

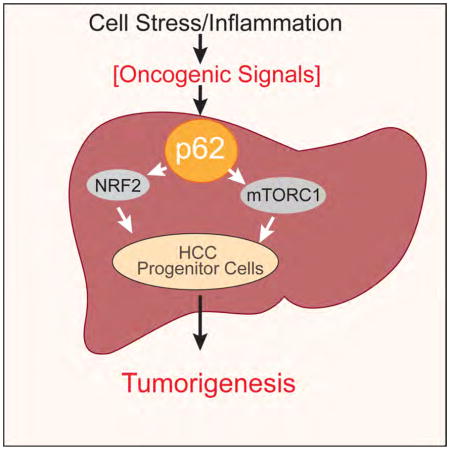
Summary
p62 is a ubiquitin-binding autophagy receptor and signaling protein that accumulates in premalignant liver diseases and most hepatocellular carcinomas (HCC). Although p62 was proposed to participate in formation of benign adenomas in autophagy-deficient livers, its role in HCC initiation was not explored. Here we show that p62 is necessary and sufficient for HCC induction in mice and that its high expression level in non-tumor human liver predicts rapid HCC recurrence after curative ablation. High p62 expression is needed for activation of NRF2 and mTORC1, induction of c-Myc and protection of HCC-initiating cells from oxidative stress-induced death.
Graphical abstract
Introduction
Despite extensive exploration, cancer initiation remains obscure. Most neoplasms form in response to environmental carcinogens that induce cancer-initiating mutations (Luch, 2005). In some cases, e.g. colorectal cancer, these mutations occur in rapidly dividing adult tissue stem cells (van de Wetering et al., 2015), but in other cases, such as hepatocellular carcinoma (HCC), cancer likely emerges from terminally differentiated cells (Font-Burgada et al., 2015). Since a typical cancer requires 2–8 driver mutations (Vogelstein et al., 2013), cancer-initiating cells should survive environmental insults, divide and give rise to a cluster of initiated cells that remain insult resistant and thus capable of accumulating additional mutations. However, how cancer-initiating cells escape death to keep accumulating driver mutations has not been fully investigated.
A common malignancy linked to chronic tissue damage, stress and environmental carcinogen exposure is HCC, most of which arise at the end stage of chronic liver disease and persistent inflammation, with HBV or HCV infections being the current leading causes (El-Serag, 2011). However, obesity and alcohol consumption, which cause hepatic steatosis (non-alcoholic steatohepatitis, NASH, or alcoholic steatohepatitis, ASH, respectively) and fibrosis, are rapidly growing in their importance as HCC risk factors (Starley et al., 2010). Mouse studies show that obesity promotes HCC through inflammation-related mechanisms (Nakagawa et al., 2014; Park et al., 2010; Wolf et al., 2014), but it is unclear how diverse forms of hepatitis and liver damage initiate HCC. Identification of a common pathogenic pathway is important for early HCC detection and improved prevention.
The heterogeneous nature of HCC, in which over 28,000 different somatic mutations have been identified (Schulze et al., 2015; Shibata and Aburatani, 2014), renders early detection and prevention challenging. Most HCCs exhibit a median of 21 silent and 64 non-silent mutations that affect 8–11 major signaling pathways and biological processes, including cellular stress responses (Schulze et al., 2015; Shibata and Aburatani, 2014). Stress responses are elicited by endoplasmic reticulum (ER) and oxidative stresses, viral hepatitis, hemochromatosis, and ASH and NASH (Malhi and Kaufman, 2011; Starley et al., 2010), and they control NASH to HCC progression in mice (Nakagawa et al., 2014). ER stress enhances oxidative stress, which is relieved by nuclear factor erythroid 2 (NRF2) by inducing expression of genes encoding antioxidant and detoxifying enzymes (Suzuki et al., 2013). NRF2 is regulated post-translationally by the E3 ubiquitin ligase Keap1, which induces NRF2 ubiquitination and proteasomal degradation. Upon accumulation of reactive oxygen species (ROS) or electrophiles, Keap1 is oxidized and unable to bind newly translated NRF2, which is stabilized and enters the nucleus to activate genes harboring anti-oxidant response elements (ARE). Because products of NRF2-regulated genes detoxify electrophiles and promote glutathione (GSH) synthesis, transient activation of this system was proposed to inhibit carcinogenesis in liver and other organs (Hayes and McMahon, 2009; Suzuki et al., 2013). Surprisingly, however, KEAP1 inactivating mutations occur in lung cancer and HCC (Ohta et al., 2008; Schulze et al., 2015; Totoki et al., 2014), and activating mutations in the gene encoding NRF2, NFE2L2, that inhibit Keap1-mediated degradation were found in lung, esophageal, skin and liver cancers (Guichard et al., 2012; Kim et al., 2010; Shibata et al., 2008). Together, NFE2L2 and KEAP1 mutations occur in 14% of HCC specimens and are considered to be driver mutations (Schulze et al., 2015; Shibata and Aburatani, 2014; Totoki et al., 2014). NRF2 was proposed to contribute to liver carcinogenesis by inhibiting senescence or death of initiated hepatocytes undergoing oxidative stress (Hayes and McMahon, 2006), but direct support for this hypothesis, first postulated by Farber (Farber, 1990), is lacking. Curiously, carcinogen-induced dysplastic lesions in rats, known as foci of altered hepatocytes (FAH), express high amounts of glutathione S transferase P1 (Gstp1), NAD(P)H:quinone oxidoreductase 1 (Nqo1) and other protective enzymes (Farber, 1990), now known as NRF2 targets (Hayes and McMahon, 2009). As a result, FAH are probably more resistant to oxidative stress and environmental toxins than the surrounding liver. Thus, antioxidants may promote rather than suppress HCC development as previously found (Maeda et al., 2005). It was also questioned whether FAH, also found in human cirrhotic and preneoplastic livers, are true HCC progenitors or reflect compensatory proliferation triggered by liver damage (Sell and Leffert, 2008). We have settled this controversy by isolating small hepatocytic cells from mouse dysplastic lesions that give rise to HCC after transplantation into a damaged liver (He et al., 2013). In addition to cancer stem cell markers, HCC progenitor cells (HcPC) exhibit elevated p62, which also accumulates in livers of high fat diet (HFD)-fed MUP-uPA mice that develop NASH and progress to HCC (Nakagawa et al., 2014). p62 is known to accumulate in most, if not all, chronic liver diseases that progress to HCC (Denk et al., 2006).
p62, encoded by SQSTM1, is an autophagy adaptor and a signaling scaffold with an N-terminal oligomerization domain (PB1) and a ubiquitin association domain (UBA) at its C-terminus (Komatsu et al., 2012). p62 also harbors LC3 (LIR) and Keap1 (KIR) interacting motifs, through which it binds LC3 on phagophore membranes and cytoplasmic Keap1, respectively. A main p62 function is to deliver polyubiquitinated proteins and organelles for autophagosomal-lysosomal degradation. Interference with autophagic flux attenuates p62 degradation (Komatsu et al., 2012). The latter results in p62 accumulation and oligomerization, aggregation of ubiquitinated proteins, Keap1 titration and NRF2 stabilization. As NRF2 can induce p62 expression (Jain et al., 2010), p62 accumulation can trigger a self-amplifying auto-regulatory loop that sustains NRF2 activation and p62 overexpression. p62 also binds TRAF6 to promote NF-κB activation (Sanz et al., 2000), and NF-κB can also induce p62 expression (Ling et al., 2012)Under nutrient-rich conditions, the p62:TRAF6 interaction also enhances mTORC1 activation (Duran et al., 2011; Linares et al., 2013). Importantly, p62 is a major component of intracellular hyaline bodies (IHB), Mallory-Denk bodies (MDB) and hybrid inclusions (Denk et al., 2006), which are hallmarks of chronic liver diseases that greatly enhance HCC risk (Zatloukal et al., 2007). p62-containing aggregates have been detected in 50% of surgical HCC specimens (Denk et al., 2006), but their significance remains unknown. Of note, whole body p62 disruption suppresses benign adenomas in autophagy-deficient livers (Takamura et al., 2011). These tumors, however, are not cancerous and thus the role of p62 and its mechanism of action in HCC initiation remain unknown although it was shown that p62 maintains malignancy of HCC cell lines (Ichimura et al., 2013; Inami et al., 2011). p62 has been suggested to act as an oncoprotein in renal cell carcinoma (RCC) by promoting mTORC1 activation (Li et al., 2013), while other studies have suggested it acts via NF-κB or oxidative stress (Mathew et al., 2009). p62 also accumulates in endometrial cancer (Iwadate et al., 2015). Yet, in no case was it investigated whether p62 accumulation in a preneoplastic tissue increases risk of cancer initiation.
