

Abstract
The lysosomal storage disorder mucolipidosis III γ is caused by defects in the γ subunit of UDP-GlcNAc:lysosomal enzyme N-acetylglucosamine-1-phosphotransferase, the enzyme that tags lysosomal enzymes with the mannose 6-phosphate lysosomal targeting signal. In patients with this disorder, most of the newly synthesized lysosomal enzymes are secreted rather than being sorted to lysosomes, resulting in increased levels of these enzymes in the plasma. Several missense mutations in GNPTG, the gene encoding the γ subunit, have been reported in mucolipidosis III γ patients. However, in most cases the impact of these mutations on γ subunit function has remained unclear. Here we report that the variants c.316G>A (p.G106S), c.376G>A (p.G126S) and c.425G>A (p.C142Y) cause misfolding of the γ subunit, while another variant, c.857C>T (p.T286M), does not appear to alter γ subunit function. The misfolded γ subunits were retained in the ER and failed to rescue the lysosomal targeting of lysosomal acid glycosidases.
Keywords: GlcNAc-1-phosphotransferase, GNPTG, misfolding, mucolipidosis III γ
Newly synthesized lysosomal acid hydrolases are modified with the mannose 6-phosphate (Man-6-P) recognition marker to enable their lysosomal delivery by Man-6-P receptors (MPRs) (Ghosh, et al., 2003). The generation of the Man-6-P recognition marker requires the sequential action of the enzymes UDP-GlcNAc:lysosomal enzyme N-acetylglucosamine-1-phosphotransferase (GlcNAc-1-phosphotransferase; EC 2.7.8.17) and “uncovering” enzyme. This process is critical for proper lysosome biogenesis, as loss-of-function mutations in the genes encoding GlcNAc-1-phosphotransferase cause serious lysosomal storage disorders.
GlcNAc-1-phosphotransferase is a heterohexameric enzyme composed of two α, β and γ subunits (Bao, et al., 1996). In the Golgi complex, it adds GlcNAc-1-phosphate to high mannose-type N-linked glycans on newly synthesized lysosomal enzymes. Failure to phosphorylate lysosomal enzymes results in their impaired targeting to lysosomes and increased secretion from the cell. As a consequence, the lysosomes accumulate undegraded material. The α and β subunits of GlcNAc-1-phosphotransferase, encoded by the gene GNPTAB, harbor the catalytic site as well as domains that mediate the recognition of the acid hydrolases (DMAP and Notch repeats) (Kudo, et al., 2005; Qian, et al., 2013; Qian, et al., 2010; Qian, et al., 2015; van Meel, et al., 2016). Depending on the residual level of GlcNAc-1-phosphotransferase activity, mutations in GNPTAB either cause the very severe lysosomal storage disorder mucolipidosis (ML) II (MIM# 252500), or result in MLIII αβ (MIM# 252600), which has an attenuated phenotype (Kudo, et al., 2006).
The γ subunit of GlcNAc-1-phosphotransferase is a soluble glycoprotein, which is encoded by the GNPTG gene (Raas-Rothschild, et al., 2000; MIM# 607838; NM_032520.3). This subunit is required for efficient phosphorylation of a subset of the lysosomal enzymes (Qian, et al., 2010). Loss of function of the γ subunit results in the least severe phenotype, MLIII γ (MIM# 252605). The clinical manifestations are variable, but typically include stiffness of the fingers and shoulders, claw-hand deformity, short stature, scoliosis, valvular heart disease and occasionally mild mental retardation. The most frequent and debilitating symptoms are bone pain and disability due to destruction of the hip joints (Braulke, et al., 2013).
To date, four missense mutations in GNPTG have been described in MLIII γ patients (Barea, et al., 2015; Liu, et al., 2014; Persichetti, et al., 2009; Raas-Rothschild, et al., 2004). While these variants were predicted to be damaging, the consequences on γ subunit function have not been studied, with the exception of the p.G126S variant, which mislocalized in the ER (Barea, et al., 2015).
