

Abstract
Surfactant micelles are dynamic entities with a rapid exchange of monomers. By “clicking” tripropargylammonium-containing surfactants with diazide cross-linkers, we obtained surface-cross-linked micelles (SCMs) that could be multifunctionalized for different applications. They triggered membrane fusion through tunable electrostatic interactions with lipid bilayers. Antenna chromophores could be installed on them to create artificial light-harvesting complexes with efficient energy migration among tens to hundreds of chromophores. When cleavable cross-linkers were used, the SCMs could break apart in response to redox or pH signals, ejecting entrapped contents quickly as a result of built-in electrostatic stress. They served as caged surfactants whose surface activity was turned on by environmental stimuli. They crossed cell membranes readily. Encapsulated fluorophores showed enhanced photophysical properties including improved quantum yields and greatly expanded Stokes shifts. Catalytic groups could be installed on the surface or in the interior, covalently attached or physically entrapped. As enzyme mimics, the SCMs enabled rational engineering of the microenvironment around the catalysts to afford activity and selectivity not possible with conventional catalysts.
1. Introduction
Applications of surfactants are intimately related to their self-assembly under different conditions. Depending on the molecular structure, concentration, temperature, and amounts of polar and nonpolar solvents present, surfactants can form a rich variety of mesophases including spherical micelles or reverse micelles, bilayer or multilayer membranes, ordered hexagonal arrays, and bicontinuous phases. Each assembly, by its unique structure and properties, enables its particular applications, be it solubilization, encapsulation, delivery, or separation.
The different self-assembled mesophases of surfactants, although ordered, are highly dynamic in nature. Every component in the assembly—surfactant, solvent (most times water), dissolved contents, any additives—undergoes exchange constantly and often rapidly. These dynamics are necessary for some applications but, for other applications, can be problematic. One well-recognized application of surfactants, for example, is in the templated synthesis of inorganic nanomaterials. Because the different domains of a self-assembled surfactant mesophase can solubilize organic and/or inorganic precursors and modulate their reactions, surfactants can strongly influence the formation of inorganic nanomaterials. However, when the surfactant mesophase itself is not only dynamic but also altered by the very new materials formed, the template evolves throughout the reaction, making templated synthesis unpredictable.1,2
Similar problems exist in other applications of surfactants. With a lipid bilayer enclosing fluid inside, liposomes or vesicles are useful in drug delivery.3 However, stabilized mainly by hydrophobic interactions, liposomes are easily destabilized through losing lipid molecules to other hydrophobic entities in the vascular system including biomembranes and plasma proteins. The destabilization frequently leads to the premature leakage of entrapped contents.
An obvious solution to the above problem is to stabilize the surfactant assemblies by covalent bonds. Indeed, there has been a long-standing interest in surfmers (i.e., polymerizible surfactants) and the covalent capture of their self-assemblies by polymerization. The polymerized assemblies, with improved stability, enabled applications not possible with their noncovalent counterparts.4−13
Micelles probably are the most common surfactant assembly experienced by any person. Even if the person has never heard of the term, he/she relies on surfactant micelles for daily cleaning tasks of all sorts. Micelles are highly dynamic structures as well, with a lifetime typically in the millisecond range.14 The polymerization of surfmers in micelles may be traced back several decades. However, because surfactant exchange between micelles is much faster than the propagation of most radicals, it is difficult to confine radical polymerization within a single micelle.15
Our group has worked on amphiphilic materials for over a decade, including conformationally tunable facial amphiphiles and membrane transporters.16−20 This Feature Article summarizes our recent efforts in polymerizing/cross-linking surfactant micelles and converting them into multifunctional nanoparticles. Our surface-cross-linked micelles (SCMs) can be tuned in multiple ways including surface functionality, internal content, and water or oil solubility. Channels and voids could be created inside. Catalytic groups on their surface or in the hydrophobic core behave differently from those in the bulk solution as a result of their unusual microenvironment. They can be made to break apart in response to specific stimuli to release entrapped molecules or deliver surface activity. Their facile synthesis and potential for multifunctionality make them an extremely versatile platform for controlled release, molecular recognition, and catalysis.
2. Design and Synthesis of SCMs
2.1. Design and Synthesis
Although free radical polymerization is the most widely used technique in the covalent capture of surfactant mesophases,4−9 we turned to a more user-friendly reaction, the Cu-catalyzed azide–alkyne cycloaddition (CuAAC),21 to cross-link micelles. Since its discovery, the so-called click reaction has caught researchers’ attention for its exceptional functional group tolerance and ease of performance.22 As will be discussed later, these features are key to the construction and various applications of SCMs.
(4-Dodecyloxybenzyl)tripropargylammonium bromide 1 could be prepared in one step from the corresponding benzyl bromide and commercially available tripropargylamine. The three propargyls in the headgroup of the surfactant place a dense layer of alkyne on the micelle surface, greatly enhancing the local concentration of the reactive group. The cationic micelle formed precipitates with the conventional CuSO4/sodium ascorbate mixture, but could be easily cross-linked by a diazide (2a–c) in the presence of CuCl2/sodium ascorbate (Scheme 1).
Scheme 1. Preparation of Alkynyl-Functionalized SCM.

2.2. Characterization
The initially prepared alkynyl-SCMs were shown to have a hydrodynamic diameter of 8–10 nm by dynamic light scattering (DLS). The size was also confirmed by transmission electron microscopy (TEM).23 These particles were larger than typical micelles and probably resulted from aggregation of the primary SCMs. In our later studies, when the surfactants were dispersed by extensive ultrasonication prior to cross-linking, particles 4 to 5 nm in size were routinely prepared.24−26 Similarly sized nanoparticles were obtained from analogous surfactants.27−31 Fortunately, as long as the cross-linked micelles maintain their individual size during aggregation, the properties derived from each unit of SCM remain largely the same.
The radius of the hydrophobic core of a micelle roughly equals the length of the fully extended hydrocarbon tail.32 The distance between the cationic nitrogen and the terminal methyl carbon is about 2.2 nm for surfactant 1. Thus, although one can never say that our SCM was a replica of the original micelle, the comparable size between the two suggests that they have similar surfactant aggregation numbers. It is possible that the extremely efficient cycloaddition, the high local concentration of alkynes on the micelle surface, and the heavy surface cross-linking all served to quickly fix the structure and confined most of the click reactions to individual micelles.
When diazide 2a was used to cross-link the micelle, the resulting SCMs could be decomposed by periodic acid to cleave the 1,2-diol in the cross-linkage. Mass spectrometric analysis of the digested SCMs showed 3 to be the major product, consistent with the click cross-linking and the 1:1 stoichiometry between 1 and 2a.23 Compound 4 was also identified, indicating that some of the surfactants underwent three cycloadditions. Similar results were obtained with disulfide-linked SCM.331H NMR spectroscopy showed the methyl protons in cross-linked 1 to have higher mobility than the methylene protons and the protons near the cross-linked headgroups to be the least mobile, consistent with the cross-linking chemistry.
3. SCMs as Multifunctionalized Nanoparticles
3.1. Surface Functionalization by Click Chemistry
Click
cross-linking worked well for other tripropargyl-functionalized surfactants,
whether in the micelle27−30,33 or in vesicle form.34 One attractive feature of alkynyl-SCM is its
extremely facile postfunctionalization by almost any azido compounds
via another round of click reaction. Surface functionalization is
typically done in one pot at room temperature by adding an azide-functionalized
ligand (e.g., 5–7) and another batch
of copper catalysts.23
A distinctive feature of the multifunctionalized nanoparticles was the high density of surface ligands. When 1:1 stoichiometry was used between the tripropargylammonium surfactant and the diazide cross-linker, the alkynyl-SCM produced was expected to have one unreacted alkyne per surfactant on average. After being postfunctionalized with PEG-N37 (MW ∼2000), the nanoparticles obtained were digested by periodic acid. 1H NMR spectroscopy revealed that each cleaved surfactant was functionalized with an average of 0.7 to 0.8 PEG chain. Considering the steric congestion of the PEGylated micellar surface, this level of functionalization was quite impressive.