Results
p62 is induced by diethylnitrosamine (DEN) and enhances its carcinogenic activity
We confirmed p62 accumulation in human patients suffering from chronic liver diseases that increase HCC risk, as well as HCC itself (Figure 1A). Consistent with this, in mice, p62 was rapidly induced in liver after challenge with a high dose of DEN (100 mg/kg), mainly around the central vein (Figure 1B), the area within which DEN undergoes metabolic activation (Kang et al., 2007). A carcinogenic DEN dose (25 mg/kg) followed by HFD feeding also led to p62 accumulation, but this time the process was slower and after 2–4 months only a few hepatocytic cell clusters were p62-high (Figures 1B and S1A). At later time points p62-high cells were mainly restricted to tumor nodules and no differences in p62 expression were noted between mice given DEN alone or DEN plus HFD. To determine p62 role in hepatocarcinogenesis we generated Sqstm1Δhep mice lacking p62 in hepatocytes (Figure S1B). Immunoblot (IB) analysis revealed complete absence of p62 protein and qPCR analysis showed reduced Nqo1 mRNA induction after DEN challenge (Figures 1C and S1C). The Sqstm1Δhep liver, however, was phenotypically normal and did not display any damage (Figure 1D). Two week old Sqstm1Δhep males given DEN alone or DEN plus HFD (Park et al., 2010) developed 55% fewer HCCs than similarly treated Sqstm1F/F mice (Figures 1E and S1D). Maximal tumor size, however, remained unchanged; suggesting that p62 mainly affects tumor initiation rather than growth. p62-deficient HCCs expressed less α-fetoprotein (AFP) than p62-proficient HCCs, but glypican 3 (GPC3) remained unchanged (Figures 1F and S1E), suggesting that p62-deficient tumors contain fewer poorly differentiated cells. All DEN-induced tumors were histologically typical HCCs, as previously found (Maeda et al., 2005). IB analysis confirmed that most HCCs in Sqstm1F/F mice displayed high p62 expression, which was also seen in non-tumor areas (Figure 1G). p62 expression generally correlated with NRF2 stabilization and elevated Nqo1 expression (Figure 1G and S1F). p62-deficient tumors expressed much less NRF2-dependent or Nqo1 (Figures 1G and S1E and S1F). p62 deletion reduced tumoral expression of NRF2 targets, including Hmox1, Gstm1 and Nqo1 (Figure S1F). c-Myc expression, which is frequently elevated in HCC (Kaposi-Novak et al., 2009; Qu et al., 2014), also correlated with elevated p62 (Figure 1G).
Figure 1. Diminished DEN-induced hepatocarcinogenesis in Sqstm1Δhep mice.

(A) p62-positive aggregates in human LC/HCC (liver cirrhosis/HCC), NASH, and ASH specimens (n=3). (B) Left: rapid p62 induction in mouse livers (n=3) after high dose DEN (100 mg/kg of body weight, BW). Right: chronic p62 expression in mouse livers (n=3) after low dose DEN (25 mg/kg BW) and HFD feeding. (C) Nqo1 liver mRNA expression 48 hr after DEN injection analyzed by qPCR. Results are mean ± S.D. (n=3). (D) Hematoxylin and eosin (H&E) staining of Sqstm1F/F and Sqstm1Δhep livers (n=4). (E) Gross appearance, tumor multiplicity and maximal sizes of DEN-induced HCCs. Results are mean ± S.E.M. (n=11–13). (F) Relative mRNA amounts of HCC and progenitor markers as well as Sqstm1 in tumors from DEN-injected mice. Results are mean ± S.D. (n=4). (G) IB analysis of p62, nuclear NRF2, HDAC1, Nqo1, c-Myc, and tubulin in tumor (T) and non-tumor (NT) liver tissue from DEN-injected mice (n=5–6). Scale bars, 100 μm. **: p < 0.01, ***: p < 0.001. See also Figure S1.
p62 is required for HCC initiation in Tsc1Δhep mice
Tsc1Δhep male mice, in which hepatocyte mTORC1 is constitutively activated, exhibit spontaneous HCC development preceded by fibrosis, liver damage and p62 accumulation (Menon et al., 2012). mTORC1 activation, however, is dispensable for DEN-induced HCC; in fact RptorΔhep mice exhibit enhanced HCC development (Umemura et al., 2014). To determine if p62 participates in mTORC1-driven hepatocarcinogenesis, we generated Tsc1Δhep mice, which developed typical HCCs as described above, and crossed them with Sqstm1Δhep mice to generate Tsc1/Sqstm1Δhep double mutants (Figures 2A and S2A). p62 ablation did not restore LC3 lipidation but reduced p70S6 kinase (S6K) phosphorylation. Remarkably, liver-specific p62 ablation completely suppressed HCC development (Figure 2B). As expected, p62 was upregulated in non-tumor and tumor areas of Tsc1Δhep liver and its upregulation correlated with enhanced NRF2, c-Myc and NRF2 target gene expression (Figures 2C and 2D). p62 ablation in Tsc1Δhep liver reduced S6K and S6 phosphorylation, suggesting decreased mTORC1 activity (Figures 2A and 2C and S2B). However, restoration of AKT phosphorylation, which is inhibited upon S6K activation (Hay and Sonenberg, 2004) was variable (Figures 2A and 2C). p62 ablation reduced expression of fibrogenic genes and the YAP targets connective tissue growth factor (Ctgf) and amphiregulin (Areg), which are upregulated in the Tsc1Δhep liver (Figure 2D). Inflammation and fibrotic changes were also reduced in the Tsc1/Sqstm1Δhep liver relative to Tsc1Δhep livers (Figure S2B). Of note, p62 ablation reduced the abundance of ROS-accumulating cells that are stained by dihydroethidine (DHE; Figure S2B). Consistent with increased S6K activation, Tsc1Δhep HCCs exhibited elevated S6 phosphorylation and c-Myc (Figure S2C).
Figure 2. p62 ablation blocks HCC development in Tsc1Δhep mice.

(A) IB analysis of liver tissues from indicated mice (n=4–6). n.s., nonspecific band. (B) Upper panel: gross liver morphology of 10 month old male mice of indicated genotypes. Lower panel: tumor numbers and maximal sizes. (n=12–16). Results are mean ± S.E.M. (C) IB analysis and quantitation of protein expression and phosphorylation in tumor (T) and non-tumor (NT) liver tissue. Results are mean ± S.D. (n=3–4). (D) qPCR analysis of relative mRNA levels of Sqstm1, NRF2 targets (Nqo1, Gstm1, Hmox1), fibrogenic markers (Col1A1, Col4A1, Acta2, Timp1) and YAP targets (Ctgf and Areg) in livers of 10 month old mice. (n=3–4 per genotype). Results are mean ± S.D. *p < 0.05, **p < 0.01, ***p < 0.001. See also Figure S2.
p62 accelerates NASH to HCC progression
Hypernutrition activates mTORC1 resulting in inhibitory ULK1/2 phosphorylation, reduced autophagic flux and p62 accumulation (Schneider and Cuervo, 2014). We developed a new mouse model of NASH-driven HCC by feeding MUP-uPA mice with HFD, which induces p62 accumulation in non-tumor liver and HCCs (Nakagawa et al., 2014). Surprisingly, HFD feeding to WT B6 mice, which do not develop NASH or HCC, did not induce p62 accumulation (Figure 3A). In contrast, HFD induced strong p62 expression in MUP-uPA mice (Figure 3A), suggesting that p62 accumulation is tightly linked to NASH and HCC development. To probe the pathogenic role of p62, we crossed MUP-uPA and Sqstm1Δhep (Sqstm1Δhep/MUP) mice and placed male progeny on HFD. Although p62 ablation did not decrease liver lipid droplets or triglycerides, it led to reduced fibrosis and nearly complete absence of cells that accumulate superoxides, that are DHE positive (Figures 3B–3D). Notably, p62 ablation substantially suppressed HCC development (Figure 3E). Both tumor number and maximal size were reduced in Sqstm1Δhep/MUP mice, and no p62 expression was seen in remaining tumors (Figure 3B).
