

Abstract
A new approach for performing Particle Mesh Ewald in ab initio QM/MM simulations with extended atomic orbital basis sets is presented. The new approach, the Ambient-Potential Composite Ewald Method (CEw), does not perform the QM/MM interaction with Mulliken charges nor electrostatically fit charges. Instead the nuclei and electron density interact directly with the MM environment, but in a manner that avoids the use of dense Fourier transform grids. By performing the electrostatics with the underlying QM density, the CEw method avoids self-consistent field instabilities that have been encountered with simple charge mapping procedures. Potential of mean force (PMF) profiles of the p-nitrophenyl phosphate dissociation reaction in explicit solvent are computed from PBE0/6-31G* QM/MM molecular dynamics simulations with various electrostatic protocols. The CEw profiles are shown to be stable with respect to real-space Ewald cutoff, whereas the PMFs computed from truncated and switched electrostatics produce artifacts. PBE0/6-311G**, AM1/d-PhoT, and DFTB2 QM/MM simulations are performed to generate two-dimensional PMF profiles of the phosphoryl transesterification reactions with ethoxide and phenoxide leaving groups. The semiempirical models incorrectly produce a concerted ethoxide mechanism, whereas PBE0 correctly produces a stepwise mechanism. The ab initio reaction barriers agree more closely to experiment than the semiempirical models. The failure of Mulliken-charge QM/MM-Ewald is analyzed.
TOC images
1 Introduction
The rigorous treatment of long-ranged electrostatics is essential for a proper modeling of biological processes in solution.1–5 One technique for including long-range electrostatics is Ewald’s method,6 which replicates a primary unit cell composed of Gaussian charges to form an infinite periodic lattice. The periodic Gaussian charge density is resolved in a plane-wave basis, whence the electrostatic potential is readily calculated. The plane-wave potential is then modified with short-ranged corrections to account for the Gaussian charge penetration and thus recover the electrostatic potential of the point charge system. The computational performance of Ewald’s method was greatly improved with the advent of the Particle Mesh Ewald7–10 (PME) method, which has become the de facto standard for molecular mechanical (MM) force field molecular dynamics (MD) simulations. Although the PME method was originally formulated for point charges, it has been extended throughout the years to handle Cartesian11–16 and solid harmonic17,18 multipoles for its application with the AMOEBA polarizable force field19 and the modified Divide-and-Conquer (mDC) quantum mechanical force field20–22 (QMFF). Before the widespread adoption of PME, electrostatic force truncation, switching, and shifting were frequently used.23,24 Electrostatic cutoff methods were later found to produce artifacts in the properties of water25–28 and the structural stability of large biomolecules.29–31 Consequently it has been suggested that new models not be parametrized using cutoff electrostatics.26
The treatment of electrostatics within quantum mechanical/molecular mechanical (QM/MM) models32 has followed one of two general prescriptions: electrostatic embedding and mechanical embedding.33,34 Mechanical embedding is a “subtractive” paradigm, whereby the quantum mechanical (QM) region is represented by a MM-analogue, the electrostatics are computed entirely with MM charges, and the QM region is introduced by removing the MM-analogue self-energy and replacing it with the gas-phase QM energy. In this sense mechanical embedding can be viewed as a type of ONIOM method.35–37 Although mechanical embedding is simple to implement, it suffers from the major drawback that the QM charge density does not directly polarize to the MM environment; therefore, the electrostatic embedding method is instead often used. Electrostatic embedding decomposes the total energy into MM/MM, QM/QM, and QM/MM “additive” components. The QM/MM interaction explicitly includes the electrostatics between the QM charge density and the MM point charges – among other interactions, including van der Waals (vdW) forces – thereby polarizing the QM electron density.
Combined QM/MM MD applications have been dominated by the use of semiempirical Hamiltonians; for example, AM1/d-PhoT,38 DFTB2,39,40 and related models,41–43 because the high cost of ab initio wavefunction methods has often precluded their ability to obtain the amount of statistical sampling necessary for making a meaningful comparison with experiment.44 Nevertheless ab initio QM/MM methods45 have found applications46 through the calculation of single point energies,47 NMR chemical shifts,48 geometry optimizations,49 adiabatic potential energy surfaces,50 nudge elastic band pathways,51 finite temperature string methods,52–54 multiple time step simulations,55 and to correct potential of mean force (PMF) free energy surfaces obtained from semiempirical QM/MM calculations.56–59
Applications of semiempirical QM/MM methods routinely employed electrostatic embedding with truncated QM/MM electrostatic cutoffs60–62 until the development of the semiempirical QM/MM Ewald presented by Nam,63 which was independently reported by Riccardi;64 both of which were influenced by the method presented several years prior by Gao.65 More recently these methods have been adapted for use with PME66 and semiempirical X-Pol models.67 The use of explicit lattice summations can also be found in the literature.68
Considering that Ewald methods have traditionally been implemented for point-charge distributions, the semiempirical QM/MM-Ewald methods have chosen to use a Mulliken charge representation of the QM region to perform the long-range interactions. When this approach was applied to ab initio QM/MM, it was found that the use of Mulliken or Löwdin charges caused self-consistent field (SCF) convergence problems when non-minimal atomic orbital (AO) basis sets were used.69,70 This has motivated the use of ChElPG71 or other electrostatic potential charge fitting procedures to produce stable ab initio QM/MM-Ewald trajectories.69,70,72–76 However, applications of ab initio QM/MM often still forego the use Ewald summations, preferring instead to model the long-range electrostatic with a reaction field method77,78 or perform real-space electrostatic truncation, shifting, or smoothing.52–54,79–84 Recent work has advocated a 22 Å real-space switched electrostatic cutoff method using the minimum image convention.85 Regrettably we’ve noticed that many authors have failed to report the size of the QM/MM nonbond cutoff that they’ve used, and other details defining how the electrostatics were performed.
Ab initio QM/MM methods will become frequently used in the near future as QMFFs80,86–88 and free energy correction methods mature and as hardware technology continues to improve. To this end, we question if the effort placed into the evaluation of the underlying ab initio calculation isn’t somewhat wasted by performing the QM/MM interaction with QM atomic partial charges rather than the nuclei and ab initio electron density. Choosing the partial charges to model the QM electrostatic potential certainly helps to alleviate this concern to the extent that those charges indeed reproduce the potential, but the effort required to perform the fit could instead have been spent on a method that avoids charge fitting altogether. After all, the reason why the community is resorting to partial charge fitting is because a tractable alternative for evaluating the Ewald sum in ab initio QM/MM simulations has yet to be realized. The principle complication encountered in a direct adaptation of the Ewald or PME methods is borne from the electron density’s rapid changes near the nuclei, which requires an unacceptably large number of plane waves to resolve. Even if one could perform Ewald’s method using no more plane waves than what is found to be acceptable in a purely MM application, the analytic evaluation of each AO product’s Fourier coefficients would still be very costly. The computational effort would be further amplified by having to re-evaluate the Ewald potential at each step of the SCF procedure. A wholly new approach is needed.84,89
In this work, we present a new ab initio QM/MM-Ewald method called the Ambient-Potential Composite Ewald Method, or Composite Ewald Method (CEw) for short. The new method does not require more plane waves than what is typically used within pure-MM applications. The analytic evaluation of AO-product Fourier coefficients are avoided by numerically integrating the Ewald reciprocal-space potential on the molecular quadrature grid normally used to compute the density functional theory (DFT) exchange-correlation functional. The long-range interactions between the QM region and its periodic replicas are computed from a truncated Taylor series that is expanded about a MM point-charge representation of the QM region. This approximation does not affect the interaction between the QM and MM regions; it only affects the interaction of the QM region with its own periodic images. As a consequence of this approximation, the Ewald contribution to the QM Fock matrix does not change during the SCF procedure, and the evaluation of the plane-wave Ewald potential becomes analogous to a one-time evaluation of a local density approximation (LDA) exchange-correlation functional.
We compare the new Ewald method to several other electrostatic protocols by performing umbrella window simulations to compute the PMF profiles of the p-nitrophenyl phosphate dissociation reaction. We show how various electrostatic protocols affect the MM-water solvation around charged QM regions. We extend and elaborate on the analysis first discussed by Holden et al.69,70 to elucidate the failure of Nam’s semiempirical QM/MM-Ewald method63 when it is applied to non-minimal basis set ab initio methods. We compare the computational cost of the new method to electrostatic embedding as a function of the number of QM atoms. Finally, we compute two-dimensional PMF profiles for phosphoryl transesterification reactions involving ethoxide and phenoxide leaving groups to compare the pathways produced by AM1/d-PhoT,38 DFTB2,39,40 and the PBE0/6-311G** hybrid functional DFT method.90,91
2 Methods
2.1 The QM/MM energy
In this work, we consider a QM/MM system that contains a localized QM region; for example, a small solute QM molecule in MM solvent, or a QM active site within a large biomolecule. Furthermore, we suppose that the calculation is performed under periodic boundary conditions, whose real- and reciprocal-space lattice vectors are a1, a2, a3 and , , , respectively. The total potential energy of the QM/MM system is
| (1) |
where R is the set of atomic coordinates and P is the single-particle density matrix (see eq. 11). The various components of the energy are defined below.
The bonded energy Ebonded, is the collection of MM terms describing the bonds, angles, and torsions between covalently linked MM atoms and those combinations of MM and QM atoms which contain at least one MM atom.
| (2) |
The k values are force constants, and Rab, θabc, and ϕabcd are bond lengths, angles, and torsion angles, respectively. The Lennard-Jones (LJ) energy ELJ, is explicitly evaluated for all pairs of nonbonded atoms within a cutoff Rcut, and a long-range correction is applied to account for the dispersion beyond the nonbond cutoff,
| (3) |
where u,v index atom types and Nu is the number of atoms of type u. The asterisk excludes pairs where a and b are both QM, Rab is assumed to be the “minimum image” distance between atoms a and b, and V = a1 · a2 × a3 is the unit cell volume. The periodic electrostatic energy Eelec, is
| (4) |
where n = n1a1 + n2a2 + n3a3 is a lattice translation and q(r) is the total charge density (see eq. 5). The MM atom charge density is a collection of static point charges (see eq. 6), and the QM charge density consists of the atomic nuclei and electrons (see eq. 7). The notation for the electrostatic energy shown in eq. 4 presumes the standard convention of excluding the infinite Coulomb self-energy of the point charges whenever those terms may appear.
Total charge density:
| (5) |
MM charge density:
| (6) |
QM charge density:
| (7) |
Total electron (number) density:
| (8) |
Spin-resolved (number) density:
| (9) |
Spin-resolved molecular orbital (MO):
| (10) |
Atomic orbital (AO) representation of the single-particle density matrix:
| (11) |
Spin-resolved density matrix:
| (12) |
where Za is a nuclear charge, χ(r) is an AO basis function, and nσ and Cσ are the spin-resolved occupation numbers and MO coefficients, respectively. The densities and MOs defined by eqs. 7–10 have been written as a function of r; however, we emphasize that these terms also depend on the atomic positions through the use of atom-centered basis functions, and this dependence must be considered when evaluating the atomic forces.
The parameters within Ebonded are chosen to implicitly account for the electrostatic interactions between the bonded atoms, but those interactions are explicitly included in eq. 4 for notational convenience. Therefore, Ebonded-elec denotes a correction that removes the explicit electrostatic interactions between the pairs of MM atoms appearing within Ebonded,
| (13) |
The remaining energy terms in eq. 1 are the electron interactions that do not directly couple to the MM environment.
The noninteracting electron kinetic energy:
| (14) |
| (15) |
The Hartree-Fock exchange energy:
| (16) |
Electron repulsion integral (ERI):
| (17) |
The density functional theory (DFT) exchange-correlation energy:
| (18) |
where , wi is a molecular quadrature weight, and exc is a linear combination of exchange and correlation functional integrands,
| (19) |
where we have simplified the exc notation for brevity. The results shown in the present work are evaluated with PBE0/6-31G* or PBE0/6-311G**. PBE0 is the Perdew-Burke-Ernzerhof hybrid functional,90,91 which uses the generalized gradient approximation functional described in Ref. 92. The PBE0 coefficients are cDFT/x = 0.75, cDFT/c = 1, and cHF/x = 0.25.