3.2. Tunable Electrostatic Interactions with Lipid Bilayers
Multifunctionalized SCMs could be used to control the fusion of lipid bilayers by their electrostatic interactions.35 Membrane fusion is an important step in many biological processes including fertilization, cell infection by enveloped viruses, and intracellular molecular trafficking.36 For two lipid membranes to fuse, they have to overcome significant steric/electrostatic repulsion. When alkynyl-SCM is functionalized with hydrophilic azides 5–7, the resulting SCM-OH, SCM-mannose, and SCM-PEG have positively charged ammonium headgroups surrounded by a layer of hydrophilic ligands. When these cationic SCMs were added to negatively charged liposomes made from a 10:1 mixture of neutral 1-palmitoyl-2-oleoyl-sn-glycero-3-phosphocholine (POPC) and negative 1-palmitoyl-2-oleoyl-sn-glycero-3-phospho-(1′-rac-glycerol) sodium salt (POPG), membrane fusion could be controlled through varying the thickness of the hydrophilic layer. SCM-PEG interacted with the liposomes negligibly as a result of the thick insulating layer of PEG in between the ammonium groups and the lipid membranes. As this layer got thinner, SCM-mannose and SCM-OH became increasingly potent at triggering membrane fusion and leakage because the electrostatic interactions between the SCMs and lipid membranes became stronger.35
For liposomes made with higher-melting DPPC/DPPG lipids (DPPC = 1,2-dipalmitoyl-sn-glycero-3-phosphocholine; DPPG = 1,2-dipalmitoyl-sn-glycero-3-phospho-(1′-rac-glycerol) sodium salt), SCM-OH was found to induce the aggregation of the liposomes at 25 °C, without any membrane fusion and leakage. The rigid, higher-melting membranes apparently were more resistant to electrostatically induced membrane fusion. On the other hand, right above the gel–liquid-crystalline transition temperature (41 °C) of the lipids, SCM-OH caused immediate fusion and leakage. The higher leakage resulted from coexisting gel and liquid-crystalline phases at the phase-transition temperature, which tend to have more packing defects at phase boundaries.37
3.3. Surface Functionalization with Light-Harvesting Chromophores
Surface functionalization of alkynyl-SCM was
not limited to hydrophilic ligands; hydrophobic azides could also
be used. 9,10-Diphenylanthracene (DPA) is a fluorophore with a high
fluorescence quantum yield. Surface functionalization occurred readily
when DPA-N38 in THF was mixed with an aqueous
solution of alkynyl-SCM and Cu(I) catalysts. After the reaction mixture was stirred overnight at room temperature the DPA-functionalized SCM (DPA-SCM) precipitated
spontaneously from the 2:1 THF/water mixture, presumably because of
the strong hydrophobicity of the product.38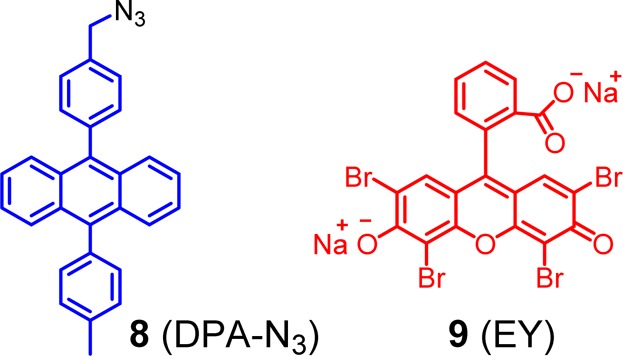
The construction of multichromophoric light-harvesting complexes often faces a dilemma: a small distance between the chromophores is needed for efficient donor–donor energy migration but easily causes undesired self-quenching and/or excimer formation of the chromophores.39 The fluorescence quantum yield of DPA-SCM was 0.80 in THF, only slightly lower than that of the monomer (0.90). One possible reason for the high quantum yield of DPA-SCM was the nonplanarity of DPA because of the 9,10-diaryl substitution that prevented chromophore stacking. Another possible reason was the limited freedom of DPA on the SCM surface due to the rigidity of the highly cross-linked micelle and the short triazole linkage between the chromophore and the SCM.38
DPA-SCM is positively charged because of the cationic cross-linkable surfactant (1). Energy acceptor 9 (Eosin Y disodium salt or EY) is attracted to the nanoparticle by electrostatic interactions. In our fluorescence titration, one acceptor was found to quench the emission of 48 ± 4 DPA chromophores. Because each SCM was estimated to contain ca. 50 surfactants and each alkynyl-SCM was estimated to contain ca. 50 unreacted alkynyl groups, assuming these alkynes were completely functionalized by DPA-N3, the result suggests that the entire DPA-SCM (with ∼50 chromophores) acted as one light-harvesting complex. In other words, no matter which DPA absorbs light initially and where EY is bound on the surface of DPA-SCM, the donor can always funnel its energy to the acceptor (Figure 1). The hypothesis was confirmed by the nearly perfect correlation between the extent of quenching and the degree of complexation of DPA-SCM by EY.38 Apparently, the DPA donors on the SCM surface were close enough for efficient donor–donor energy migration to occur but too far for self-quenching/excimer formation between the donors, an ideal situation for a light-harvesting system. What should be emphasized is that the highly sophisticated multichromophoric light-harvesting complex was synthesized by simply mixing the various building blocks and catalysts at room temperature, with no purification other than simple washing at the end of the synthesis. The synthetic ease was derived from combined self-assembly and covalent capture. As in natural light-harvesting systems, self-assembly affords order and efficiency in structural formation, and covalent construction ensures the stability of the final material.
Figure 1.
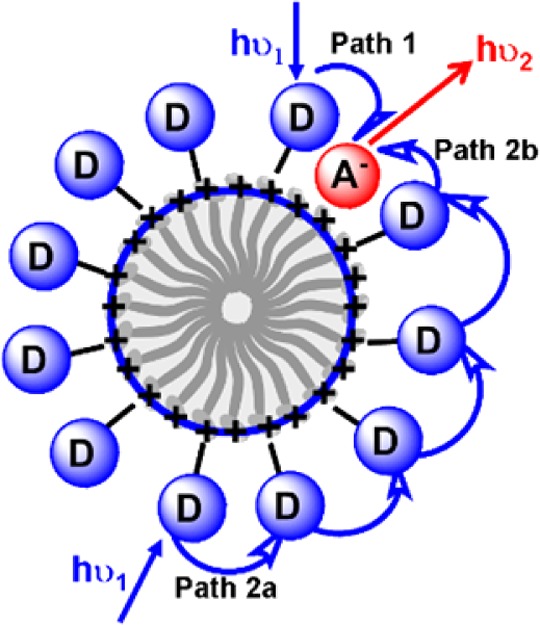
Two main pathways involved in the light-harvesting system: (1) direct energy transfer from donor to acceptor (path 1); (2) energy migration from donor to donor (path 2a) and then energy transfer to acceptor (path 2b). D and A represent the donor and acceptor, respectively. (Reprinted with permission from ref (38). Copyright 2012, Wiley-VCH Verlag GmbH.)
3.4. Light-Harvesting SCMs with Internal Chromophores
The conversion of light energy to chemical or electrical potential is the fundamental process in photosynthesis, photocatalysis, and photovoltaics.40−42 SCM could be used to prepare not only individual light-harvesting complexes as described above but also supercomplexes mimicking those found in higher plants.42 Construction of the latter type of supercomplex is very challenging because energy migration needs to occur efficiently first within individual light-harvesting complexes and then among self-assembled complexes, without significant self-quenching or excimer formation.
Cross-linkable surfactant 10 is similar to 1 except for the fluorescent dansyl-like
chromophore in between the hydrophobic C12 chain and the tripropargylammonium
headgroup. It may be used as the only cross-linkable surfactant or
together with 1 to prepare fluorescent SCMs with internal
chromophores.26 Normally, close contact
between fluorophores causes severe quenching. In our case, the quantum
yield of 10 was 1.6% in water in the un-cross-linked
form and ranged from 5.3 to 17% in SCM as the ratio of [10]/[1] varied from 100:0 to 20:80 (Table 1). The emission of dansyl is known to be
strongly polarity-dependent and increases when the probe enters a
nonpolar environment such as a micelle.43,44 Apparently,
dansyl self-quenching was more than compensated for by the environmental
enhancement by the SCM, making the fluorescent surfactant more emissive
after surface cross-linking.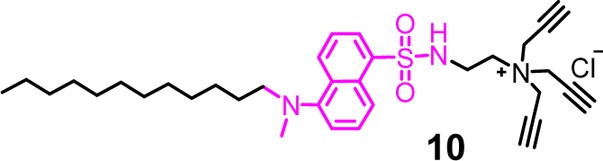
Table 1. Characterization of Dansyl-SCM Prepared from 1 and 10b.
| SCMa | [1]/[10] | DLS diameter (nm) | QY (%)b |
|---|---|---|---|
| SCM1 | 80:20 | 3.7 | 17 |
| SCM2 | 60:40 | 4.3 | 10 |
| SCM3 | 40:60 | 6.7 | 8.3 |
| SCM4 | 20:80 | 6.2 | 7.2 |
| SCM5 | 0:100 | 5.3 | 5.3 |
The quantum yield for un-cross-linked surfactant 2 in water below the CMC was 1.6%.