Figure 3. p62 ablation prevents HCC development but not steatosis in MUP-uPA mice.

(A) IB analysis of liver tissue lysates from BL6 (WT) and MUP-uPA mice of indicated ages kept on LFD or HFD. (n=2–3). (B) Histochemistry of livers from 5 month old mice kept on HFD. Lower panel shows p62 staining in tumors of 10 month old mice on HFD. Arrows indicate tumor portion. Scale bars, 100 μm. Graphs indicate % areas occupied by stained cells. Results are mean ± S.D. (n=3–5). (C) Liver triglyceride (TG) content of 5 month old mice on HFD. Results are mean ± S.D. (n=4). (D) Relative mRNA amounts of fibrogenic markers in mice of indicated genotypes. Results are mean ± S.D. (n=4) (E) Gross liver morphology (upper), and tumor numbers and maximal sizes (lower) in 10 month old mice kept on HFD. Results are mean ± S.E.M. (n=7–11). *p < 0.05, **p < 0.01, ***p < 0.001. See also Figure S3.
NASH development and HCC progression is also seen in the STZ-HFD model, based on induction of type I diabetes (β cell destruction) with streptozotocin (STZ) followed by HFD feeding (Kawai et al., 2012). STZ-HFD mice develop HCC, whose histological features were similar to HCC in MUP-uPA mice including p62 accumulation. Although p62 deletion in the STZ-HFD model did not suppress fibrosis, steatosis or triglyceride accumulation, it strongly suppressed HCC development (Figures S3A–D).
Ectopic p62 expression induces HCC
We examined whether ectopic p62 expression can induce HCC without carcinogen administration, hypernutrition or deliberate tissue injury. We generated plasmid vectors expressing GFP and Flag-tagged p62 proteins, either full-length (FL), KIR-deficient (KIR−) or UBA-deleted (ΔUBA) (Figure 4A). Transfected mouse hepatocytes exhibited efficient expression of all 3 p62 variants (Figures 4B and 4C). Whereas p62 FL and p62 KIR− formed aggregates, the ΔUBA construct did not (Figure 4C). p62 FL and p62 ΔUBA, but not p62 KIR−, induced endogenous NRF2 targets, including Nqo1 and Hmox1 (Figures 4B and S4A). We transferred the different constructs into adeno-associated virus (AAV) 8 vectors, which exhibit strong liver tropism and stably express ectopic proteins and confirmed that GFP was efficiently expressed in hepatocytes for at least 12 months after AAV infection (Figure S4B). Ectopic p62 was expressed in cells expressing the Flag epitope (Figure S4C). Notably, p62 FL and p62 ΔUBA induced tumors with either steatotic or typical HCC morphologies at 12 months after infection of WT male mice (Figures 4D and S4D). Tumor multiplicity was 50% of that seen in HFD-fed MUP-uPA mice. No significant differences between tumor multiplicity were observed between p62 FL and p62 ΔUBA infected mice, although tumors in the latter were somewhat smaller, and no tumors were observed in mice receiving p62 KIR− (Figure 4D). Only one mouse infected with AAV-GFP and kept on HFD developed a tumor at 12 months. Although AAV injection into mice was reported to induce occasional liver tumors, these tumors were found at 14 to 25 months post-infection (Chandler et al., 2015), well beyond the time point at which we examined our mice. Some p62-induced tumors exhibited lipid droplets, ballooning deformity, and MDB, which are common features of steatohepatitic HCCs (Figure S4D). All p62-induced HCCs expressed exogenous and endogenous p62 and Flag epitope, together with elevated NRF2 and Nqo1 (Figure 4E). S6K phosphorylation and c-Myc were elevated, but the LC3-II:LC3-I ratio remained high (Figure 4E). This was markedly different from Atg7Δhep liver in which p62 was strongly induced and autophagy was completely blocked, but phospho-S6K was not elevated. p62-induced tumors were positive for HCC markers, including GPC3, AFP, and YAP (Figures 4F and S4E). These tumors also included a small number of Sox9 positive cells. Tumor nodules were positive for p62 and the Flag epitope, as well as phospho-S6, but were negative for nuclear RelA (Figures 4F and S4E).
Figure 4. Ectopic p62 expression induces HCC.

(A) Schematic representation of p62 constructs (FL: full-length p62; KIR−: KIR mutated p62; ΔUBA: UBA-deleted p62. Control: Flag-only). (B) IB analysis of Flag, p62 and Nqo1 in murine hepatocytes transfected with plasmid expressing p62 or its variants. (n=3). (C) Expression patterns of p62 constructs in mouse hepatocytes analyzed by IF microscopy (left panel: Blue, DAPI; Green, p62; Red, Flag; Scale bars, 10 μm). Higher magnification of Flag-positive aggregates (right panel). Representative images of three independent experiments are shown. (D) Representative images of tumor-bearing livers from 12 month old mice infected with indicated AAV8 vectors and kept on either low fat (LFD) or high fat (HFD) diet. Tumor numbers and maximal sizes are shown to the right. p62 FL (n=13); p62 ΔUBA p62 (n=11); p62 KIR− (5 mice were infected, but only 2 were of same BL6 genotype as other mice in this figure); Control (n=10). Data are mean ± S.E.M. n.s., not significant. (E) IB analysis of lysates from BL6 and Atg7Δhep livers, three p62-induced HCCs (#1 from LFD and #2 and #3 from HFD), and p62 FL infected mouse hepatocytes (NT). Endo, endogenous p62. (F) IHC analysis of p62-induced HCCs. (n=5). (Scale bars, 100 μm). *p < 0.05, **p < 0.01, ****p < 0.0001. See also Figure S4.
Liver p62 controls NRF2 and mTORC1/c-Myc signaling
We performed RNAseq on Sqstm1Δhep and Sqstm1F/F livers to identify differentially expressed genes and signaling pathways that are strongly influenced by p62. Ingenuity Pathway Analysis (IPA) revealed that the NRF2-mediated oxidative stress response was the highest-ranking downregulated pathway in the Sqstm1Δhep liver (Figure 5A). Heatmap depiction shows downregulated “NRF2-mediated Oxidative Stress Response” related genes, including Cbr1, Gstm5, Abcc4, Nqo1 and Gpx2 in Sqstm1Δhep liver (Figure 5B). qPCR analysis of additional NRF2 genes revealed marked reduction of this pathway (Figure S5A). The products of these genes are involved in glutathione metabolism, antioxidant defenses and electrophile detoxification. c-Fos which was implicated as a key contributor to HCC pathogenesis (Min et al., 2012), was also downregulated in Sqstm1Δhep liver. We conducted similar analyses on Tsc1Δhep and Tsc1/Sqstm1Δhep livers. Again, p62 ablation reduced expression of NRF2-regulated genes (Figure 5C). In this case, however, GSEA of RNAseq data indicated that p62 ablation also reduced expression of c-Myc- and mTORC1-responsive genes (Figures 5D and 5E), in agreement with its effects on S6K phosphorylation and c-Myc expression.
Figure 5. p62-dependence of the NRF2-induced antioxidant response and mTORC1 signaling.

(A) Ingenuity Pathway Analysis of genes differentially represented in RNAseq data from Sqstm1Δhep vs. Sqstm1F/F livers (n=3). Top canonical pathways are ranked by –log (p value) with threshold p value=0.05. Highest-ranking categories are displayed along the x axis in a decreased order of significance. Blue bars: pathways with z-score<0 (downregulated pathways); grey bars: no activity pattern available, white bars: z-score=0. (B) Heatmap representation of downregulated genes in Sqstm1Δhep relative to Sqstm1F/F livers (n=3) associated with “NRF2-mediated Oxidative Stress Response” including Log2FC and adjusted p value. (C) Heatmap representation of downregulated genes in Tsc1/Sqstm1Δhep relative to Tsc1Δhep livers (n=3) including Log2FC and adjusted p value. (D) GSEA analysis showing that p62 ablation abrogates mTORC1 signaling and reduces expression of c-Myc targets that are induced upon TSC1 ablation. Top-five hallmark genesets sorted by NES comparing Tsc1/Sqstm1Δhep vs Tsc1Δhep mice (n=3). In red, genesets related to c-Myc and mTORC1. (E) Enrichment graph of mTORC1_SIGNALING dataset (n=3). See also Figure S5.