The summation appearing in eq. 18 is a numerical integration of the DFT functional performed on a molecular quadrature grid. The molecular quadrature grid is a union of atomic quadrature grids, and an atomic grid is a series of concentric discretized spheres. That is, each atomic quadrature point i is an element within a set of angular points that form a spherical shell of radius Rrad which is tethered to atom a. The atomic grid point locations and weights are and watomic,i = wrad,iwΩ,i, respectively, where the notation i ∈ a denotes the grid point i within the set of points tethered to atom a. Many types of radial quadrature rules have been developed, including those based on Gauss-Chebyshev, Gauss-Legendre, Euler-Maclauren schemes.93–96 In the present work, we use Gauss-Laguerre and Lebedev97 rules for the radial and angular quadratures to form atomic grids consisting of 5580 points per heavy atom and 4296 points per hydrogen. In principle, each atomic grid integrates all-space; however, each atomic grid only samples the integrand adequately near their respective centers. Therefore, the molecular quadrature weights wi ≡ w(ri∈a,R) = Γa(ri,R)watomic,i, introduce a “spatial partition function” Γa, to avoid an over-counting of the integrand when two-or-more atomic grids sample the same spatial area. Specifically, we use the “fuzzy Voronoi” partitioning scheme proposed by Becke,98 which is summarized by eqs. 20–26. The relative size of the Voronoi are biased according to the atom’s Bragg-Slater radius, RBS,a.
| (20) |
| (21) |
| (22) |
| (23) |
| (24) |
| (25) |
| (26) |
Formally, one would need to notationally account for periodicity within eqs. 15–18. If the electron density extends no more than half the box length, then one can evaluate these energy terms as written while assuming a minimum image convention. This is typically implemented by translating the QM region to the center of the simulation box, and then wrapping the MM atoms around it.
For a given set of coordinates, one must nonlinearly minimize the energy with respect to the MO coefficients in a SCF procedure, under the constraint that the MOs remain orthonormal to each other,
| (27) |
where S is the AO overlap matrix,
| (28) |
and Iij = δij is the identity matrix. Under these constraints, the optimal set MO coefficients can be shown to obey the Roothaan-Hall equation,
| (29) |
where Eσ are the spin-resolved orbital eigenvalues and Fσ is the AO representation of the spin-resolved Fock matrix,
| (30) |
Equation 29 is a generalized eigenvalue problem, which can be reduced to standard form by introducing a transformation matrix,
| (31) |
where U and s are the eigenvectors and eigenvalues of the overlap matrix, respectively; S = U · s · UT. It follows that XT · S · X = I, and
| (32) |
where Fσ,OAO is the orthonormal atomic orbital (OAO) basis representation of the Fock matrix,
| (33) |
and Cσ,OAO are the corresponding MO coefficients,
| (34) |
for the OAO basis defined by the transformation:
| (35) |
The AO Fock matrix elements are the partial derivatives shown in eq. 30 applied to eq. 1:
| (36) |
where σ′ denotes the spin that is antiparallel to σ.
Upon reaching SCF convergence, the atomic gradients ∂/∂Xa, are readily obtained from elementary chain-rule differentiation of the energy.
MM atom gradients (a ∈ mm):
| (37) |
QM atom gradients (a ∈ qm):
| (38) |
Gradient of the exchange-correlation energy:
| (39) |
The ∂xi/∂Xa derivative has a value of one only if the quadrature point i is tethered to atom a. Furthermore, the density is a linear combination of AO products, whose gradients satisfy ∇rχ(r − R) = −∇Rχ(r − R); therefore, the last term on the first line of eq. 7 reduces to:
| (40) |
For brevity, we refer the reader to Ref. 99 for additional simplifications of eq. 39. The appendix of Ref. 99 also contains explicit expressions for the quadrature weight derivative, ∂w/∂Xa. Algorithms for computing the ERIs and ERI gradients are found in Refs. 100 and 101, and the one electron Gaussian integrals can be found in the seminal work by Obara and Saika.102
When the QM/MM boundary severs a covalent bond, we use the link atom approach described in Ref. 66. In brief, “dangling bonds” are capped with a hydrogen QM atom. The link atom bond length is fixed, and its orientation is colinear with the severed QM/MM bond. The atomic forces of the link atom are propagated to the real QM and MM atoms via elementary chain rule derivatives. For completeness, we note that other treatments of the QM/MM boundary can be found in the literature, including the Generalized Hybrid Orbital method,103–106 Effective Fragment Potential method,107–109 and Local Self-Consistent Field method110–112 to name just a few.113
2.2 The Ambient-Potential Composite Ewald Method
It is worthwhile to begin with some clarifying remarks regarding eq. 4 which may cause confusion for some readers because the lattice translation n, occurs in one (but not both) of the charge densities. In other words, one might have expected the Coulomb self-energy of a periodic density Σnq(r + n), to be
| (41) |
because the Coulomb self-energy of an aperiodic density q(r), is
| (42) |
However, eq. 41 is formally infinite if q(r) is anywhere nonzero114 because each cell then contains some amount of self-energy and there are an infinite number of cells in the lattice. When performing an inner-product of two functions that are each periodic (the density and the electrostatic potential are both periodic in eq. 41), the desired quantity is actually the inner-product’s average per unit cell. All cells in the lattice are identical, so the calculation of the average energy per unit cell merely requires one to change the range of integration in eq. 41 from “all space” to “the volume of one cell”.
| (43) |
Equation 4 is recovered from eq. 43 by exploiting the periodicity of the electrostatic potential. Although the aperiodic density is not necessarily confined within a unit cell, the combined effluence of density produced from the construction of Σnq(r + n) causes each cell within the lattice to contain one instance of q(r) that appears to have been “wrapped” to the cell boundary. Equation 4 differs from eq. 43 only by “unwrapping” the density and making a corresponding adjustment to the integration limits.
Another possible source of confusion may arise from the prevalence of expressions in the literature that place lattice translations in the denominator rather than the numerator; that is,
| (44) |
Equations 4 and 44 are equivalent. Equation 4 is recovered from eq. 44 by performing a u-substitution that replaces r′ → u + n and d3r′ → d3u within eq. 44 and then changing the dummy integration variable from u to r′. One could also write eq. 44 with a denominator of |r − r′ − n|−1, because the lattice summation considers all unique cell translations.
Our description of the Ambient-Potential Composite Ewald Method makes frequent use of Dirac notation, which is now summarized. A function is written as a “ket”, f(r) = 〈r|f〉. A complex conjugate is a “bra”, f∗(r) = 〈f|r〉. An inner-product is a “braket”, ∫ f ∗(r)g(r)d3r = 〈f|g〉. If both functions are periodic, then the inner-product’s integration is performed over the unit cell volume, ∫Vd3r. From these definitions, one can immediately write the aperiodic (or “primary”) charge density:
| (45) |
and the periodic charge density:
| (46) |
One can further define an “ambient charge density”:
| (47) |
which consists of all translated copies of the density enclosing the primary image.
The act of producing the various forms of the density described above is aided through the use of specialized electrostatic operators. Let us define the “primary electrostatic operator”:
| (48) |
the “periodic electrostatic operator”:
| (49) |
and the “ambient electrostatic operator”:
| (50) |
These operators act upon an aperiodic charge density to produce the “primary electrostatic potential” (the electrostatic potential of the aperiodic density):
| (51) |
the “periodic electrostatic potential” (the electrostatic potential of the periodic density):
| (52) |
and the “ambient electrostatic potential”:
| (53) |
The ambient electrostatic potential is produced solely from the periodic surroundings. That is, it is how the electrostatic potential is altered upon introducing periodicity to the system.
The electrostatic energy of the periodic system (eq. 4) can be decomposed into QM/QM, MM/MM, and QM/MM interactions as follows:
| (54) |
The last term in eq. 54 is the “QM ambient energy”. It is the Coulomb interaction between the primary image’s QM region with the QM regions located in the periodic surrounding. The QM ambient energy is inconvenient to evaluate because the character of the QM ambient potential changes at each SCF step. We shall introduce an approximation that avoids this inconvenience. To begin, note that the QM region’s ambient energy can be expressed as a Taylor series expansion about a reference charge density, .
QM charge density evaluated about the reference:
| (55) |
QM ambient energy evaluated about the reference:
| (56) |
where .
Obviously, if , then the last term in eq. 56 is small. More importantly, the ambient electrostatic operator only interacts the QM region with those located in different periodic cells. If the unit cell was larger than the sphere which circumscribes the QM charge density, then this energy could, in principle, be performed via multipole moment expansions of qqm(r). In other words, the last term in eq. 56 is also negligible when the multipole moments of reasonably approximate those of qqm(r). Therefore, an appropriate choice of is one which satisfies
| (57) |
where Clμ(r) is a regular solid harmonic.115 There are many potential choices which could satisfy this condition; however, considering that force fields have already developed their partial charges to reasonably model the electrostatics, the most convenient choice would be to reuse the underlying MM atomic charges for . Complicating the form of by using, for example, atomic multipoles or diffuse auxiliary basis functions, would only increase the accuracy of the method insofar as those complications could improve the overall description of the QM region’s multipole moments. Alternatively, one could improve the multipole moments by simply adjusting the underlying MM partial charges if it was found to be necessary, thus rendering additional complications moot. Following this logic, the approach taken in the present work is to use a set of static point charges to approximate the ambient QM charge density,
| (58) |
and then truncate the Taylor series to first-order,
| (59) |
| (60) |
such that the ambient QM energy becomes a composite interaction between qqm(r) with , and with itself. After simplification, the energy becomes ECEw[q] ≈ Eelec[q],
| (61) |
In this manner, the periodic potentials only involve static point charge distributions, which can be computed once before the SCF procedure begins.
Equation 61 is an approximation, but our formulation was designed to reduce the error’s magnitude for typical QM/MM applications. It should be pointed out that the truncated Taylor series expansion only effects the interaction of the QM region with its images; it does not approximate the interaction between the QM and MM regions nor the MM region with itself. Nevertheless, if the interaction between the QM region with its periodic images was such that the Taylor series could not reasonably be truncated, then one could directly evaluate the QM ambient energy in eq. 54 by evaluating the multipole moments of qqm(r) at each SCF step and then use the point-multipole particle mesh Ewald (PME) method described in Ref. 17; however, the small size of – and, therefore, intercellular distance between – QM regions should make this added layer of complexity unnecessary in most applications.
The periodic electrostatic potential of the point charge distributions appearing in eq. 61 can be computed with either the Ewald or PME methods. A detailed theoretical development of these methods can be found in Ref. 17, whose notation we adopt henceforth. Specifically, we reserve
| (62) |
to index the angular wave numbers of a plane wave basis:
| (63) |
Furthermore, we shall refer to B-spline weights,
| (64) |
that are constructed from order–n Cardinal B-spline functions,
| (65) |
and whose discrete Fourier transform coefficients,
| (66) |
are evaluated from a uniformly spaced grid. The grid consists of N = N1N2N3 points that are indexed by t = (t1,t2,t3) and positioned at Rt.
| (67) |
We will continue by summarizing the necessary equations for PME, written below for a generic point charge distribution, qpt(r).
| (68) |
In brief, the PME method computes the periodic potential of a model Gaussian density
| (69) |
and then corrects for the short-range difference between the point and Gaussian potentials. The electrostatic potential of the periodic Gaussian density is performed analytically upon fitting it to a plane wave basis. Therefore, the PME potential (see eq. 70) decomposes into a plane-wave potential (see eq. 75), a short-range correction that removes the Gaussian potential (see eq. 73), a corresponding short-range “near-field” potential that reintroduces the point-charges (see eq. 71), and, for charged systems, a uniform background potential (see eq. 76). Several of these potentials can be grouped together, when convenient, into “far-field” (see eq. 72), “real-space” (see eq. 74), and “reciprocal-space” (see eq. 77) potentials. The difference between the Gaussian and point-charge electrostatic potentials is negligible at large distances, so one need only evaluate the near-field and Gaussian potentials within a real-space Ewald cutoff Rcut around r while assuming a minimum image convention. When applied to eq. 61, one must evaluate the potential at all r values where qqm(r) is non-negligible. In other words, the near-field and Gaussian potentials must be evaluated for all imaged point charges within Rcut of qqm(r).
PME potential:
| (70) |
Near-field potential; that is, the potential caused by everything within the real-space Ewald cutoff Rcut around r:
| (71) |
Far-field potential; that is, the potential caused by everything outside Rcut:
| (72) |
Gaussian potential:
| (73) |
Real-space potential:
| (74) |
Plane-wave potential (interpolated from a regular grid):
| (75) |
Uniform background potential:
| (76) |
Reciprocal-space potential:
| (77) |
PME potential interpolation control points:
| (78) |
PME structure factor; that is, the forward discrete Fourier transform coefficients of the B-spline interpolated point charges:
| (79) |
Having now introduced the near and far-field potentials, we show how they are used to simplify the second line in eq. 61,
| (80) |
through the cancellation of terms,
which are equivalent because is necessarily within Rcut of qqm(r). For the same reason, the last line of eq. 61 merely removes of the QM reference density’s near-field energy, assuming that the QM region’s size is less than half the box length.
| (81) |
The energy expression for the Ambient-Potential Composite Ewald method (see eq. 83) is obtained upon replacing the second and third lines of eq. 61 with the last line of eq. 80 and eq. 81, respectively,
| (82) |
and then expanding the far field potentials using the second line of eq. 72.
| (83) |
Explicit expressions for the energy terms appearing in eq. 83 are summarized by eqs. 84–96.
| (84) |
| (85) |
| (86) |
| (87) |
| (88) |
| (89) |
| (90) |
| (91) |
| (92) |
| (93) |
| (94) |
| (95) |
| (96) |
Equation 88 is performed numerically using a molecular quadrature grid consisting of quadrature points ri and partitioned quadrature weights wi. In other words, one integrates in a manner analogous to a LDA exchange-correlation potential. Specifically, the reciprocal-space potential is computed at the FFT grid points, and the B-splines are used to interpolate the potential onto the molecular quadrature grid. If β (see eq. 69) was large, then the numerical integration of the reciprocal space potential shown in eq. 88 would be inaccurate unless additional atomic quadrature grids were also placed around the near-field MM atoms; that is, the reciprocal space potential has “lumps” at the atomic positions, and those lumps look increasingly like q/r as β → ∞. In practice, the β values used in typical QM/MM simulations are sufficiently small that the use of “MM quadrature centers” is unnecessary. For the sake of argument, even if this were an issue, one would eliminate the lumps by numerically integrating eq. 90 along with the reciprocal-space potential, rather than introducing additional quadrature centers.
