

Abstract
Co-exposure to opiates and HIV/HIV proteins results in enhanced CNS morphological and behavioral deficits in HIV+ individuals and in animal models. Opiates with abuse liability, such as heroin and morphine, bind preferentially to and have pharmacological actions through μ-opioid-receptors (MORs). The mechanisms underlying opiate-HIV interactions are not understood. Exposure to the HIV-1 transactivator of transcription (Tat) protein causes neurodegenerative outcomes that parallel many aspects of the human disease. We have also observed that in vivo exposure to Tat results in apparent changes in morphine efficacy, and thus have hypothesized that HIV proteins might alter MOR activation. To test our hypothesis, MOR-mediated G-protein activation was determined in neuroAIDS-relevant forebrain regions of transgenic mice with inducible CNS expression of HIV-1 Tat. G-protein activation was assessed by MOR agonist-stimulated [35S]guanosine-5′-O-(3-thio)triphosphate ([35S]GTPγS) autoradiography in brain sections, and in concentration-effect curves of MOR agonist-stimulated [35S]GTPγS binding in membranes isolated from specific brain regions. Comparative studies were done using the MOR-selective agonist DAMGO ([D-Ala2, N-MePhe4, Gly-ol]-enkephalin) and a more clinically relevant agonist, morphine. Tat exposure reduced MOR-mediated G-protein activation in an agonist, time, and regionally dependent manner. Levels of the GPCR regulatory protein β-arrestin-2, which is involved in MOR desensitization, were found to be elevated in only one affected brain region, the amygdala; amygdalar β-arrestin-2 also showed a significantly increased association with MOR by co-immunoprecipitation, suggesting decreased availability of MOR. Interestingly, this correlated with changes in anxiety and fear-conditioned extinction, behaviors that have substantial amygdalar input. We propose that HIV-1 Tat alters the intrinsic capacity of MOR to signal in response to agonist binding, possibly via a mechanism involving altered expression and/or function of β-arrestin-2.
Keywords: Amygdala, Caudate–putamen, GPCR, neuroAIDS, Nucleus accumbens, DAMGO, Morphine, Autoradiography, Desensitization, Drug abuse, Addiction, Hippocampus, Prefrontal cortex
1. Introduction
Interactions between opiates and HIV/viral proteins that enhance neuro-acquired immune deficiency syndrome (neuroAIDS) pathology are well documented, both in animal models and in HIV+ individuals (Anthony et al., 2008; Bell et al., 2002; Bokhari et al., 2011; Bruce-Keller et al., 2008; Byrd et al., 2011; Dougherty et al., 2002; Fitting et al., 2014; Fitting et al., 2010; Hauser and Knapp, 2014; Pitcher et al., 2014). The prototypical opiate used in animal studies has been morphine, which is also a major bioactive metabolite of heroin (diacetylmorphine). Interactions between HIV and morphine may also have important consequences for patients using prescribed opiates, because morphine is routinely administered in clinical settings for pain management. Co-administration of morphine and HIV/viral proteins enhances central nervous system (CNS) inflammation and the synaptodendritic damage that is the presumed substrate of HIV-associated neurocognitive disorders (HAND), a spectrum of cognitive and motor deficits that are seen in many HIV patients despite antiretroviral therapy (Antinori et al., 2007; Heaton et al., 2010; Robertson et al., 2007). Morphine binds preferentially to μ-opioid-receptors (MOR), through which most of its major pharmacological actions are directed. While findings from our work and others have generally shown that morphine augments the neurodegenerative actions of the HIV-1 transactivator of transcription (Tat) protein, it is unknown whether Tat affects MOR function. We have hypothesized that some morphine–HIV interactive effects might be due to an effect of Tat protein on functional activation of the MOR, since an apparent decline in morphine efficacy has been seen in previous studies. To test this hypothesis, MOR-mediated G-protein activation was determined in neuro-AIDS-relevant forebrain regions of transgenic mice inducibly expressing the HIV-1 tat transgene in the CNS. G-protein activation was examined by both agonist-stimulated [-35S]guanosine-5′-O-(3-thio)triphosphate ([35S]GTPγS) autoradiography in brain sections, and concentration-effect curves of agonist-stimulated [35S]GTPγS binding in isolated membranes, using both the selective MOR agonist [D-Ala2, N-MePhe4, Gly-ol]-enkephalin (DAMGO) and the clinically relevant opioid agonist morphine. In addition to MOR selectivity, these ligands also differ in relative efficacy for G-protein activation, in that DAMGO is a full agonist whereas morphine is a partial agonist relative to DAMGO, as reflected in different Emax values in GTPγS binding assays (Selley et al., 1998; Selley et al., 1997).
Our results generally show a Tat exposure time-related and brain region-dependent reduction in MOR-mediated G-protein activation, with the greatest magnitude of reductions observed in the striatum and amygdala. These findings suggest that either MOR expression is reduced or coupling to G-proteins is desensitized, either of which could contribute to the apparent reduction in agonist efficacy. Interestingly, levels of β-arrestin-2 (β-Arr2), a regulatory protein important in G-protein coupled receptor (GPCR) desensitization and internalization (including the MOR) and which has been shown to play a role in morphine tolerance (Bohn et al., 2000; Dang and Christie, 2012; Shenoy and Lefkowitz, 2011) was elevated only in the amygdala of Tat(+) mice. Co-immunoprecipitation showed significantly increased binding of MOR to β-Arr2 specifically in the amygdala. These findings correlated with significant alterations in behaviors related to amygdalar function, such as fear conditioning and anxiety (open field and elevated plus maze), in the absence of any changes in baseline motor behaviors. We propose that exposure to HIV-1 Tat decreases MOR agonist efficacy to activate G-proteins, possibly via a mechanism involving altered expression and/or function of β-Arr2.
2. Materials and methods
Animal studies were approved by the Institutional Animal Care and Use Committee at Virginia Commonwealth University.
2.1. Subjects and housing
The inducible Tat transgenic mice used here are a well-accepted model for neuroAIDS since their neuropathology and behavioral deficits tend to mirror those observed in HIV patients with HAND. These include structural abnormalities in neurons/dendrites including reduced spine density and changes in synaptic proteins (Fitting et al., 2010; Hahn et al., 2015), disrupted hippocampal circuitry (Fitting et al., 2013), as well as glial abnormalities including microglial activation and micro/astrogliosis (Bruce-Keller et al., 2008; Hahn et al., 2015). Importantly, these mice and a related Tattransgenic mouse also develop changes in learning/memory and motor behaviors relevant for HIV patients (Carey et al., 2012; Fitting et al., 2013; Mediouni et al., 2015; Paris et al., 2014a; Paris et al., 2014b), although some of these effects develop only after chronic Tat expression (Hahn et al., 2015).
Adult, male, Tat-transgenic mice that express Tat1–86, a major CNS variant of the full-length Tat1–101 protein, were generated as previously described (Bruce-Keller et al., 2008; Fitting et al., 2010; Hauser et al., 2009). Tat transgene activity is inducibly controlled via the reverse tetracycline transactivator (rtTa) system driven by a glial fibrillary acidic protein (GFAP) promoter, which restricts Tat protein expression in the CNS to astroglia. Mice were genotyped to confirm that tat and rtTA transgenes were present. Chronic CNS Tat expression was induced starting at 2.5–3 months of age by feeding chow that contains doxycycline (DOX) (Harlan Laboratories, Inc., Indianapolis, IN; 6 g/kg) ad libitum. Tat protein expression in striatum and whole brain has previously been documented in this and a similar inducible transgenic model by our lab (Fitting et al., 2010) and others (Carey et al., 2012) using both immunostaining and western blot. Control mice (Tat−/rtta+) received the same chow to control for potential off-target DOX effects. Autoradiography and membrane binding assays were conducted after 4-day and/or 30-day DOX exposure, to capture changes at early times of exposure when we have seen in vivo inflammatory changes (Bruce-Keller et al., 2008) and at a more chronic stage of the neurodegenerative process. Motor behavioral studies were conducted weekly for 4 weeks. Anxiety and fear-conditioned responses were tested after 4 weeks of DOX exposure. To specifically assess MOR agonist effects on anxiety, some Tat(−) and Tat(+) mice (n = 6 and n = 10, respectively) were administered an injection of saline and assessed in an open field 15 min later, and then administered an acute injection of morphine (10 mg/kg, i.p.) and assessed in an open field after 15 min. Open field activity was assessed 15 min post-injection given that we have observed this time-point to be the peak of morphine-stimulated locomotion, consistent with reports using some rodent models (Kalinichev et al., 2002; Steidl and Yeomans, 2009).
2.2. Agonist-stimulated [35S]GTPγS autoradiography
Agonist-stimulated [35S]GTPγS autoradiography was performed on triplicate serial sections as described previously (Sim et al., 1995) with minor modifications. Coronal brain sections (20 μm) were cut on a cryostat maintained at −20 °C and thaw-mounted onto gelatin-coated slides. Sections were collected at levels that included 1) prefrontal cortex (PFC), 2) striatum, including nucleus accumbens (NAc) and caudate–putamen (CPu), and, 3) hippocampus and amygdala. Slides were stored desiccated at −80 °C until use, then brought to room temperature, and incubated in 50 mM Tris–HCl buffer (pH 7.4) with 3 mM MgCl2, 0.2 mM EGTA and 100 mM NaCl (TME buffer) for 10 min at 25 °C. Slides were transferred to TME buffer +0.5% bovine serum albumin (BSA) with 2 mM GDP and 10 mU/ml adenosine deaminase for 15 min at 25 °C. Slides were then incubated in TME Buffer +0.5% BSA containing 0.04 nM [35S]GTPγS, 2 mM GDP, and 10 mU/ml adenosine deaminase with/without agonist for 2 h at 25 °C. Basal binding was determined in the absence of agonist and MOR-stimulated [35S]GTPγS binding was measured using maximally effective concentrations (20 μM) of DAMGO or morphine. After final incubation, slides were rinsed twice in 50 mM Tris buffer (pH 7.4) at 4 °C and then briefly in deionized water (4 °C). Slides were dried and exposed to Kodak Biomax MR film with [14C] microscales for 18 h. Films were digitized at 8-bits per pixel with a Sony XC-77 video camera. Brain regions of interest were determined using The Mouse Brain Atlas (Franklin and Paxinos, 1997). Images were analyzed using NIH Image J software, and resulting values are expressed as nanocuries (nCi) of [35S] per gram of tissue [net stimulation (nCi/g) = (agonist-stimulated − basal)] as previously published (Sim et al., 1996).