We also investigated SQSTM1 mRNA expression in different HCC datasets (Oncomine and TCGA), 6 of which showed higher SQSTM1 mRNA in HCC than in normal tissue (Figure S5B). We chose the largest and most completely annotated TCGA dataset and performed GSEA of correlation profiles. Analyses with the C3 compilation of MsigDB revealed that among genes directly correlated with SQSTM1, there is a striking enrichment for genes controlled by NRF2, including a strong positive correlation with NQO1 (Figure S5C–E). Further analysis of TCGA dataset revealed a significant tendency towards co-occurrence between SQSTM1 overexpression and NQO1 and GPX2 (Figure S5F). GSEA of these data also revealed good correlation between SQSTM1 and MYC- and mTORC1-responsive genes (Figures S5G and S5H). Furthermore, silencing of p62 in an established human HCC cell line, confirmed the relationship between elevated SQSTM1 and increased MYC and NQO1 expression and S6K phosphorylation (Figures S5I and S5J).
p62 accumulation correlates with HCC recurrence
We investigated the relationship between p62 accumulation and human HCC risk by analyzing non-tumor liver tissue of early stage HCC patients undergoing curative ablation. The results were graded from 0 to 3 depending on average number of p62-positive aggregates per field (Figure 6A). Remarkably, 79 out of 121 specimens were p62 positive. We next analyzed disease-free survival (in years) in the same patients (Figure 6B). Disease-free survival was much worse in patients with high-grade p62 in non-tumor liver than patients with low or no p62 staining. Moreover, the p62 staining index positively correlated with fibrosis score and liver stiffness assessed by histology and transient elastography, respectively (Figure 6C). These results suggest that p62 is a useful biomarker for risk of HCC recurrence that can be used to identify patients who should be closely monitored after curative HCC ablation. Kaplan-Meier survival analysis of patients stratified according to SQSTM1 expression, whose data was included in the Oncomine and TCGA datasets, revealed that high SQSTM1 expression correlated with significantly worse patient survival (Figures S6A and S6B).
Figure 6. p62-positive aggregates in human livers correlate with HCC recurrence after curative ablation.

(A) Representative images of p62 IHC of non-tumor liver biopsies from human patients with resectable HCC. Biopsies were collected when HCCs were treated curatively. Staining patterns were categorized into four grades (G0–G3) based on number of p62-positive aggregates (Scale bars, 100 μm). (B) Disease free survival curves of all 4 groups (upper panel), and G0–2 vs G3 groups (lower panel). (C) Correlation between p62 expression and fibrotic changes in background liver from above HCC patients were analyzed by histological assessment (left panel) and fibroscan (right panel). Box-and-whisker plots showing median (horizontal line), interquartile range (box) and 10th–90th percentiles (whiskers) of the data. Dots indicate outliers. Data of liver stiffness (right panel) were not available in some patients: G0 (n=32), G1 (n=41), G2 (n=22) and G3 (n=8). See also Figure S6.
Discussion
Cancer often develops in tissues, such as liver, with very low inherent cell division rates. Injury and inflammation promote such cancers by enhancing proliferation of initiated cells (Finkin et al., 2015; Grivennikov et al., 2010; Kuraishy et al., 2011), but mechanisms that increase cancer risk and operate prior to acquisition of oncogenic mutations are obscure. It is not even clear how the progeny of a single initiated cell can acquire multiple mutations needed for malignant transformation. Studying HCC, a cancer that most likely originates from differentiated hepatocytes (Font-Burgada et al., 2015), we find that accumulation of p62, a marker of chronic liver disease (Denk et al., 2006), is an early event that sets up a “field effect” within which HCC evolves. “Field cancerization” is a poorly understood process that occurs prior to emergence of neoplasia, during which a histologically invisible change renders part of a tissue highly likely to develop malignant tumors (Rubin, 2011). We show that p62 accumulation precedes HCC development in mice and humans and suggest it is responsible for increasing cancer risk by supporting survival and accumulation of ROS-containing HcPC, the cells in which HCC-driver mutations occur.
Although p62 was upregulated in autophagy-deficient livers and p62 ablation attenuated the genesis of benign hepatic adenomas (Takamura et al., 2011) and inhibited proliferation and growth of established HCC cell lines (Ichimura et al., 2013; Inami et al., 2011), the role of p62 in HCC initiation within a pre-cancerous liver was not investigated. We now show that in human patients with early HCC p62 accumulation in non-tumor liver correlates with much faster post-therapeutic recurrence and reduced disease-free survival. Hence, patients with high p62 expression in non-tumor tissue need closer surveillance and additional intervention. Using four distinct mouse HCC models of different etiologies, we show that p62 is needed for initiation of malignant tumors with classical HCC features. The most dramatic dependence on p62 was exhibited by Tsc1Δhep mice, in which HCC development depends on mTORC1 (Menon et al., 2012), which is also activated by hypernutrition, causing attenuation of autophagy and p62 accumulation (Schneider and Cuervo, 2014). Nonetheless, HFD feeding causes p62 accumulation in MUP-uPA and STZ but not in WT mice, and is not linked to the development of steatotic changes. Rather, p62 is strictly required for NASH to HCC progression. Our results also indicate that p62 accumulation can initiate HCC development, suggesting it is a critical pre-neoplastic event that leads to non-mutational activation of three important cancer drivers: NRF2, mTORC1 and c-Myc.
Pathogenic p62 accumulation in premalignant liver tissue is regulated transcriptionally and post-transcriptionally. The major activators of p62 transcription NRF2 and NF-κB (Jain et al., 2010; Ling et al., 2012) are activated during chronic liver inflammation and oxidative stress (Figure 7). In turn, p62 can activate NF-κB (Sanz et al., 2000) and NRF2 (Ichimura et al., 2013), constituting two self-amplifying autoregulatory loops. In our experimental system, exogenous p62 activated NRF2 and this may have induced endogenous p62, thereby explaining preferential p62 accumulation within tumor nodules. Even when initial p62 expression is relatively uniform, small fluctuations can generate foci of high p62 concentrations. Given the ability of p62 to activate NRF2, these are probably the preneoplastic foci previously detected by virtue of Nqo1 or Gstp1 (a marker of rat, but not mouse, FAH) expression (Farber, 1990; Hayes and McMahon, 2006).
Figure 7. p62 expression and signaling during HCC development.

p62 gene transcription is induced by NRF2 and NF-κB, which are activated by oxidative stress and inflammation, respectively. Newly synthesized p62 protein binds ubiquitinated proteins and organelles and LC3 on phagophore membranes to promote autophagy and lysosomal degradation. mTORC1 activation and ER stress promote p62 accumulation by interfering with initiation of autophagy, whereas alcohol and HCV affect p62 by interfering with termination of autophagy and can also lead to IKK/NF-κB activation. Once p62 accumulates it leads to IKK/NF-κB activation via TRAF6 binding and NRF2 activation through titration of Keap1. p62 accumulation also activates mTORC1/c-Myc signaling to alter metabolism and promote proliferation. mTORC1-mediated p62 phosphorylation enhances NRF2 activation.
Elevated SQSTM1 mRNA is insufficient for formation of p62-containng aggregates, because a major factor that dictates p62 protein amounts is autosomal-lysosomal proteolysis (Komatsu et al., 2012). Dramatic p62 upregulation is seen after ATG5 or ATG7 ablation, although the complete absence of either protein in the liver prevents malignant HCC development. Therefore, only partial inhibition of autophagy is compatible with progression of p62-expressing FAH to malignant HCC. Such conditions occur during NASH development or in Tsc1Δhep mice, where autophagy is partially inhibited due to mTORC1 activation. However, modest mTORC1 activation induced by HFD feeding does not cause p62 accumulation and malignant progression. Other conditions that lead to p62 accumulation are inflammation and oxidative stress (Figure 7), which are general features of chronic liver disease along with reduced autophagic flux (Czaja, 2011; Schneider and Cuervo, 2014).
