

Abstract
Key points
Increase in endothelial cell (EC) calcium activates calcium‐sensitive intermediate and small conductance potassium (IK and SK) channels, thereby causing hyperpolarization and endothelium‐dependent vasodilatation.
Endothelial cells express inward rectifier potassium (Kir) channels, but their role in endothelium‐dependent vasodilatation is not clear.
In the mesenteric arteries, only ECs, but not smooth muscle cells, displayed Kir currents that were predominantly mediated by the Kir2.1 isoform.
Endothelium‐dependent vasodilatations in response to muscarinic receptor, TRPV4 (transient receptor potential vanilloid 4) channel and IK/SK channel agonists were highly attenuated by Kir channel inhibitors and by Kir2.1 channel knockdown.
These results point to EC Kir channels as amplifiers of vasodilatation in response to increases in EC calcium and IK/SK channel activation and suggest that EC Kir channels could be targeted to treat endothelial dysfunction, which is a hallmark of vascular disorders.
Abstract
Endothelium‐dependent vasodilators, such as acetylcholine, increase intracellular Ca2+ through activation of transient receptor potential vanilloid 4 (TRPV4) channels in the plasma membrane and inositol trisphosphate receptors in the endoplasmic reticulum, leading to stimulation of Ca2+‐sensitive intermediate and small conductance K+ (IK and SK, respectively) channels. Although strong inward rectifier K+ (Kir) channels have been reported in the native endothelial cells (ECs) their role in EC‐dependent vasodilatation is not clear. Here, we test the idea that Kir channels boost the EC‐dependent vasodilatation of resistance‐sized arteries. We show that ECs, but not smooth muscle cells, of small mesenteric arteries have Kir currents, which are substantially reduced in EC‐specific Kir2.1 knockdown (EC‐Kir2.1 −/−) mice. Elevation of extracellular K+ to 14 mm caused vasodilatation of pressurized arteries, which was prevented by endothelial denudation and Kir channel inhibitors (Ba2+, ML‐133) or in the arteries from EC‐Kir2.1 −/− mice. Potassium‐induced dilatations were unaffected by inhibitors of TRPV4, IK and SK channels. The Kir channel blocker, Ba2+, did not affect currents through TRPV4, IK or SK channels. Endothelial cell‐dependent vasodilatations in response to activation of muscarinic receptors, TRPV4 channels or IK/SK channels were reduced, but not eliminated, by Kir channel inhibitors or EC‐Kir2.1 −/−. In angiotensin II‐induced hypertension, the Kir channel function was not altered, although the endothelium‐dependent vasodilatation was severely impaired. Our results support the concept that EC Kir2 channels boost vasodilatory signals that are generated by Ca2+‐dependent activation of IK and SK channels.
Key points
Increase in endothelial cell (EC) calcium activates calcium‐sensitive intermediate and small conductance potassium (IK and SK) channels, thereby causing hyperpolarization and endothelium‐dependent vasodilatation.
Endothelial cells express inward rectifier potassium (Kir) channels, but their role in endothelium‐dependent vasodilatation is not clear.
In the mesenteric arteries, only ECs, but not smooth muscle cells, displayed Kir currents that were predominantly mediated by the Kir2.1 isoform.
Endothelium‐dependent vasodilatations in response to muscarinic receptor, TRPV4 (transient receptor potential vanilloid 4) channel and IK/SK channel agonists were highly attenuated by Kir channel inhibitors and by Kir2.1 channel knockdown.
These results point to EC Kir channels as amplifiers of vasodilatation in response to increases in EC calcium and IK/SK channel activation and suggest that EC Kir channels could be targeted to treat endothelial dysfunction, which is a hallmark of vascular disorders.