Quantum yields were determined using quinine sulfate in 0.05 M H2SO4 as a standard, with excitation at 343 nm. The quantum yields were calculated according to Φ = ΦS × (I/IS) × (ODS/OD) × (η2/ηS2), in which Φ is the quantum yield, Qs = 0.577 for quinine sulfate, I is the integrated intensity, η is the refractive index (η2 = ηS2 because water was used for both systems), and OD is the optical density. Subscript S refers to the standard.
Most interestingly, the number of donor molecules quenched by EY was found to increase steadily as the amount of 10 in the fluorescent SCM increased, up to 540 when 10 was the only cross-linkable surfactant in the synthesis. Because each dansyl-SCM contained ∼74 cross-linked surfactants according to DLS, 7 nanoparticles could funnel its excitation energy to the acceptor.26 Our subsequent study showed that EY, being negatively charged, induced the aggregation of dansyl-SCM in water by neutralizing its surface charge. The aggregates were confirmed by DLS to be ∼30 nm in size and equivalent to tens of individual SCMs. Therefore, intermicellar energy transfer happened in the aggregates among neighboring dansyl-SCMs, not over the entire aggregates.
4. SCMs with Cleavable Cross-Linkages for Controlled Release
4.1. Rapid Controlled Release from SCMs
Nearly half of the potential drug candidates identified in high-throughput screening are denied a further chance of development because of solubility problems.45,46 Although surfactant micelles have been proposed to solubilize hydrophobic drugs in water, their high critical micelle concentration (CMC), low thermodynamic stability, and highly dynamic nature hamper their use in drug delivery.
One way to overcome the above problem is to employ polymeric amphiphiles that aggregate into much more stable micelles, at concentrations that are orders of magnitude lower than for small-molecule surfactants.47 A hydrophobic drug may be physically trapped48 inside the hydrophobic core of a polymeric micelle or covalently attached to it.49 The latter approach is more amenable to the controlled release of drugs and more effective at preventing premature drug release than physical entrapment. Meanwhile, however, covalent conjugation puts severe constraints on the structure of the drug and the delivery vehicle and adds considerable complexity to the production of the entire package.
When cleavable cross-linkers are used, the SCMs prepared could break apart in response to specific chemical signals. To understand the potential of SCMs in drug delivery, we trapped pyrene as a mock hydrophobic drug. Its five vibronic bands of emission respond to the environmental polarity differently and can reveal the location of the probe.50 Being hydrophobic, it can be easily solubilized in water by 1 and physically trapped within the SCM (Figure 2).18
Figure 2.
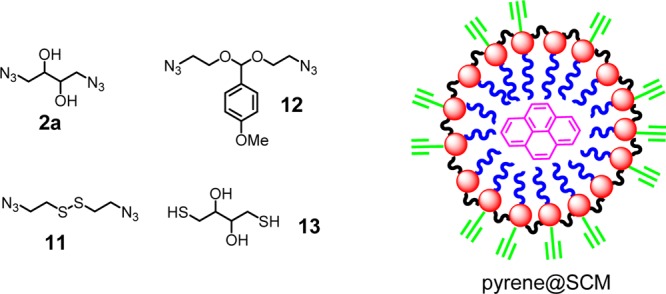
Cleavable cross-linkers (2a, 11, and 12) used in the preparation of pyrene-containing SCM. Dithiol 13 was used to cleave the disulfide bond in 11.
Pyrene encapsulated in SCM showed no change in emission over 6 months of storage, suggesting that the probe was physically trapped and that SCM was stable during storage. Although the release of the probe was fully expected after cleaving the cross-linkages, the rate of release was surprising. For both periodic acid (to cleave the 1,2-diol in 2a) and 13 (to cleave the disulfide in 11), the release of pyrene was complete in less than 1 min, after the addition of 1 equiv of cleaving reagent followed by gentle vortex mixing (Figure 3). Another surprise was the sensitivity of the cleavable SCMs toward the stimuli. The cleavage of disulfide bonds in cross-linked polymers, for example, often takes hours to days to complete and requires millimolar concentrations of reducing thiol,51,52 in contrast to 20 μM in our case.
Figure 3.

Change in pyrene I3/I1 ratio after the addition of 0 (△), 1 (□), 10 (◇), and 100 equiv ( ×) of cleaving agent to the pyrene-containing SCMs in deionized water at ambient temperature. (a) Cross-linker = 2a, cleaving agent = HIO4. (b) Cross-linker = 11, cleaving agent = 13. [1] = 20 μM. (Reprinted with permission from ref (18). Copyright 2010, American Chemical Society, Washington, DC.)
Why did the cleavable SCMs release their contents so differently from the way in which conventional highly cross-linked polymers released theirs? Ionic micelles are formed with two opposing forces: attractive hydrophobic interactions among the tails and repulsive Coulombic interactions among the headgroups. Below the CMC of the surfactant, the integrity of the SCM is maintained by the covalent cross-linkages whereas the entire system is under stress from the electrostatic repulsion among the headgroups. As soon as the covalent constraint is removed, the nanoparticle may burst open like an electrostatic bomb. Of course, not all cross-linkages have to be cleaved all at once; a partial rupture of the structure might be enough to expel the entrapped pyrene. The same electrostatic stress may be responsible for the enhanced sensitivity of the SCM toward the cleaving agent and may have accelerated the cleaving reaction. After all, any stress in the ground state of a reaction, whether steric, conformational, or electrostatic in this case, should raise the free energy of the system and lower the activation energy.
One might wonder why pyrene, a hydrophobic probe, did not reside within the partially cleaved SCMs. According to DLS (vide infra), the nanoparticles broke into smaller fragments instead of individual surfactants that are too small to scatter light below the CMC. As some surfactant molecules became free and escaped from the SCM, pyrene could be shielded by the remaining surfactants if the remaining (cross-linked) surfactants could rearrange themselves around the hydrophobic probe. As long as the binding affinity was sufficiently high between the partially cleaved SCM and pyrene, the guest would stay inside the hydrophobic particle. Such rearrangement is expected to be quite easy for un-cross-linked structures but difficult for a cross-linked material. Consistent with the above explanation, the release of pyrene indeed seemed to correlate with the initial cross-linking density of the SCM.18
Acid-triggered release is important to endocytic delivery because endosomes are more acidic than cytosols.53 Cancerous and inflammatory tissues are also known to be more acidic than normal tissues.54,55 Pyrene@SCMs prepared with acid-sensitive 12 as the cross-linker, however, showed no release of pyrene over 96 h at pH 5. Recognizing that pyrene emission was an indirect indicator of breakage, we turned to DLS to monitor the size change of the SCMs directly. As shown in Figure 4, SCMs prepared with 2a (△) and 11 (□) as the cross-linkers broke immediately upon the addition of the corresponding cleaving agent. The SCMs prepared with acetal-functionalized 12, however, broke apart gradually at pH 5 over 30 min or so (◇) and were largely unchanged at pH 7 over the same period of time (×).
Figure 4.

Relative intensity of scattered light for the pyrene-containing SCMs upon different stimulation: 1 equiv of HIO4 for SCMs cross-linked with 2a (△), 1 equiv of 13 for SCMs cross-linked with 11 (□), and pH 5 (◇) and 7 (×) acetate buffer at 37 °C for SCMs cross-linked with 12. [1] = 20 μM. (Reprinted with permission from ref (18). Copyright 2010, American Chemical Society, Washington, DC.)
Why did the three breakages have different profiles? One likely reason was the different charge character of the cleaving reagent. Anionic periodate is electrostatically attracted to cationic SCM, dithiol 13 is neutral, and protons for the hydrolysis of acetal are repelled by the nanoparticles. Another reason might be related to the carbocation intermediate for the acid-triggered release. The carbocation is not expected to be stable on a polycationic nanoparticle, especially when it is located in a relatively hydrophobic region of the SCM as a result of the hydrophobicity of the acetal.
Our entrapment–release strategy combines the ease of physical entrapment and the precision of chemical ligation and requires no covalent modification of the entrapped agents. This could be very useful in the delivery and controlled release of pharmaceutical agents. In biological studies, delivery vehicles are frequently labeled with fluorophores, which allow the delivery to be monitored by fluorescence imaging. In a follow-up study, we demonstrated that the physical entrapment strategy could be extended to solubilize hydrophobic fluorophores and improve their photophysical properties.