The contribution of ECEw to the QM spin-resolved Fock matrix is the derivative with respect to the density matrix:
| (97) |
Similarly, the electrostatic energy contribution to the atomic gradients is the derivative ∂E/∂Xa|P. Most of the gradient terms reduce to expressions involving standard nuclear-nuclear, electron-nuclear, and electron-repulsion integrals; for brevity, eqs. 98–101 summarizes only those expressions which involve the plane wave basis and/or numerical quadrature grid.
| (98) |
| (99) |
| (100) |
where
| (101) |
Equation 101 requires the evaluation of the QM electron density on the molecular quadrature grid. Each quadrature grid point density is then B-spline interpolated onto the FFT grid. The FFT-grid representation of the QM charge density then undergoes a forward FFT to produce a set of structure factors. Equation 101 does not introduce a new approximation; it naturally arises from standard chain-rule differentiation of the energy. The numerical integration of the reciprocal-space potential avoids the explicit evaluation of the AO-product Fourier coefficients.
2.3 Other QM/MM electrostatic protocols
The Ambient-Potential Composite Ewald method (denoted by “CEw”, see eq. 83) will be compared to three other electrostatic models:
Electrostatic embedding with truncated electrostatics for both the MM/MM and QM/MM interactions (denoted by “cut”, see eq. 102).
The QM/MM Mulliken charge Ewald method introduced by Nam63 (denoted by “Ewq”, see eq. 104).
A model-MM Ewald energy with a smooth, short-range QM/MM correction (denoted by “MMEw”, see eq. 108). The MMEw model is a mixture of mechanical and electrostatic embedding. We define a nonbond cutoff radius around the QM region, outside of which the QM/MM interactions occur with mechanical embedding (including the long-range Ewald interaction), and inside of which the interactions are performed with electrostatic embedding. This approach is generalized in the MMEw method by introducing a switch that smoothly transitions between the two limits to yield a continuous potential energy. In the limit that the switching width becomes zero, the transition occurs discontinuously.
Cutoff-based electrostatics (Ecut[q] ≈ Eelec[q]):
| (102) |
Ecut contribution to the Fock matrix:
| (103) |
Mulliken charge QM/MM Ewald method (EEwq[q] ≈ Eelec[q]):
| (104) |
| (105) |
Mulliken charge:
| (106) |
EEwq contribution to the Fock matrix:
| (107) |
Model-MM Ewald energy with a short-range QM/MM correction (EMMEw[q] ≈ Eelec[q]):
| (108) |
The smooth, short-range incorporation of the QM/MM interactions:
| (109) |
The switched, short-range model-MM interactions:
| (110) |
The “switch off” function:
| (111) |
The distance between MM atom b and the nearest QM atom:
| (112) |
EMMEw contribution to the Fock matrix:
| (113) |
The expression for the Lennard Jones energy (eq. 3) and the ECEw (eq. 83), Ecut (eq. 102), EEwq (eq. 104), and EMMEw (eq. 108) electrostatic methods all employ the symbol “Rcut”. In all cases, this distance is the “nonbond cutoff” radius used by the MM program to generate neighbor lists. We proceed by summarizing how the nonbond cutoff is used within different methods. In doing so, we use the phrases “real-space Ewald cutoff”, “electrostatic cutoff”, and “switched cutoff” to aid the reader’s ability to recall how the various methods behave at the nonbond cutoff. If an atom is inside the nonbond cutoff, then the pairwise Lennard-Jones energy is computed; whereas, if it is outside the nonbond cutoff, then its interaction is modeled through a long-range tail correction. The electrostatic methods use a single neighbor list for the QM region, as a whole, which is constructed from the union of the individual QM atom neighbor lists. Therefore, an MM atom is within the nonbond cutoff if its distance to any QM atom is less than Rcut. The Rcut appearing in the ECEw and EEwq methods are “real-space Ewald cutoffs”, which merely denotes the distance where the point charge and PME Gaussian potentials are sufficiently similar that additional real-space corrections would have a negligible effect. The Rcut appearing in the Ecut method is an “electrostatic cutoff” that marks the distance outside of which the electrostatic interactions are strictly (and discontinuously) ignored. The Rcut appearing in the EMMEw method is the outer edge of a “switched cutoff” that extends from Rcut − wsw to Rcut. Inside the switching region (R ≤ Rcut − wsw), the QM/MM electrostatic interaction explicitly involve the atomic nuclei and AO products. Outside of the switched cutoff (R ≥ Rcut), the QM/MM electrostatic interaction (including the long-range Ewald-component of the energy) is performed using a static, MM point charge representation of the QM atoms. The EMMEw switching region (Rcut −wsw > R > Rcut) smoothly transitions between these two limits. Therefore, the EMMEw QM atom forces are influenced by the Ewald potential, but the QM electron density explicitly polarizes only to the nearby MM atoms within the switched cutoff.
It has previously been noted69,70 that the use of Mulliken charges within EEwq can result in SCF convergence instability when applied to standard, all-electron AO basis sets. In order to improve the SCF stability, our implementation of EEwq holds the Mulliken charges within the current MD step fixed to the SCF converged Mulliken charges from the previous MD step. In other words, the charge derivatives appearing in eq. 107 are computed once at the beginning of the SCF procedure.
2.4 Computational details
The ab initio code and all electrostatic methods described in the previous sections were implemented from scratch within a development version of AmberTools 15 and interfaced to the Sander MD program, which was used to perform all simulations described below.116
The notation Ecut R, EMMEw RwD, ECEw R, EEwq R is used to distinguish the electrostatic protocols described in the previous sections and their associated nonbond cutoffs. For example, Ecut 14 denotes the use of eq. 102 with Rcut = 14 Å, and EMMEw 14w4 is eq. 108 with Rcut = 14 Å and wsw = 4 Å. Similarly, ECEw 14 and EEwq 14 refer to eq. 83 and eq. 104, respectively, with Rcut = 14 Å.
All Ewald reciprocal-space calculations are performed using a 1 Å−3 grid density, and the Ewald coefficient was chosen from the value of Rcut to reproduce a direct sum tolerance of 10−6 au.
Figures 1–6 display simulation results for a system composed of a p-nitrophenyl phosphate (pNPP) QM region in a truncated octahedron filled with 4563 TIP4P/Ew water molecules. Salt ions were not added to the system. The total charge of the system is 2-. The Ewald methods use a neutralizing uniform background potential (eq. 76) to account for the net charge. The pNPP Lennard-Jones and qref parameters were obtained from the Antechamber program included in AmberTools 15, which chose the LJ parameters and charges from the GAFF force field and AM1-BCC protocol, respectively.116–119 The system volume was equilibrated using the DFTB2 semiempirical Hamiltonian and the QM/MM Ewald method described in Ref. 63 in the isothermal-isobaric ensemble (NPT) for 100 ps (1 fs/step) at 298 K and 1 atm. The Berendsen barostat was used to control the pressure with a relaxation time of 2 ps, and the Langevin thermostat controlled the temperature with a collision frequency of 5 ps−1. The simulation cell’s equilibrated real-space lattice vectors are 56.42 Å. The equilibrated volume and coordinates were then used for the microcanonical (NVE) and canonical (NVT) ensemble simulations described below.
Figure 1.
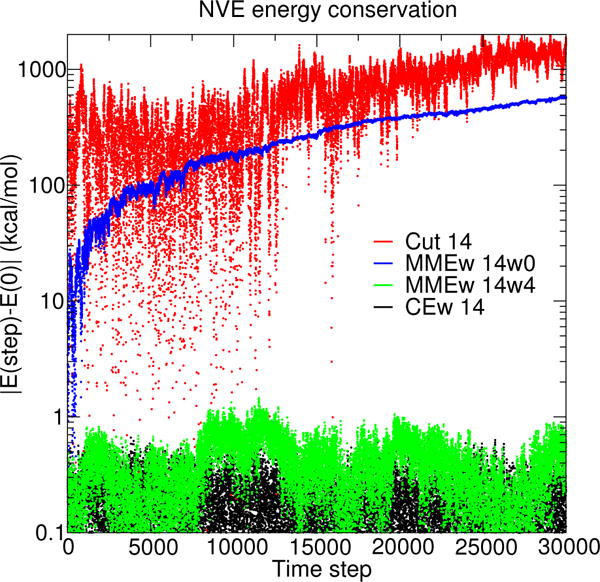
NVE simulation energy conservation as a function of time step (0.001 ps/step) using various electrostatic protocols. The system is a pNPP solute computed with PBE0/6-31G* in a truncated octahedron of TIP4P/Ew waters. See the text in Computational Details for a description of the notation used to distinguish the various electrostatic protocols.
Figure 6.
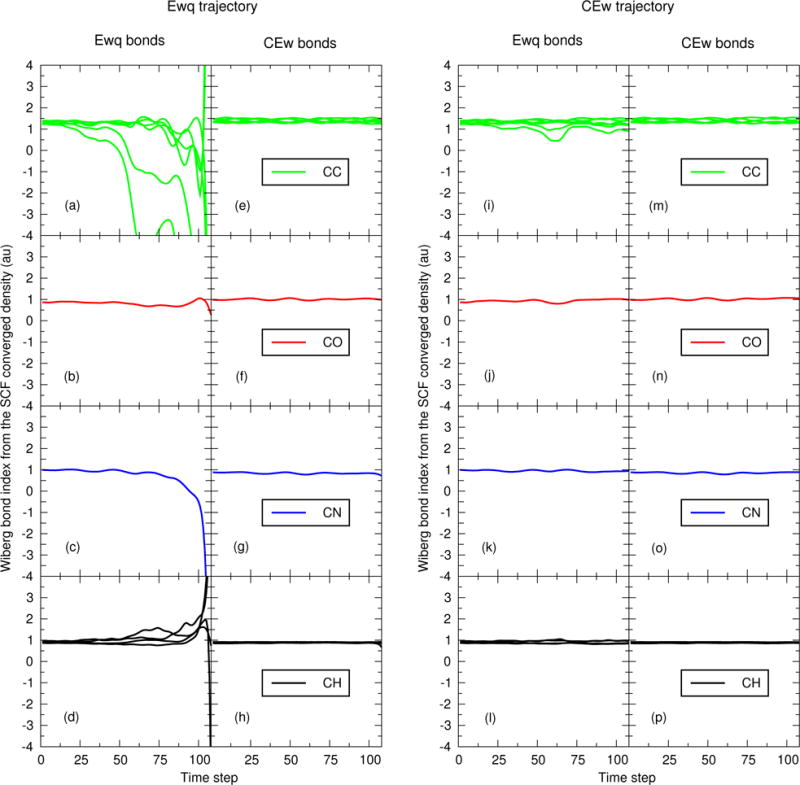
Wiberg bond order indices of the EEwq 14 and ECEw 14 SCF converged electron densities as a function of time step. The two left-most columns are evaluated at the atomic positions generated by the EEwq 14 trajectory. The two right-most columns are evaluated at the atomic positions generated by the ECEw 14 trajectory. The EEwq 14 and ECEw 14 trajectories as those shown in Figure 4.
Figure 1 shows the PBE0/6-31G* QM/MM simulation total energy, relative to the first step, for a series of NVE simulations that differ only by their choice of electrostatic protocol. Each simulation was run for 30 ps (30 000 steps), and they started from the same initial conditions.
Figure 2 displays PBE0/6-31G* QM/MM potential of mean force (PMF) profiles of the pNPP dissociation reaction: , where the reaction coordinate is the P-O distance. Each PMF was generated from 96 umbrella window NVT simulations that spanned from RPO = 1.4 Å to 6.1 Å in steps of 0.1 Å. The umbrella harmonic potentials used a force constant of 50 kcal mol−1 Å−2. The PMFs were generated from the distribution of RPO values using the variational free energy profile (vFEP) method.120 Each NVT simulation was performed at 298 K for 12 ps (Fig. 2a), 24 ps (Fig. 2b and Fig. 2c), or 36 ps (Fig. 2d). The total amount of sampling for each curve was chosen to achieve aesthetically pleasing figures; that is, the PMFs shown in Fig. 2d are all sufficiently similar to one another that additional sampling was added to remove small numerical noise in the region where the PMF is relatively “flat”, so that the curves could be more easily distinguished. Alternatively, the PMFs appearing in Fig. 2a are identifiably different such that sampling beyond 12 ps was not considered to be a prudent use of resources. We note that the EMMEw 14w1 PMF appearing in Fig. 2b is terminated at RPO ≈ 4 Å because many of the EMMEw 14w1 simulation windows beyond RPO > 4 fail to complete due to large velocity warnings. This is expected because the switching width in this case is so small that any waters within the switching layer experience a large force, because the potential energy approaches a discontinuity as the width nears zero. The dashed, horizontal lines in Fig. 2a–2d mark the experimental barrier in solution121 (29.6 kcal/mol).