The subcellular localization of the variants was first analyzed by confocal immunofluorescence microscopy. Human GNPTG-FLAG in pcDNA3.1(+) (Qian, et al., 2015) was modified by QuikChange site-directed mutagenesis to generate the variants p.G106S, p.G126S, p.C142Y and p.T286M. See Supp. Materials and Methods and Supp. Table S1 for details. We made use of HeLa cells that transiently overexpressed the WT αβ subunits of GlcNAc-1-phosphotransferase together with WT or mutant γ subunit. While the WT γ subunit localized to the Golgi complex as shown by co-localization with the cis-Golgi marker GM130, two of the variants, p.G106S and p.C142Y γ, showed little Golgi localization (Fig. 1A, B). Instead, they overlapped with the molecular chaperone calnexin in the ER. The p.T286M variant localized normally to the Golgi complex (Fig. 1A).
Figure 1. The γ subunit variants p.G106S, p.G126S and p.C142Y form aggregates in the ER.

A-B. Confocal immunofluorescence miscroscopy of HeLa cells transiently transfected with the WT αβ subunits of GlcNAc-1-phosphotransferase (not shown) and WT or variant γ subunit (anti-γ, green). WT and p.T286M γ co-localize with the cis-Golgi marker GM130 (red), 48h after transfection. The variants p.G106S and p.C142Y γ are retained in the ER, as shown by co-localization with calnexin (red). Scale bars, 10 μm. C-D. Western blot analysis (anti-FLAG antibody, showing the γ subunit) of transfected HEK293 cell lysates transiently transfected with the various γ subunit variants and the WT αβ subunits (not shown). The lysates were run in the absence (C) or the presence of reducing reagent (D). Note that the p.G106S γ variant migrates slower than the WT γ subunit. All variants formed dimers (C, arrowhead), while the variants p.G106S, p.G126S and p.C142Y also formed higher molecular weight aggregates (arrows). E. Western blots (anti-FLAG antibody) of the cell lysates from C treated for 3h at 37°C with Endo Hf (H) or PNGase F (F) show a similar shift as compared to the untreated lysates (-), indicating the presence of high mannose-type N-linked glycans. Neuraminidase (N) treatment had no apparent effect. The p.G106S variant behaved similar to WT, suggesting that the slower migration was not due to altered glycosylation.
The γ variants were next analyzed in the presence of the WT αβ subunits, by SDS-PAGE and immunoblotting under non-reducing conditions. This revealed a single band around 75 kDa in the case of the WT and p.T286M mutant γ subunits, representing the γ dimer (Fig. 1C). The three ER localized variants (p.G106S, p.C142Y γ and the previously reported p.G126S variant (Barea, et al., 2015)) also formed dimers, but in addition showed higher molecular weight bands around 150 and 250 kDa. These likely represent aggregates that form due to misfolding in the ER.
Curiously, p.G106S γ migrated slightly slower than the WT protein. This was more apparent in the reducing gel that showed the γ monomer (Fig. 1D). To determine whether this effect could be explained by altered N-linked glycan processing, the lysates were treated with endoglycosidase Hf or N-glycosidase F (PNGase F), which cleave high mannose-type or both high mannose and complex-type N-glycans, respectively. The WT protein showed an equal shift in migration after both endoglycosidase Hf and PNGase F treatment (Fig. 1E), which is in agreement with previous observations that the γ subunit contains two high mannose-type N-linked glycans (Encarnacao, et al., 2011). Treatment of the p.G106S mutant had a similar effect, but it still exhibited a slightly slower migration. In both WT and p.G106S γ no apparent effect was observed after neuraminidase treatment, which removes sialic acids. Therefore, the altered migration of this mutant is most likely due to a direct effect of the amino acid substitution or possibly some other type of post-translational modification.