4.2. Dye-Containing SCMs for Cellular Imaging
Coumarin derivative 14 emitted at 470 nm in water and 442 nm in SCM. Its fluorescence quantum
yield went from 0.05 in water to 0.30 in SCM. For BODIPY derivatives 15, a red shift of 14 nm was observed upon incorporation into
SCM, and the quantum yield increased from 0.034 to 0.29. BODIPY derivative 16 emitted at 575 and 652 nm, corresponding to the fluorescence
of BODIPY and distyryl BODIPY functionalities in the structure, respectively.
SCM encapsulation enhanced the peak at 575 nm relative to that at
652 nm.56
With a hydrophobic radius of slightly greater than 2 nm, an SCM can trap two nonpolar dye molecules within its FRET (Fluorescence Resonance Energy Transfer) distance. When 14 and 15 were coencapsulated within the same SCM, strong FRET was observed with an energy-transfer efficiency of 79% (Figure 5), translating to a donor–acceptor distance of 3.0 nm. The energy transfer also increased the Stokes shift to 160 nm, in comparison to 20 nm in donor 14 alone. A large Stokes shift is desirable in fluorescence imaging because it minimizes interference from the excitation. Nevertheless, common planar, conjugated organic probes have only small Stokes shifts in the range of 10–20 nm.57,58
Figure 5.
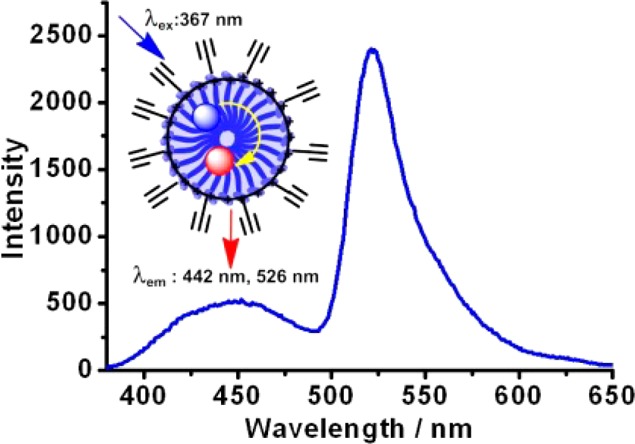
Emission spectrum of SCM with encapsulated 14 and 15. ([SCM] = 5.0 μM, λex = 367 nm.) (Reprinted with permission from ref (56). Copyright 2013, Royal Society of Chemistry.)
Another pleasant finding was that the cationic SCMs showed very low cytotoxicity to Hela cells, even at 10 μM.56 Although a detailed mechanism is not currently available, they were also found to permeate cell membranes readily and accumulate in the cytoplasm (Figure 6). Thus, good water solubility and excellent membrane permeability, two seemingly contradictory properties, were obtained simultaneously for the SCM-encapsulated fluorophores. For conventional fluorophores, water solubility is achieved by installing polar functional groups such as sulfonates. However, such covalent modification may not be compatible with some structures. Another issue is that the resulting water-soluble dyes often have difficulty crossing hydrophobic barriers such as lipid membranes.
Figure 6.
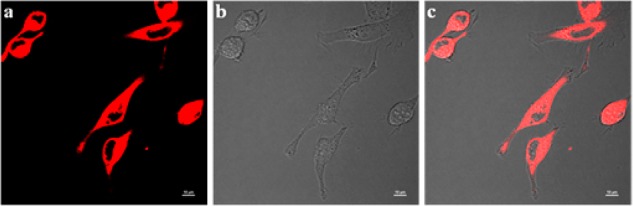
(a) Fluorescence, (b) transmission, and (c) overlapping fluorescence/transmission images of Hela cells observed by confocal laser scanning microscopy. The cells were incubated with SCM-F3 at a concentration of 1 μM for 30 min at 37 °C under a humidified atmosphere containing 5% CO2. The excitation wavelength was fixed at 561 nm, and the fluorescence signals were collected between 570 and 620 nm. (Reprinted with permission from ref (56). Copyright 2013, Royal Society of Chemistry.)
4.3. Controlled Release of Surface Activity
To further explore the application of cleavable SCMs in drug delivery, we took advantage of their controlled release in surface activity. A biological example of such release is found in the influenza virus. After it enters a host cell through endocytosis, the virus uses lower pH to trigger the exposure of buried hydrophobic fusion peptides. The fusion peptides, once activated through this conformational change, insert into the endosomal membrane of the host cell and ultimately cause the viral and host membranes to fuse.59
Our idea was based on the fact that an SCM has all of its hydrophobic tails buried inside the cross-linked nanoparticle and thus possesses very little surface activity. As its surface cross-linkages are cleaved, the hydrophobic tails will be exposed and make the resulting material surface-active. If the release can be controlled temporally and spatially, then we can use the released amphiphiles to induce localized destabilization in the lipid membranes.
To demonstrate
the concept, we prepared SCMs from surfactant 1 and disulfide
cross-linker 11 and postfunctionalized the SCMs with
azido PEG 7 to afford SCM-(S-S)-PEG. Fluorinated surfactants
are known for their outstanding surface activity.60 We thus prepared fluoro-SCM-(S-S)-PEG from surfactant 18 in a similar manner.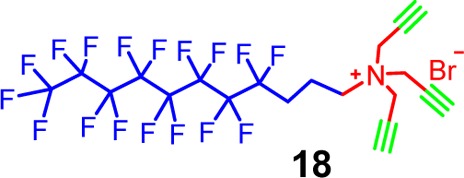
Figure 7a shows that SCM-(S-S)-PEG displayed little surface activity: its aqueous solution had a surface tension of ∼60 mN/m. The addition of 1 mM dithiothreitol (DTT) triggered a small but noticeable decrease in surface tension when SCM-(S-S)-PEG contained 1 to 2 μM cross-linked surfactant. A larger and precipitous drop in surface tension was observed at higher concentrations of SCMs. The fast drop in surface tension was consistent with the electrostatically activated release mechanism as described earlier. Meanwhile, the scattering intensity of SCM-(S-S)-PEG dropped sharply within 2 min after DTT addition (Figure 7b), and the hydrodynamic radius (R) of the particles decreased from ∼90 nm to 55–60 nm. Note that these particles were much larger than the parent SCMs as a result of the surface PEGylation. The particle size at the end of 30 min was ca. 50 nm, much larger than a single polymer. The nanoparticles thus must have fragmented into pieces consisting of multiple surfactants, similar to the parent SCMs.18 (Complete cleavage required a large excess of DTT and a much longer incubation time (ca. 48).)33 In our hands, fluoro-SCM-(S-S)-PEG showed similar breakage. Expectedly, the reduction in surface tension was larger; it more than doubled what was observed with SCM-(S-S)-PEG.
Figure 7.

(a) Surface tension of aqueous solutions of SCM-(S-S)-PEG upon the addition of DTT. [Surfactant in SCM] = 1 (□), 2 (△), 5 (◇), and 10 μM (+) from top to bottom. [DTT] = 1 mM. (b) Relative scattering intensity (△) and hydrodynamic radius (red ■) of SCM-PEG after the addition of DTT. [Surfactant in SCM] = 0.6 mM. [DTT] = 6 mM. (Reprinted with permission from ref (33). Copyright 2012, American Chemical Society, Washington, DC.)
The carboxyfluorescein (CF) leakage assay was employed to monitor how the cleaved SCMs disrupted liposome membranes.3 CF is a water-soluble fluorescent dye that self-quenches above 50 mM. Liposomes were prepared from POPC and POEPC (1-palmitoyl-2-oleoyl-sn-glycero-3-ethylphosphocholine) with 50 mM CF trapped in the internal water pool. The cationic POEPC lipid was added to keep the liposomes positively charged and repulsive to the SCMs. If the released surfactants insert into the membranes by hydrophobic interactions, then the membranes should be destabilized. Once CF escapes from the liposomes, it will be diluted and will emit more strongly.
Figure 8a shows the CF leakage of liposomes induced by different concentrations of DTT when the SCMs present had 15 μM cross-linked or caged surfactants. Our data shows that CF leakage increased steadily with increasing concentrations of DTT. The leakage rate could also be controlled by the amount of surfactant released. In general, leakage became noticeable with as little as 1 μM caged surfactant in the mixture and increased steadily when more cleavable SCMs were present (Figure 8b). The thiol-triggered release has practical implications for intracellular delivery because the concentration of reducing thiol (mostly glutathione) is typically 0.5–10 mM in cytosol but only 2–20 μM in plasma.61,62
Figure 8.