Figure 2.
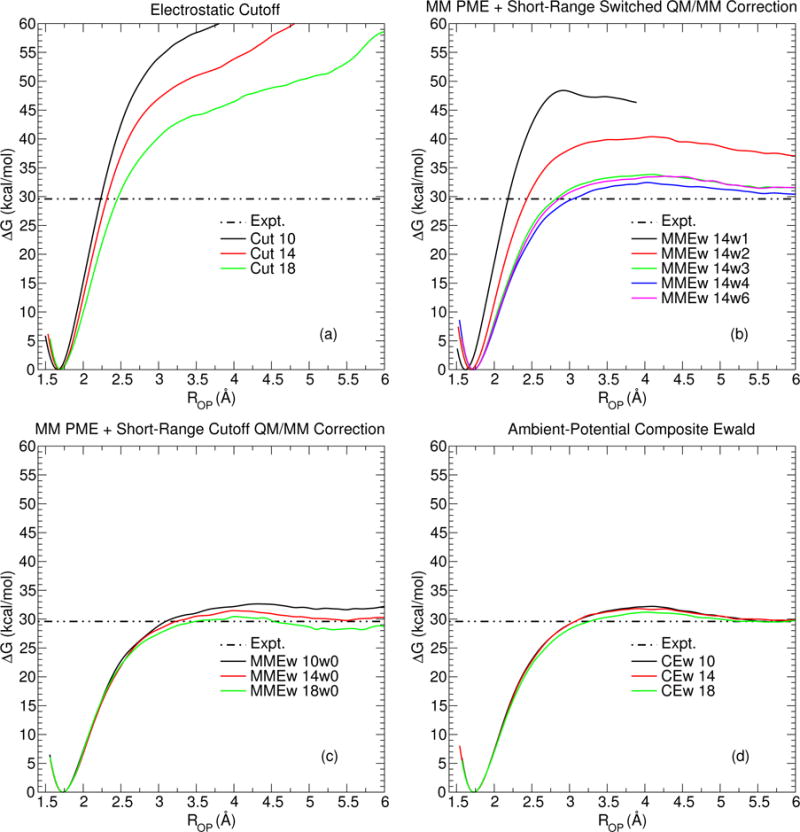
The effect of various electrostatic protocols on the potential of mean force profiles of the pNPP dissociative reaction.
Figure 3 compares radial distribution functions (RDFs) between the phosphorous and water-oxygens observed within PBE0/6-31G* QM/MM pNPP simulations performed using several electrostatic protocols. Figures 3a and 3b are RDFs obtained from simulations whose umbrella potential is centered about RPO = 1.7 Å (the approximate minimum of the PMF), and Figs. 3c and 3d were obtained using an umbrella window centered about RPO = 4.0 Å (the approximate transition state of the PMF). Figures 3b and 3d are zoomed areas of 3a and 3c, respectively, as marked by the black boxes. The RDFs were generated from 500 ps of NVT simulations at 298 K, whose trajectory was written every 50 steps.
Figure 3.
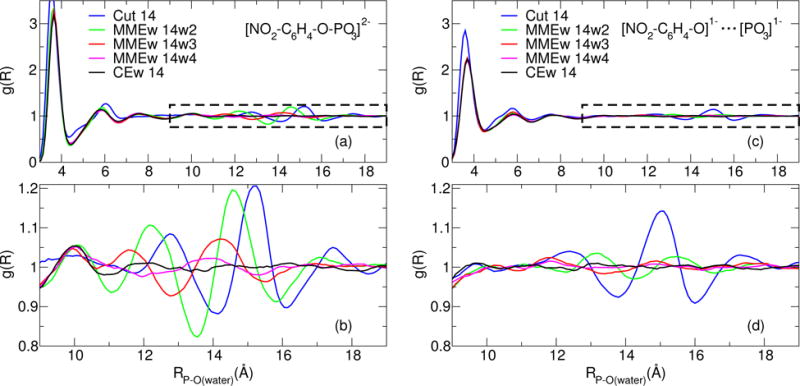
The pairwise radial distribution function between phosphorous and the water-oxygens. The metaphosphate RPO separation in panes (a) and (c) are 1.70 Å and 4.00 Å, respectively. Panes (b) and (d) are zoomed areas of (a) and (c), as indicated by the dashed boxes.
Figure 4 shows the potential, kinetic, and total energies from PBE0/6-31G* QM/MM NVE simulations of pNPP performed using the EEwq 14 (Fig. 4a) and ECEw 14 (Fig. 4b) electrostatic protocols as a function of time step (1 fs/step). Both trajectories start from the same coordinates and initial velocities, and an umbrella potential at RPO = 1.7 Å is applied. The EEwq 14 trajectory fails to SCF converge after step 108. Therefore, the EEwq 14 trajectory is restarted at step 108 from the coordinates and velocities produced by the ECEw 14 trajectory at step 108. The EEwq 14 trajectory again fails to SCF converge after step 197. The EEwq 14 SCF failures are marked in Fig. 4a by “Implosion #1” and “Implosion #2.” Several snapshots of the pNPP solute from the EEwq 14 and ECEw 14 trajectories are shown immediately below their respective energy profiles.
Figure 4.
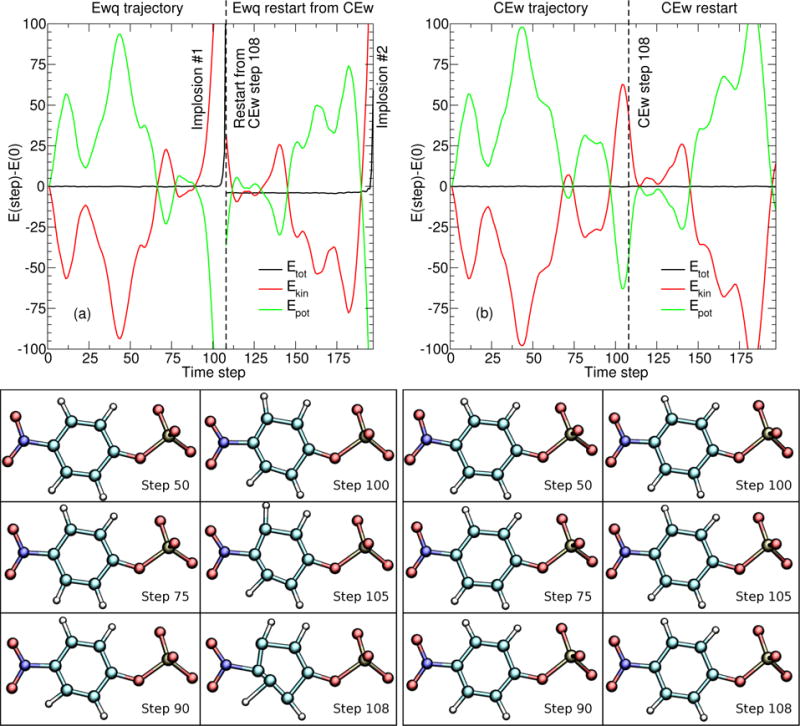
QM/MM NVE simulations of the pNPP molecule (near the minimum of the PMF; that is, the RPO = 1.7Å window) using the EEwq 14 (left) and ECEw 14 (right) methods. The plots (top) decompose the total simulation energy into kinetic (red) and potential (green) energy contributions, relative to the initial condition. Both simulations start from the same initial condition. The molecules beneath each plot are snapshots of the QM solute along their respective trajectory. Each time step is 0.001 ps.
Figures 5 and 6 re-analyze the PBE0/6-31G* Mulliken charges and Wiberg bond orders of the first 108 steps from each trajectory shown in Fig. 4. Specifically, Figs. 5a–5d are the SCF converged Mulliken charges produced by the EEwq 14 method using the atomic coordinates from the first 108 steps of Fig. 4a. Figures 5e–5h are the SCF converged Mulliken charges produced by the ECEw 14 method using the atomic coordinates from the first 108 steps of Fig. 4a. Similarly, Figs. 5i–5l and 5m–5p are the EEwq 14 and ECEw 14 Mulliken charges, respectively, evaluated from the first 108 steps of Fig. 4b. The format of Fig. 6 is analogous; it differs from Fig. 5 only by displaying the Wiberg bond orders rather than Mulliken charges. For clarity, the Mulliken charges and Wiberg bond orders are evaluated using the coordinates of the entire QM/MM system.
Figure 5.
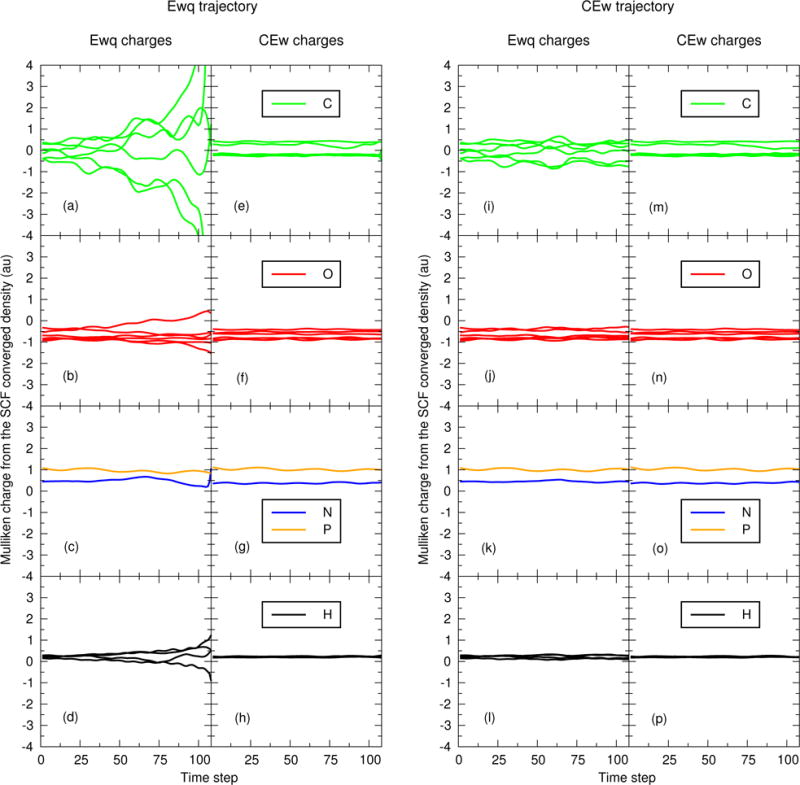
Mulliken charge analysis of the EEwq 14 and ECEw 14 SCF converged electron densities as a function of time step. The two left-most columns are evaluated at the atomic positions generated by the EEwq 14 trajectory. The two right-most columns are evaluated at the atomic positions generated by the ECEw 14 trajectory. The EEwq 14 and ECEw 14 trajectories as those shown in Figure 4.
Table 1 compares the QM/MM simulation rates acheived using ECEw 14 and EMMEw 14w4 when applied to the solvated 72 atom alanine chain shown in Fig. 7. The alanine chain is enclosed within a truncated octohedron containing 5083 TIP4P/Ew waters. The timings are listed as the average simulation rate (ps/day) reported by Sander after 1 ps (1000 steps) of PBE0/6-31G* NVT simulation at 298 K. The simulation rates are reported as a function of central processing unit (CPU) core count and QM system size, including link atoms. The QM region consists of all atoms to the left of the cuts shown in Fig. 7 and the hydrogen link-atoms crossing the boundaries. The timings were performed on the stampede supercomputing cluster at the Texas Advanced Computing Center. Each node contains two Intel Xeon E5-2680 processors (8 cores/processor), and the nodes are interconnected with Mellanox FDR InfiniBand technology. Although Intel Phi coprocessors and/or Nvidia graphics cards were available on the compute nodes, our code currently only runs on the CPU.
Table 1.