To obtain a more quantitative measure of the consequences of the various missense mutations on GlcNAc-1-phosphotransferase function, we analyzed the variants in γ subunit deficient cells. GNPTG-/- HeLa cells have greatly reduced levels of many lysosomal acid hydrolases compared to the parental HeLa cells and display a lysosomal storage phenotype (van Meel, et al., 2016). In these cells, transient transfection with WT γ resulted in a marked increase in the intracellular activities of a panel of lysosomal glycosidases 3 days after transfection, with the maximum increase set to 100% rescue. The misfolded γ mutants showed only low levels of rescue, in most cases below 20% of WT, while being expressed at similar or even increased levels as the WT protein (Fig. 2A-B). To our surprise, the p.T286M variant completely rescued the lysosomal targeting of the glycosidases measured (Fig. 2A). Thus, it seems likely that this mutation does not affect GlcNAc-1-phosphotransferase function and represents a benign variant.
Figure 2. Effect of the γ variants on lysosomal enzyme targeting.
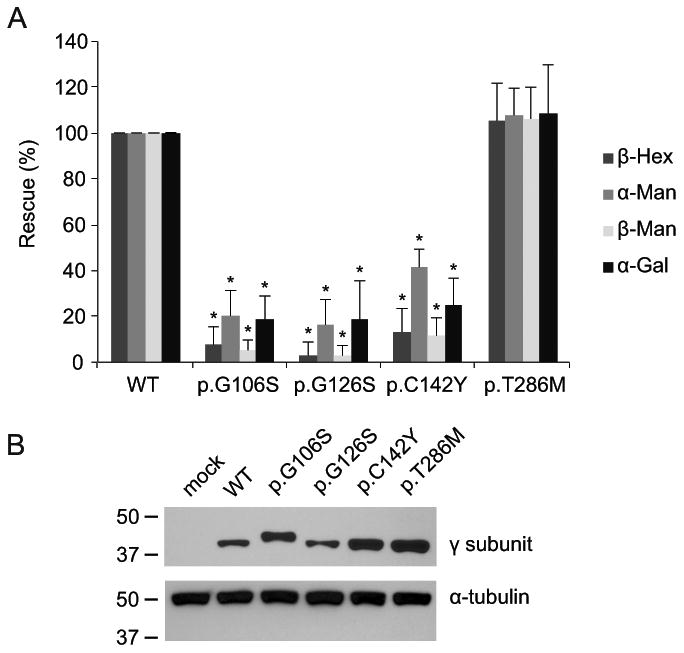
A. The intracellular activities of a panel of lysosomal acid glycosidases were measured in GNPTG-/- HeLa cells ∼72h after transfection with WT or mutant γ subunit. The values are represented as percentages of those obtained with the WT γ subunit, which were set to 100% rescue. The values are averages of four independent experiments, ± standard deviations. * p-value < 0.01. B. Representative Western blot (anti-FLAG upper panel) showing the expression levels of the various mutants to be similar to or higher than the level of the WT γ subunit. Anti-α-tubulin (lower panel) was used as a loading control.
In summary, we have analyzed the consequences of the missense mutations in the γ subunit of GlcNAc-1-phosphotransferase reported to date in patients with the lysosomal storage disorder mucolipidosis III γ. Three of the four variants, p.G106S, p.G126S and p.C142Y, caused misfolding of the γ subunit, while one variant, p.T286M, was normal in all assays.
The γ subunit contains two distinct domains, a Man-6-P receptor homology (MRH) domain, which binds high mannose-type N-glycans on the lysosomal enzymes to enable their phosphorylation by the αβ subunits, and a DMAP interaction domain (van Meel, et al., 2016). The amino acid residues G106, G126 and C142 are all located in the MRH domain. While they do not represent residues that are directly involved in mannose binding, they are highly conserved among various species. The residue C142 was previously implicated in the formation of intramolecular disulfide bonds (Encarnacao, et al., 2011). Consistent with this report, our data show that the variant p.C142Y did not impair intermolecular disulfide linkages, as γ dimer formation was intact. It is probable that the inability to form an intramolecular disulfide bond, involving residue C142, results in misfolding of the enzyme.