(a) Percent leakage of CF from POPC/POEPC LUVs triggered by SCM-(S-S)-PEG for different concentrations of DTT. [Surfactant in SCM] = 15 μM. [DTT] = 0 (□), 0.025 (△), 0.05 (◇), 0.1 (+), 0.2 (*), and 1.0 mM (○) from bottom to top. (b) Percent leakage of CF from POPC/POEPC LUVs triggered by SCM-(S-S)-PEG and DTT. [Surfactant in SCM] = 0 (□), 1 (△), 2 (◇), 3 (+), and 5 μM (*) from bottom to top. [DTT] = 1 mM. The error in the leakage experiments was generally within 10%. The 100% leakage at 60 min was induced by the addition of 1% Triton X-100. (Reprinted with permission from ref (33). Copyright 2012, American Chemical Society, Washington, DC.)
5. Catalytic SCMs as Enzyme Mimics
5.1. SCMs as Mimics of Hydrolytic Enzymes
To achieve efficient catalysis, enzymes need to place appropriate catalytic groups around the substrate bound in the active site. These functional groups are obviously critical to the catalysis. Another important factor, maybe less obvious, is the environmental effect that determines how differently these catalytic functional groups behave in the unique environment of the active site. Acids and bases, for example, are among the most common catalysts in organic chemistry but strong acids and bases are not available in typical biological systems. Many enzymes, not surprisingly, have developed remarkable capabilities to alter the pKa of acidic or basic groups used for catalysis.63
SCMs are hydrophobic nanoparticles with a layer of hydrophilic surface groups. They resemble water-soluble proteins in size (4–5 nm or 50 000–60 000 MW) and topology. Because they can be functionalized both on the surface and in the interior, we can use them as enzyme mimics for various catalytic applications.
Micelles have been studied as enzyme mimics for a long time;64−66 SCMs, however, behave very differently from their non-cross-linked counterparts. In comparison to a micelle, an obvious advantage of SCMs is the lack of CMC, which means that the materials could be used at both high and low concentrations. Using several fluorescent probes, we found that SCMs could better shield noncovalently bound guests from solvent exposure and have higher surface basicity than CTAB (cetyltrimethylammonium bromide) micelles. The basicity enabled the parent SCM to catalyze the hydrolysis of an activated phosphate ester.67
To enhance the hydrolytic
activity, we installed imidazole groups on SCMs using azido derivatives 19 and 20. The catalytic imidazoles of SCM(19) should be close to the micellar surface but have limited
exposure to water because the C16 chain of 19 has to
stay in the hydrophobic core and acts as a hydrophobic anchor. Imidazoles
in SCM(20), on the other hand, were introduced after
cross-linking and should be located on the micellar surface, likely
fully exposed to water.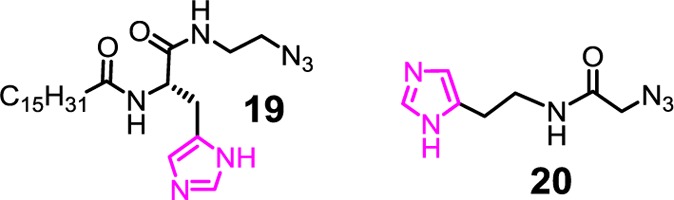
The different location of imidazoles strongly influenced their activity. In the catalytic hydrolysis of activated ester, SCM(19) consistently outperformed SCM(20) over pH 4–8.24 The lower environmental polarity makes it more difficult to protonate the imidazoles in SCM(19) than those on SCM(20). Because only deprotonated imidazoles could catalyze the hydrolysis, SCM(19) is expected to be more active than SCM(20). Consistent with this model, the largest difference in catalytic activity between the two was observed under the most acidic condition (pH 4). The environmentally derived resistance to protonation, although quite simple in concept, allowed us to perform nucleophilic/basic catalysis under acidic conditions, a quite unusual feature.
We went on to prepare more potent hydrolytic catalysts by installing derivatives of 4-dimethylaminopyridine (DMAP) on the SCMs, also on the surface and internally. Surface-functionalized DMAP-SCM(21) was obtained by the postfunctionalization of alkynyl-SCM using azide 21 following our standard procedures (Figure 9). For internally functionalized DMAP-SCM, we developed a completely different procedure.
Figure 9.

Comparison between DMAP-SCM(21) with catalytic groups on the surface and DMAP-SCM(23) with internal catalytic groups.
Surfactant 22 has two orthogonal cross-linking groups: a tripropargylammonium headgroup for the click reaction and a methacrylate at the tail for free radical polymerization. As shown in Figure 9, we first used mixed micelles of 22 and CTAB to solubilize 23 (a polymerizible DMAP derivative), xylene, and DMPA (2,2-dimethoxy-2-phenylacetophenone, a photoinitiator) in water. After surface cross-linking with diazide 2a and surface-functionalization with 24 by the click reaction, we initiated free radical photopolymerization in the hydrophobic core of the mixed micelle. In this synthesis, CTAB and xylene were both temporary space holders: although they were solubilized in the micelle, they participated in neither the surface nor the core cross-linking. Once they are removed at the end of the synthesis, they should leave behind channels/voids in the micellar core. These channels/voids are expected to not only recruit a hydrophobic substrate to the SCM but also make the internal DMAP groups more accessible to the substrate during catalysis. We varied the amounts of CTAB (25–75 mol % with respect to 22) and xylene (2–6 equiv) in the preparation and discovered that DMAP-SCM(23) made with 50% CTAB and 2 equiv of xylene had the best water solubility.25
We then studied the hydrolysis of para-nitrophenyl hexanoate (PNPH), para-nitrophenyl acetate (PNPA), and 2-hydroxylpropyl-4-nitrophenyl phosphate
(HPNPP). The catalytic effect was very weak for HPNPP. The low activity
was attributed to the weak binding between the SCM and the hydrophilic
substrate. PNPA and PNPH, on the other hand, exhibited very interesting
behavior. In aqueous buffer (pH 8), PNPA hydrolyzed 7 times faster
than did PNPH. When catalyzed by the two DMAP-SCMs, however, PNPH
became 2–4 times faster than PNPA. The reversed reactivity
could be explained by the stronger binding of PNPH by the SCMs as
a result of its higher hydrophobicity. Very impressively, the two
DMAP-SCMs were thousands or tens of thousands of times more active
than molecular DMAP and even maintained most activity at a solution
pH of 5, at which molecular DMAP (pKa =
9.7) was completely inactive. Consistent with the environmentally
derived resistance to protonation, DMAP-SCM(23) consistently
displayed higher hydrolytic activity than DMAP-SCM(21). This work demonstrated that the environmental effect of catalysts
is substrate-dependent, easily overriding the inherent chemical reactivity
of the substrates.
5.2. SCMs with Entrapped Transition-Metal Catalysts
Water-soluble transition-metal catalysts are typically obtained by installing water-solubilizing groups such as sulfonate on the ligands.68 Not only does such modification significantly complicate the ligand synthesis, but the resulting water-soluble catalysts are also rarely useful for highly nonpolar substrates because the substrates have difficulty accessing the catalysts located in the aqueous phase.69
Instead of fluorophores, SCMs could encapsulate nonpolar transition-metal catalysts. The resulting nanoparticles resemble artificial metalloenzymes with a hydrophobic core and a hydrophilic exterior. As a proof of concept, we trapped commercially available bisphosphine rhodium(I) complex (25) in the SCM using 1 as the cross-linkable surfactant.70 ICP-MS showed that each SCM contained 0.92 ± 0.03 rhodium, in agreement with our preparation. To demonstrate physical entrapment, we layered an aqueous solution of SCM(25) on chloroform. After hand shaking, the mixture quickly separated into two layers for the SCM-entrapped sample, whereas the rhodium complex solubilized by CTAB formed an emulsion (Figure 10). When the mixture finally settled, the yellow rhodium complex migrated to the lower chloroform layer in the CTAB case but remained in water when trapped inside the SCM.
Figure 10.
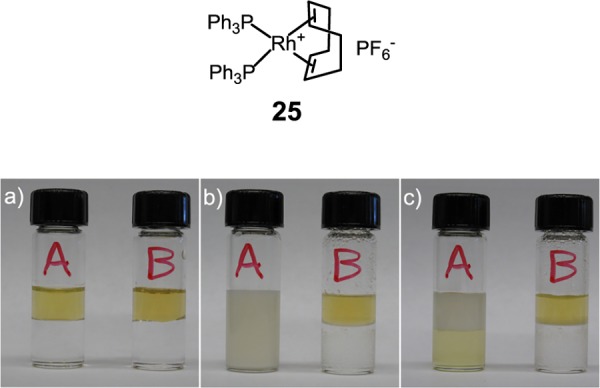
Comparison of rhodium complex 25 protected by CTAB (A) and SCM (B) in the presence of CHCl3 (a) before hand shaking and standing, (b) after 2 min of hand shaking and 1 min of standing, and (c) after standing overnight at room temperature. (Reprinted with permission from ref (70). Copyright 2012, Royal Society of Chemistry.)