Simulation rates (ps/day; 1 fs/step) using ECEw 14 and EMMEw 14w4 as a function of CPU core-count (Ncore) and QM region size (Nqm, the number of QM atoms) for the solvated alanine chain shown in Fig. 7.
| Ncore |
Nqm, ECEw 14
|
Nqm, EMMEw 14w4
|
||||||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
| 12 | 22 | 32 | 42 | 52 | 62 | 72 | 12 | 22 | 32 | 42 | 52 | 62 | 72 | |
| 1 | 7.4 | 1.6 | 0.66 | 0.36 | 0.23 | 0.16 | 0.12 | 8.3 | 1.7 | 0.71 | 0.38 | 0.24 | 0.17 | 0.12 |
| 2 | 14.4 | 3.0 | 1.3 | 0.71 | 0.45 | 0.30 | 0.22 | 16.5 | 3.3 | 1.4 | 0.76 | 0.48 | 0.31 | 0.23 |
| 4 | 27.3 | 5.7 | 2.4 | 1.3 | 0.84 | 0.56 | 0.41 | 31.1 | 6.2 | 2.6 | 1.4 | 0.88 | 0.60 | 0.42 |
| 8 | 51.2 | 10.6 | 4.3 | 2.4 | 1.5 | 0.99 | 0.72 | 59.6 | 11.6 | 4.6 | 2.5 | 1.6 | 1.0 | 0.75 |
| 16 | 92.4 | 19.8 | 8.1 | 4.5 | 2.8 | 1.9 | 1.3 | 110.3 | 21.8 | 8.6 | 4.7 | 3.0 | 2.0 | 1.4 |
| 32 | 157.3 | 35.3 | 14.6 | 7.8 | 4.9 | 3.3 | 2.4 | 187.9 | 38.0 | 15.3 | 8.2 | 5.1 | 3.5 | 2.5 |
| 64 | 237.4 | 59.4 | 24.4 | 13.3 | 8.3 | 5.5 | 3.9 | 289.0 | 64.0 | 25.4 | 13.8 | 8.6 | 5.7 | 4.1 |
| 128 | 337.5 | 90.4 | 37.3 | 19.9 | 12.6 | 8.3 | 5.8 | 418.5 | 97.7 | 39.1 | 20.8 | 13.0 | 8.6 | 6.0 |
| 256 | 428.4 | 122.7 | 51.9 | 28.1 | 17.0 | 11.0 | 7.9 | 536.4 | 135.1 | 54.5 | 28.5 | 17.4 | 11.4 | 8.1 |
| 512 | 493.0 | 152.4 | 62.2 | 34.5 | 20.8 | 13.4 | 9.4 | 622.5 | 167.1 | 66.0 | 34.7 | 21.1 | 13.7 | 9.6 |
Figure 7.
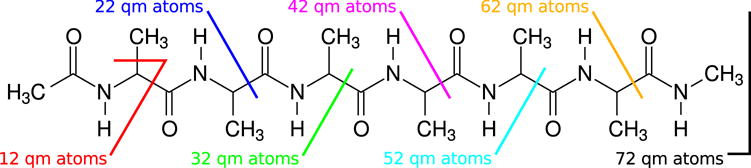
The QM/MM simulation rates are performed using a 72 atom alanine chain solvated by 5083 TIP4P/Ew waters in a truncated octahedron simulation cell. The timings were performed with different QM regions, which correspond to the various cuts in the covalent bonds shown in the figure. The severed bonds are capped with hydrogen link atoms, which are included in the count for the number of QM atoms.
Figure 8 is a schematic of the transesterification of hydroxyalkyl phosphate esters. Of particular interest in this work are the reactions involving ethoxide (EtO) or phenoxide (PhO), which are representative of “poor” and “enhanced” leaving groups, respectively. The “R1” and “R2” bond labels appearing in Fig. 8 are reaction coordinates used to perform the two-dimensional (2D) umbrella window simulations shown in Fig. 9. Figures 9a–9c are 2D PMFs of the EtO phosphoryl transesterification and Figs. 9d–9f are 2D PMFs of the PhO phosphoryl transesterification. Figures 9a and 9d were performed with AM1/d-PhoT (abbreviated as AM1/d); Figs. 9b and 9e were performed with DFTB2; and Figs. 9b and 9e were performed with PBE0/6-311G**. The EtO simulations consisted of 134 windows that form a grid from (R1,R2) = (1.4 Å,1.4 Å) to (5.6 Å,2.2 Å) and from (1.4 Å,2.1 Å) to (2.4 Å,3.0 Å) using a 0.2 Å spacing in either direction. The PhO simulations consisted of 109 windows from (R1,R2) = (1.4 Å,1.4 Å) to (5.6 Å,2.2 Å). The umbrella windows had a force constant of 85 kcal mol−1 Å−2, and the QM solute was enclosed within an truncated octohedron containing 4204 (EtO) or 5183 (PhO) TIP4P/Ew waters. The density of the system was equilibrated with DFTB2 in a NPT simulation in a manner analogous to the pNPP equilibration protocol described above, and production was performed in the NVT ensemble at 298 K. The AM1/d-PhoT and DFTB2 semiempirial models were sampled and analyzed for 100 ps/window, whereas the PBE0/6-311G** simulations were run for 30 ps/window. Analysis of the first 20 ps changes the PBE0/6-311G** barriers by less than 0.2 kcal/mol; therefore, sampling was suspended at 30 ps. The vFEP method was used to generate the 2D PMFs from the distributions of R1 and R2 values observed in the simulations.120 The circle and X marks appearing in Fig. 9 are minima and transition states of the 2D surfaces, and the colored lines connecting the stationary points are a nudge elastic band minimum free energy path.122,123 The 1D PMFs appearing in Fig. 9 are the free energy profiles along the minimum free energy path.
Figure 8.
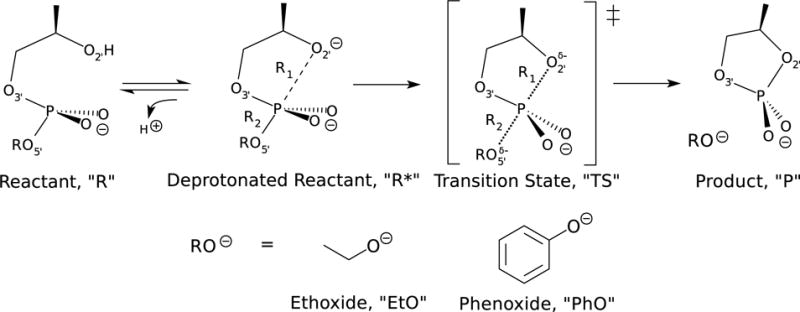
Schematic of the phosphoryl transfer (model RNA transesterification) reactions with ethoxide and phenoxide leaving groups.
Figure 9.
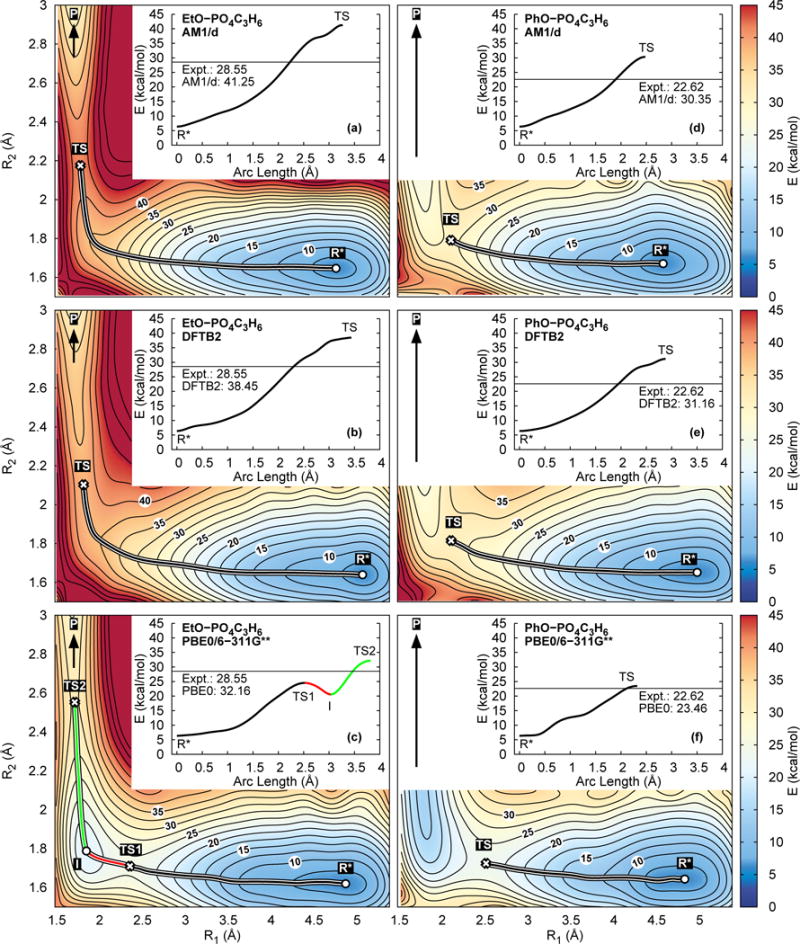
Free energy profiles of the transesterification reactions shown in Fig. 8. Ethoxide leaving group: (a)–(c). Phenoxide leaving group: (d)–(f). The inset, one-dimensional plots are the free energies along the nudge elastic band path connecting the minimum to the rate-limiting transition state. The horizontal line in the inset plots mark the experimental barrier.
Our QM/MM simulations assume that the 2′-O has already been deprotonated. That is, we explicitly model the reaction barrier from R* to TS (see Fig. 8), whereas the experimental barrier corresponds to the reaction from R to TS. Therefore, our calculated free energies are corrected to account for the free energy difference between R and R*. The rate of the reaction shown in Fig. 8 was experimentally determined to be k = 1.57×107 and 7.40×1010 mol−1s−1 for the EtO and PhO leaving groups, respectively, at 80 C in a pH 8 buffer solution.124 The transition state free energies at standard state concentration are computed from the Eyring equation, ΔG‡ = −RT ln(kh/kBT), where h and kB are Planck’s constant and Boltmann’s constant, respectively. Thus the experimental barriers for EtO and PhO are 28.55 and 22.62 kcal/mol, respectively. To correct our free energies, we must consider the pKa of 2′-OH at pH 8. Experimental measurements of the 2′-OH pKa in small molecule models of RNA have ranged between 12.35–13.9, depending on the structure and experimental technique.125–128 Of the available experimental pKa data, the model most closely resembling those shown in Fig. 8 is the adenosine 3′-ethyl phosphate used in Ref. 127, whose 2′-OH pKa was found to be 12.68 ± 0.06. Assuming a pKa of 12.68, the free energy of the deprotonated 2′-O at pH 8 and 298 K is estimated to be ΔGR→R* = −2.303RT(pH − pKa) = 6.38 kcal/mol. Therefore, 6.38 kcal/mol have been added to all computed free energies shown in Fig. 9.
3 Results
3.1 Comparison of electrostatic protocols
Energy conservation
The drift in total energy observed within NVE simulations of pNPP are shown in Figure 1. The Ecut 14 and EMMEw 14w0 methods produce strong energy drifts because their potential energy surfaces contain discontinuities. The Ecut 14 energy drift is larger than EMMEw 14w0 because the Ecut 14 method excludes the long-range interactions entirely. The EMMEw 14w4 and ECEw 14 methods conserve the total energy. Both models have continuous potential energies and model the long-range interactions, albeit in different manners. The EEwq electrostatic method first developed by Nam et al.63 is not shown in Fig. 1 because the QM solute “implodes” after 100 steps. The instability of EEwq trajectories will be discussed in detail below. In brief, the EEwq simulations do conserve the total energy for the length of time that they can be run.
The conservation of total energy is important for maintaining a stable simulation temperature.129 Although the temperature can be corrected through velocity rescaling, the lack of total energy conservation can, over time, manifest itself by dampening high-frequency motion and transforming it to low-frequency motions.130 Furthermore, the treatment of long-range forces has also been shown to dramatically affect the structural stability of proteins30 even when switching functions are introduced.131 The simulations performed in the remainder of this manuscript are not performed on a time scale that is long enough to directly observe these previously observed artifacts.
pNPP free energy profiles
Simulations of reactions involving charged species are ubiquitous in biology. Among the most important are phosphoryl transfer reactions, which are vital for many celluler processes; for example, gene regulation, cell signalling, and energy conversion.132–134 Phosphoryl transfer reactions have been studied extensively by computational methods,135 and mechanistic insights have been gained by studying model systems that provide an interpretation of linear free-energy relations136 and kinetic isotope effects.137 In this section, we examine how various electrostatic protocols affect the the phosphoryl transfer reaction barrier of pNPP (see Figure 2).
In the vicinity of ROP ≈ 1.7 Å, the metaphosphate is covalently bound to nitrophenyl such that it is a single molecule with a −2 charge. At a separation of ROP ≳ 4 Å, the solute is better described as two polyatomic monoanions. One can gain insight into the role of solvation in these two situations by refering to a Born ion model which, for water, approximately translates to: ΔGsolv ≈ −Q2/R, where R is the radius of the ion. In other words, there is a greater degree of solvent stabilization near the PMF minimum than there is near the transition state. Although various electrostatic methods appear to increase the barrier, the differences are related to the solvent stabilization near the PMF minimum, which has been chosen to define the zero of free energy in all cases. Had the system consisted of a pair of counter ions, such as the or systems examined in Ref. 63, then the solvent stability of the QM region would be enhanced as the ions were separated, because the separation of ions produces an increasing dipole moment.