The variant p.T286M was reported in a single patient, who was heterozygous for the mutation, with a yet undetected mutation in the second allele (Persichetti, et al., 2009). The authors reported that the nucleotide change (c.857C>T) was not found in a screen of 50 healthy Italian control subjects, neither was it listed as a polymorphism in dbSNP. While the patient exhibited a mild clinical phenotype together with significantly decreased levels of lysosomal enzymes in fibroblasts, consistent with the diagnosis ML III γ, this variant is unlikely to be responsible for the phenotype, since no effect could be found on γ subunit function in our assays. Importantly, expression of this variant in γ subunit deficient cells rescued the lysosomal targeting of lysosomal acid glycosidases equally well as WT γ. Residue T286 is found in the C-terminal region of the γ subunit, which has no reported function. In addition, this residue is not well conserved among vertebrates. As parental heterozygosity was not formally demonstrated due to the unavailability of the parental DNA, it is possible that a different mutation in GNPTG or GNPTAB is responsible for the patient's disease. This finding underscores the importance of functional testing of newly reported variants.
Supplementary Material
Acknowledgments
We thank our colleagues for helpful discussions. This work was supported by the National Institutes of Health [CA-008759 to S.K.] and the Yash Gandhi Foundation.
Grants: National Institutes of Health CA-008759 to S.K. and the Yash Gandhi Foundation
References
- Bao M, Booth JL, Elmendorf BJ, Canfield WM. Bovine UDP-N-acetylglucosamine:lysosomal-enzyme N-acetylglucosamine-1-phosphotransferase. I. Purification and subunit structure. J Biol Chem. 1996;271(49):31437–45. doi: 10.1074/jbc.271.49.31437. [DOI] [PubMed] [Google Scholar]
- Barea JJ, van Meel E, Kornfeld S, Bird LM. Tuberous sclerosis, polycystic kidney disease and mucolipidosis III gamma caused by a microdeletion unmasking a recessive mutation. Am J Med Genet A. 2015;167(11):2844–6. doi: 10.1002/ajmg.a.37213. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Braulke T, Raas-Rothschild A, Kornfeld S. I-cell disease and pseudo-hurler polydystrophy: disorders of lysosomal enzyme phosphorylation and localization. In: Valle D, Vogelstein B, Kinzler K, Antonarakis S, Ballabio A, Gibson K, Mitchell G, editors. The Online Metabolic & Molecular Bases of Inherited Disease. New York: McGraw-Hill Inc; 2013. [Google Scholar]
- Encarnacao M, Kollmann K, Trusch M, Braulke T, Pohl S. Post-translational modifications of the gamma-subunit affect intracellular trafficking and complex assembly of GlcNAc-1-phosphotransferase. J Biol Chem. 2011;286(7):5311–8. doi: 10.1074/jbc.M110.202382. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ghosh P, Dahms NM, Kornfeld S. Mannose 6-phosphate receptors: new twists in the tale. Nat Rev Mol Cell Biol. 2003;4(3):202–12. doi: 10.1038/nrm1050. [DOI] [PubMed] [Google Scholar]
- Kudo M, Bao M, D'Souza A, Ying F, Pan H, Roe BA, Canfield WM. The alpha- and beta-subunits of the human UDP-N-acetylglucosamine:lysosomal enzyme N-acetylglucosamine-1-phosphotransferase [corrected] are encoded by a single cDNA. J Biol Chem. 2005;280(43):36141–9. doi: 10.1074/jbc.M509008200. [DOI] [PubMed] [Google Scholar]