2.3. Agonist-stimulated [35S]GTPγS membrane binding assay
Agonist-stimulated [35S]GTPγS binding was conducted as previously described (Selley et al., 2004). Each brain region was dissected as we have described (Lazenka et al., 2014; Lazenka et al., 2015), flash frozen and stored at −80 °C. On the day of each assay, samples were thawed, homogenized in ice-cold membrane buffer (50 mM Tris–HCl, pH 7.4, 3 mM MgCl2, 1 mM EGTA and 100 mM NaCl) and centrifuged at 48,000 ×g at 4 °C for 10 min. Pellets were resuspended in assay buffer (100 mM NaCl, 3 mM MgCl2, 0.2 mM EGTA, 50 mM Tris–HCl, pH 7.4), and the concentration of membrane protein was determined using the Bradford assay. Concentration-effect curves were generated by incubating the appropriate concentration of membrane protein (5–10 μg, depending on region of interest) in assay buffer containing, 1% w/v BSA, 30 μM GDP, 0.1 nM [35S]GTPγS, 0.6 mU/ml adenosine deaminase, and varying concentrations of DAMGO or morphine in a 500 μl total volume. Basal binding was assessed in the absence of agonist, and nonspecific binding was measured in the presence of 20 μM GTPγS. The assay was incubated for 2 h at 30 °C and terminated by rapid filtration under vacuum through Whatman GF/B glass fiber filters (Brandel, Inc., Gaithersburg, MD, USA). Bound radioactivity was determined by liquid scintillation spectrophotometry at 95% efficiency after overnight extraction in ScintiSafe Econo 1 scintillation fluid (Research Products International, Mt. Prospect, IL).
2.4. Western blotting and co-immunoprecipitation
Brain tissue samples were homogenized in Pierce™ IP lysis buffer (1:10 w/v, 25 mM Tris–HCl pH 7.4, 150 mM NaCl, 1% NP-40, 1 mM EDTA, 5% glycerol; Thermo Scientific, Pittsburgh, PA), containing protease and phosphatase inhibitor (Roche, Indianapolis, IN). Homogenized lysates were incubated at 4 °C for 40 min with end-over-end mixing. Cell debris was removed by centrifugation at ~13,000 ×g for 10 min at 4 °C and cleared supernatants were transferred to a new tube and stored at −80 °C until use. Protein concentrations were quantified using the BCA protein assay (Pierce, Rockford, IL). For immunoprecipitation, equal amounts of lysate (200 μg, amygdala) from each sample were incubated with anti-MOR antibody (10 μg per reaction; target sequence KSCMDRGMRNLLPDDGPRQE, Antibodies Inc., Davis, CA) using the Pierce Co-IP kit (Life Technologies™, Frederick, MD) following manufacturer's directions. Immunoprecipitated samples were analyzed by western blot as described previously (Hahn et al., 2015). Briefly, the samples were resuspended and boiled for 5 min in 4× Laemmli sample buffer (Bio-Rad Laboratories, Hercules, CA) and then loaded per well onto 4–20% Criterion™ TGX Stain-Free™ Gel (Bio-Rad). Precision Plus Protein™ WesternC™ Standards (Bio-Rad; MW range: 10–250 kDa) were used to determine whole protein transfer and molecular weight. Proteins were transferred to Immuno-Blot® Low Fluorescence PVDF membrane (Bio-Rad). The immunoblots were probed with either rabbit (1:1000, C16D9, Cell Signaling Technology, Danvers, MA) or mouse antibodies to β-Arr2 (1:200, H-9, target sequence, GEKP GTRVFKKSSPNCKLTVYLGKRDFVDHLD, amino acids 2–33 at the N-terminus, Santa Cruz Biotechnology, Dallas, TX) or rabbit-antibody to MOR (1:1000, Antibodies Inc.). Co-immunoprecipitation (Co-IP) was performed with rabbit anti-MOR and mouse anti-β-Arr2. Protein loading in all blots was normalized to glyceraldehyde 3-phosphate dehydrogenase (GAPDH, 1:2500, ab8245, Abcam, Cambridge, UK). Host-matched Alexa Fluor® (Life Technologies™, Frederick, MD) and HRP-conjugated secondary antibodies were applied to visualize the bands of co-IP proteins and GAPDH. Protein bands were detected on a ChemiDoc™ MP Imaging System and intensity was analyzed by Image Lab 5.1 (Bio-Rad).
2.5. Behavioral assessments
Prior to all behavioral testing, mice were acclimated to the testing room for 1 h. All behaviors were recorded and digitally-encoded by an ANY-maze behavioral tracking system (Stoelting Co., Wood Dale, IL, USA).
2.5.1. Open field
The open field test assesses anxiety-like behavior and ataxia (Hall, 1932). Mice were placed in the lower left corner of a square Plexiglas box (40 × 40 × 35 cm; Stoelting Co., Wood Dale, IL, USA) and allowed to explore for 5 min. A longer latency to enter the brightly-lit (inner 20 cm square) center, less time spent in the center, and a greater amount of time spent in the corners of the field were each considered an index of greater anxiety-like behavior. The total distance (in centimeters) traveled, as well as the frequency and time spent rearing, were utilized as indices of exploratory behavior (Bailey and Crawley, 2009).
2.5.2. Elevated plus maze
The elevated plus maze assesses anxiety-like and exploratory behavior (Lister, 1987). Mice were placed in the center of a maze (Columbus Instruments, Columbus, OH, USA) comprised of four arms (two open arms: 30 × 5 × 0 cm each; two closed arms: 30 × 5 × 15.25 cm each) elevated 40 cm off the ground and allowed to explore for 5 min. A greater amount of time spent in the closed, vs. open, arms was used as an index for greater anxiety-like behavior. The number of arm transitions and the total distance traveled were used as indices of exploratory behavior (File et al., 2005).
2.5.3. Tone-paired conditioned fear and extinction
A tone-paired conditioned fear test was implemented as previously described (Kamprath and Wotjak, 2004). All aspects of testing were carried out in a sound-attenuated chamber that contained its own white light source. On the first day (day 0), mice were placed in an olfactory-paired, transparent, Plexiglas experimental chamber (47.5 × 41 × 22 cm) with a shock grid in the chamber floor. After a 3 min acclimation period, 3 tones of 20 s duration (80 dB, 30 s interval) were presented and the tone co-terminated with a scrambled, 2 s electric foot shock (0.7 mA, alternating current). Mice were returned to their home cage 1 min later. 24 h after conditioning, extinction testing was applied in different experimental chambers that were paired with a new olfactory cue and lacked shock grids. In extinction sessions, mice were placed in the extinction context for 3 min and presented with the original tone for 200 s (80 dB, on days 1 to 6). Freezing behavior (% of 200 s) was recorded and mice were removed from the apparatus and returned to their respective home cages 1 min later. Tone-paired fear is quickly extinguished in normal mice, which discriminates between the place and olfactory contexts.
2.5.4. Rotarod
Locomotor behavior was assessed on an accelerated rotarod as previously described (Paris et al., 2013). Briefly, mice were trained to balance on an immobile rotarod (3 cm in diameter and suspended 44.5 cm high; Columbus Instruments, Columbus, OH, USA) for 30 s. Mice were then trained to navigate the task across two 30 s fixed speed trials (10 rpm) and two 180 s fixed speed trials (10 rpm). Lastly, mice were tested on two accelerated speed trials (180 s maximum latency at 0–20 rpm). The mean latency to fall from the rotarod across the two accelerated trials was utilized as an index for locomotor performance. Decreased latencies to fall on the accelerated test indicate an impaired motor phenotype.
2.6. Statistical analyses
[35S]GTPγS experiments: All samples were assayed in triplicate, and data are reported as mean values ± SEM from at least five mice per group. Net stimulation is defined as agonist-stimulated minus basal [35S]GTPγS binding. Differences between Tat(−) and Tat(+) mice in [35S]GTPγS autoradiography were determined within each region by a two-tailed Student's t-test, with p < 0.05 considered significant. Agonist concentration-effect curves were first analyzed by two-way ANOVA comparing each agonist concentration-effect curve in each region between genotypes, such that genotype and agonist concentration were the main factors, with α set at 0.05. Agonist concentration-effect curves were then analyzed by nonlinear regression to determine Emax and EC50 values. Differences in the resulting Emax and EC50 values between genotypes were determined for each agonist in each region using the two-tailed Student's t-test, with p < 0.05 considered significant. Biochemistry and behavior experiments: Tone-paired conditioned fear and extinction were analyzed with two-way ANOVA and Dunnett's post-hoc tests. All data from two-group comparisons in immunoblotting, Co-IP, and behavioral studies were conducted using Student's t-test. Effects were considered significant when p < 0.05. Data from all experiments were analyzed with Prism® version 5 (GraphPad Software, San Diego, CA) and Statistica 8 (StatSoft, Inc., Tulsa, OK, USA).
3. Results
3.1. Net DAMGO and morphine-stimulated [35S]GTPγS autoradiography was reduced in the forebrain of HIV-1 Tat-expressing mice
To determine the effect of Tat expression on MOR-mediated G-protein activation, we first examined DAMGO- and morphine-stimulated [35S]GTPγS autoradiography in forebrain sections from Tat(−) and Tat(+) mice exposed to DOX for 4 days. The anatomical distribution of MOR-stimulated [35S]GTPγS binding was similar to that reported in previous studies in rodents, including high levels of stimulation in NAc, CPu, and amygdala, moderate levels in PFC and cingulate cortex (CC) and lower levels in hippocampus (Sim et al., 1995; Sim et al., 1996), and did not noticeably differ between Tat(−) and Tat(+) mice. DAMGO stimulated a higher level of [35S]GTPγS binding than morphine in Tat(−) and Tat(+) mice, consistent with the finding that morphine is a partial agonist for activation of G-proteins in brain (Selley et al., 1997). Visual inspection revealed that DAMGO-stimulated [35S]GTPγS binding appeared to be decreased in several brain regions of Tat(+) mice. Densitometric analysis was then conducted to compare DAMGO- and morphine-stimulated [35S]GTPγS binding between DOX-treated Tat(−) and Tat(+) mice to determine whether HIV-1 Tat1–86 expression in the CNS altered MOR-mediated G-protein activity. DAMGO-stimulated [35S]GTPγS binding was significantly lower in both the NAc and CPu (p < 0.05) in Tat(+) mice compared to Tat(−) mice (Fig. 1A; Table 1). No significant differences in DAMGO-stimulated [35S]GTPγS binding were found between Tat(−) and Tat(+) mice in the other regions examined. In contrast, net morphine-stimulated [35S]GTPγS binding did not significantly differ between Tat(−) and Tat(+) mice in the striatum or any other region examined (Fig. 1B; Table 1). To further examine the effect of Tat on MOR-mediated G-protein activation in the NAc, densitometric analysis was conducted in the core versus shell. Results (Fig 1C, D; Table 1) showed that both DAMGO- and morphine-stimulated [35S]GTPγS binding were significantly reduced in the shell (p < 0.05), but not in the core, although there was a trend toward significance with DAMGO in the core (p = 0.0528). Thus, HIV-1 Tat expression reduced MOR-mediated G-protein activation in both the ventral and dorsal striatum, with the most robust difference seen in the NAc shell.
Fig. 1.

The effect of HIV-1 Tat1–86 expression on DAMGO- and morphine-stimulated [35S]GTPγS autoradiography in multiple brain regions. A. Tat(+) mice treated with DOX for 4 days exhibited significantly less DAMGO-stimulated net [35S]GTPγS binding in the CPu and NAc as compared to those regions in Tat(−) mice. B. In contrast, morphine-stimulated net [35S]GTPγS binding was not changed in any region examined. C & D. Closer examination of NAc core and shell subregions showed that the shell was preferentially affected. Data are presented as means (nCi/g) ± SEM (n = 8 mice per group, *p < 0.05, one-way ANOVA and Duncan's post hoc tests). PFC; prefrontal cortex, CC; cingulate cortex, NAc; nucleus accumbens, CPu; caudate–putamen, Amyg; amygdala, Hipp; hippocampus.