The series of Ecut profiles shown in Fig. 2a differ strikingly from the other electrostatic protocols. The PMFs continually increase as the metaphosphate dissociates. The truncated electrostatic forces cause the waters to become structured at the QM/MM electrostatic cutoff (see Fig. 3), which acts to create an artificial solvation shell. As one decreases the electrostatic cutoff from 18 Å to 10 Å, the artificial solvation shell forms closer to the solute and thus increases the stability of the PMF minimum. The profiles do not contain a transition state below ROP < 6 Å for two reasons:
The length of the electrostatic cutoff prevents the solvation shells of each fragement from dissociating from each other in this range.
The water around phosphorous continues to display an artifically structured RDF even when the metaphosphate is separated (see Figs. 3c–3d), so the radius of the “effective Born ion” increases as the metaphosphate dissociates.
The EMMEw method’s treatment of the long-range QM/MM electrostatics is fundamentally different from Ecut, and this difference causes the EMMEw PMFs to exhibit transition states (see Fig. 2b). As the width of the switching region is reduced, the dissociation barrier increases. This observation appears to be related to how the switching width affects the solvent structure in the vicinity of the switching region. Figures 3a–3b and 3c–3d illustrate the solvent structure of EMMEw 14w2, EMMEw 14w3, and EMMEw 14w4 at ROP = 1.70 Å and ROP = 4.00 Å, respectively. As the switching width becomes smaller, the transition between the QM/MM and MM-analogue/MM interactions occurs more abruptly, and the waters experience a greater force in the switching region. The degree of water structure at the switching region is greater at ROP = 1.70 Å because the solute more closely resembles a dianion. Unlike the Ecut method, the artificial solvation shell dissipates as the metaphosphate dissociates. The series of EMMEw profiles shown in Fig. 2c use a switching width of zero. Thus, the width is too small for the waters to experience a force associated with the transition, and the difference between MM-analogue/MM and QM/MM interactions is too subtle to cause a dramatic difference in the transition state barrier. Increasing the real-space Ewald cutoff from 10 to 18 Å lowers the barrier from 32.4 (EMMEw 10w0) to 30.2 (EMMEw 18w0) kcal/mol. Relative to Figs. 2a–2b, the EMMEw Rw0 profiles shown in Fig. 2c are relatively stable and agree with experiment much more closely; however, the reader should take note that the electrostatic protocols shown in Fig. 2c do not conserve the total energy in NVE simulations.
The series of ECEw profiles shown in Fig. 2d exhibit the greatest degree of similarity as a function of nonbond cutoff. The barriers are: 32.2 kcal/mol (ECEw 10), 31.8 kcal/mol (ECEw 14), 31.2 kcal/mol (ECEw 18). Unlike the other electrostatic protocols appearing in Fig. 2a–2c, the ECEw method allows the QM Hamiltonian to explicitly polarize to both the short- and long-range electrostatics. As a consequence, ECEw 14 does not induce an artificially structured solvation shell, as can be seen in Fig. 3.
Electronic polarization
Figures 4–6 are used to discuss the symptoms which arise when attempting to use the semiempirical-style QM/MM-Ewald method63 with an ab initio Hamiltonian. To analyze the behavior of EEwq 14 and ECEw 14, we construct NVE trajectories of each (Fig. 4a and 4b, respectively), starting from the same initial conditions. The EEwq 14 total energy is conserved until step 106, at which point the atom velocities are too large to adequately propagate with a 1 fs time step. Furthermore, we are unable to SCF converge the EEwq 14 QM region after step 108. The solute exhibits unusual changes in the hydrogen covalent angles after the first 75 steps, a dramatic deformation of the phenyl ring after the first 90 steps, and wildly nonphysical bond lengths in the last few steps – at which point, the solute can only be described as having undergone an “implosion”. The ECEw 14 trajectory (Fig. 4b) does not encounter any of these symptoms. To demonstrate that the EEwq 14 implosion is not a fluke, we restarted the EEwq 14 simulation from the coordinates taken from step 108 of the ECEw 14 trajectory. The EEwq 14 trajectory again implodes after an additional 80 steps.
The instability of EEwq trajectories has previously been described by Holden et al.,69,70 which motivated them to concoct a point-charge representation of the QM charge density using a ChElPG charge-fitting procedure.71 The ECEw method described in the present work interacts the QM region with its surroundings without resorting to an auxiliary representation. Nevertheless, we agree with Holden et al. that the underlying reason for the instability of EEwq trajectories arises from the manner in which the Mulliken potentials contribute to the Fock matrix. In other words, the EEwq 14 trajectory in Fig. 4a becomes nonphysical because the electrostatic protocol spuriously polarizes the density matrix to the environment.
Figures 5–6 compare how the EEwq 14 and ECEw 14 protocols affect the polarization of the QM electron density by monitoring the Mulliken charges and Wiberg bond orders at each step of the trajectories. Figure 5a shows the EEwq 14 carbon Mulliken charges evaluated using the first 108 steps of the EEwq 14 trajectory. The carbon charges diverge as the trajectory reaches the first implosion. At first glance, a reader might be tempted to flippantly dismiss Fig. 5a because Mulliken charges are known to exhibit a sensitivity to basis set.138,139 However, the ECEw 14 carbon Mulliken charges shown in Fig. 5e are evaluated at the same coordinates as those used in Fig. 5a, and yet the ECEw 14 charges are remarkably stable – even as the solute implodes. The carbon charges are recomputed along the ECEw 14 trajectory in Figs. 5m and Fig. 5i. Again, the EEwq 14 charges (Fig. 5i) are more sensitive, but they do not diverge because the ECEw 14 trajectory does not implode. The EEwq 14 Mulliken charges of the other atoms (O,N,P,H) are relatively stable in comparison to carbon. What we conclude from Fig. 5 is:
The EEwq 14 Mulliken charges of carbon are particularly sensitive to geometry, and
The EEwq 14 electrostatics must be polarizing the underlying density matrix in a suspicious manner, because the ECEw 14 Mulliken charges are stable when evaluated with the same coordinates.
The C-C, C-O, C-N, and C-H Wiberg bond indices shown in Fig. 6 are a second means for analyzing the electron polarization. The C-C bond orders in Fig. 6a, 6e, 6i, and 6m should be about 1.5 because of the conjugation within the phenyl ring. However, some of the C-C bonds in Fig. 6a show strong antibonding behavior. The spurious EEwq 14 C-C bonding pattern significantly alters carbon’s bonding to the other atoms as one approaches the implosion.
The EEwq method’s polarization is sensitive because:
There is a large amount of C-C AO overlap, and
The Mulliken potentials produce multiple, inconsistent views of the external environment’s electronic chemical potential.
There is a large amount of C-C AO overlap because the “C3s” function – that is, the primitive s-function in 6-31G* – has a small Gaussian exponent and there are 6 carbons in close proximity to each other. To be more precise, there is a large amount of C3s-C3s AO overlap. The strong C3s AO overlap produces a near-linear dependence and a correspondingly small AO overlap matrix eigenvalue. Consequently, the first column (assuming the eigenvalues are sorted) of X (eq. 31) is a series of numbers that are large in magnitude, oscillate in sign, and are dominated by C3s-character. Alternatively stated, the C3s AOs are transformed in the OAO basis (eq. 35) to become oscillatory and delocalized across the phenyl ring. The near-linear dependence that we’ve just described is expected and routine. Most applications of ab initio methods with small molecules encounter far smaller AO overlap eigenvalues than what is found here. The degree of near-linear dependence in the present example only becomes a problem once the chemical potential is inconsistently viewed. The electronic chemical potential produced by the external environment (the MM atoms and long-range electrostatics) is
| (114) |
and it polarizes the electron density through its contribution to the Fock matrix.
| (115) |
By performing the interaction through Mulliken charges, the EEwq method produces different chemical potentials for each AO product.
| (116) |
Each of these chemical potentials is a constant throughout space, but every AO-pair experiences a different constant. When eq. 115 is transformed into the OAO basis, the spatial inconsistencies of μEwq,µν(r) and the multicenter delocalization of eq. 35 malforms the chemical potential relative to the other OAO basis functions to produce irregularities in the electron polarization. The ECEw method presented in this work does not produce errant polarizations because it uses a single, consistent view of the external environment’s chemical potential.
Computational efficiency
The ECEw method is more expensive than EMMEw because ECEw polarizes the QM region to both the short- and long-range interactions. Specifically, ECEw integrates the Ewald Gaussian potential via molecular quadrature, and the ECEw gradients (eq. 100) require a forward Fourier transform of the QM charge density from the molecular quadrature grid. As shown in Table 1, this additional layer of computation slows the ECEw simulations by only a small amount because the dominant effort continues to involve the calculation of standard Gaussian integrals required by the underlying ab initio Hamiltonian. For a 52 atom QM region, the difference in timings between ECEw and EMMEw is 1% to 4%, depending on how many CPU cores are used.
The choice of real-space Ewald cutoff affects the relative cost between the real- and reciprocal-space evaluations, and it thus could be tuned to optimize the overall cost. Specifically, reducing the size of the real-space Ewald cutoff necessitates the use of larger Ewald coefficients which in turn requires a larger number of plane waves to resolve. The optimal choice is obtained from empirical observation. The strategy used in Amber, which we have adopted in this work, is to use a 1 point/Å3 FFT grid density, and then choose the Ewald coefficient such that erf(βRcut) = εRcut, where ε is a “direct sum tolerance”. One could argue that larger values of Rcut may be necessary when using ab initio methods because the electron density extends several Ångstroms beyond the nucleus. By employing a 14 Å real-space Ewald cutoff, we are attempting to pre-emptively diffuse that argument. Having said that, we have not yet encountered a scenerio where a 10 Å real-space Ewald cutoff produces a questionable result. In practice, the pNPP simulations employing a 14 Å real-space Ewald cutoff slowed the calculation by 7 to 8% relative to the 10 Å real-space Ewald cutoff.
3.2 Comparison between ab initio and semiempirical free energy profiles
Semiempirical theory has seen widespread use for generating potential of mean force free energy profiles from QM/MM simulations. Considering that semiempirical models are approximately 100 times faster than many ab initio methods, one may verily question whether the accuracy of ab initio approaches is worth their added effort. Figure 9 compares the AM1/d-PhoT and DFTB2 semiempirical model PMFs with those produced by PBE0/6-311G** for the transesterification reaction shown in Fig. 8. The most striking difference occurs with the EtO leaving group (Figs. 9a–9c). The semiempirical models predict a single, late transition state, whereas PBE0 produces two transition states (the late transition state is rate controlling) and an intermediate. In other words, the AM1/d-PhoT and DFTB2 semiempirical models predict a “concerted” mechanism, and PBE0 predicts a “stepwise” mechanism. The experimental rates of this reaction do not provide insight into whether or not an intermediate exists; however, we have used the Gaussian program140 to perform geometry optimizations and transition state searches using PBE0/6-31G*, PBE0/6-311G**, B3LYP/6-31G*, and B3LYP/6-311G** with polarized continuum model (PCM) implicit solvent.141 All of the PCM calculations that we performed predict the existence of an intermediate and two transition states, in agreement with our PBE0 explicit solvent QM/MM PMF. Previous studies of transesterification reactions with various leaving groups have found that poor (EtO) and enhanced (PhO) leaving groups generally follow a stepwise and concerted mechanisms, respectively.136 Indeed, the transesterification reaction PMFs involving the PhO leaving group (Figs. 9d–9f) are predicted to undergo a concerted mechanism for all 3 methods.
The PBE0/6-311G** reaction barriers agree more closely to experiment than either AM1/d-PhoT or DFTB2. The semiempirical methods predict EtO barriers that are 10 kcal/mol larger than experiment, whereas PBE0 is only 3.6 kcal/mol larger. Similarly, the semiempirical models produce PhO barriers that are 8 kcal/mol larger than experiment, and PBE0 differs from experiment by only 0.8 kcal/mol.
4 Conclusion
In this work, we presented a new, composite Ewald method (CEw) for QM/MM simulations. Unlike the semiempirical-QM/MM Ewald method introduced by Nam,63 the new method produces stable MD trajectories when evaluated with non-minimal Gaussian AO basis sets. The CEw method differs from the recently described QM/MM-LREC approach85 by polarizing the QM region to the long-range electrostatics using an Ewald summation. It further differs from the approach described by Holden et al.69,70 by interacting the QM region with the external environment directly through the QM charge density, as opposed to resorting to a point-charge auxiliary representation.
We compared how various electrostatic protocols affect the PMF of the p-nitrophenyl phosphate dissociation reaction. It was shown that the CEw PMFs are less sensitive to the division of short- and long-range interactions than the other methods that we compared to. For example, the difference in free energy barriers between using a real-space Ewald cutoff of 10 Å and 18 Å was found to be 1 kcal/mol when using the composite Ewald method. We compared CEw to a subtractive Ewald scheme (MMEw) and found that CEw avoided the structural artifacts associated with switching the QM/MM interaction between electrostatic and mechanical embedding.