In comparison to conventional water-soluble transition-metal catalysts, our physical entrapment method employs unmodified commercially available hydrophobic catalysts and is extremely easy to perform. Also, the SCM provides a local hydrophobic microenvironment, allowing even very nonpolar substrates to access the catalyst. To aid mass transfer to the catalyst, we created channels in SCM(25) using dodecanol as a temporary space holder. To our delight, terminal alkenes with 6–10 carbons underwent catalytic hydrogenation in quantitative yield under our experimental conditions. Interestingly, an increase of two additional carbons reduced the yield to 21% for 1-dodecene. Most likely, the cross-linked micelle, limited by the chain length of the hydrophobic tail, could accommodate only hydrocarbons with a certain chain length. 1-Dodecene was probably too long to fit within the hydrophobic sites of the SCM (Figure 11). Hydrophilic or internal alkenes displayed low reactivity, consistent with the location of the catalyst.
Figure 11.
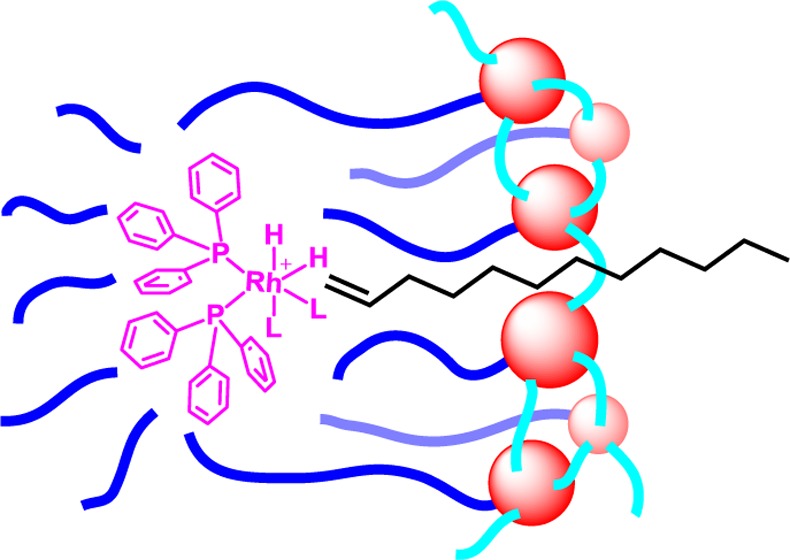
Schematic representation of the hypothesized chain-length selectivity. (Reprinted with permission from ref (70). Copyright 2012, Royal Society of Chemistry.)
Catalytic rhodium(I) species deactivates easily in homogeneous solution through dimerization. Such deactivation can be inhibited when the SCM contains only one complex, allowing the catalysts to be recycled. Gratifyingly, SCM(25) could be reused many times in biphasic catalytic hydrogenation, and a significant decrease in yield occurred only at the eighth cycle. Given the harsh treatment of the samples in between reactions (extraction with methylene chloride, followed by solvent evaporation at 50 °C for ∼2 min, all in open air), the SCM-enabled stability of the catalyst is quite remarkable.
6. Summary and Perspective
SCMs are readily synthesized nanoparticles with tremendous tunability. The clickable cross-linkable surfactant makes both the synthesis and postfunctionalization of the materials extremely easy to perform. The modular synthesis allows us to modify almost every aspect of the material—water or organic solubility, multivalent surface decoration, encapsulated guests with different functions, high stability or sensitivity to prescribed stimuli, surface cross-linking, and surface–core double cross-linking, to name a few. These features, when combined in different ways, create a powerful platform for diverse applications in chemistry and biology.
This Feature Article summarizes our efforts over the last 5 to 6 years in using SCMs for controlled release, light harvesting, and catalysis. We believe that the most promising applications of the materials, at least in the near future, might be in controlled release and biomimetic catalysis. As potential drug delivery vehicles, SCMs already display a number of attractive features, including easy preparation, simple functionalization, facile fluorescent labeling, controllable release, and membrane permeability. Drugs can be physically entrapped with potential long-term stability during storage and yet can be released quickly on demand. The release profile can be tuned by the type and density of surface cross-linkage. Although these studies are largely proof-of-concept in nature and much needs to be learned to understand how SCMs interact with cells, their tunability and versatility are highly desirable features for a biomaterial. As for their applications in catalysis, SCMs can have catalysts entrapped or covalently attached at different locations. The large structural tunability has already enabled activity and selectivity that are difficult to achieve with conventional catalysts. Environmental control of the SCM, in particular, has enabled remarkable features such as efficient nucleophilic/basic catalysis under acidic conditions, unusual chain length selectivity, and excellent reusability of otherwise easily decomposed transition-metal catalysts. Similar strategies have been used by enzymes with huge success in nature; an adventure along the same path using multifunctional synthetic nanoparticles is expected to be equally fruitful.
Acknowledgments
I thank the NSF (CHE-1303764, DMR-1464927) and NIGMS (R01GM113883) for the financial support of the research and all of the students whose names appear in the cited papers for their contributions.
Biography

Yan Zhao received his B.S. in chemistry from Lanzhou University in 1992 and his Ph.D. from Northwestern University in 1996 (Prof. Joseph B. Lambert). After a postdoctoral stay at the University of Illinois (Prof. Steven C. Zimmerman), he worked for the Procter & Gamble Company from 1998 to 2002 and is currently a professor of chemistry at Iowa State University. His areas of interest include the synthesis of molecules capable of controllable conformational changes and their use as “smart” sensors, materials, molecular transporters, and catalysts; self-assembly in water; biomimetic chemistry in materials synthesis and catalysis; and the design and construction of nanoscale structures.
The author declares no competing financial interest.