Abbreviations
- AKAP150
A‐kinase anchoring protein 150
- Ang II
angiotensin II
- Apa
apamin
- CCh
carbachol
- ChTx
charybdotoxin
- EC
endothelial cell
- EC‐Kir2.1−/−
endothelial cell‐specific Kir2.1 channel knockdown
- EDH
endothelium‐dependent hyperpolarization
- HT
hypertensive
- IK
potassium current
- IK channel
intermediate conductance potassium channel
- IP3R
inositol trisphosphate receptor
- Kir channel
inward rectifier potassium channel
- MEP
myoendothelial projection
- NT
normal or normotensive
- PSS
physiological saline solution
- SHR
spontaneously hypertensive rat
- SK channel
small conductance potassium channel
- SMC
smooth muscle cell
- TRPV4
transient receptor potential vanilloid 4
Introduction
Endothelial cells (ECs) line all arteries in the body and are uniquely positioned to respond to neurohumoral and mechanical stimuli. Endothelial cells regulate the contractile state of the surrounding smooth muscle, which determines blood flow to the target organs and vascular resistance. Intracellular Ca2+ in ECs regulates the elaboration of vasodilatory signals that are mediated by endothelial nitric oxide synthase (Fleming & Busse, 1999) and phospholipase A2 (Bogatcheva et al. 2005). Another major vasodilatory pathway, which is of particular importance in resistance arteries, is the activation of EC intermediate (IK) or small (SK) conductance Ca2+‐activated K+ channels by Ca2+ signals from transient receptor potential vanilloid 4 (TRPV4) channels and inositol trisphosphate receptors (IP3Rs; Taylor et al. 2003; Köhler et al. 2006; Marrelli et al. 2007; Ledoux et al. 2008; Zhang et al. 2009; Sonkusare et al. 2012) and referred to as ‘endothelium‐dependent hyperpolarization’ (EDH). Endothelium‐dependent hyperpolarization is spread electronically to the adjacent smooth muscle cells (SMCs) through gap junctions in specialized EC membrane structures [myoendothelial projections (MEPs); Yamamoto et al. 1999; Busse et al. 2002; Isakson & Duling, 2005; Mather et al. 2005; Garland et al. 2011; Sandow et al. 2012], which make direct contact with the smooth muscle. Membrane potential hyperpolarization of the SMCs deactivates L‐type voltage‐dependent Ca2+ channels, leading to relaxation of SMCs and vasodilatation.
The vascular endothelium can be viewed as a sensory organ, detecting subtle changes in mechanical forces or vasoactive substances and translating them into changes in blood flow through modulation of smooth muscle tone. The mechanisms by which local changes in vasoactive substances are amplified to cause endothelium‐dependent vasodilatation remain a fundamental and unresolved issue. We have recently shown that an important mechanism for the amplification of actions of a classic endothelium‐dependent vasodilator, acetylcholine, is through activation of Ca2+‐permeable TRPV4 channels exclusively at MEPs. Strong co‐operative gating of TRPV4 channels at MEPs elevates local Ca2+ influx about two‐ to threefold, which in turn activates nearby IK and SK channels to cause membrane potential hyperpolarization. Given the exceedingly low levels of baseline TRPV4 channel activity (open probability < 0.01) even at the MEPs, we reasoned that there might be another step to amplify the hyperpolarization to activation of EC IK and SK channels. Strong inward rectifier K+ channels (Kir2) are activated by membrane potential hyperpolarization and by external K+ and are therefore likely candidates to boost endothelium‐dependent vasodilatations that are driven by IK/SK activation (Jackson, 2005; Longden & Nelson, 2015). Endothelial cells and some types of vascular smooth muscles have been shown to express functional Kir channels (von Beckerath et al. 1996; Romanenko et al. 2002; Crane et al. 2003; Yang et al. 2003; Fang et al. 2005, 2006; Jackson, 2005; Climent et al. 2011).
The Kir channel family consists of seven subfamilies (Kir1–7) and a total of 15 subunit isoforms (Kir1.1, Kir2.1–4, Kir3.1–4, Kir4.1–2, Kir5.1, Kir6.1–2 and Kir7.1; Longden & Nelson, 2015). Notably, the Kir2 channels are expressed in arteries of the cerebral circulation and are involved in the regulation of membrane potential and arterial diameter. Activation of Kir2 channels in this vascular bed leads to a rapid and profound hyperpolarization that causes vasodilatation (Quayle et al. 1993, 1997; Filosa et al. 2006; Longden & Nelson, 2015).