Table 1.
Densitometric analysis of basal, net DAMGO- and net morphine-stimulated [35S]GTPγS autoradiography in forebrain regions of Tat(−) and Tat(+) mice.
| Region | Net stimulation (nCi/g) |
|||||
|---|---|---|---|---|---|---|
| Tat(−) |
Tat(+) |
|||||
| Basal | DAMGO | Morphine | Basal | DAMGO | Morphine | |
| PFC | 505 ± 31 | 287 ± 32 | 114 ± 25 | 487 ± 17 | 235 ± 24 | 96 ± 19 |
| CC | 491 ± 25 | 219 ± 21 | 139 ± 32 | 469 ± 17 | 187 ± 31 | 140 ± 18 |
| NAc | 529 ± 23 | 375 ± 11 | 108 ± 29 | 540 ± 19 | 281 ± 27* | 104 ± 12 |
| CPu | 530 ± 25 | 301 ± 21 | 117 ± 22 | 559 ± 19 | 210 ± 21* | 125 ± 14 |
| Amygdala | 517 ± 19 | 307 ± 32 | 171 ± 15 | 528 ± 12 | 247 ± 18 | 145 ± 22 |
| Hippocampus | 430 ± 22 | 77 ± 10 | 110 ± 26 | 426 ± 16 | 55 ± 11 | 146 ± 35 |
Basal, net DAMGO- and netmorphine-stimulated [35S]GTPγS bindingwere measured in brain sections from Tat(−) and Tat(+) mice following 4-day administration of DOX (n = 8). Brain sections were assayed as described in Materials andmethods and autoradiograms were analyzed using densitometry. Data are mean ± SEM of basal and net agonist-stimulated (agoniststimulated − basal) [35S]GTPγS binding (nCi/g).
p < 0.05 different from the corresponding condition in Tat(−) mice.
3.2. Tat expression decreases maximal stimulation of [35S]GTPγS binding by MOR agonists in membranes from the forebrain
To determine the concentration-effect relationship of MOR agonist-stimulated [35S]GTPγS binding, we examined DAMGO- and morphine-stimulated [35S]GTPγS binding in membrane preparations from the NAc and CPu of Tat(−) and Tat(+) mice treated with DOX for 4 days. These studies used a full range of agonist concentrations and permitted determination of Emax and EC50 values. In the NAc, there was an overall decrease in stimulation of [35S]GTPγS binding by both DAMGO and morphine in Tat(+) compared to Tat(−) mice (data not shown). Two-way ANOVA of the concentration-effect curves (genotype × agonist concentration) revealed a main effect of genotype on both DAMGO- and morphine-stimulated [35S]GTPγS binding (DAMGO: [F = 4.075, p = 0.0478, df = 1]; morphine: [F = 13.51, p = 0.0005, df = 1]. Nonlinear regression analysis of the concentration-effect curves revealed a significantly greater Emax value of morphine (p < 0.05) in the NAc of Tat(−) (Emax = 80.7 ± 4.3 fmol/mg) compared to Tat(+) (Emax = 61.4 ± 6.0 fmol/mg) mice. However, there was no significant effect of genotype on DAMGO Emax values (107.2 ± 13.6 fmol/mg in Tat(−) versus 80.8 ± 13.9 fmol/mg in Tat(+) mice). Likewise, EC50 values did not differ significantly between genotypes with either morphine (306 ± 64 nM in Tat(−) versus 382 ± 99 nM in Tat(+) mice) or DAMGO (124 ± 6 nM in Tat(−) versus 78 ± 12 nM in Tat(+) mice).
In the CPu, both DAMGO and morphine concentration-effect curves appeared to be similar between genotypes (data not shown), and there was no significant effect of genotype on either the DAMGO [F = 2.123, p = 0.1489, df = 1] or morphine [F = 0.6604, p = 0.4184, df = 1] curves. Likewise, neither the Emax nor EC50 values of DAMGO or morphine differed significantly between genotypes (data not shown). These results are in general agreement with the autoradiographic studies showing a decrease in MOR-mediated G-protein activation in the NAc of Tat(+) relative to Tat(−) mice, and further suggest that this difference is mainly attributable to a decrease in agonist Emax value, at least with morphine. However, in the CPu, DAMGO concentration-effect curves showed no significant difference between genotypes, unlike the results of the autoradiographic experiments.
The finding of decreased agonist-stimulated [35S]GTPγS binding in the striatum of Tat(+) compared to Tat(−) mice after 4-day DOX treatment suggests a suppressive effect of transgene expression on MOR-mediated G-protein activation. However, there were some discrepancies between the results of the autoradiographic versus membrane [35S]GTPγS binding assays. Therefore, an additional cohort of mice were treated for 30 days with DOX to induce a longer duration of transgene expression, and concentration-effect curves of DAMGO-and morphine-stimulated [35S]GTPγS binding were examined in membranes prepared from the PFC, NAc, CPu, amygdala, and hippocampus. Results showed decreased MOR-mediated G-protein activation by DAMGO and/or morphine in all regions examined (Fig. 2). In the PFC (Fig. 2A), there was no significant effect of genotype on DAMGO-stimulated [35S]GTPγS binding, as revealed by two-way ANOVA. However, there was a significant decrease in morphine-stimulated [35S]GTPγS binding in Tat(+) compared to Tat(−) mice. Accordingly, nonlinear regression analysis showed this difference to be attributable to a 52% lower Emax value of morphine in Tat(−) relative to Tat(+) mice (Table 2), with no significant difference in morphine EC50 values. Neither the Emax nor EC50 values of DAMGO differed between genotypes in this region, although there was a trend toward a lower Emax value in Tat(+) mice (p = 0.0704).
Fig. 2.

Concentration-effect curves of MOR agonist-stimulated [35S]GTPγS binding to membranes from multiple brain regions of Tat(+) and Tat(−) mice after 30-day DOX treatment. Stimulation was generally reduced in Tat(+) compared to Tat(−) mice. Data are mean ± SEM (n = 4–8) of net-stimulated [35S]GTPγS binding by DAMGO or morphine in the prefrontal cortex (A), nucleus accumbens (B), caudate-putamen (C) and amygdala (D) of Tat(−) versus Tat(+) mice. Emax and EC50 values derived from curve-fits of these data are given in Table 2. Hippocampus was also examined in these studies (data not shown here; Emax and EC50 values are included in Table 1). Two-way ANOVA results (all concentration-effect curves had a significant main effect of ligand concentration): (A) main effect of Tat: DAMGO [F = 0.6823, p = 0.08, df = 1], morphine [F = 20.42, p < 0.0001, df = 1]; (B) main effect of Tat: DAMGO [F = 16.55, p < 0.001, df = 1], morphine [F = 12.94, p = 0.005, df = 1]; (C) main effect of Tat: DAMGO [F = 20.3, p < 0.0001, df = 1], morphine [F = 22.8, p < 0.0001, df = 1], interaction of Tat x morphine concentration [F = 5.056, p = 0.0002, df = 6]; (D) main effect of Tat: DAMGO [F = 19.91, p < 0.0001, df = 1], morphine [F = 29.55, p < 0.0001, df = 1], interaction of Tat x morphine concentration [F = 4.132, p = 0.0014, df = 6]; Hippocampus (not shown): main effect of Tat: DAMGO [F = 0.2285, p = 0.6343, df = 1], morphine [F = 0.3326, p = 0.0005, df = 1].
Table 2.
EC50, Emax, and basal binding values from DAMGO- and morphine-stimulated [35S]GTPγS binding in brain regions of Tat(−) and Tat(+) mice.
| Region | Genotype | DAMGO |
Morphine |
Basal (fmol/mg) | ||
|---|---|---|---|---|---|---|
| Emax (fmol/mg) | EC50 (nM) | Emax (fmol/mg) | EC50 (nM) | |||
| Prefrontal cortex | Tat(−) | 52.2 ± 5.3 | 181.4 ± 26.4 | 49.6 ± 5.8 | 501.7 ± 121.0 | 68.6 ± 5.0 |
| Tat(+) | 36.9 ± 4.4 | 146.4 ± 53.5 | 23.6 ± 4.4* | 578.2 ± 376.8 | 62.8 ± 6.7 | |
| Nucleus accumbens | Tat(−) | 88.2 ± 12.9 | 132.8 ± 12.2 | 65.4 ± 9.1 | 389.0 ± 80.0 | 75.8 ± 17.7 |
| Tat(+) | 56.4 ± 8.4 | 106.8 ± 13.3 | 50.0 ± 5.0* | 347.1 ± 87.0 | 55.2 ± 5.0 | |
| Caudate–putamen | Tat(−) | 122.2 ± 8.4 | 119.7 ± 13.9 | 85.9 ± 3.8 | 311.8 ± 34.3 | 95.6 ± 11.1 |
| Tat(+) | 89.5 ± 1.7* | 91.6 ± 11.6 | 58.7 ± 2.5** | 252.4 ± 103.5 | 81.7 ± 8.9 | |
| Amygdala | Tat(−) | 122.4 ± 13.5 | 125.7 ± 22.3 | 91.4 ± 14.3 | 333.2 ± 68.1 | 213.3 ± 29.9 |
| Tat(+) | 79.4 ± 13.4* | 98.2 ± 11.2 | 50.0 ± 6.5* | 365.0 ± 100.4 | 117.5 ± 18.3* | |
| Hippocampus | Tat(−) | 42.8 ± 3.8 | 91.8 ± 21.6 | 29.9 ± 3.9 | 67.5 ± 39.5 | 94.9 ± 13.9 |
| Tat(+) | 43.5 ± 5.3 | 105.2 ± 49.9 | 22.9 ± 4.4 | 67.2 ± 21.2 | 101.1 ± 14.7 | |
Data are mean ± SEM of basal and net agonist-stimulated (agonist-stimulated − basal) [35S]GTPγS binding (nCi/g).
p < 0.05 different from the corresponding condition in Tat(−) mice.
p < 0.001 different from the corresponding condition in Tat(−) mice.
The Tat(+) genotype was associated with significant reductions in MOR-mediated G-protein activation in both the ventral (NAc) and dorsal (CPu) striatum. In the NAc (Fig. 2B), there was a significant effect of genotype on the concentration-effect curves of both DAMGO- and morphine-stimulated [35S]GTPγS binding. The morphine Emax value was 24% lower in Tat(−) relative to Tat(+) mice, with no significant effect of genotype on morphine EC50 values (Table 2). There were no significant differences in DAMGO Emax or EC50 values between genotypes, but the Emax value showed a trend toward a decrease in Tat(+) mice (p = 0.0585) (Table 2). In the CPu (Fig. 2C), there was a significant effect of genotype on DAMGO- and morphine-stimulated [35S]GTPγS binding. There was also a significant interaction between genotype and concentration for morphine-stimulated [35S]GTPγS binding, and post hoc analysis with Bonferroni's test showed significantly lower stimulation in Tat(+) relative to Tat(−) mice at 1 (p < 0.01), 3 and 10 (p < 0.001) μM morphine. The Emax value of DAMGO was 27% lower and that of morphine was 32% lower in Tat(+) compared to Tat(−) mice, and there were no significant effects of genotype on DAMGO or morphine EC50 values (Table 2).