Previous work attributed pro-tumorigenic p62 activity to mTORC1 (Li et al., 2013; Linares et al., 2015). Elevated S6K phosphorylation, indicative of mTORC1 activation, is also seen in HCC nodules induced by ectopic p62, whereas p62 ablation in Tsc1Δhep mice reduces S6K phosphorylation. However, AKT T308 or S473 phosphorylation, which is suppressed upon S6K activation, is only weakly and inconsistently restored by p62 ablation, suggesting that other signaling pathways contribute to hepatic insulin resistance. One likely contributor is the JNK pathway, which is activated during HCC development (Nakagawa et al., 2014; Sakurai et al., 2006) and inhibits insulin-activated AKT (Solinas et al., 2006). These data support the notion that p62-dependent liver tumorigenesis may be mediated in part via mTORC1. Dependence on mTORC1, however, does not apply to the DEN model, where Raptor ablation enhances, rather than suppresses, HCC development (Umemura et al., 2014). Another oncogenic mediator induced by p62 is c-Myc, a known player in HCC development (Shibata and Aburatani, 2014). As seen with p70S6, c-Myc expression and activity in mouse and human HCC correlates with p62 and is decreased upon its ablation, results that support earlier findings regarding the ability of p62-mTORC1 signaling to induce c-Myc (Valencia et al., 2014).
Another critical mediator of p62-induced hepatocarcinogenesis is NRF2 whose activation by p62 depends on Keap1 binding. A point mutation in the p62 KIR domain, which prevents Keap1 binding (Suzuki et al., 2013), blocks NRF2 activation and abolishes p62 oncogenic activity. Importantly, NRF2 is mutationally activated in approximately 14% of human HCCs (Schulze et al., 2015; Shibata and Aburatani, 2014). NRF2 is also mutationally activated also in chemically-induced HCC in rats, where Nfe2l2(Nrf2) or Keap1 mutations occur in 70% of early lesions (Zavattari et al., 2015). Given the dependence of NRF2 activation on p62 in the models we studied, the main driver of NRF2 activation in mouse HCC and human HepG2 cells is p62 rather than mutations. Furthermore, no NRF2 or KEAP1 mutations were observed in human dysplastic lesions (Nault et al., 2014). NRF2 ablation in Atg5Δhep mice inhibits development of adenomas (Ni et al., 2014), but these are benign tumors that emerge in autophagy-deficient livers and their relationship to HCC is questionable.
NRF2 activation and upregulation of its targets are common in human and rodent HCCs, where they occur in the majority of FAH/dysplastic lesions (Farber, 1990; Hayes and McMahon, 2006). It was proposed, but never proven, that NRF2 activation and consequent induction of antioxidant and detoxifying enzymes allow these lesions to survive in chronically stressed livers, where ROS accumulation is readily detected (Nakagawa et al., 2014; Sakurai et al., 2008). Curiously, however, chronic upregulation of these protective enzymes does not inhibit ROS-mediated mutagenesis, as human HCCs contain multiple mutations in addition to those that activate NRF2. On the contrary, persistent NRF2 activation promotes survival of HcPC and allows them to accumulate additional mutations. Congruent with this notion, we find that p62 is required for survival of hepatocytes that accumulate high amounts of superoxides and stain positively with DHE. Although p62 ablation reduces expression of antioxidant enzymes, it decreases rather than increases the number of DHE-positive, ROS-containing cells. Most likely, without NRF2-dependent defenses, cells undergoing extensive ROS accumulation die and cannot serve as HCC progenitors. Although exogenous p62 may function by inducing endogenous p62, both this function and activation of the antioxidant response require a functional KIR domain. NRF2 was suggested to induce metabolic reprogramming that favors cell proliferation (DeNicola et al., 2015; Mitsuishi et al., 2012). However, this effect was mainly detected in cancer cell lines that exhibit sustained AKT activation, which is certainly not the case in livers of Tsc1Δhep or HFD-fed MUP-uPA and STZ-HFD mice, where AKT is not activated due to hepatic insulin resistance.
Nonetheless, constitutive activation of liver NRF2 does not induce HCC (Kohler et al., 2014). It is plausible that NRF2 only increases HCC risk in the presence of activated mTORC1, as occurs in Tsc1Δhep livers and during NASH-driven HCC development. Although NRF2 induces Sqstm1 transcription and p62 activates NRF2 (Figure 7), the latter effect requires p62 phosphorylation by mTORC1 (Ichimura et al., 2013). Future experiments should examine the role of this phosphorylation event in p62-induced tumorigenesis. The ability of p62 to oligomerize and aggregate ubiquitinated proteins was suggested to promote tumorigenesis by inducing oxidative stress (Mathew et al., 2009) and p62 ablation in Atg7Δhep liver was reported to reduce oxidative stress (Komatsu et al., 2007), results that are not entirely consistent with our current understanding of p62 importance in maintaining liver antioxidant defenses..
Our results illustrate the existence of a common pathway through which diverse etiological factors, including HBV and HCV infections, NASH and ASH trigger premalignant changes that progress to HCC by inducing p62 accumulation and subsequent NRF2, mTORC1 and c-Myc activation. The p62-Keap1-NRF2 pathway allows HcPC with high ROS content to survive in the chronically stressed liver environment and accumulate numerous mutations that commit them to the malignant fate under conditions where the metabolic and proliferative functions of mTORC1 and c-Myc are also activated. Elevated p62 in human liver predicts rapid recurrence of resectable HCC. Thus, small molecules that interfere with p62 binding to Keap1 and/or TRAF6, which is needed for mTORC1 activation, may be useful for preventing progression of chronic liver disease to HCC as well as attenuate recurrence of resectable HCC.
Experimental Procedures
Mice
Sqstm1F/F and Tsc1F/F mice (Mori et al., 2009; Muller et al., 2013) were backcrossed to C57BL/6 mice for at least 7 generations, and then bred with Alb-Cre mice (also C57BL/6) to generate Sqstm1F/F; Alb-Cre (Sqstm1Δhep) and Tsc1F/F; Alb-Cre (Tsc1Δhep) mice, respectively. Sqstm1Δhep mice were crossed to Tsc1Δhep and MUP-uPA mice to generate Tsc1/Sqstm1Δhep and Sqstm1Δhep/MUP mice, respectively. All studies used male mice, and were in accordance with NIH guidelines and approved by the UCSD Institutional Animal Care and Use Committee. Mice were maintained in filter-topped cages on autoclaved chow diet (low-fat diet; LFD, composed of 12% fat, 23% protein, 65% carbohydrates based on caloric content) or high-fat diet (HFD, composed of 59% fat, 15% protein, 26% carbohydrates based on caloric content; Bio-Serv) and water.
Human Liver Samples
Human tissue specimens were retrospectively obtained from liver biopsies or liver resections, and fixed in buffered formalin and embedded in paraffin using standard methods. Corresponding clinical data were obtained from medical records and de-identified. The study was approved by the institutional review board and research and development committees of the University of Tokyo and Kyoto Prefectural University of Medicine.
Supplementary Material
Significance.
p62 is the main component of Mallory Denk Bodies (MDB) and hyaline granules that are present in the majority of premalignant liver diseases and HCC. Our results indicate that p62 accumulation is required for progression from premalignancy to malignancy, most likely by preventing oxidative stress-induced death of HCC-initiating cells, thereby allowing such cells to accumulate multiple oncogenic mutations. Hepatocyte-specific p62 expression promotes c-Myc induction and NRF2 and mTORC1 activation. Therefore, p62 levels serve as a strong prognostic indicator of rapid HCC recurrence after curative ablation.
Highlights.
p62 in preneoplastic lesions is important for HCC development regardless of etiology.
Ectopic p62 expression is sufficient for HCC induction in autophagy-competent liver.
p62 exerts its oncogenic activity via NRF2 and mTORC1 but not via ubiquitin binding.
p62 promotes survival and expansion of ROS-containing HCC initiating cells.