We examined the computational cost of the new method and found that the incorporation of Ewald sums into the QM calculation only slowed the simulation rate by 1% to 4% for a 52 atom QM region.
We confirmed Holden’s explanation69,70 for the instability encountered with Mulliken charge-based QM/MM Ewald methods.63 The use of Mulliken charges produces inconsistent representations of the electronic chemical potential which, in the orthogonalized atomic orbital representation of the Fock matrix, become deformed and thus errantly polarizes the QM electron density. The new CEw method polarizes the QM density to a globally-consistent representation of the chemical potential.
We performed two-dimensional PMFs of phosphoryl transesterification reactions with ethoxide and phenoxide leaving groups to compare PBE0/6-311G** with the AM1/d-PhoT and DFTB2 semiempirical models. We found that the semiempirical models produced concerted reaction mechanisms for the ethoxide leaving group, whereas PBE0 simulations produced a stepwise mechanism, in agreement with implicit solvation calculations and previous work.136,137 Furthermore, the PBE0/6-311G** reaction barriers were found to more closely match experiment than the semiempirical methods by 6 to 7 kcal/mol for both leaving groups.
Acknowledgments
The authors are grateful for financial support provided by the National Institutes of Health (No. GM107485). Computational resources were provided by the National Institutes of Health under Grant No. S10OD012346 and by the Extreme Science and Engineering Discovery Environment (XSEDE), which is supported by National Science Foundation Grant No. OCI-1053575, with Project No. TG-MCB110101.
Notes and References
- 1.York DM, Darden T, Pedersen LG. J Chem Phys. 1993;99:8345–8348. [Google Scholar]
- 2.York DM, Yang W, Lee H, Darden T, Pedersen LG. J Am Chem Soc. 1995;117:5001–5002. [Google Scholar]
- 3.Cheatham TE, III, Young MA. Biopolymers. 2000;56:232–256. doi: 10.1002/1097-0282(2000)56:4<232::AID-BIP10037>3.0.CO;2-H. [DOI] [PubMed] [Google Scholar]
- 4.Cisneros GA, Karttunen M, Ren P, Sagui C. Chem Rev. 2014;114:779–814. doi: 10.1021/cr300461d. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Cisneros GA, Babin V, Sagui C. Biomolecular Simulations: Methods and Protocols. In: Monticelli L, Salonen E, editors. Methods in Molecular Biology. Vol. 924. Springer Science+Business Media; New York: 2013. pp. 243–270. [DOI] [PubMed] [Google Scholar]
- 6.Ewald PP. Ann Phys. 1921;64:253–268. [Google Scholar]
- 7.Darden T, York D, Pedersen L. J Chem Phys. 1993;98:10089–10092. [Google Scholar]
- 8.Essmann U, Perera L, Berkowitz ML, Darden T, Hsing L, Pedersen LG. J Chem Phys. 1995;103:8577–8593. [Google Scholar]
- 9.Cerutti DS, Duke RE, Darden TA, Lybrand TP. J Chem Theory Comput. 2009;5:2322–2338. doi: 10.1021/ct9001015. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Cerutti DS, Case DA. J Chem Theory Comput. 2010;6:443–458. doi: 10.1021/ct900522g. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Toukmaji A, Sagui C, Board J, Darden T. J Chem Phys. 2000;113:10913–10927. [Google Scholar]
- 12.Nymand TM, Linse P. J Chem Phys. 2000;112:6152–6160. [Google Scholar]
- 13.Sagui C, Pedersen LG, Darden TA. J Chem Phys. 2004;120:73–87. doi: 10.1063/1.1630791. [DOI] [PubMed] [Google Scholar]
- 14.Schnieders MJ, Fenn TD, Pande VS, Brunger AT. Acta Cryst. 2009;D65:952–965. doi: 10.1107/S0907444909022707. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Cisneros GA, Piquemal J, Darden TA. J Chem Phys. 2006;125:184101. doi: 10.1063/1.2363374. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Darden TA. International Tables for Crystallography, Reciprocal Space. 3rd. Springer; AA Dordrecht, The Netherlands: 2008. pp. 458–483. (Vol. B; Chapter Extensions of the Ewald Method for Coulomb Interactions in Crystals). [Google Scholar]
- 17.Giese TJ, Panteva MT, Chen H, York DM. J Chem Theory Comput. 2015;11:436–450. doi: 10.1021/ct5007983. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Simmonett AC, Pickard FC, IV, Schaefer HF, III, Brooks BR. J Chem Phys. 2014;140:184101. doi: 10.1063/1.4873920. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Simmonett AC, Pickard FC, IV, Shao Y, Cheatham TE, III, Brooks BR. J Chem Phys. 2015;143:074115. doi: 10.1063/1.4928530. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Giese TJ, Panteva MT, Chen H, York DM. J Chem Theory Comput. 2015;11:451–461. doi: 10.1021/ct500799g. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Giese TJ, Chen H, Huang M, York DM. J Chem Theory Comput. 2014;10:1086–1098. doi: 10.1021/ct401035t. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Giese TJ, Huang M, Chen H, York DM. Acc Chem Res. 2014;47:2812–20. doi: 10.1021/ar500103g. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Fennell C, Gezelter JD. J Chem Phys. 2006;124:234104. doi: 10.1063/1.2206581. [DOI] [PubMed] [Google Scholar]
- 24.Steinbach PJ, Brooks BR. J Comput Chem. 1994;15:667–683. [Google Scholar]
- 25.Horn HW, Swope WC, Pitera JW, Madura JD, Dick TJ, Hura GL, Head-Gordon T. J Chem Phys. 2004;120:9665–9678. doi: 10.1063/1.1683075. [DOI] [PubMed] [Google Scholar]
- 26.Rick SW. J Chem Phys. 2004;120:6085–6093. doi: 10.1063/1.1652434. [DOI] [PubMed] [Google Scholar]
- 27.van der Spoel D, van Maaren PJ. J Chem Theory Comput. 2006;2:1–11. doi: 10.1021/ct0502256. [DOI] [PubMed] [Google Scholar]
- 28.Lee FS, Warshel A. J Chem Phys. 1992;97:3100–3107. [Google Scholar]
- 29.York DM, Wlodawer A, Pedersen LG, Darden T. Proc Natl Acad Sci USA. 1994;91:8715–8718. doi: 10.1073/pnas.91.18.8715. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Cheatham TE, III, Miller JL, Fox T, Darden TA, Kollman PA. J Am Chem Soc. 1995;117:4193–4194. [Google Scholar]
- 31.Cheatham TE, III, Kollman PA. J Mol Biol. 1996;259:434–444. doi: 10.1006/jmbi.1996.0330. [DOI] [PubMed] [Google Scholar]
- 32.Warshel A, Levitt M. J Mol Biol. 1976;103:227–249. doi: 10.1016/0022-2836(76)90311-9. [DOI] [PubMed] [Google Scholar]
- 33.Bakowies D, Thiel W. J Phys Chem. 1996;100:10580–10594. [Google Scholar]
- 34.Lin H, Truhlar DG. Theor Chem Acc. 2007;117:185–199. [Google Scholar]
- 35.Maseras F, Morokuma K. J Comput Chem. 1995;16:1170. [Google Scholar]
- 36.Svensson M, Humbel S, Froese RDJ, Matsubara T, Sieber S, Morokuma K. J Phys Chem. 1996;100:19357–19363. [Google Scholar]
- 37.Froese RDJ, Musaev DG, Morokuma K. J Am Chem Soc. 1998;120:1581–1587. [Google Scholar]
- 38.Nam K, Cui Q, Gao J, York DM. J Chem Theory Comput. 2007;3:486–504. doi: 10.1021/ct6002466. [DOI] [PubMed] [Google Scholar]
- 39.Elstner M, Porezag D, Jungnickel G, Elsner J, Haugk M, Frauenheim T, Suhai S, Seifert G. Phys Rev B. 1998;58:7260–7268. [Google Scholar]
- 40.Han W-G, Elstner M, Jalkanen KJ, Frauenheim T, Suhai S. Int J Quantum Chem. 2000;78:459–479. [Google Scholar]
- 41.Dewar MJ, Thiel W. J Am Chem Soc. 1977;99:4899–4907. [Google Scholar]
- 42.Dewar MJS, Zoebisch E, Healy EF, Stewart JJP. J Am Chem Soc. 1985;107:3902–3909. [Google Scholar]
- 43.Field MJ, Bash PA, Karplus M. J Comput Chem. 1990;11:700–733. [Google Scholar]
- 44.Gao J, York D. Theor Chem Acc. 2003;109:99. [Google Scholar]
- 45.Stanton RV, Hartsough DS, Merz KM., Jr J Phys Chem. 1993;97:11868–11870. [Google Scholar]
- 46.Schlegel HB. J Comput Chem. 2003;24:1514–1527. doi: 10.1002/jcc.10231. [DOI] [PubMed] [Google Scholar]
- 47.Zhu X, Yethiraj A, Cui Q. J Chem Theory Comput. 2007;3:1538–1549. doi: 10.1021/ct600352e. [DOI] [PubMed] [Google Scholar]
- 48.He X, Wang B, Merz KM., Jr J Phys Chem B. 2009;113:10380. doi: 10.1021/jp901992p. [DOI] [PubMed] [Google Scholar]
- 49.Pentikäinen U, Shaw KE, Senthilkumar K, Woods CJ, Mulholland AJ. J Chem Theory Comput. 2009;5:396–410. doi: 10.1021/ct800135k. [DOI] [PubMed] [Google Scholar]
- 50.Mlýnský V, Banáš P, Šponer J, van der Kamp MW, Mulholland AJ, Otyepka M. J Chem Theory Comput. 2014;10:1608–1622. doi: 10.1021/ct401015e. [DOI] [PubMed] [Google Scholar]
- 51.Elsässer B, Fels G, Weare JH. J Am Chem Soc. 2014;136:927–936. doi: 10.1021/ja406122c. [DOI] [PubMed] [Google Scholar]
- 52.Zhang S, Ganguly A, Goyal P, Bingaman JL, Bevilacqua PC, Hammes-Schiffer S. J Am Chem Soc. 2015;137:784–798. doi: 10.1021/ja510387y. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Thaplyal P, Ganguly A, Hammes-Schiffer S, Bevilacqua PC. Biochemistry. 2015;54:2160–2175. doi: 10.1021/acs.biochem.5b00190. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Ganguly A, Thaplyal P, Rosta E, Bevilacqua PC, Hammes-Schiffer S. J Am Chem Soc. 2014;136:1483–1496. doi: 10.1021/ja4104217. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Nam K. J Chem Theory Comput. 2014;10:4175–4183. doi: 10.1021/ct5005643. [DOI] [PubMed] [Google Scholar]
- 56.Plotnikov NV. J Chem Theory Comput. 2014;10:2987–3001. doi: 10.1021/ct500109m. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Plotnikov NV, Warshel A. J Phys Chem B. 2012;116:10342–10356. doi: 10.1021/jp304678d. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Wong K-Y, Gao J. Biochemistry. 2007;46:13352–13369. doi: 10.1021/bi700460c. [DOI] [PubMed] [Google Scholar]
- 59.Woods CJ, Manby FR, Mulholland AJ. J Chem Phys. 2008;128:14109. doi: 10.1063/1.2805379. [DOI] [PubMed] [Google Scholar]
- 60.Garcia-Viloca M, Gao J, Karplus M, Truhlar DG. Science. 2004;303:186–195. doi: 10.1126/science.1088172. [DOI] [PubMed] [Google Scholar]
- 61.Gregersen BA, Lopez X, York DM. J Am Chem Soc. 2003;125:7178–7179. doi: 10.1021/ja035167h. [DOI] [PubMed] [Google Scholar]
- 62.Elstner M, Frauenheim T, Suhai S. J Mol Struct (Theochem) 2003;632:29–41. [Google Scholar]
- 63.Nam K, Gao J, York DM. J Chem Theory Comput. 2005;1:2–13. doi: 10.1021/ct049941i. [DOI] [PubMed] [Google Scholar]
- 64.Riccardi D, Schaefer P, Cui Q. J Phys Chem B. 2005;109:17715–17733. doi: 10.1021/jp0517192. [DOI] [PubMed] [Google Scholar]
- 65.Gao J, Alhambra C. J Chem Phys. 1997;107:1212–1217. [Google Scholar]