- Kudo M, Brem MS, Canfield WM. Mucolipidosis II (I-cell disease) and mucolipidosis IIIA (classical pseudo-hurler polydystrophy) are caused by mutations in the GlcNAc-phosphotransferase alpha / beta -subunits precursor gene. Am J Hum Genet. 2006;78(3):451–63. doi: 10.1086/500849. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lee WS, Payne BJ, Gelfman CM, Vogel P, Kornfeld S. Murine UDP-GlcNAc:lysosomal enzyme N-acetylglucosamine-1-phosphotransferase lacking the gamma-subunit retains substantial activity toward acid hydrolases. J Biol Chem. 2007;282(37):27198–203. doi: 10.1074/jbc.M704067200. [DOI] [PubMed] [Google Scholar]
- Liu S, Zhang W, Shi H, Meng Y, Qiu Z. Three novel homozygous mutations in the GNPTG gene that cause mucolipidosis type III gamma. Gene. 2014;535(2):294–8. doi: 10.1016/j.gene.2013.11.010. [DOI] [PubMed] [Google Scholar]
- Persichetti E, Chuzhanova NA, Dardis A, Tappino B, Pohl S, Thomas NS, Rosano C, Balducci C, Paciotti S, Dominissini S, et al. Identification and molecular characterization of six novel mutations in the UDP-N-acetylglucosamine-1-phosphotransferase gamma subunit (GNPTG) gene in patients with mucolipidosis III gamma. Hum Mutat. 2009;30(6):978–84. doi: 10.1002/humu.20959. [DOI] [PubMed] [Google Scholar]
- Qian Y, Flanagan-Steet H, van Meel E, Steet R, Kornfeld SA. The DMAP interaction domain of UDP-GlcNAc:lysosomal enzyme N-acetylglucosamine-1-phosphotransferase is a substrate recognition module. Proc Natl Acad Sci U S A. 2013;110(25):10246–51. doi: 10.1073/pnas.1308453110. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Qian Y, Lee I, Lee WS, Qian M, Kudo M, Canfield WM, Lobel P, Kornfeld S. Functions of the alpha, beta, and gamma subunits of UDP-GlcNAc:lysosomal enzyme N-acetylglucosamine-1-phosphotransferase. J Biol Chem. 2010;285(5):3360–70. doi: 10.1074/jbc.M109.068650. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Qian Y, van Meel E, Flanagan-Steet H, Yox A, Steet R, Kornfeld S. Analysis of mucolipidosis II/III GNPTAB missense mutations identifies domains of UDP-GlcNAc:lysosomal enzyme GlcNAc-1-phosphotransferase involved in catalytic function and lysosomal enzyme recognition. J Biol Chem. 2015;290(5):3045–56. doi: 10.1074/jbc.M114.612507. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Raas-Rothschild A, Bargal R, Goldman O, Ben-Asher E, Groener JE, Toutain A, Stemmer E, Ben-Neriah Z, Flusser H, Beemer FA, et al. Genomic organisation of the UDP-N-acetylglucosamine-1-phosphotransferase gamma subunit (GNPTAG) and its mutations in mucolipidosis III. J Med Genet. 2004;41(4):e52. doi: 10.1136/jmg.2003.015222. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Raas-Rothschild A, Cormier-Daire V, Bao M, Genin E, Salomon R, Brewer K, Zeigler M, Mandel H, Toth S, Roe B, et al. Molecular basis of variant pseudo-hurler polydystrophy (mucolipidosis IIIC) J Clin Invest. 2000;105(5):673–81. doi: 10.1172/JCI5826. [DOI] [PMC free article] [PubMed] [Google Scholar]
- van Meel E, Lee WS, Liu L, Qian Y, Doray B, Kornfeld S. Multiple domains of GlcNAc-1-phosphotransferase mediate recognition of lysosomal enzymes. J Biol Chem. 2016 doi: 10.1074/jbc.M116.714568. in press. [DOI] [PMC free article] [PubMed] [Google Scholar]
- van Meel E, Qian Y, Kornfeld SA. Mislocalization of phosphotransferase as a cause of mucolipidosis III alphabeta. Proc Natl Acad Sci U S A. 2014;111(9):3532–7. doi: 10.1073/pnas.1401417111. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.