Acknowledgments
We thank E.P. Sandgren and K.L. Guan for MUP-uPA and Tsc1F/F mice, respectively, X. Xiao for the AAV vector, H. Nakai and A. Miyanohara for advice and AAV generation, R. Loomba, D.E. Kleiner, T. Kisseleva and D.A. Brenner for suggestions and constructive criticism, Karin lab members for helpful discussions and Cell Signaling Technology, Santa Cruz Biotechnology and Abcam for antibodies. A.U., H.N., K.Y., and Y.I. were supported by JSPS KAKENHI Grant (A.U.; #15H06547, H.N.; #15K19313, K.Y.; #25461005, Y.I.; #15K09018), Kanae Foundation for the Promotion of Medical Science (A.U., H.N.), and a Global Grant Scholarship from The Rotary Foundation (A.U.), the Japanese Society of Gastroenterology (H.N.) and Research Program on Hepatitis from Japan Agency for Medical Research and Development, AMED (H.N., K.K.). K.T., J.F.-B. and Z.Z. were supported by Postdoctoral Fellowship for Research Abroad, Research Fellowship for Young Scientists from the JSPS, and a Uehara Memorial Foundation Fellowship (K.T.); CIRM Training Grant II (TG2-01154; J.F.-B.) and Cancer Research Institute (CRI) Irvington postdoctoral fellowship (Z.Z.). M.K. holds the Ben and Wanda Hildyard Chair for Mitochondrial and Metabolic Diseases. Research was supported by the NIH (R01CA163798, R01CA118165 and P01DK098108 to M.K.; R01CA172025 and R01DK108743 to J.M., R01CA192642 to M.T.D.-M., 5P30CA030199 to M.T.D-M. and J.M.), the Superfund Basic Research Program (P42ES010337 to M.K. and E.S.), and the American Association for Study of Liver Diseases/American Liver Foundation (E.S).
Footnotes
Accession Numbers
The GEO accession number for RNAseq data reported in this paper is GSE77323.
Author Contributions
A.U. designed and performed the main experiments and wrote the paper with M.K., who conceived and supervised the project. F.H., K.T., S.Y., J.F.-B., Z.Z, S.S., S.R., A.D., M.R.-C., S.U. and K.Y. provided technical assistance for main experiments. A.U., H.N. and K.Y. analyzed human specimens. J.F.-B. and J.F.L. generated p62 mutants. E.S., K.K. and Y.I. discussed and interpreted results. M.T.D.-M., and J.M. discussed and interpreted the results, analyzed DEN-injected mice, RNA seq data and HCC datasets, provided Sqstm1F/F mice, and revised the manuscript. M.A.V. provided pathological diagnosis.
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final citable form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
References
- Chandler RJ, LaFave MC, Varshney GK, Trivedi NS, Carrillo-Carrasco N, Senac JS, Wu W, Hoffmann V, Elkahloun AG, Burgess SM, et al. Vector design influences hepatic genotoxicity after adeno-associated virus gene therapy. J Clin Invest. 2015;125:870–880. doi: 10.1172/JCI79213. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Czaja MJ. Functions of autophagy in hepatic and pancreatic physiology and disease. Gastroenterology. 2011;140:1895–1908. doi: 10.1053/j.gastro.2011.04.038. [DOI] [PMC free article] [PubMed] [Google Scholar]
- DeNicola GM, Chen PH, Mullarky E, Sudderth JA, Hu Z, Wu D, Tang H, Xie Y, Asara JM, Huffman KE, et al. NRF2 regulates serine biosynthesis in non-small cell lung cancer. Nat Genet. 2015;47:1475–1481. doi: 10.1038/ng.3421. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Denk H, Stumptner C, Fuchsbichler A, Muller T, Farr G, Muller W, Terracciano L, Zatloukal K. Are the Mallory bodies and intracellular hyaline bodies in neoplastic and non-neoplastic hepatocytes related? J Pathol. 2006;208:653–661. doi: 10.1002/path.1946. [DOI] [PubMed] [Google Scholar]
- Duran A, Amanchy R, Linares JF, Joshi J, Abu-Baker S, Porollo A, Hansen M, Moscat J, Diaz-Meco MT. p62 is a key regulator of nutrient sensing in the mTORC1 pathway. Mol Cell. 2011;44:134–146. doi: 10.1016/j.molcel.2011.06.038. [DOI] [PMC free article] [PubMed] [Google Scholar]
- El-Serag HB. Hepatocellular carcinoma. The New England journal of medicine. 2011;365:1118–1127. doi: 10.1056/NEJMra1001683. [DOI] [PubMed] [Google Scholar]
- Farber E. Clonal adaptation during carcinogenesis. Biochemical pharmacology. 1990;39:1837–1846. doi: 10.1016/0006-2952(90)90599-g. [DOI] [PubMed] [Google Scholar]
- Finkin S, Yuan D, Stein I, Taniguchi K, Weber A, Unger K, Browning JL, Goossens N, Nakagawa S, Gunasekaran G, et al. Ectopic lymphoid structures function as microniches for tumor progenitor cells in hepatocellular carcinoma. Nature immunology. 2015;16:1235–1244. doi: 10.1038/ni.3290. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Font-Burgada J, Shalapour S, Ramaswamy S, Hsueh B, Rossell D, Umemura A, Taniguchi K, Nakagawa H, Valasek MA, Ye L, et al. Hybrid Periportal Hepatocytes Regenerate the Injured Liver without Giving Rise to Cancer. Cell. 2015;162:766–779. doi: 10.1016/j.cell.2015.07.026. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Grivennikov SI, Greten FR, Karin M. Immunity, inflammation, and cancer. Cell. 2010;140:883–899. doi: 10.1016/j.cell.2010.01.025. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Guichard C, Amaddeo G, Imbeaud S, Ladeiro Y, Pelletier L, Maad IB, Calderaro J, Bioulac-Sage P, Letexier M, Degos F, et al. Integrated analysis of somatic mutations and focal copy-number changes identifies key genes and pathways in hepatocellular carcinoma. Nature genetics. 2012;44:694–698. doi: 10.1038/ng.2256. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hay N, Sonenberg N. Upstream and downstream of mTOR. Genes Dev. 2004;18:1926–1945. doi: 10.1101/gad.1212704. [DOI] [PubMed] [Google Scholar]
- Hayes JD, McMahon M. The double-edged sword of Nrf2: subversion of redox homeostasis during the evolution of cancer. Mol Cell. 2006;21:732–734. doi: 10.1016/j.molcel.2006.03.004. [DOI] [PubMed] [Google Scholar]
- Hayes JD, McMahon M. NRF2 and KEAP1 mutations: permanent activation of an adaptive response in cancer. Trends Biochem Sci. 2009;34:176–188. doi: 10.1016/j.tibs.2008.12.008. [DOI] [PubMed] [Google Scholar]
- He G, Dhar D, Nakagawa H, Font-Burgada J, Ogata H, Jiang Y, Shalapour S, Seki E, Yost SE, Jepsen K, et al. Identification of liver cancer progenitors whose malignant progression depends on autocrine IL-6 signaling. Cell. 2013;155:384–396. doi: 10.1016/j.cell.2013.09.031. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ichimura Y, Waguri S, Sou YS, Kageyama S, Hasegawa J, Ishimura R, Saito T, Yang Y, Kouno T, Fukutomi T, et al. Phosphorylation of p62 activates the Keap1-Nrf2 pathway during selective autophagy. Mol Cell. 2013;51:618–631. doi: 10.1016/j.molcel.2013.08.003. [DOI] [PubMed] [Google Scholar]
- Inami Y, Waguri S, Sakamoto A, Kouno T, Nakada K, Hino O, Watanabe S, Ando J, Iwadate M, Yamamoto M, et al. Persistent activation of Nrf2 through p62 in hepatocellular carcinoma cells. J Cell Biol. 2011;193:275–284. doi: 10.1083/jcb.201102031. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Iwadate R, Inoue J, Tsuda H, Takano M, Furuya K, Hirasawa A, Aoki D, Inazawa J. High Expression of p62 Protein Is Associated with Poor Prognosis and Aggressive Phenotypes in Endometrial Cancer. The American journal of pathology. 2015;185:2523–2533. doi: 10.1016/j.ajpath.2015.05.008. [DOI] [PubMed] [Google Scholar]