Other limbic forebrain regions also showed reduced MOR-mediated G-protein activation in Tat(+) mice, with amygdala exhibiting much greater sensitivity to this effect than hippocampus. In the amygdala (Fig. 2D), there was a significant effect of genotype on DAMGO-and morphine-stimulated [35S]GTPγS binding. There was also a significant interaction between genotype and concentration for morphine-stimulated [35S]GTPγS binding, and post-hoc analysis with Bonferroni's test showed significantly lower stimulation in Tat(+) relative to Tat(−) mice at 1 (p < 0.01), 3 (p < 0.05) and 10 (p < 0.001) μM morphine. The DAMGO Emax value was 35% lower and the morphine Emax value was 45% lower in Tat(+) relative to Tat(−) mice, and there were no significant effects of genotype on DAMGO or morphine EC50 values (Table 2). In hippocampus (data not shown), there was no significant effect of genotype on [35S]GTPγS binding stimulated by DAMGO, but there was a significant effect of genotype on morphine-stimulated [35S]GTPγS binding. There were no significant effects of genotype on the Emax or EC50 values of DAMGO or morphine in this region (Table 2).
Basal [35S]GTPγS binding ranged from approximately 55 fmol/mg in nucleus accumbens of Tat(+) to approximately 213 fmol/mg in amygdala of Tat(−) mice (Table 2). There were no significant effects of genotype on basal [35S]GTPγS binding in any region examined, with the exception of amygdala, which exhibited 45% lower basal [35S]GTPγS binding in Tat(+) relative to Tat(−) mice. This result implies that lower net MOR-stimulated [35S]GTPγS binding in amygdala could have occurred within the context of an overall decrease in inhibitory G-protein activity. Therefore, to normalize for the difference in basal [35S]GTPγS binding in this region, we also normalized the DAMGO concentration-effect data to the basal activity in each genotype to obtain the % stimulation (net-stimulated − basal / basal [35S]GTPγS binding) by DAMGO. This calculation of the data (not shown) revealed a significant effect of genotype on the DAMGO concentration-effect curve [F = 15.5, p = 0.0002, df = 1]. Altogether, these results demonstrate a suppressive effect of Tat transgene expression on MOR-mediated G-protein activation in all brain regions examined, with the striatum and amygdala exhibiting the greatest sensitivity, and the PFC and hippocampus exhibiting lower sensitivity to this effect of the transgene. In fact, a significant reduction in G-protein activation was only detected with morphine, the agonist with lower intrinsic efficacy, in the hippocampus.
3.3. Tat expression increases β-arrestin-2 expression and association with MOR in the amygdala
As an initial attempt to determine the mechanism(s) underlying Tat-induced reductions in MOR-mediated G-protein activation, we first determined by immunoblot whether MOR protein expression levels were altered by 30-day DOX exposure in Tat(+) and Tat(−) mice. MOR levels were unchanged by Tat induction in any region examined, and were similar to levels observed in ICR mouse brain homogenate (Fig. 3E–F).
Fig. 3.

β-Arr2 level is specifically increased in the amygdala of Tat mice and shows increased binding to μ-opioid receptors. (A, B) Lysates from multiple brain regions of DOX-treated Tat mice were evaluated by immunoblot using both rabbit (A) and mouse (B) anti-β-Arr2 antibodies. Representative blots show two different molecular sizes of β-Arr2 bands [~50 kDa (A, B) and ~125 kDa (B)] in different brain regions and enhanced expression in the amygdala of Tat(+) mice. Anti-GAPDH was used to normalize protein loading. Data are presented as the mean ± SEM. (N = 6–7, *p ≤ 0.05, one-way ANOVA, post-hoc Duncan's test). (C, D) Association of MORs with β-Arr2 was assessed via Co-IP and immunoblot. Representative blot shows that β-Arr2 was detected following Co-IP with rabbit anti-μ-opioid receptor on amygdala lysates and subsequent probing with mouse anti β-Arr2 antibody. Independent anti-GAPDH immunoblotting was performed on the spared lysates after Co-IP to examine equal protein loading (C). Data are presented as the mean ± SEM. (N = 3, *p ≤ 0.05, one-way ANOVA, post-hoc Duncan's test) (D). (E, F) The expression of MOR was detected by western blot. Lysates from 30 day DOX-treated Tat(−) and Tat(+) mice were evaluated by immunoblot using rabbit-anti-MOR antibody used for immunoprecipitation. Lanes represent lysates (40 μg/well) from different brain regions. Whole brain lysate from ICR outbred mice was included as a positive control. All Tat(−) and Tat(+) lysates demonstrate a strong band detected ~48 kDa (E). Expression of MOR did not significantly differ between brain regions nor was it significantly influenced by DOX treatment or Tat-genotype (F). Data are presented as the mean ± SEM (N = 3, *p ≤ 0.05, one-way ANOVA, post-hoc Duncan's test); prefrontal cortex (PFC), caudate–putamen (CPu), nucleus accumbens (NAc), amygdala (Amyg), and hippocampus (Hipp).
Since no differences in MOR expression were detected, we next examined whether changes in the MOR agonist-stimulated [35S]GTPγS binding caused by Tat exposure might be related to changes in β-Arr2 levels and/or the degree of association between MOR and β-Arr2. β-Arrestin-1 and -2 are involved in agonist-mediated desensitization and recycling of GPCRs and the control of downstream signaling pathways (Gainetdinov et al., 2004; Nguyen et al., 2012). MOR is thought to associate predominantly with β-Arr2 compared to β-Arr1 when occupied by morphine, and genetic knockout of β-Arr2 is associated with reduced morphine-induced MOR desensitization and antinociceptive tolerance (Bohn et al., 2000; Groer et al., 2011). In western blots of brain tissue lysates, a rabbit β-Arr2 antibody (Cell Signaling Technology) detected significantly increased protein expression (~50 kDa band) in the amygdala of Tat(+) compared to Tat(−) mice whereas no differences were noted in CPu or NAc (Fig. 3A). A second mouse β-Arr2 antibody (Santa Cruz) also detected several higher molecular weight bands in amygdalar lysates from Tat(+) mice (~125 kDa), as well as showing significantly enhanced expression of β-Arr2 (N = 6–7, Fig. 3B). We speculated that these higher molecular weight bands might indicate increased β-Arr2 protein binding to membrane GPCRs. To quantify MOR association with β-Arr2, we performed Co-IP with amygdalar lysates using a rabbit antibody to MOR (Antibodies Inc.) and immunoblots with mouse anti-β-Arr2 (Santa Cruz). Amygdalar lysates from DOX-treated Tat(+) mice showed an increased association of MOR and β-Arr2 compared to lysates from DOX-treated Tat(−) mice (Fig. 3C–D).
3.4. Tat expression in the CNS alters extinction of a conditioned freezing response
Tat expression increased β-Arr2 levels and its association with MOR in the amygdala, and we therefore evaluated fear-conditioned freezing and subsequent extinction, behaviors associated with amygdalar function (Dunsmoor and Paz, 2015; Keifer et al., 2015; Skelly et al., 2015). Tat(+) and Tat(−) mice evaluated after 30-day DOX treatment exhibited a similar magnitude of freezing behavior to a tone-cued foot-shock on the day of conditioning (day 0) (Fig. 4). On 6 subsequent days of testing, mice were presented with a similar tone, but in a different context and without foot-shock. Tat(−) mice exhibited significantly decreased freezing behavior across the 200 s tone session on days 1 through 6; there was no difference in freezing time across the 6-day extinction period. In contrast, Tat(+) mice showed delayed extinction of freezing behavior, and were not equivalent to Tat(−) mice until day 3. From day 3 to day 6, the two genotypes were indistinguishable. The delayed extinction in a new context was interpreted as increased fear/anxiety behavior in Tat(+) mice (Schneider et al., 2015; Sotres-Bayon et al., 2004).
Fig. 4.
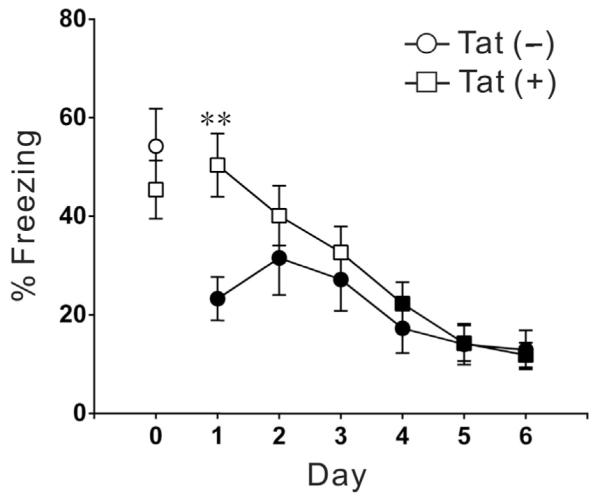
HIV-1 Tat induction results in impaired extinction of a conditioned freezing response. Both groups of mice were exposed to paired tone and foot shock on the training day (day 0) after 30 day DOX treatment, and there was no difference in freezing time between genotypes on the training day. Mice were then re-evaluated for tone-induced freezing behavior in a new environment. Tone-induced freezing behavior was significantly reduced in Tat(−) mice on all test days (1–6; black squares and circles indicate that freezing time is significantly decreased compared to the same genotype mice on training day 0; N = 9, p < 0.001, one-way ANOVA, post-hoc Dunnett's test). In contrast, freezing time remained at training day levels in Tat(+) mice on test days 1–3 (open squares or circles indicate equivalence to training day 0). ** indicates significant different in percent of freezing time between Tat(−) and Tat(+) groups on that day (N = 9, **p < 0.01, one-way ANOVA, post-hoc Dunnett's multiple comparisons test).
3.5. Tat expression in the CNS increases anxiety-like behavior and suppresses morphine-induced anxiolysis
To evaluate the affective influence of HIV-1 Tat expression in the CNS, Tat(−) and Tat(+) mice were placed on DOX chow and assessed in an open field once per week for 4 weeks. Differences in locomotor behavior were not observed over repeated testing sessions (Fig. 5A); however, anxiety-like behavior as assessed by time spent in the brightly-lit center of the open field was significantly greater among Tat(+) mice by 4 weeks of DOX exposure, as compared to Tat(−) control mice [t(14) = 2.19, p < 0.05] (Fig. 5B). As further validation, separate groups of Tat(−) and Tat(+) mice were more extensively characterized for anxiety-like and locomotor behavior after 4 weeks of DOX administration. Compared to Tat(−) controls, Tat(+) mice demonstrated an increased latency to enter the brightly-lit center of an open field [t(24) = 2.35, p < 0.05] (Fig. 5C), spent significantly less time in the center [t(24) = 2.63, p < 0.05] (Fig. 5D) and spent more time in the peripheral corners [t(24) = 2.83, p < 0.05] (Table 3) of the open field, and spent significantly less time on the open arms of an elevated plus maze [t(15) = 2.31, p < 0.05] (Fig. 5E). Tat(+) mice exhibited less exploratory behavior than Tat(−) controls, traveling less distance [t(24) = 3.85, p < 0.05] and demonstrating less frequency [t(24) = 2.61, p < 0.05] and time [t(24) = 2.08, p < 0.05] spent rearing (Table 3). Importantly, this is not thought to be due to differences in overall motor activity, which was similar in the two groups. Tat(+) mice did not differ from Tat(−) mice on the number of arm transitions or distance traveled within an elevated plus maze (Table 3). Nor were differences observed in the latency to fall from an accelerated rotarod (Fig. 3F) or the maximum speed achieved on the rotarod (Fig. 5F, inset) between Tat(−) and Tat(+) mice. Thus, induction of central HIV-1 Tat produced anxiety-like behavior and reduced open field exploration, independent of changes in locomotor activity.