We hypothesized that Kir channels serve as ‘end‐stage’ boosters to cause vasodilatation through Ca2+‐dependent activation of IK and SK channels. We found that the ECs, but not SMCs, from resistance‐sized mesenteric arteries exhibit strong Kir currents. Consistent with the findings that Kir2.1 is the major channel subtype in cerebral arteries (Bradley et al. 1999; Zaritsky et al. 2000), EC Kir currents in the mesenteric arteries were substantially reduced in EC‐specific Kir2.1 channel knockdown (EC‐Kir2.1 −/−) mice. An increase in extracellular K+ concentration caused vasodilatation that was dependent on endothelial Kir2.1 channels, which was in agreement with the external K+‐dependent nature of Kir2 channel activity (Leech & Stanfield, 1981). The vasodilatations in response to endothelium‐dependent vasodilators acting through the TRPV4–IK/SK pathway were also inhibited by Kir channel inhibitors and by EC‐Kir2.1 −/−. Our studies reveal an important role for endothelial Kir2.1 channels in amplifying the vasodilatory response to physiological stimuli in small, resistance‐sized arteries. Although EC Kir2.1 channels are required for maximal EDH response, endothelial dysfunction in angiotensin II (Ang II)‐induced hypertension is not associated with a Kir channel dysfunction.
Methods
Animal procedures
All animal procedures used in this study were approved by the Institutional Animal Care and Use Committee of the University of Vermont and performed in accord with the National Research Council's Guide for the Care and Use of Laboratory Animals (8th edition, 2011). Male, 12‐ to 14‐week‐old C57BL6 (Jackson Laboratory, Bar Harbor, ME, USA), EC‐specific Kir2.1 channel knockdown (EC‐Kir2.1 −/−, described in the next subsection) and GCaMP2Cx40 mice were used in this study. The GCaMP2Cx40 mice express the circularly permutated Ca2+ sensor, GCaMP2, under the control of the connexin40 (Cx40) promoter (Tallini et al. 2006, 2007), which limits expression of GCaMP2 to ECs in the vascular wall. Adult (3‐ to 4‐month‐old) male mice were killed by i.p. injection of sodium pentobarbital (Wilcox Pharmacy, Rutland, VT, USA; 150 mg kg−1) followed by decapitation. Third‐order branches of mesenteric arteries (∼100 μm internal diameter at 80 mmHg) were isolated into Hepes‐buffered physiological saline solution (PSS) of the following composition (mm): 10 Hepes, 134 NaCl, 6 KCl, 1 MgCl2, 2 CaCl2 and 7 glucose, adjusted to pH 7.4 with NaOH. The investigators understand the ethical principles under which the journal operates, and this work complies with the animal ethics checklist.
Generation of EC‐Kir2.1 −/− mice
Heterozygous Kir2.1 floxed mice were provided by inGenious Targeting Laboratory, which were then bred to be homozygous for the LoxP expression. The second exon of the Kir2.1 gene in these mice is flanked by LoxP sites, and a downstream Neo cassette is flanked by FRT sites. The Neo cassette was deleted by breeding to an FLP‐deleter mouse (Jackson Laboratory). To generate EC‐Kir2.1 −/−, the floxed Kir2.1 mice were bred with Tie2‐Cre mice (Jackson Laboratory), which have endothelium‐specific expression of Cre recombinase directed by receptor tyrosine kinase (Tek or Tie2) promoter. The following primers were used for detecting Neo cassette deletion: NDEL1, CTGACTGAACACACAGGTCCAGGG; and NDEL2, GGGACCATCAAGCCCTGGTAATGG; and for distal loxP sites: loxP forward, AGCGGAGGTACCTCATCTATGTTC; and loxP reverse, ACCAATGTACTTTAGATTGAATTGTGC.
Diameter studies in pressurized arteries
Measurements of diameter in pressurized arteries were performed as previously described (Sonkusare et al. 2012, 2014 b). In brief, mesenteric arteries were dissected free of surrounding tissue and mounted on similar‐sized glass pipettes in an arteriograph chamber (Instrumentation and Model Facility, University of Vermont, Burlington, VT, USA). The arteries were then pressurized to 80 mmHg for at least 45 min in 37°C PSS of the following composition (mm): 119 NaCl, 4.7 KCl, 1.2 KH2PO4, 1.2 MgCl2, 2 CaCl2, 7 glucose and 24 NaHCO3, at pH 7.4 and constantly equilibrated with bioair (20% O2 and 5% CO2 in N2). Internal diameter was continuously monitored with a CCD camera and edge‐detection software (IonOptix, Milton, MA, USA). All compounds were added to the perfusate (PSS), which was continuously recirculated through the arteriograph chamber. Arteries were treated with Ca2+‐free PSS (mm: 119 NaCl, 4.7 KCl,1.2 KH2PO4,1.2 MgCl2, 7 glucose, 24 NaHCO3 and 5 EGTA, pH 7.4) at the conclusion of each experiment to obtain maximal diameter. For some of the experiments, the endothelium was denuded by passing an air bubble through the artery for ∼45 s. Endothelial denudation was confirmed by treating the arteries with NS309 (an IK/SK channel opener).