- Jain A, Lamark T, Sjottem E, Larsen KB, Awuh JA, Overvatn A, McMahon M, Hayes JD, Johansen T. p62/SQSTM1 is a target gene for transcription factor NRF2 and creates a positive feedback loop by inducing antioxidant response element-driven gene transcription. J Biol Chem. 2010;285:22576–22591. doi: 10.1074/jbc.M110.118976. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kang JS, Wanibuchi H, Morimura K, Gonzalez FJ, Fukushima S. Role of CYP2E1 in diethylnitrosamine-induced hepatocarcinogenesis in vivo. Cancer research. 2007;67:11141–11146. doi: 10.1158/0008-5472.CAN-07-1369. [DOI] [PubMed] [Google Scholar]
- Kaposi-Novak P, Libbrecht L, Woo HG, Lee YH, Sears NC, Coulouarn C, Conner EA, Factor VM, Roskams T, Thorgeirsson SS. Central role of c-Myc during malignant conversion in human hepatocarcinogenesis. Cancer research. 2009;69:2775–2782. doi: 10.1158/0008-5472.CAN-08-3357. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kawai D, Takaki A, Nakatsuka A, Wada J, Tamaki N, Yasunaka T, Koike K, Tsuzaki R, Matsumoto K, Miyake Y, et al. Hydrogen-rich water prevents progression of nonalcoholic steatohepatitis and accompanying hepatocarcinogenesis in mice. Hepatology. 2012;56:912–921. doi: 10.1002/hep.25782. [DOI] [PubMed] [Google Scholar]
- Kim YR, Oh JE, Kim MS, Kang MR, Park SW, Han JY, Eom HS, Yoo NJ, Lee SH. Oncogenic NRF2 mutations in squamous cell carcinomas of oesophagus and skin. The Journal of pathology. 2010;220:446–451. doi: 10.1002/path.2653. [DOI] [PubMed] [Google Scholar]
- Kohler UA, Kurinna S, Schwitter D, Marti A, Schafer M, Hellerbrand C, Speicher T, Werner S. Activated Nrf2 impairs liver regeneration in mice by activation of genes involved in cell-cycle control and apoptosis. Hepatology. 2014;60:670–678. doi: 10.1002/hep.26964. [DOI] [PubMed] [Google Scholar]
- Komatsu M, Kageyama S, Ichimura Y. p62/SQSTM1/A170: physiology and pathology. Pharmacol Res. 2012;66:457–462. doi: 10.1016/j.phrs.2012.07.004. [DOI] [PubMed] [Google Scholar]
- Komatsu M, Waguri S, Koike M, Sou YS, Ueno T, Hara T, Mizushima N, Iwata J, Ezaki J, Murata S, et al. Homeostatic levels of p62 control cytoplasmic inclusion body formation in autophagy-deficient mice. Cell. 2007;131:1149–1163. doi: 10.1016/j.cell.2007.10.035. [DOI] [PubMed] [Google Scholar]
- Kuraishy A, Karin M, Grivennikov SI. Tumor promotion via injury- and death-induced inflammation. Immunity. 2011;35:467–477. doi: 10.1016/j.immuni.2011.09.006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Li L, Shen C, Nakamura E, Ando K, Signoretti S, Beroukhim R, Cowley GS, Lizotte P, Liberzon E, Bair S, et al. SQSTM1 is a pathogenic target of 5q copy number gains in kidney cancer. Cancer cell. 2013;24:738–750. doi: 10.1016/j.ccr.2013.10.025. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Linares JF, Duran A, Reina-Campos M, Aza-Blanc P, Campos A, Moscat J, Diaz-Meco MT. Amino Acid Activation of mTORC1 by a PB1-Domain-Driven Kinase Complex Cascade. Cell reports. 2015;12:1339–1352. doi: 10.1016/j.celrep.2015.07.045. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Linares JF, Duran A, Yajima T, Pasparakis M, Moscat J, Diaz-Meco MT. K63 polyubiquitination and activation of mTOR by the p62-TRAF6 complex in nutrient-activated cells. Mol Cell. 2013;51:283–296. doi: 10.1016/j.molcel.2013.06.020. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ling J, Kang Y, Zhao R, Xia Q, Lee DF, Chang Z, Li J, Peng B, Fleming JB, Wang H, et al. KrasG12D-induced IKK2/beta/NF-kappaB activation by IL-1alpha and p62 feedforward loops is required for development of pancreatic ductal adenocarcinoma. Cancer Cell. 2012;21:105–120. doi: 10.1016/j.ccr.2011.12.006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Luch A. Nature and nurture - lessons from chemical carcinogenesis. Nature reviews Cancer. 2005;5:113–125. doi: 10.1038/nrc1546. [DOI] [PubMed] [Google Scholar]
- Maeda S, Kamata H, Luo JL, Leffert H, Karin M. IKKbeta couples hepatocyte death to cytokine-driven compensatory proliferation that promotes chemical hepatocarcinogenesis. Cell. 2005;121:977–990. doi: 10.1016/j.cell.2005.04.014. [DOI] [PubMed] [Google Scholar]
- Malhi H, Kaufman RJ. Endoplasmic reticulum stress in liver disease. J Hepatol. 2011;54:795–809. doi: 10.1016/j.jhep.2010.11.005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mathew R, Karp CM, Beaudoin B, Vuong N, Chen G, Chen HY, Bray K, Reddy A, Bhanot G, Gelinas C, et al. Autophagy suppresses tumorigenesis through elimination of p62. Cell. 2009;137:1062–1075. doi: 10.1016/j.cell.2009.03.048. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Menon S, Yecies JL, Zhang HH, Howell JJ, Nicholatos J, Harputlugil E, Bronson RT, Kwiatkowski DJ, Manning BD. Chronic activation of mTOR complex 1 is sufficient to cause hepatocellular carcinoma in mice. Sci Signal. 2012;5:ra24. doi: 10.1126/scisignal.2002739. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Min L, Ji Y, Bakiri L, Qiu Z, Cen J, Chen X, Chen L, Scheuch H, Zheng H, Qin L, et al. Liver cancer initiation is controlled by AP-1 through SIRT6-dependent inhibition of survivin. Nat Cell Biol. 2012;14:1203–1211. doi: 10.1038/ncb2590. [DOI] [PubMed] [Google Scholar]
- Mitsuishi Y, Taguchi K, Kawatani Y, Shibata T, Nukiwa T, Aburatani H, Yamamoto M, Motohashi H. Nrf2 redirects glucose and glutamine into anabolic pathways in metabolic reprogramming. Cancer Cell. 2012;22:66–79. doi: 10.1016/j.ccr.2012.05.016. [DOI] [PubMed] [Google Scholar]
- Mori H, Inoki K, Munzberg H, Opland D, Faouzi M, Villanueva EC, Ikenoue T, Kwiatkowski D, MacDougald OA, Myers MG, Jr, et al. Critical role for hypothalamic mTOR activity in energy balance. Cell Metab. 2009;9:362–374. doi: 10.1016/j.cmet.2009.03.005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Muller TD, Lee SJ, Jastroch M, Kabra D, Stemmer K, Aichler M, Abplanalp B, Ananthakrishnan G, Bhardwaj N, Collins S, et al. p62 links beta-adrenergic input to mitochondrial function and thermogenesis. J Clin Invest. 2013;123:469–478. doi: 10.1172/JCI64209. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nakagawa H, Umemura A, Taniguchi K, Font-Burgada J, Dhar D, Ogata H, Zhong Z, Valasek MA, Seki E, Hidalgo J, et al. ER stress cooperates with hypernutrition to trigger TNF-dependent spontaneous HCC development. Cancer Cell. 2014;26:331–343. doi: 10.1016/j.ccr.2014.07.001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nault JC, Calderaro J, Di Tommaso L, Balabaud C, Zafrani ES, Bioulac-Sage P, Roncalli M, Zucman-Rossi J. Telomerase reverse transcriptase promoter mutation is an early somatic genetic alteration in the transformation of premalignant nodules in hepatocellular carcinoma on cirrhosis. Hepatology. 2014;60:1983–1992. doi: 10.1002/hep.27372. [DOI] [PubMed] [Google Scholar]