Fig. 5.

(A) Distance traveled and (B) time spent in the brightly-lit center of an open field among Tat(−) or Tat(+) mice (n = 8/group) following 0–4 weeks of exposure to DOX to induce HIV-1 Tat in the CNS. In a separate experiment, different groups of mice were exposed to 4 weeks of DOX and assessed for: (C) latency to enter and (D) time spent in the center of an open field (n = 12–14/group), (E) time spent on the open arms of an elevated plus maze (n = 8–9/group) and (F) latency to fall from an accelerated rotarod (inset depicts maximum RPM achieved; n = 8–12/group). In a separate experiment to assess whether Tat affects anxiety related to MOR activation, (G) the distance traveled and (H) the time spent in the brightly-lit center of an open field significantly differed between Tat(−) or Tat(+) mice (n = 6–10/group after 30 days of DOX treatment) following an injection of saline and an acute injection of morphine (10 mg/kg, i.p., −15 min). Data are presented as a percent of saline-induced behavior. * indicates significant difference from Tat(−) control mice, p < 0.05, Student's t-test. Dashed line indicates control level in G and H.
Table 3.
Performance ofmale Tat(−) or Tat(+)mice (n=8–14/group) in an open field or elevated plus maze.
| Behavioral measure | Tat(−) | Tat(+) |
|---|---|---|
| Open field | ||
| Frequency of central entries | 7.2 ± 1.3 | 2.3 ± 0.8* |
| Time spent in corners (s) | 178 ± 10 | 222 ± 11* |
| Frequency of rearing | 32 ± 6 | 16 ± 3* |
| Time spent rearing (s) | 21 ± 4 | 11 ± 2* |
| Distance explored (m) | 11.3 ± 1.2 | 7.4 ± 0.7* |
| Elevated plus maze | ||
| Total arm entries | 19 ± 4 | 20 ± 1 |
| Distance explored (m) | 6.3 ± 1.1 | 7.4 ± 0.4 |
Data presented as mean ± SEM.
p < 0.05 difference fromTat(−) control mice, Student's t-test (s, seconds; m, meters).
To begin to address the functional effects of interactions between Tat and MOR activation, we assessed locomotor and anxiety-like behavior in both Tat(−) and Tat(+) mice in an open field 15 min after an injection of saline, and 15 min after an acute injection of morphine (10 mg/kg, i.p.). Morphine-stimulated locomotion was significantly reduced among Tat(+), compared to Tat(−), mice (Fig. 5G) [t(14) = 3.26, p < 0.05]. As well, anxiolysis produced by acute morphine was attenuated among Tat(+) mice, compared to controls, as indicated by a significant lack of morphine-stimulated increase in the amount of time spent in the brightly-lit center of an open field (Fig. 5B) [t(14) = 2.12, p < 0.05]. Thus, Tat by itself enhanced anxiety, but Tat also reduced the motor and affective outcomes of acute MOR activation.
4. Discussion
The pathology and cognitive deficits associated with neuroAIDS are enhanced by exposure to opiates both in experimental animal models and in patients (Anthony et al., 2008; Bell et al., 2002; Bokhari et al., 2011; Bruce-Keller et al., 2008; Byrd et al., 2011; Dougherty et al., 2002; Fitting et al., 2014; Fitting et al., 2010; Hauser and Knapp, 2014; Pitcher et al., 2014). Experimental evidence from relatively acute, in vitro studies suggests that opiate contributions to these deficits represent a combination of direct and indirect effects on both neurons and glia (Masvekar et al., 2014; Zou et al., 2011). The balance of cellular inputs in vivo is likely more nuanced, and is also likely to shift over the course of more chronic exposure/disease. A number of outcomes of combined opiate-HIV exposure have been described. For example, by several measures, CNS inflammation is enhanced by HIV-morphine interactions in vivo (Bokhari et al., 2009; Bruce-Keller et al., 2008; Dutta and Roy, 2015; Hollenbach et al., 2014), and undoubtedly plays a significant role in enhancing neuropathology. The present work examined a more proximal step in the cascade of events culminating in opiate-HIV pathology. We hypothesized that HIV, and specifically the expression of the HIV-1 Tat protein, might affect the functional activation of the MOR. Among the several viral proteins produced by HIV-infected cells, Tat may be particularly relevant to patients receiving antiretroviral therapy because it is produced by infected cells in HIV patients despite viral suppression (Johnson et al., 2013).
4.1. HIV-1 Tat exposure decreases MOR-mediated G-protein signaling
Quantitative [35S]GTPγS autoradiography and membrane binding studies showed that maximal MOR-mediated G-protein activation was reduced in multiple brain regions of transgenic mice inducibly expressing HIV-1 Tat1–86. Given the low MOR expression levels in glia, these measurements largely reflect activity in neurons. While moderate differences in net morphine- and DAMGO-stimulated [35S]GTPγS binding between genotypes were seen in CPu and NAc after only 4 days of Tat exposure (Fig. 1; Table 1), these initial effects were restricted to the striatum and were most prominent in the NAc shell. However, more chronic, 30-day Tat exposure showed effects on signaling in additional forebrain regions. These decreases in [35S]GTPγS binding resulted mainly from a decrease in Emax value for both agonists, rather than a loss of potency (increased EC50 value). These results suggest that Tat expression does not interfere with ligand binding affinity to the MOR, but rather decreases the maximal signaling capacity of the MOR population resulting in decreased agonist efficacy. These decreases in maximal agonist-stimulated MOR activation were not due to decreased levels of MOR protein, as revealed by immunoblot, suggesting instead Tat-induced uncoupling of MOR from G-protein activation.
Regional differences could involve the density of MOR expression on the cell types affected and the relative production of Tat in each area, as well as differential Tat binding to various membranes in a region. In our mouse model, Tat titer depends on local numbers of astroglia, while in patients it depends on the local infiltration of infected cells. In general, striatal regions, especially the NAc shell, and amygdala were more sensitive to the suppressive effects of Tat expression on MOR-mediated G-protein activation compared to either PFC or hippocampus. Regional differences in the efficiency of MOR-mediated G-protein activation, measured as the ratio of Bmax of net DAMGO-stimulated [35S]GTPγS binding to MOR Bmax value, calculated from [3H]naloxone saturation analysis, have previously been determined in rats (Maher et al., 2000). Interestingly, the percent decrease in DAMGO-stimulated [35S]GTPγS binding in Tat(+) relative to Tat(−) mice in the present study correlated inversely with MOR efficiency from the Maher et al. study (Maher et al., 2000) as a function of region (r2 = 0.94, p < 0.05). Likewise, regional differences in desensitization of MOR-mediated G-protein activation by prolonged morphine administration have been reported in rats and mice (Sim-Selley et al., 2007; Sim et al., 1996). Although mechanisms underlying regional differences in MOR signaling efficiency and desensitization are unknown, it is not surprising that regional differences in MOR sensitivity to Tat-induced attenuation in signaling were also observed in the present study. Such effects could be due to regional differences in MOR regulatory mechanisms in addition to regional variations in Tat expression.
Morphine is a prototypical narcotic drug, and is also the main bioactive metabolite of heroin. Morphine appears to be a full MOR agonist in most antinociceptive tests, but is actually a high efficacy partial agonist for MOR-mediated G-protein activation (Selley et al., 1998; Selley et al., 1997), as seen in the present study where maximal stimulation of [35S]GTPγS binding by morphine was lower in magnitude than that of DAMGO. Most physiological actions of morphine are attributed to binding at MORs, although morphine can also bind κ- and δ-opioid receptors with much lower affinity (Suarez-Roca and Maixner, 1992; Suarez-Roca and Maixner, 1993; Thompson et al., 2004). The classic pharmacological actions of morphine are absent in MOR-null mice (Kieffer, 1999; Matthes et al., 1996). DAMGO is a full MOR agonist that is highly selective for the MOR. Although there were some differences in results obtained using DAMGO versus morphine, in all instances when Tat exposure affected agonist-stimulated [35S]GTPγS binding, the stimulation was decreased. Some discrepancies between DAMGO and morphine in the [35S]GTPγS binding studies could reflect their different selectivities. Morphine actions at the single, maximally effective concentration used in [35S]GTPγS autoradiography might reflect additional activity at δ-and/or κ-opioid receptors, which could be unaffected or oppositely modulated by Tat expression, but would not be detected with the MOR-selective agonist, DAMGO. Nonetheless, the concentration-effect studies in 30-day DOX-exposed mice showed that morphine-stimulated G-protein activity was generally reduced to a greater extent than DAMGO-stimulated activity, as would be expected from the fact that morphine has lower intrinsic efficacy at the MOR than DAMGO (Elliott et al., 1997; Selley et al., 1998; Selley et al., 1997). Although the general findings were similar between [35S]GTPγS autoradiography and membrane binding studies, some specific differences were noted. For example, decreased DAMGO-stimulated G-protein activation was detected in the CPu of 4-day DOX-exposed Tat(+) mice using [35S]GTPγS autoradiography, but was not detected in the agonist concentration-effect curves in isolated membranes. We have previously reported that greater Δ9-THC-induced desensitization of cannabinoid receptor-mediated G-protein activation was measured using autoradiography as compared to [35S]GTPγS binding assays in isolated membranes (Breivogel et al., 1999). This might be because cellular structure is preserved in brain sections (autoradiography) as compared to membrane homogenates.
How might Tat reduce agonist-stimulated [35S]GTPγS binding? The present findings show no effect of Tat exposure on MOR expression levels in several brain regions. Tat could decrease expression of G-protein subunits that are activated by the MOR, which would be consistent with the lower basal [35S]GTPγS binding observed in the amygdala of Tat(+) mice. It is also possible that Tat affects the cellular trafficking of MORs, such that a greater proportion of the MOR population is internalized to intracellular sites with reduced access to G-proteins. Another, related possibility is that Tat increases MOR desensitization in response to endogenous agonist or other stimuli, a finding consistent with the enhanced MOR association with β-Arr2 observed in the amygdala. Reductions in agonist-stimulated [35S]GTPγS binding might also originate from increased levels of the inflammatory chemokine CCL5/RANTES and activation of CCR5. CCL5/RANTES is markedly elevated in the striata of our transgenic mice following Tat induction (Fitting et al., 2010), and CCR5 and MOR can undergo heterologous, bidirectional cross-desensitization in the CNS (Chen et al., 2004; Song et al., 2011). Regardless of the mechanism, our results overall demonstrate the important finding that HIV-1 Tat reduces MOR-dependent G-protein activation in a regionally dependent manner, which has implications for the overall function of the endogenous opioid system.