- 66.Walker RC, Crowley MF, Case DA. J Comput Chem. 2008;29:1019–1031. doi: 10.1002/jcc.20857. [DOI] [PubMed] [Google Scholar]
- 67.Zhang P, Truhlar DG, Gao J. Phys Chem Chem Phys. 2012;14:7821–7829. doi: 10.1039/c2cp23758j. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 68.Gao D, Svoronos P, Wong PK, Maddalena D, Hwang J, Walker H. J Phys Chem A. 2005;109:10776–10785. doi: 10.1021/jp053996e. [DOI] [PubMed] [Google Scholar]
- 69.Holden ZC, Richard RM, Herbert JM. J Chem Phys. 2013;139:244108. doi: 10.1063/1.4850655. [DOI] [PubMed] [Google Scholar]
- 70.Holden ZC, Richard RM, Herbert JM. J Chem Phys. 2015;142:059901. doi: 10.1063/1.4907623. [DOI] [PubMed] [Google Scholar]
- 71.Breneman CM, Wiberg KB. J Comput Chem. 1990;11:361–373. [Google Scholar]
- 72.Jacobson LD, Herbert JM. J Chem Phys. 2011;134:094118. doi: 10.1063/1.3560026. [DOI] [PubMed] [Google Scholar]
- 73.Herbert JM, Jacobson LD, Laoa KU, Rohrdanza MA. Phys Chem Chem Phys. 2012;14:7679–7699. doi: 10.1039/c2cp24060b. [DOI] [PubMed] [Google Scholar]
- 74.Hu H, Lu Z, Yang W. J Chem Theory Comput. 2007;3:390–406. doi: 10.1021/ct600240y. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 75.Hu H, Lu Z, Parks JM, Burger SK, Yang W. J Chem Phys. 2008;128:034105. doi: 10.1063/1.2816557. [DOI] [PubMed] [Google Scholar]
- 76.Zeng X, Hu H, Hu X, Cohen AJ, Yang W. J Chem Phys. 2008;128:124510. doi: 10.1063/1.2832946. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 77.Alper H, Levy RM. J Chem Phys. 1993;99:1–6. [Google Scholar]
- 78.Adams DJ, Adams EM. J Mol Phys. 1979;38:387–400. [Google Scholar]
- 79.Woodcock HL, III, Hodoscek M, Gilbert AT, Gill PM, Schaefer HF, III, Brooks BR. J Comput Chem. 2007;28:1485–1502. doi: 10.1002/jcc.20587. [DOI] [PubMed] [Google Scholar]
- 80.Xie W, Orozco M, Truhlar DG, Gao J. J Chem Theory Comput. 2009;5:459–467. doi: 10.1021/ct800239q. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 81.Cui Q, Karplus M. J Am Chem Soc. 2001;123:2284–2290. doi: 10.1021/ja002886c. [DOI] [PubMed] [Google Scholar]
- 82.Cisneros GA, Liu H, Zhang Y, Yang W. J Am Chem Soc. 2003;125:10384–10393. doi: 10.1021/ja029672a. [DOI] [PubMed] [Google Scholar]
- 83.Liu H, Zhang Y, Yang W. J Am Chem Soc. 2000;122:6560–6570. [Google Scholar]
- 84.Liu J, Zhu T, Wang X, He X, Zhang JZH. J Chem Theory Comput. 2015;11:5897–5905. doi: 10.1021/acs.jctc.5b00558. [DOI] [PubMed] [Google Scholar]
- 85.Fang D, Duke RE, Cisneros GA. J Chem Phys. 2015;143:044103. doi: 10.1063/1.4926652. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 86.Xie W, Gao J. J Chem Theory Comput. 2007;3:1890–1900. doi: 10.1021/ct700167b. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 87.Xie W, Song L, Truhlar DG, Gao J. J Chem Phys. 2008;128:234108. doi: 10.1063/1.2936122. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 88.Song L, Han J, lin Lin Y, Xie W, Gao J. J Phys Chem A. 2009;113:11656–11664. doi: 10.1021/jp902710a. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 89.Hu H, Yang W. J Mol Struct THEOCHEM. 2009;898:17–30. doi: 10.1016/j.theochem.2008.12.025. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 90.Perdew JP, Ernzerhof M, Burke K. J Chem Phys. 1996;105:9982–9985. [Google Scholar]
- 91.Adamo C, Scuseria GE. J Chem Phys. 1999;111:2889–2899. [Google Scholar]
- 92.Perdew JP, Burke K, Ernzerhof M. Phys Rev Lett. 1996;77:3865–3868. doi: 10.1103/PhysRevLett.77.3865. [DOI] [PubMed] [Google Scholar]
- 93.Murray CW, Handy N, Laming G. Mol Phys. 1993;78:997–1014. [Google Scholar]
- 94.Treutler O, Ahlrichs R. J Chem Phys. 1995;102:346–354. [Google Scholar]
- 95.Mura ME, Knowles PJ. J Chem Phys. 1996;104:9848–9858. [Google Scholar]
- 96.Lindh R, Malmqvist P, Gagliardi L. Theor Chem Acc. 2001;106:178–187. [Google Scholar]
- 97.Lebedev VI. Sib Math J. 1977;18:99–107. [Google Scholar]
- 98.Becke AD. J Chem Phys. 1988;88:2547–2553. [Google Scholar]
- 99.Johnson BG, Gill PMW, Pople JA. J Chem Phys. 1993;98:5612–5626. [Google Scholar]
- 100.Head-Gordon M, Pople JA. J Chem Phys. 1988;89:5777–5786. [Google Scholar]
- 101.Hamilton TP, Schaefer HF., III Chem Phys. 1991;150:163–171. [Google Scholar]
- 102.Obara S, Saika A. J Chem Phys. 1986;84:3963–3974. [Google Scholar]
- 103.Gao J, Amara P, Alhambra C, Field MJ. J Phys Chem A. 1998;102:4714–4721. [Google Scholar]
- 104.Amara P, Field MJ, Alhambra C, Gao J. Theor Chem Acc. 2000;104:336–343. [Google Scholar]
- 105.Garcia-Viloca M, Gao J. Theor Chem Acc. 2004;111:280–286. [Google Scholar]
- 106.Pu J, Gao J, Truhlar DG. J Phys Chem A. 2004;108:5454–5463. [Google Scholar]
- 107.Day PN, Jensen JH, Gordon MS, Webb SP, Stevens WJ, Krauss M, Garmer D, Basch H, Cohen D. J Chem Phys. 1996;105:1968–1986. [Google Scholar]
- 108.Kairys V, Jensen JH. J Phys Chem A. 2000;104:6656–6665. [Google Scholar]
- 109.Gordon MS, Freitag MA, Bandyopadhyay P, Jensen JH, Kairys V, Stevens WJ. J Phys Chem A. 2001;105:293–307. [Google Scholar]
- 110.Thery V, Rinaldi D, Rivail J-L. J Comput Chem. 1994;15:269–282. [Google Scholar]
- 111.Monard G, Loos M, Thery V, Baka K, Rivail JL. Int J Quantum Chem. 1996;58:153–159. [Google Scholar]
- 112.Assfeld X, Rivail J-L. Chem Phys Lett. 1996;263:100–106. [Google Scholar]
- 113.Senn HM, Thiel W. Angew Chem Int Ed. 2009;48:1198–1229. doi: 10.1002/anie.200802019. [DOI] [PubMed] [Google Scholar]
- 114.Equation 41 is formally +∞ if q(r) is anywhere nonzero, but it could be −∞ if the system consisted of point charges and their self-energies are excluded from the energy.
- 115.If the QM region was positioned at the center of the box, then one should also center the solid harmonic to avoid biasing the importance of the high-order moments in a truncated multipole expansion.
- 116.Case DA, Berryman JT, Betz RM, Cerutti DS, Cheatham TE, III, Darden TA, Duke RE, Giese TJ, Gohlke H, Goetz AW, Homeyer N, Izadi S, Janowski P, Kaus J, Kovalenko A, Lee TS, LeGrand S, Li P, Luchko T, Luo R, Madej B, Merz KM, Monard G, Needham P, Nguyen H, Nguyen HT, Omelyan I, Onufriev A, Roe DR, Roitberg A, Salomon-Ferrer R, Simmerling CL, Smith W, Swails J, Walker RC, Wang J, Wolf RM, Wu X, York DM, Kollman PA. AMBER 2015. University of California; San Francisco: 2015. [Google Scholar]
- 117.Wang J, Wang W, Kollman PA, Case DA. J Mol Graph Model. 2006;25:247–260. doi: 10.1016/j.jmgm.2005.12.005. [DOI] [PubMed] [Google Scholar]
- 118.Wang J, Wolf RM, Caldwell JW, Kollman PA, Case DA. J Comput Chem. 2004;25:1157–1174. doi: 10.1002/jcc.20035. [DOI] [PubMed] [Google Scholar]
- 119.Jakalian A, Jack DB, Bayly CI. J Comput Chem. 2002;23:1623–1641. doi: 10.1002/jcc.10128. [DOI] [PubMed] [Google Scholar]
- 120.Lee T-S, Radak BK, Pabis A, York DM. J Chem Theory Comput. 2013;9:153–164. doi: 10.1021/ct300703z. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 121.Kirby AJ, Jencks WP. J Am Chem Soc. 1965;87:3209–3216. [Google Scholar]
- 122.Henkelman G, Uberuaga BP, Hannes Jónsson. J Chem Phys. 2000;113:9901–9904. [Google Scholar]
- 123.Henkelman G, Jónsson H. J Chem Phys. 2000;113:9978–9985. [Google Scholar]
- 124.Brown DM, Usher DA. J Chem Soc. 1965;87:6558–6564. [Google Scholar]
- 125.Izatt RM, Hansen LD, Rytting JH, Christensen JJ. J Am Chem Soc. 1965;87:2760–2761. doi: 10.1021/ja01090a044. [DOI] [PubMed] [Google Scholar]
- 126.Izatt RM, Rytting JH, Hansen LD, Christensen JJ. J Am Chem Soc. 1966;88:2641–2645. doi: 10.1021/ja00964a003. [DOI] [PubMed] [Google Scholar]
- 127.Velikyan I, Acharya S, Trifonova A, Földesi A, Chattopadhyaya J. J Am Chem Soc. 2001;123:2893–2894. doi: 10.1021/ja0036312. [DOI] [PubMed] [Google Scholar]
- 128.Li Y, Breaker RR. J Am Chem Soc. 1999;121:5364–5372. [Google Scholar]
- 129.Kitchen DB, Hirata F, Westbrook JD, Levy R. J Comput Chem. 1990;11:1169–1180. [Google Scholar]
- 130.Harvey SC, Tan RKZ, Cheatham TE., III J Comp Chem. 1998;19:726–740. [Google Scholar]
- 131.Loncharich RJ, Brooks BR. Proteins. 1989;6:32–45. doi: 10.1002/prot.340060104. [DOI] [PubMed] [Google Scholar]
- 132.Hengge AC. Acc Chem Res. 2002;35:105–112. doi: 10.1021/ar000143q. [DOI] [PubMed] [Google Scholar]
- 133.Cochrane JC, Strobel SA. Acc Chem Res. 2008;41:1027–1035. doi: 10.1021/ar800050c. [DOI] [PubMed] [Google Scholar]
- 134.Lassila JK, Zalatan JG, Herschlag D. Annu Rev Biochem. 2011;80:669–702. doi: 10.1146/annurev-biochem-060409-092741. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 135.Kamerlin SCL, Sharma PK, Prasad RB, Warshel A. Q Rev Biophys. 2013;46:1–132. doi: 10.1017/S0033583512000157. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 136.Huang M, York DM. Phys Chem Chem Phys. 2014;16:15846–15855. doi: 10.1039/c4cp01050g. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 137.Chen H, Giese TJ, Huang M, Wong K-Y, Harris ME, York DM. Chem Eur J. 2014;20:14336–14343. doi: 10.1002/chem.201403862. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 138.Åstrand P-O, Ruud K, Mikkelsen KV, Helgaker T. J Phys Chem A. 1998;102:7686–7691. [Google Scholar]
- 139.Reed AE, Weinstock RB, Weinhold F. J Chem Phys. 1985;83:735–746. [Google Scholar]
- 140.Frisch MJ, Trucks GW, Schlegel HB, Scuseria GE, Robb MA, Cheeseman JR, Scalmani G, Barone V, Mennucci B, Petersson GA, Nakatsuji H, Caricato M, Li X, Hratchian HP, Izmaylov AF, Bloino J, Zheng G, Sonnenberg M, Hada M, Ehara M, Toyota K, Fukuda R, Hasegawa J, Ishida M, Nakajima T, Honda Y, Kitao O, Nakai H, Vreven T, Montgomery JAJ, Peralta JE, Ogliaro F, Bearpark M, Heyd JJ, Brothers E, Kudin KN, Straverov VN, Kobayashi R, Normand J, Raghavachari K, Rendell A, Burant JC, Iyengar SS, Tomasi J, Cossi M, Rega N, Millam JM, Klene M, Knox JE, Cross JB, Bakken V, Adamo C, Jaramillo J, Gomperts R, Stratmann RE, Yazyev O, Austin AJ, Cammi R, Pomelli C, Ochterski JW, Martin RL, Morokuma K, Zakrzewski VG, Voth GA, Salvador P, Dannenberg JJ, Dapprich S, Daniels AD, Farkas O, Foresman JB, Ortiz JV, Cioslowski J, Fox DJ. Gaussian 09, Revision A.02. Gaussian, Inc; Wallingford, CT: 2009. [Google Scholar]
- 141.Scalmani G, Frisch MJ. J Chem Phys. 2010;132:114110–114124. doi: 10.1063/1.3359469. [DOI] [PubMed] [Google Scholar]