- Ni HM, Woolbright BL, Williams J, Copple B, Cui W, Luyendyk JP, Jaeschke H, Ding WX. Nrf2 promotes the development of fibrosis and tumorigenesis in mice with defective hepatic autophagy. J Hepatol. 2014;61:617–625. doi: 10.1016/j.jhep.2014.04.043. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ohta T, Iijima K, Miyamoto M, Nakahara I, Tanaka H, Ohtsuji M, Suzuki T, Kobayashi A, Yokota J, Sakiyama T, et al. Loss of Keap1 function activates Nrf2 and provides advantages for lung cancer cell growth. Cancer research. 2008;68:1303–1309. doi: 10.1158/0008-5472.CAN-07-5003. [DOI] [PubMed] [Google Scholar]
- Park EJ, Lee JH, Yu GY, He G, Ali SR, Holzer RG, Osterreicher CH, Takahashi H, Karin M. Dietary and genetic obesity promote liver inflammation and tumorigenesis by enhancing IL-6 and TNF expression. Cell. 2010;140:197–208. doi: 10.1016/j.cell.2009.12.052. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Qu A, Jiang C, Cai Y, Kim JH, Tanaka N, Ward JM, Shah YM, Gonzalez FJ. Role of Myc in hepatocellular proliferation and hepatocarcinogenesis. J Hepatol. 2014;60:331–338. doi: 10.1016/j.jhep.2013.09.024. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rubin H. Fields and field cancerization: the preneoplastic origins of cancer: asymptomatic hyperplastic fields are precursors of neoplasia, and their progression to tumors can be tracked by saturation density in culture. BioEssays: news and reviews in molecular, cellular and developmental biology. 2011;33:224–231. doi: 10.1002/bies.201000067. [DOI] [PubMed] [Google Scholar]
- Sakurai T, He G, Matsuzawa A, Yu GY, Maeda S, Hardiman G, Karin M. Hepatocyte necrosis induced by oxidative stress and IL-1 alpha release mediate carcinogen-induced compensatory proliferation and liver tumorigenesis. Cancer Cell. 2008;14:156–165. doi: 10.1016/j.ccr.2008.06.016. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sakurai T, Maeda S, Chang L, Karin M. Loss of hepatic NF-kappa B activity enhances chemical hepatocarcinogenesis through sustained c-Jun N-terminal kinase 1 activation. Proc Natl Acad Sci U S A. 2006;103:10544–10551. doi: 10.1073/pnas.0603499103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sanz L, Diaz-Meco MT, Nakano H, Moscat J. The atypical PKC-interacting protein p62 channels NF-kappaB activation by the IL-1-TRAF6 pathway. Embo J. 2000;19:1576–1586. doi: 10.1093/emboj/19.7.1576. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schneider JL, Cuervo AM. Liver autophagy: much more than just taking out the trash. Nature reviews Gastroenterology & hepatology. 2014;11:187–200. doi: 10.1038/nrgastro.2013.211. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schulze K, Imbeaud S, Letouze E, Alexandrov LB, Calderaro J, Rebouissou S, Couchy G, Meiller C, Shinde J, Soysouvanh F, et al. Exome sequencing of hepatocellular carcinomas identifies new mutational signatures and potential therapeutic targets. Nat Genet. 2015;47:505–511. doi: 10.1038/ng.3252. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sell S, Leffert HL. Liver cancer stem cells. J Clin Oncol. 2008;26:2800–2805. doi: 10.1200/JCO.2007.15.5945. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shibata T, Aburatani H. Exploration of liver cancer genomes. Nature reviews Gastroenterology & hepatology. 2014;11:340–349. doi: 10.1038/nrgastro.2014.6. [DOI] [PubMed] [Google Scholar]
- Shibata T, Ohta T, Tong KI, Kokubu A, Odogawa R, Tsuta K, Asamura H, Yamamoto M, Hirohashi S. Cancer related mutations in NRF2 impair its recognition by Keap1-Cul3 E3 ligase and promote malignancy. Proceedings of the National Academy of Sciences of the United States of America. 2008;105:13568–13573. doi: 10.1073/pnas.0806268105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Solinas G, Naugler W, Galimi F, Lee MS, Karin M. Saturated fatty acids inhibit induction of insulin gene transcription by JNK-mediated phosphorylation of insulin-receptor substrates. Proceedings of the National Academy of Sciences of the United States of America. 2006;103:16454–16459. doi: 10.1073/pnas.0607626103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Starley BQ, Calcagno CJ, Harrison SA. Nonalcoholic fatty liver disease and hepatocellular carcinoma: a weighty connection. Hepatology. 2010;51:1820–1832. doi: 10.1002/hep.23594. [DOI] [PubMed] [Google Scholar]
- Suzuki T, Motohashi H, Yamamoto M. Toward clinical application of the Keap1-Nrf2 pathway. Trends in pharmacological sciences. 2013;34:340–346. doi: 10.1016/j.tips.2013.04.005. [DOI] [PubMed] [Google Scholar]
- Takamura A, Komatsu M, Hara T, Sakamoto A, Kishi C, Waguri S, Eishi Y, Hino O, Tanaka K, Mizushima N. Autophagy-deficient mice develop multiple liver tumors. Genes Dev. 2011;25:795–800. doi: 10.1101/gad.2016211. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Totoki Y, Tatsuno K, Covington KR, Ueda H, Creighton CJ, Kato M, Tsuji S, Donehower LA, Slagle BL, Nakamura H, et al. Trans-ancestry mutational landscape of hepatocellular carcinoma genomes. Nature genetics. 2014;46:1267–1273. doi: 10.1038/ng.3126. [DOI] [PubMed] [Google Scholar]
- Umemura A, Park EJ, Taniguchi K, Lee JH, Shalapour S, Valasek MA, Aghajan M, Nakagawa H, Seki E, Hall MN, et al. Liver damage, inflammation, and enhanced tumorigenesis after persistent mTORC1 inhibition. Cell Metab. 2014;20:133–144. doi: 10.1016/j.cmet.2014.05.001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Valencia T, Kim JY, Abu-Baker S, Moscat-Pardos J, Ahn CS, Reina-Campos M, Duran A, Castilla EA, Metallo CM, Diaz-Meco MT, et al. Metabolic reprogramming of stromal fibroblasts through p62-mTORC1 signaling promotes inflammation and tumorigenesis. Cancer Cell. 2014;26:121–135. doi: 10.1016/j.ccr.2014.05.004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- van de Wetering M, Francies HE, Francis JM, Bounova G, Iorio F, Pronk A, van Houdt W, van Gorp J, Taylor-Weiner A, Kester L, et al. Prospective derivation of a living organoid biobank of colorectal cancer patients. Cell. 2015;161:933–945. doi: 10.1016/j.cell.2015.03.053. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Vogelstein B, Papadopoulos N, Velculescu VE, Zhou S, Diaz LA, Jr, Kinzler KW. Cancer genome landscapes. Science. 2013;339:1546–1558. doi: 10.1126/science.1235122. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wolf MJ, Adili A, Piotrowitz K, Abdullah Z, Boege Y, Stemmer K, Ringelhan M, Simonavicius N, Egger M, Wohlleber D, et al. Metabolic activation of intrahepatic CD8+ T cells and NKT cells causes nonalcoholic steatohepatitis and liver cancer via cross-talk with hepatocytes. Cancer Cell. 2014;26:549–564. doi: 10.1016/j.ccell.2014.09.003. [DOI] [PubMed] [Google Scholar]
- Zatloukal K, French SW, Stumptner C, Strnad P, Harada M, Toivola DM, Cadrin M, Omary MB. From Mallory to Mallory-Denk bodies: what, how and why? Experimental cell research. 2007;313:2033–2049. doi: 10.1016/j.yexcr.2007.04.024. [DOI] [PubMed] [Google Scholar]
- Zavattari P, Perra A, Menegon S, Kowalik MA, Petrelli A, Angioni MM, Follenzi A, Quagliata L, Ledda-Columbano GM, Terracciano L, et al. Nrf2, but not beta-catenin, mutation represents an early event in rat hepatocarcinogenesis. Hepatology. 2015;62:851–862. doi: 10.1002/hep.27790. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.