4.2. MOR signaling dysfunction and β-arrestin-2
Agonist binding at GPCRs, including MOR, induces phosphorylation of the receptor by G-protein receptor kinase-2 (GRK2), which drives subsequent binding with β-Arr2 (Zhang et al., 1998). For many MOR agonists, β-Arr2 binding triggers clathrin-mediated receptor endocytosis and eventual sorting/recycling of MOR to the cell surface. However, in the case of morphine and related alkaloid agonists, β-Arr2 binding results in very limited endocytosis, unless GRK2 is driven to supraphysiological levels (Zhang et al., 1998). Although the tolerance that develops to the classic, antinociceptive effect of morphine is clearly diminished in β-Arr2 knockout mice (Bohn et al., 2000; Martini and Whistler, 2007), the specifics of the MOR desensitization–resensitization cycle differ among neuron subpopulations (Gainetdinov et al., 2004; Noble and Cox, 1996) and perhaps even to regions of a neuron (Haberstock-Debic et al., 2003). Recent evidence also establishes that MOR dephosphorylation and resensitization do not require endocytosis (Dang and Christie, 2012). Our studies have shown that desensitization of MOR-mediated G-protein activity induced by morphine or heroin varies by brain region (Sim-Selley et al., 2000; Sim et al., 1996), supporting the idea that region-specific reductions in MOR-stimulated [35S]GTPγS binding by Tat exposure might involve regulatory proteins such as β-Arr2.
To understand the observed changes in agonist-stimulated [35S]GTPγS binding in the striatum and amygdala, we asked whether Tat expression would alter the relationship between MOR and β-Arr2. Immunoblots with two validated antibodies showed an increase in overall expression levels of β-Arr2 protein specifically in the amygdala of Tat(+) mice (Fig. 3A, B), and Co-IP studies showed an enhanced association between MOR-β-Arr2 (Fig. 3C) in the Tat(+) versus Tat(−) mice in this region, suggesting an overall decreased availability of functional MOR. Regulatory mechanisms involved in MOR desensitization, including association with β-Arr2 and subsequent endocytosis, are usually examined in the context of chronic agonist/morphine administration, and might not be directly comparable to our finding of changes in basal MOR/β-Arr2 association. However, it is possible that increased β-Arr2 expression could result in increased β-Arr2 recruitment to the MOR in response to endogenous opioid agonists, resulting in augmented MOR desensitization. In principle, this finding would agree with evidence of a decreased response to morphine in assays of antinociception and physical dependence after Tat induction (Fitting et al., 2012; Fitting et al, 2016). Increased MOR association with β-Arr2 might also occur in regions where overall β-Arr2 expression did not change, a possibility to be investigated in future studies.
4.3. Altered fear and anxiety-like behaviors parallel changes in amygdalar β-arrestin-2
The amygdala is a critical region for processing information that, along with coordinated input from other regions such as the PFC and hippocampus, mediates behaviors such as fear, fear extinction, and anxiety (Castro-Gomes et al., 2015; Dunsmoor and Paz, 2015; Keifer et al., 2015; Skelly et al., 2015). Although this region has received little attention in HIV patients, the amygdala shows structural abnormalities that correspond with affective deficits (Clark et al., 2012; Clark et al., 2015), and abnormalities are also seen in experimental models (Carey et al., 2013; Nesil et al., 2015). Studies with transgenic mice deficient in individual opioid receptors have shown that global loss of δ-opioid receptors tends to enhance anxiety, while similar loss of MORs tends to reduce anxiety, albeit more subtly (Filliol et al., 2000). Their opposing roles suggest that the balance between MOR and δ-opioid receptor signaling may influence anxiety levels. Global knockout studies are not informative about individual regions, and some pharmacological studies show contrary findings. For example, endogenous MOR ligands (endomorphins) were anxiolytic and reduced conditioned defeat in rodents (Asakawa et al., 1998; Whitten et al., 2001), and morphine lowered anxiety triggered by acute restraint stress (Joshi et al., 2015). When morphine was injected into the ventral tegmental area, anxiety was enhanced (Calenco-Choukroun et al., 1991), but morphine or DAMGO injections into the amygdala, have in general alleviated fear/anxiety (File et al., 2005; Good and Westbrook, 1995). The changes we measured in MOR-mediated G-protein activity, β-Arr2 levels, and Co-IP with MOR correlated with altered amygdalar function as assessed by increased levels of fear and anxiety-like behavior after 30 days of Tat induction (Fig. 4, Fig. 5B–E). And while Tat by itself enhanced anxiety, Tat reduced the motor and affective outcomes of acute MOR stimulation (Fig. 5G, H) in the absence of measurable changes in MOR levels (Fig. 3E, F), attesting to a complex interaction between Tat exposure and the effectiveness of MOR receptor engagement. Whether Tat's effects on anxiety-like behavior involve actions at MOR in the opiatenaïve state is not known. Importantly, fear conditioning, open field, and elevated plus-maze deficits all occurred in the absence of any observed motor deficits that may have confounded their interpretation (Fig. 5). Fear and anxiety-like behaviors were also unrelated to genetic sex since all animals were male, although sex was shown to influence behavior after 3 months of Tat induction (Hahn et al., 2015).
Contextual extinction is known to be dependent on the capacity to learn a new inhibitory association in place of a previously paired association (Bouton, 2002; Davis and Myers, 2002). Subregions of the medial PFC (infralimbic and prelimbic cortices) project to the amygdala (with the basal nucleus projecting back to the prelimbic cortex) and likely contribute to inhibitory learning in this region (Rosenkranz and Grace, 2001). However, this circuit may be disrupted in HIV-1 Tat-expressing animals given demonstrated microglial activation in medial prefrontal and amygdalar subregions that occurred concurrent with an increased startle response and an impaired capacity to inhibit such behavior (Paris et al., 2015). Moreover, we have previously observed spatial learning deficits in Tat(+), compared to Tat(−) mice, concurrent with a loss of hippocampal LTP (Fitting et al., 2013), a region (along with the subiculum) that may influence contextual fear extinction via strong innervation within mPFC infralimbic and prelimbic cortices (Conde et al., 1995; Sotres-Bayon et al., 2004). Lastly, it must be considered that circuits involved in fear conditioning outcomes in mice and other rodents may not entirely mimic circuitry involved in humans.
Our studies in membrane homogenates (binding, western blot, Co-IP) included the entire amygdalar complex, although outcomes might differ among the various nuclei; understanding the subregions involved may be critical for unraveling the functional importance of our findings. For example, intercalated inhibitory neurons that modulate the relay of information between amygdalar regions and play a role in fear extinction are targeted by DAMGO activation, which reduces GABAergic transmission between medial and central nuclei (Blaesse et al., 2015). MOR activation by DAMGO microinjected into the central amygdala enhanced anxiety in the elevated plus maze, but decreased anxiety in response to predator odors, suggesting the importance of context in the behavioral response (Wilson and Junor, 2008).
5. Conclusions
The findings presented here are the first to show that exposure to an HIV protein can change the intrinsic response of the MOR to selective or therapeutically-relevant agonists. Whether or not the mechanism involves the changes in MOR-β-Arr2 association shown here, a reduced MOR-dependent G-protein activation suggests that HIV patients might require greater amounts of opiates to attain a response commensurate with that seen in uninfected individuals. The reduced effectiveness of MOR ligands has great clinical significance given the extensive literature suggesting that opiate exposure worsens neuropathology in HIV patients. In terms of HIV-opiate drug abuse interactions, it is intriguing that effects were specifically observed in the CPu, NAc, and amygdala. These areas have robust expression of MORs whose responses to endogenous opioid peptides are involved in such basic physiological functions as reward, learning, and fear/anxiety. These regions are also of interest from the perspective of cognitive and affective changes that have been observed in HIV patients, including those exposed to opiate drugs of abuse (Becker et al., 2011; Ortega et al., 2015). We noted a higher sensitivity of the NAc shell versus core to the effects of Tat. As the NAc shell and core are reported to play differential and perhaps opposing roles in limbic information processing (Ito and Hayen, 2011), response to reward cues (Ambroggi et al., 2011; Ghitza et al., 2004), impulsivity (Feja et al., 2014), and other behaviors observed in HIV patients and animal models, the ability of Tat to change the balance of these two outputs is potentially of great clinical importance.
Acknowledgments
We gratefully acknowledge NIH support: DA034231 (PEK and KFH); DA037096 (KFH and DES); DA018633 and DA027374 (KFH); DA039791 (JJP).