References
- Pileni M. P. Nanosized particles made in colloidal assemblies. Langmuir 1997, 13, 3266–3276. 10.1021/la960319q. [DOI] [Google Scholar]
- Pileni M. P. The role of soft colloidal templates in controlling the size and shape of inorganic nanocrystals. Nat. Mater. 2003, 2, 145–150. 10.1038/nmat817. [DOI] [PubMed] [Google Scholar]
- New R. R. C.Liposomes: A Practical Approach; IRL Press: Oxford, 1990. [Google Scholar]
- Mueller A.; O’Brien D. F. Supramolecular materials via polymerization of mesophases of hydrated amphiphiles. Chem. Rev. 2002, 102, 727–757. 10.1021/cr000071g. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gin D. L.; Gu W.; Pindzola B. A.; Zhou W. J. Polymerized lyotropic liquid crystal assemblies for materials applications. Acc. Chem. Res. 2001, 34, 973–980. 10.1021/ar000140d. [DOI] [PubMed] [Google Scholar]
- Eastoe J.; Summers M.; Heenan R. K. Control over phase curvature using mixtures of polymerizable surfactants. Chem. Mater. 2000, 12, 3533–3537. 10.1021/cm001096z. [DOI] [Google Scholar]
- Yan F.; Texter J. Polymerization of and in mesophases. Adv. Colloid Interface Sci. 2006, 128–130, 27–35. 10.1016/j.cis.2006.11.014. [DOI] [PubMed] [Google Scholar]
- Tajima K.; Aida T. Controlled polymerizations with constrained geometries. Chem. Commun. 2000, 2399–2412. 10.1039/b007618j. [DOI] [Google Scholar]
- Summers M.; Eastoe J. Applications of polymerizable surfactants. Adv. Colloid Interface Sci. 2003, 100-102, 137–152. 10.1016/S0001-8686(02)00058-1. [DOI] [Google Scholar]
- Gin D. L.; Lu X. Y.; Nemade P. R.; Pecinovsky C. S.; Xu Y. J.; Zhou M. J. Recent advances in the design of polymerizable lyotropic liquid-crystal assemblies for heterogeneous catalysis and selective separations. Adv. Funct. Mater. 2006, 16, 865–878. 10.1002/adfm.200500280. [DOI] [Google Scholar]
- Zhang S.; Zhao Y. Facile Preparation of Organic Nanoparticles by Interfacial Cross-Linking of Reverse Micelles and Template Synthesis of Subnanometer Au–Pt Nanoparticles. ACS Nano 2011, 5, 2637–2646. 10.1021/nn102666k. [DOI] [PubMed] [Google Scholar]
- Lee L.-C.; Zhao Y. Size-Selective Phase-Transfer Catalysis with Interfacially Cross-Linked Reverse Micelles. Org. Lett. 2012, 14, 784–787. 10.1021/ol203319w. [DOI] [PubMed] [Google Scholar]
- Lee L.-C.; Zhao Y. Metalloenzyme-Mimicking Supramolecular Catalyst for Highly Active and Selective Intramolecular Alkyne Carboxylation. J. Am. Chem. Soc. 2014, 136, 5579–5582. 10.1021/ja501277j. [DOI] [PubMed] [Google Scholar]
- Aniansson E. A. G.; Wall S. N.; Almgren M.; Hoffmann H.; Kielmann I.; Ulbricht W.; Zana R.; Lang J.; Tondre C. Theory of the kinetics of micellar equilibria and quantitative interpretation of chemical relaxation studies of micellar solutions of ionic surfactants. J. Phys. Chem. 1976, 80, 905–922. 10.1021/j100550a001. [DOI] [Google Scholar]
- Hamid S.; Sherrington D. Polymerized micelles: fact or fancy?. J. Chem. Soc., Chem. Commun. 1986, 936–938. 10.1039/c39860000936. [DOI] [Google Scholar]
- Zhao Y. Facial amphiphiles in molecular recognition: From unusual aggregates to solvophobically driven foldamers. Curr. Opin. Colloid Interface Sci. 2007, 12, 92–97. 10.1016/j.cocis.2007.05.001. [DOI] [Google Scholar]
- Zhao Y.; Zhong Z.; Ryu E.-H. Preferential solvation within hydrophilic nanocavities and its effect on the folding of cholate foldamers. J. Am. Chem. Soc. 2007, 129, 218–225. 10.1021/ja0671159. [DOI] [PubMed] [Google Scholar]
- Zhang S.; Zhao Y. Rapid Release of Entrapped Contents from Multi-Functionalizable, Surface Cross-Linked Micelles upon Different Stimulation. J. Am. Chem. Soc. 2010, 132, 10642–10644. 10.1021/ja103391k. [DOI] [PubMed] [Google Scholar]
- Zhang S.; Zhao Y. Oligocholate Foldamers as Carriers for Hydrophilic Molecules across Lipid Bilayers. Chem. - Eur. J. 2011, 17, 12444–12451. 10.1002/chem.201101510. [DOI] [PubMed] [Google Scholar]
- Zhao Y.; Cho H.; Widanapathirana L.; Zhang S. Conformationally Controlled Oligocholate Membrane Transporters: Learning through Water Play. Acc. Chem. Res. 2013, 46, 2763–2772. 10.1021/ar300337f. [DOI] [PubMed] [Google Scholar]
- Kolb H. C.; Finn M. G.; Sharpless K. B. Click chemistry: Diverse chemical function from a few good reactions. Angew. Chem., Int. Ed. 2001, 40, 2004–2021. . [DOI] [PubMed] [Google Scholar]
- Iha R. K.; Wooley K. L.; Nystrom A. M.; Burke D. J.; Kade M. J.; Hawker C. J. Applications of Orthogonal ″Click″ Chemistries in the Synthesis of Functional Soft Materials. Chem. Rev. 2009, 109, 5620–5686. 10.1021/cr900138t. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhang S.; Zhao Y. Facile Synthesis of Multivalent Water-Soluble Organic Nanoparticles via “Surface Clicking” of Alkynylated Surfactant Micelles. Macromolecules 2010, 43, 4020–4022. 10.1021/ma100497k. [DOI] [Google Scholar]
- Chadha G.; Zhao Y. Histidine-functionalized water-soluble nanoparticles for biomimetic nucleophilic/general-base catalysis under acidic conditions. Org. Biomol. Chem. 2013, 11, 6849–6855. 10.1039/c3ob41485j. [DOI] [PubMed] [Google Scholar]
- Chadha G.; Zhao Y. Environmental control of nucleophilic catalysis in water. Chem. Commun. 2014, 50, 2718–2720. 10.1039/c3cc49593k. [DOI] [PubMed] [Google Scholar]
- Chadha G.; Yang Q.-Z.; Zhao Y. Self-assembled light-harvesting supercomplexes from fluorescent surface-cross-linked micelles. Chem. Commun. 2015, 51, 12939–12942. 10.1039/C5CC04377H. [DOI] [PubMed] [Google Scholar]
- Awino J. K.; Zhao Y. Protein-Mimetic, Molecularly Imprinted Nanoparticles for Selective Binding of Bile Salt Derivatives in Water. J. Am. Chem. Soc. 2013, 135, 12552–12555. 10.1021/ja406089c. [DOI] [PubMed] [Google Scholar]
- Awino J. K.; Zhao Y. Molecularly Imprinted Nanoparticles as Tailor-Made Sensors for Small Fluorescent Molecules. Chem. Commun. 2014, 50, 5752–5755. 10.1039/c4cc01516a. [DOI] [PubMed] [Google Scholar]
- Awino J. K.; Zhao Y. Water-Soluble Molecularly Imprinted Nanoparticles (MINPs) with Tailored, Functionalized, Modifiable Binding Pockets. Chem. - Eur. J. 2015, 21, 655–661. 10.1002/chem.201404919. [DOI] [PubMed] [Google Scholar]
- Awino J. K.; Zhao Y. Polymeric Nanoparticle Receptors as Synthetic Antibodies for Nonsteroidal Anti-Inflammatory Drugs (NSAIDs). ACS Biomater. Sci. Eng. 2015, 1, 425–430. 10.1021/acsbiomaterials.5b00042. [DOI] [PubMed] [Google Scholar]
- Awino J. K.; Hu L.; Zhao Y. Molecularly Responsive Binding through Co-occupation of Binding Space: A Lock–Key Story. Org. Lett. 2016, 18, 1650–1653. 10.1021/acs.orglett.6b00527. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rosen M. J.Surfactants and Interfacial Phenomena, 2nd ed.; Wiley: New York, 1989; pp 108–168. [Google Scholar]
- Li X.; Zhao Y. Protection/Deprotection of Surface Activity and Its Applications in the Controlled Release of Liposomal Contents. Langmuir 2012, 28, 4152–4159. 10.1021/la2050702. [DOI] [PubMed] [Google Scholar]
- Zhang S.; Zhao Y. Controlled Release from Cleavable Polymerized Liposomes upon Redox and pH Stimulation. Bioconjugate Chem. 2011, 22, 523–528. 10.1021/bc1003197. [DOI] [PubMed] [Google Scholar]
- Li X.; Zhao Y. Tunable fusion and aggregation of liposomes triggered by multifunctional surface-cross-linked micelles. Bioconjugate Chem. 2012, 23, 1721–1725. 10.1021/bc300082b. [DOI] [PubMed] [Google Scholar]
- Jahn R.; Lang T.; Südhof T. C. Membrane Fusion. Cell 2003, 112, 519–533. 10.1016/S0092-8674(03)00112-0. [DOI] [PubMed] [Google Scholar]
