA deadenylase in Saccharomyces cerevisiae. Cell. 2001;104:377–386. doi: 10.1016/s0092-8674(01)00225-2. [DOI] [PubMed] [Google Scholar]
- van der Laden J, Soppa U, Becker W. Effect of tyrosine autophosphorylation on catalytic activity and subcellular localisation of homeodomain-interacting protein kinases (HIPK) Cell Commun Signal. 2015;13:3. doi: 10.1186/s12964-014-0082-6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Vousden KH. Outcomes of p53 activation–spoilt for choice. J Cell Sci. 2006;119:5015–5020. doi: 10.1242/jcs.03293. [DOI] [PubMed] [Google Scholar]
- Zekri L, Kuzuoglu-Ozturk D, Izaurralde E. GW182 proteins cause PABP dissociation from silenced miRNA targets in the absence of deadenylation. EMBO J. 2013;32:1052–1065. doi: 10.1038/emboj.2013.44. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhang Q, Nottke A, Goodman RH. Homeodomain-interacting protein kinase-2 mediates CtBP phosphorylation and degradation in UV-triggered apoptosis. Proc Natl Acad Sci USA. 2005;102:2802–2807. doi: 10.1073/pnas.0409373102. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.