Abbreviations
- Amyg
Amygdala
- β-Arr2
β-arrestin-2
- BSA
bovine serum albumin
- CNS
central nervous system
- Co-IP
co-immunoprecipitation
- CPu
caudate–putamen
- [D-Ala2, N-MePhe4, Gly-ol]-enkephalin
DAMGO
- DOX
doxycycline
- EC50
ligand concentration causing half-maximal effect
- Emax
maximal effect at full receptor occupancy
- GAPDH
glyceraldehyde 3-phosphate dehydrogenase
- GDP
guanosine diphosphate
- GFAP
glial fibrillary acidic protein
- GPCR
G-protein coupled receptor
- GRK2
G-protein receptor kinase-2
- GTPγS
guanosine-5′-O-(3-thio)triphosphate
- HAND
HIV-associated neurocognitive disorders
- HIV
human immunodeficiency virus
- MOR
μ-opioid-receptor
- NAc
nucleus accumbens
- neuroAIDS
neuro-acquired immune deficiency syndrome
- PFC
prefrontal cortex
- rtTa
reverse tetracycline transactivator
- Tat
transactivator of transcription
References
- Ambroggi F, et al. Roles of nucleus accumbens core and shell in incentive-cue responding and behavioral inhibition. J. Neurosci. 2011;31:6820–6830. doi: 10.1523/JNEUROSCI.6491-10.2011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Anthony IC, et al. The effects of illicit drugs on the HIV infected brain. Front. Biosci. 2008;13:1294–1307. doi: 10.2741/2762. [DOI] [PubMed] [Google Scholar]
- Antinori A, et al. Updated research nosology for HIV-associated neurocognitive disorders. Neurology. 2007;69:1789–1799. doi: 10.1212/01.WNL.0000287431.88658.8b. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Asakawa A, et al. Endomorphins have orexigenic and anxiolytic activities in mice. Neuroreport. 1998;9:2265–2267. doi: 10.1097/00001756-199807130-00022. [DOI] [PubMed] [Google Scholar]
- Bailey KR, Crawley JN. Anxiety-related behaviors in mice. In: Buccafusco JJ, editor. Methods of Behavior Analysis in Neuroscience. Boca Raton (FL): 2009. [Google Scholar]
- Becker JT, et al. Subcortical brain atrophy persists even in HAART-regulated HIV disease. Brain Imaging Behav. 2011;5:77–85. doi: 10.1007/s11682-011-9113-8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bell JE, et al. HIV and drug misuse in the Edinburgh cohort. J. Acquir. Immune Defic. Syndr. 2002;31(Suppl. 2):S35–S42. doi: 10.1097/00126334-200210012-00003. [DOI] [PubMed] [Google Scholar]
- Blaesse P, et al. Mu-opioid receptor-mediated inhibition of intercalated neurons and effect on synaptic transmission to the central amygdala. J. Neurosci. 2015;35:7317–7325. doi: 10.1523/JNEUROSCI.0204-15.2015. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bohn LM, et al. Mu-opioid receptor desensitization by beta-arrestin-2 determines morphine tolerance but not dependence. Nature. 2000;408:720–723. doi: 10.1038/35047086. [DOI] [PubMed] [Google Scholar]
- Bokhari SM, et al. Morphine potentiates neuropathogenesis of SIV infection in rhesus macaques. J. NeuroImmune Pharmacol. 2011;6:626–639. doi: 10.1007/s11481-011-9272-9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bokhari SM, et al. Morphine enhances Tat-induced activation in murine microglia. J. Neurovirol. 2009:1–10. doi: 10.1080/13550280902913628. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bouton ME. Context, ambiguity, and unlearning: sources of relapse after behavioral extinction. Biol. Psychiatry. 2002;52:976–986. doi: 10.1016/s0006-3223(02)01546-9. [DOI] [PubMed] [Google Scholar]
- Breivogel CS, et al. Chronic delta9-tetrahydrocannabinol treatment produces a time-dependent loss of cannabinoid receptors and cannabinoid receptor-activated G proteins in rat brain. J. Neurochem. 1999;73:2447–2459. doi: 10.1046/j.1471-4159.1999.0732447.x. [DOI] [PubMed] [Google Scholar]
- Bruce-Keller AJ, et al. Morphine causes rapid increases in glial activation and neuronal injury in the striatum of inducible HIV-1 tat transgenic mice. Glia. 2008;56:1414–1427. doi: 10.1002/glia.20708. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Byrd DA, et al. Neurocognitive impact of substance use in HIV infection. J. Acquir. Immune Defic. Syndr. 2011;58:154–162. doi: 10.1097/QAI.0b013e318229ba41. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Calenco-Choukroun G, et al. Opioid delta agonists and endogenous enkephalins induce different emotional reactivity than mu agonists after injection in the rat ventral tegmental area. Psychopharmacology. 1991;103:493–502. doi: 10.1007/BF02244249. [DOI] [PubMed] [Google Scholar]
- Carey AN, et al. Conditional Tat protein expression in the GT-tg bigenic mouse brain induces gray matter density reductions. Prog. Neuro-Psychopharmacol. Biol. Psychiatry. 2013;43:49–54. doi: 10.1016/j.pnpbp.2012.12.018. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Carey AN, et al. Expression of HIV-Tat protein is associated with learning and memory deficits in the mouse. Behav. Brain Res. 2012;229:48–56. doi: 10.1016/j.bbr.2011.12.019. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Castro-Gomes V, et al. A dendritic organization of lateral amygdala neurons in fear susceptible and resistant mice. Neurobiol. Learn. Mem. 2015;127:64–71. doi: 10.1016/j.nlm.2015.11.010. [DOI] [PubMed] [Google Scholar]
- Chen C, et al. Heterodimerization and cross-desensitization between the muopioid receptor and the chemokine CCR5 receptor. Eur. J. Pharmacol. 2004;483:175–186. doi: 10.1016/j.ejphar.2003.10.033. [DOI] [PubMed] [Google Scholar]
- Clark US, et al. Effects of HIV and early life stress on amygdala morphometry and neurocognitive function. J. Int. Neuropsychol. Soc. 2012;18:657–668. doi: 10.1017/S1355617712000434. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Clark US, et al. Facial emotion recognition impairments are associated with brain volume abnormalities in individuals with HIV. Neuropsychologia. 2015;70:263–271. doi: 10.1016/j.neuropsychologia.2015.03.003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Conde F, et al. Afferent connections of the medial frontal cortex of the rat. II. Cortical and subcortical afferents. J. Comp. Neurol. 1995;352:567–593. doi: 10.1002/cne.903520407. [DOI] [PubMed] [Google Scholar]
- Dang VC, Christie MJ. Mechanisms of rapid opioid receptor desensitization, resensitization and tolerance in brain neurons. Br. J. Pharmacol. 2012;165:1704–1716. doi: 10.1111/j.1476-5381.2011.01482.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Davis M, Myers KM. The role of glutamate and gamma-aminobutyric acid in fear extinction: clinical implications for exposure therapy. Biol. Psychiatry. 2002;52:998–1007. doi: 10.1016/s0006-3223(02)01507-x. [DOI] [PubMed] [Google Scholar]
- Dougherty RH, et al. Progression of HIV-associated dementia treated with HAART. AIDS Read. 2002;12:69–74. [PubMed] [Google Scholar]
- Dunsmoor JE, Paz R. Fear generalization and anxiety: behavioral and neural mechanisms. Biol. Psychiatry. 2015;78:336–343. doi: 10.1016/j.biopsych.2015.04.010. [DOI] [PubMed] [Google Scholar]
- Dutta R, Roy S. Chronic morphine and HIV-1 Tat promote differential central nervous system trafficking of CD3+ and Ly6C+ immune cells in a murine streptococcus pneumoniae infection model. J. Neuroinflammation. 2015;12:120. doi: 10.1186/s12974-015-0341-5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Elliott J, et al. Tolerance to mu-opioid agonists in human neuroblastoma SH-SY5Y cells as determined by changes in guanosine-5′-O-(3-[35S]-thio)triphosphate binding. Br. J. Pharmacol. 1997;121:1422–1428. doi: 10.1038/sj.bjp.0701253. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Feja M, et al. Nucleus accumbens core and shell inactivation differentially affects impulsive behaviours in rats. Prog. Neuro-Psychopharmacol. Biol. Psychiatry. 2014;54:31–42. doi: 10.1016/j.pnpbp.2014.04.012. [DOI] [PubMed] [Google Scholar]
- File SE, et al. Animal tests of anxiety. Curr Protoc. Pharmacol. 2005 doi: 10.1002/0471141755.ph0538s27. (Chapter 5, Unit 5 38) [DOI] [PubMed] [Google Scholar]
- Filliol D, et al. Mice deficient for delta- and mu-opioid receptors exhibit opposing alterations of emotional responses. Nat. Genet. 2000;25:195–200. doi: 10.1038/76061. [DOI] [PubMed] [Google Scholar]
- Fitting S, et al. Interactive comorbidity between opioid drug abuse and HIV-1 Tat: chronic exposure augments spine loss and sublethal dendritic pathology in striatal neurons. Am. J. Pathol. 2010;177:1397–1410. doi: 10.2353/ajpath.2010.090945. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fitting S, et al. Morphine efficacy is altered in conditional HIV-1 Tat transgenic mice. Eur. J. Pharmacol. 2012;689:96–103. doi: 10.1016/j.ejphar.2012.05.029. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fitting S, et al. Synaptic dysfunction in the hippocampus accompanies learning and memory deficits in human immunodeficiency virus type 1 transactivator of transcription transgenic mice. Biol. Psychiatry. 2013;73:443–453. doi: 10.1016/j.biopsych.2012.09.026. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fitting S, et al. Interactive HIV-1 Tat and morphine-induced synaptodendritic injury is triggered through focal disruptions in Na(+) influx, mitochondrial instability, and Ca(2)(+) overload. J. Neurosci. 2014;34:12850–12864. doi: 10.1523/JNEUROSCI.5351-13.2014. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fitting S. Morphine tolerance and physical dependence are altered in conditional HIV-1 Tat transgenic mice. J. Pharmacol. Exp. Ther. 2016;356:96–105. doi: 10.1124/jpet.115.226407. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Franklin KBJ, Paxinos G. The Mouse Brain in Stereotaxic Coordinates. Academic Press; San Diego: 1997. [Google Scholar]
- Gainetdinov RR, et al. Desensitization of G protein-coupled receptors and neuronal functions. Annu. Rev. Neurosci. 2004;27:107–144. doi: 10.1146/annurev.neuro.27.070203.144206. [DOI] [PubMed] [Google Scholar]
- Ghitza UE, et al. Differences between accumbens core and shell neurons exhibiting phasic firing patterns related to drug-seeking behavior during a discriminative-stimulus task. J. Neurophysiol. 2004;92:1608–1614. doi: 10.1152/jn.00268.2004. [DOI] [PubMed] [Google Scholar]
- Good AJ, Westbrook RF. Effects of a microinjection of morphine into the amygdala on the acquisition and expression of conditioned fear and hypoalgesia in rats. Behav. Neurosci. 1995;109:631–641. doi: 10.1037//0735-7044.109.4.631. [DOI] [PubMed] [Google Scholar]
- Groer CE, et al. Agonist-directed interactions with specific beta-arrestins determine mu-opioid receptor trafficking, ubiquitination, and dephosphorylation. J. Biol. Chem. 2011;286:31731–31741. doi: 10.1074/jbc.M111.248310. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Haberstock-Debic H, et al. Morphine acutely regulates opioid receptor trafficking selectively in dendrites of nucleus accumbens neurons. J. Neurosci. 2003;23:4324–4332. doi: 10.1523/JNEUROSCI.23-10-04324.2003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hahn YK, et al. Effects of chronic HIV-1 Tat exposure in the CNS: heightened vulnerability of males versus females to changes in cell numbers, synaptic integrity, and behavior. Brain Struct. Funct. 2015;220:605–623. doi: 10.1007/s00429-013-0676-6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hall C. A Study of the Rat's Behavior in a Field, a Contribution to Method in Comparative Psychology. Calvin Hall and E. University of California Press, L. Ballachey; 1932. [Google Scholar]