- Mills J. K.; Needham D. Lysolipid incorporation in dipalmitoylphosphatidylcholine bilayer membranes enhances the ion permeability and drug release rates at the membrane phase transition. Biochim. Biophys. Acta, Biomembr. 2005, 1716, 77–96. 10.1016/j.bbamem.2005.08.007. [DOI] [PubMed] [Google Scholar]
- Peng H.-Q.; Chen Y.-Z.; Zhao Y.; Yang Q.-Z.; Wu L.-Z.; Tung C.-H.; Zhang L.-P.; Tong Q.-X. Artificial Light-Harvesting System Based on Multifunctional Surface-Cross-Linked Micelles. Angew. Chem., Int. Ed. 2012, 51, 2088–2092. 10.1002/anie.201107723. [DOI] [PubMed] [Google Scholar]
- Peng H.-Q.; Niu L.-Y.; Chen Y.-Z.; Wu L.-Z.; Tung C.-H.; Yang Q.-Z. Biological Applications of Supramolecular Assemblies Designed for Excitation Energy Transfer. Chem. Rev. 2015, 115, 7502–7542. 10.1021/cr5007057. [DOI] [PubMed] [Google Scholar]
- Krause G. H.; Weis E. Chlorophyll Fluorescence and Photosynthesis: The Basics. Annu. Rev. Plant Physiol. Plant Mol. Biol. 1991, 42, 313–349. 10.1146/annurev.pp.42.060191.001525. [DOI] [Google Scholar]
- Nelson N.; Ben-Shem A. The complex architecture of oxygenic photosynthesis. Nat. Rev. Mol. Cell Biol. 2004, 5, 971–982. 10.1038/nrm1525. [DOI] [PubMed] [Google Scholar]
- Scholes G. D.; Fleming G. R.; Olaya-Castro A.; van Grondelle R. Lessons from nature about solar light harvesting. Nat. Chem. 2011, 3, 763–774. 10.1038/nchem.1145. [DOI] [PubMed] [Google Scholar]
- Li Y. H.; Chan L. M.; Tyer L.; Moody R. T.; Himel C. M.; Hercules D. M. Study of Solvent Effects on Fluorescence of 1-(Dimethylamino)-5-Naphthalenesulfonic Acid and Related Compounds. J. Am. Chem. Soc. 1975, 97, 3118–3126. 10.1021/ja00844a033. [DOI] [Google Scholar]
- Zhao Y.; Zhong Z. Detection of Hg2+ in aqueous solutions with a foldamer-based fluorescent sensor modulated by surfactant micelles. Org. Lett. 2006, 8, 4715–4717. 10.1021/ol061735x. [DOI] [PubMed] [Google Scholar]
- Lipinski C. A. Drug-like properties and the causes of poor solubility and poor permeability. J. Pharmacol. Toxicol. Methods 2000, 44, 235–249. 10.1016/S1056-8719(00)00107-6. [DOI] [PubMed] [Google Scholar]
- Lipinski C. A.; Lombardo F.; Dominy B. W.; Feeney P. J. Experimental and computational approaches to estimate solubility and permeability in drug discovery and development settings. Adv. Drug Delivery Rev. 2001, 46, 3–26. 10.1016/S0169-409X(00)00129-0. [DOI] [PubMed] [Google Scholar]
- Kwon G. S.; et al. Block copolymer micelles as vehicles for hydrophobic drugs. Colloids Surf., B 1994, 2, 429. 10.1016/0927-7765(94)80007-3. [DOI] [Google Scholar]
- Kabanov A. V.; Chekhonin V. P.; Alakhov V.; Batrakova E. V.; Lebedev A. S.; Melik-Nubarov N. S.; Arzhakov S. A.; Levashov A. V.; Morozov G. V.; Severin E. S.; et al. The neuroleptic activity of haloperidol increases after its solubilization in surfactant micelles. Micelles as microcontainers for drug targeting. FEBS Lett. 1989, 258, 343–345. 10.1016/0014-5793(89)81689-8. [DOI] [PubMed] [Google Scholar]
- Yokoyama M.; Kwon G. S.; Okano T.; Sakurai Y.; Seto T.; Kataoka K. Preparation of micelle-forming polymer-drug conjugates. Bioconjugate Chem. 1992, 3, 295–301. 10.1021/bc00016a007. [DOI] [PubMed] [Google Scholar]
- Kalyanasundaram K.; Thomas J. K. Environmental Effects of Vibronic Band Intensities in Pyrene Monomer Fluorescence and Their Application in Studies of Micellar Systems. J. Am. Chem. Soc. 1977, 99, 2039–2044. 10.1021/ja00449a004. [DOI] [Google Scholar]
- Koo A. N.; Lee H. J.; Kim S. E.; Chang J. H.; Park C.; Kim C.; Park J. H.; Lee S. C. Disulfide-cross-linked PEG-poly(amino acid)s copolymer micelles for glutathione-mediated intracellular drug delivery. Chem. Commun. 2008, 6570–6572. 10.1039/b815918a. [DOI] [PubMed] [Google Scholar]
- Zhang L.; Liu W.; Lin L.; Chen D.; Stenzel M. H. Degradable Disulfide Core-Cross-Linked Micelles as a Drug Delivery System Prepared from Vinyl Functionalized Nucleosides via the RAFT Process. Biomacromolecules 2008, 9, 3321–3331. 10.1021/bm800867n. [DOI] [PubMed] [Google Scholar]
- Mellman I.; Fuchs R.; Helenius A. Acidification of the Endocytic and Exocytic Pathways. Annu. Rev. Biochem. 1986, 55, 663–700. 10.1146/annurev.bi.55.070186.003311. [DOI] [PubMed] [Google Scholar]
- Engin K.; Leeper D. B.; Cater J. R.; Thistlethwaite A. J.; Tupchong L.; Mcfarlane J. D. Extracellular Ph Distribution in Human Tumors. Int. J. Hyperthermia 1995, 11, 211–216. 10.3109/02656739509022457. [DOI] [PubMed] [Google Scholar]
- Trevani A. S.; Andonegui G.; Giordano M.; Lopez D. H.; Gamberale R.; Minucci F.; Geffner J. R. Extracellular acidification induces human neutrophil activation. J. Immunol. 1999, 162, 4849–4857. [PubMed] [Google Scholar]
- Chen Y.-Z.; Chen P.-Z.; Peng H.-Q.; Zhao Y.; Ding H.-Y.; Wu L.-Z.; Tung C.-H.; Yang Q.-Z. Water-soluble, membrane-permeable organic fluorescent nanoparticles with large tunability in emission wavelengths and Stokes shifts. Chem. Commun. 2013, 49, 5877–5879. 10.1039/c3cc41959b. [DOI] [PubMed] [Google Scholar]
- Wang L.; Tan W. Multicolor FRET Silica Nanoparticles by Single Wavelength Excitation. Nano Lett. 2006, 6, 84–88. 10.1021/nl052105b. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ueno Y.; Jose J.; Loudet A.; Pérez-Bolívar C.; Anzenbacher P.; Burgess K. Encapsulated Energy-Transfer Cassettes with Extremely Well Resolved Fluorescent Outputs. J. Am. Chem. Soc. 2011, 133, 51–55. 10.1021/ja107193j. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Harrison S. C. Viral membrane fusion. Nat. Struct. Mol. Biol. 2008, 15, 690–698. 10.1038/nsmb.1456. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kissa E.Fluorinated Surfactants and Repellents, 2nd ed.; Marcel Dekker: New York, 2001. [Google Scholar]
- Heffernan M. J.; Murthy N. Disulfide-Crosslinked Polyion Micelles for Delivery of Protein Therapeutics. Ann. Biomed. Eng. 2009, 37, 1993–2002. 10.1007/s10439-009-9734-x. [DOI] [PubMed] [Google Scholar]
- Meng F. H.; Hennink W. E.; Zhong Z. Reduction-sensitive polymers and bioconjugates for biomedical applications. Biomaterials 2009, 30, 2180–2198. 10.1016/j.biomaterials.2009.01.026. [DOI] [PubMed] [Google Scholar]
- Westheimer F. H. Coincidences, decarboxylation, and electrostatic effects. Tetrahedron 1995, 51, 3–20. 10.1016/0040-4020(94)00865-R. [DOI] [Google Scholar]
- Menger F. M.; Whitesell L. G. A Protease Mimic with Turnover Capabilities. J. Am. Chem. Soc. 1985, 107, 707–708. 10.1021/ja00289a034. [DOI] [Google Scholar]
- Menger F. M.; Gan L. H.; Johnson E.; Durst D. H. Phosphate ester hydrolysis catalyzed by metallomicelles. J. Am. Chem. Soc. 1987, 109, 2800–2803. 10.1021/ja00243a038. [DOI] [Google Scholar]
- Dwars T.; Paetzold E.; Oehme G. Reactions in micellar systems. Angew. Chem., Int. Ed. 2005, 44, 7174–7199. 10.1002/anie.200501365. [DOI] [PubMed] [Google Scholar]
- Chadha G.; Zhao Y. Properties of surface-cross-linked micelles probed by fluorescence spectroscopy and their catalysis of phosphate ester hydrolysis. J. Colloid Interface Sci. 2013, 390, 151–157. 10.1016/j.jcis.2012.09.042. [DOI] [PubMed] [Google Scholar]
- Li C.-J.; Chan T.-H.. Comprehensive Organic Reactions in Aqueous Media, 2nd ed.; Wiley-Interscience: Hoboken, NJ, 2007. [Google Scholar]
- Cornils B.; Herrmann W. A.. Aqueous-Phase Organometallic Catalysis: Concepts and Applications, 2nd ed.; Wiley-VCH: Weinheim, 2004. [Google Scholar]
- Zhang S.; Zhao Y. Artificial metalloenzymes via encapsulation of hydrophobic transition-metal catalysts in surface-crosslinked micelles (SCMs). Chem. Commun. 2012, 48, 9998–10000. 10.1039/c2cc33012a. [DOI] [PubMed] [Google Scholar]