- Hauser KF, et al. HIV-1 Tat and morphine have interactive effects on oligodendrocyte survival and morphology. Glia. 2009;57:194–206. doi: 10.1002/glia.20746. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hauser KF, Knapp PE. Interactions of HIV and drugs of abuse: the importance of glia, neural progenitors, and host genetic factors. Int. Rev. Neurobiol. 2014;118:231–313. doi: 10.1016/B978-0-12-801284-0.00009-9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Heaton RK, et al. HIV-associated neurocognitive disorders persist in the era of potent antiretroviral therapy: CHARTER study. Neurology. 2010;75:2087–2096. doi: 10.1212/WNL.0b013e318200d727. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hollenbach R, et al. Effect of morphine and SIV on dendritic cell trafficking into the central nervous system of rhesus macaques. J. Neurovirol. 2014;20:175–183. doi: 10.1007/s13365-013-0182-x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ito R, Hayen A. Opposing roles of nucleus accumbens core and shell dopamine in the modulation of limbic information processing. J. Neurosci. 2011;31:6001–6007. doi: 10.1523/JNEUROSCI.6588-10.2011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Johnson TP, et al. Induction of IL-17 and nonclassical T-cell activation by HIV-Tat protein. Proc. Natl. Acad. Sci. U. S. A. 2013;110:13588–13593. doi: 10.1073/pnas.1308673110. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Joshi JC, et al. Effects of morphine on stress induced anxiety in rats: role of nitric oxide and Hsp70. Physiol. Behav. 2015;139:393–396. doi: 10.1016/j.physbeh.2014.11.056. [DOI] [PubMed] [Google Scholar]
- Kalinichev M, et al. Early neonatal experience of Long–Evans rats results in long-lasting changes in reactivity to a novel environment and morphine-induced sensitization and tolerance. Neuropsychopharmacology. 2002;27:518–533. doi: 10.1016/S0893-133X(02)00326-3. [DOI] [PubMed] [Google Scholar]
- Kamprath K, Wotjak CT. Nonassociative learning processes determine expression and extinction of conditioned fear in mice. Learn. Mem. 2004;11:770–786. doi: 10.1101/lm.86104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Keifer OP, Jr., et al. The physiology of fear: reconceptualizing the role of the central amygdala in fear learning. Physiology (Bethesda) 2015;30:389–401. doi: 10.1152/physiol.00058.2014. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kieffer BL. Opioids: first lessons from knockout mice. Trends Pharmacol. Sci. 1999;20:19–26. doi: 10.1016/s0165-6147(98)01279-6. [DOI] [PubMed] [Google Scholar]
- Lazenka MF, et al. DeltaFosB induction correlates inversely with CB(1) receptor desensitization in a brain region-dependent manner following repeated delta(9)-THC administration. Neuropharmacology. 2014;77:224–233. doi: 10.1016/j.neuropharm.2013.09.019. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lazenka MF, et al. Role of dopamine type 1 receptors and dopamine- and cAMP-regulated phosphoprotein Mr 32 kDa in delta9-tetrahydrocannabinol-mediated induction of deltaFosB in the mouse forebrain. J. Pharmacol. Exp. Ther. 2015;354:316–327. doi: 10.1124/jpet.115.224428. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lister RG. The use of a plus-maze to measure anxiety in the mouse. Psychopharmacology. 1987;92:180–185. doi: 10.1007/BF00177912. [DOI] [PubMed] [Google Scholar]
- Maher CE, et al. Relationship of mu opioid receptor binding to activation of G-proteins in specific rat brain regions. Biochem. Pharmacol. 2000;59:1395–1401. doi: 10.1016/s0006-2952(00)00272-0. [DOI] [PubMed] [Google Scholar]
- Martini L, Whistler JL. The role of mu opioid receptor desensitization and endocytosis in morphine tolerance and dependence. Curr. Opin. Neurobiol. 2007;17:556–564. doi: 10.1016/j.conb.2007.10.004. [DOI] [PubMed] [Google Scholar]
- Masvekar RR, et al. Morphine enhances HIV-1SF162-mediated neuron death and delays recovery of injured neurites. PLoS One. 2014;9:e100196. doi: 10.1371/journal.pone.0100196. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Matthes HW, et al. Loss of morphine-induced analgesia, reward effect and withdrawal symptoms in mice lacking the mu-opioid-receptor gene. Nature. 1996;383:819–823. doi: 10.1038/383819a0. [DOI] [PubMed] [Google Scholar]
- Mediouni S, et al. Didehydro-cortistatin A inhibits HIV-1 Tat mediated neuroin-flammation and prevents potentiation of cocaine reward in Tat transgenic mice. Curr. HIV Res. 2015;13:64–79. doi: 10.2174/1570162x13666150121111548. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nesil T, et al. Nicotine attenuates the effect of HIV-1 proteins on the neural circuits of working and contextual memories. Mol Brain. 2015;8:43. doi: 10.1186/s13041-015-0134-x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nguyen PT, et al. Beta-arrestin2 regulates cannabinoid CB1 receptor signaling and adaptation in a central nervous system region-dependent manner. Biol. Psychiatry. 2012;71:714–724. doi: 10.1016/j.biopsych.2011.11.027. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Noble F, Cox BM. Differential desensitization of mu- and delta-opioid receptors in selected neural pathways following chronic morphine treatment. Br. J. Pharmacol. 1996;117:161–169. doi: 10.1111/j.1476-5381.1996.tb15169.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ortega M, et al. Effects of HIV and combination antiretroviral therapy on corticostriatal functional connectivity. AIDS. 2015;29:703–712. doi: 10.1097/QAD.0000000000000611. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Paris JJ, et al. Effects of conditional central expression of HIV-1 tat protein to potentiate cocaine-mediated psychostimulation and reward among male mice. Neuropsychopharmacology. 2014a;39:380–388. doi: 10.1038/npp.2013.201. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Paris JJ, et al. Central administration of angiotensin IV rapidly enhances novel object recognition among mice. Neuropharmacology. 2013;70:247–253. doi: 10.1016/j.neuropharm.2013.01.025. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Paris JJ, et al. Exposure to HIV-1 Tat in brain impairs sensorimotor gating and activates microglia in limbic and extralimbic brain regions of male mice. Behav. Brain Res. 2015;291:209–218. doi: 10.1016/j.bbr.2015.05.021. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Paris JJ, et al. Anxiety-like behavior of mice produced by conditional central expression of the HIV-1 regulatory protein. Tat. Psychopharmacology (Berl) 2014b;231:2349–2360. doi: 10.1007/s00213-013-3385-1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pitcher J, et al. Neuronal ferritin heavy chain and drug abuse affect HIV-associated cognitive dysfunction. J. Clin. Invest. 2014;124:656–669. doi: 10.1172/JCI70090. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Robertson KR, et al. The prevalence and incidence of neurocognitive impairment in the HAART era. AIDS. 2007;21:1915–1921. doi: 10.1097/QAD.0b013e32828e4e27. [DOI] [PubMed] [Google Scholar]
- Rosenkranz JA, Grace AA. Dopamine attenuates prefrontal cortical suppression of sensory inputs to the basolateral amygdala of rats. J. Neurosci. 2001;21:4090–4103. doi: 10.1523/JNEUROSCI.21-11-04090.2001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schneider BL, et al. Increased cortical GABA precedes incomplete extinction of conditioned fear and increased hippocampal excitatory tone in a mouse model of mild traumatic brain injury. J. Neurotrauma. 2015 doi: 10.1089/neu.2015.4190. [DOI] [PubMed] [Google Scholar]
- Selley DE, et al. Long-term administration of Delta9-tetrahydrocannabinol desensitizes CB1-, adenosine A1-, and GABAB-mediated inhibition of adenylyl cyclase in mouse cerebellum. Mol. Pharmacol. 2004;66:1275–1284. doi: 10.1124/mol.104.000604. [DOI] [PubMed] [Google Scholar]
- Selley DE, et al. Signal transduction correlates of mu opioid agonist intrinsic efficacy: receptor-stimulated [35S]GTP gamma S binding in mMOR-CHO cells and rat thalamus. J. Pharmacol. Exp. Ther. 1998;285:496–505. [PubMed] [Google Scholar]
- Selley DE, et al. Mu-opioid receptor-stimulated guanosine-5′-O-(gamma-thio)-triphosphate binding in rat thalamus and cultured cell lines: signal transduction mechanisms underlying agonist efficacy. Mol. Pharmacol. 1997;51:87–96. doi: 10.1124/mol.51.1.87. [DOI] [PubMed] [Google Scholar]
- Shenoy SK, Lefkowitz RJ. Beta-arrestin-mediated receptor trafficking and signal transduction. Trends Pharmacol. Sci. 2011;32:521–533. doi: 10.1016/j.tips.2011.05.002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sim-Selley LJ, et al. Region-dependent attenuation of mu opioid receptor-mediated G-protein activation in mouse CNS as a function of morphine tolerance. Br. J. Pharmacol. 2007;151:1324–1333. doi: 10.1038/sj.bjp.0707328. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sim-Selley LJ, et al. Chronic heroin self-administration desensitizes mu opioid receptor-activated G-proteins in specific regions of rat brain. J. Neurosci. 2000;20:4555–4562. doi: 10.1523/JNEUROSCI.20-12-04555.2000. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sim LJ, et al. In vitro autoradiography of receptor-activated G proteins in rat brain by agonist-stimulated guanylyl 5′-[gamma-[35S]thio]-triphosphate binding. Proc. Natl. Acad. Sci. U. S. A. 1995;92:7242–7246. doi: 10.1073/pnas.92.16.7242. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sim LJ, et al. Effects of chronic morphine administration on mu opioid receptor-stimulated [35S]GTPgammaS autoradiography in rat brain. J. Neurosci. 1996;16:2684–2692. doi: 10.1523/JNEUROSCI.16-08-02684.1996. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Skelly MJ, et al. Behavioral and neurophysiological evidence that lateral paracapsular GABAergic synapses in the basolateral amygdala contribute to the acquisition and extinction of fear learning. Neurobiol. Learn. Mem. 2015;127:10–16. doi: 10.1016/j.nlm.2015.11.006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Song C, et al. Protein kinase Cζ mediates μ-opioid receptor-induced cross-desensitization of chemokine receptor CCR5. J. Biol. Chem. 2011;286:20354–20365. doi: 10.1074/jbc.M110.177303. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sotres-Bayon F, et al. Emotional perseveration: an update on prefrontal-amygdala interactions in fear extinction. Learn. Mem. 2004;11:525–535. doi: 10.1101/lm.79504. [DOI] [PubMed] [Google Scholar]
- Steidl S, Yeomans JS. M5 muscarinic receptor knockout mice show reduced morphine-induced locomotion but increased locomotion after cholinergic antagonism in the ventral tegmental area. J. Pharmacol. Exp. Ther. 2009;328:263–275. doi: 10.1124/jpet.108.144824. [DOI] [PubMed] [Google Scholar]
- Suarez-Roca H, Maixner W. Morphine produces a multiphasic effect on the release of substance P from rat trigeminal nucleus slices by activating different opioid receptor subtypes. Brain Res. 1992;579:195–203. doi: 10.1016/0006-8993(92)90051-a. [DOI] [PubMed] [Google Scholar]
- Suarez-Roca H, Maixner W. Activation of kappa opioid receptors by U50488H and morphine enhances the release of substance P from rat trigeminal nucleus slices. J. Pharmacol. Exp. Ther. 1993;264:648–653. [PubMed] [Google Scholar]
- Thompson CM, et al. Activation of G-proteins by morphine and codeine congeners: insights to the relevance of O- and N-demethylated metabolites at mu- and delta-opioid receptors. J. Pharmacol. Exp. Ther. 2004;308:547–554. doi: 10.1124/jpet.103.058602. [DOI] [PubMed] [Google Scholar]
- Whitten RD, et al. The effects of endomorphin-1 on conditioned defeat in Syrian hamsters (Mesocricetus auratus) Brain Res. 2001;914:74–80. doi: 10.1016/s0006-8993(01)02775-5. [DOI] [PubMed] [Google Scholar]
- Wilson MA, Junor L. The role of amygdalar mu-opioid receptors in anxiety-related responses in two rat models. Neuropsychopharmacology. 2008;33:2957–2968. doi: 10.1038/sj.npp.1301675. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhang J, et al. Role for G protein-coupled receptor kinase in agonist-specific regulation of mu-opioid receptor responsiveness. Proc. Natl. Acad. Sci. U. S. A. 1998;95:7157–7162. doi: 10.1073/pnas.95.12.7157. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zou S, et al. Morphine potentiates neurodegenerative effects of HIV-1 Tat through actions at mu-opioid receptor-expressing glia. Brain. 2011;134:3616–3631. doi: 10.1093/brain/awr281. [DOI] [PMC free article] [PubMed] [Google Scholar]