Myogenic tone was calculated as follows:
Mesenteric artery dilatation was expressed as follows:
where Diameterdilated is the diameter after addition of carbachol (CCh; a muscarinic receptor agonist), GSK101 (a TRPV4 channel agonist), NS309 (an IK/SK channel opener) or K+.
Endothelial cell Ca2+ imaging
Ca2+ signals in the ECs were imaged using a Revolution Andor confocal system (Andor Technology, Belfast, UK) that consisted of an upright Nikon microscope with a ×60, water‐dipping objective (NA 1.0) and an electron‐multiplying CCD camera, as previously described (Sonkusare et al. 2012, 2014 b). In brief, images were recorded with Andor Revolution TL acquisition software (Andor Technology) at 30 frames s−1. Bound Ca2+ was detected by exciting at 488 nm with a solid‐state laser and collecting emitted fluorescence using a 527.5–49 nm bandpass filter. Experiments were performed at 36°C. The TRPV4 Ca2+ sparklets were analysed within a region of interest defined by a 1.7 μm2 box (5 × 5 pixels) positioned at a point corresponding to peak TRPV4 Ca2+ sparklet amplitude and analysed using custom software, written by Dr A. D. Bonev (Sonkusare et al. 2012, 2014 b). In the presence of 3 nm GSK101, images were recorded before and 5 min after treatment with the Kir channel blocker barium (Ba2+). Changes in Ba2+‐induced activity were expressed as the fold change relative to 3 nm GSK101 alone.
Endothelial cell patch clamp
Endothelial cells and SMCs were freshly isolated from third‐order branches of mesenteric arteries as previously described (Sonkusare et al. 2012, 2014 b). The currents were recorded using a perforated patch configuration at a holding potential of −50 mV and 400 ms ramps from −140 to +50 mV. The composition of external bathing solution was (mm): 10 Hepes, 134 NaCl, 6 KCl, 1 MgCl2, 2 CaCl2 and 10 glucose. The pipette solution was of the following composition (mm): 10 Hepes, 30 KCl, 10 NaCl, 110 potassium aspartate and 1 MgCl2 (adjusted to pH 7.2 with NaOH). The Kir channel currents were activated by increasing the extracellular K+ concentration to 6 and 60 mm (replacing Na+) and blocked using Ba2+ (100 μm) and ML‐133 (20 μm). The IK and SK channel currents were activated by NS309 (1 μm); the IK channel current was determined from the decrease in current after charybdotoxin (200 nm), whereas the SK channel current was determined from a further decrease in current with subsequent addition of apamin (300 nm). Sampled at 2 kHz, currents were acquired in voltage‐clamp mode with an Axopatch 200A (Axon Instruments, Sunny Vale, CA, USA) and analysed using the pClamp suite (Axon Instruments).
Angiotensin II‐induced hypertension
For measurements of systolic blood pressure using tail‐cuff plethysmography (Columbus Instruments, Columbus, OH, USA), 12‐ to 14‐week‐old mice were trained for 1 week (twice daily) before recording of baseline systolic blood pressure on 2 days consecutively. For Ang II‐induced hypertension, osmotic minipumps (ALZET 2004; Durect Corporation, Cupertino, CA, USA) were implanted subcutaneously in mice anaesthetized by inhalation of 5% isoflurane and maintained with 2% isoflurane in 100% O2. The minipumps were filled with Ang II (Bachem Americas, Inc., Torrance, CA, USA) in 0.9% saline, and the infusion rate was 1.4 mg kg−1 day−1. The minipumps in control, normotensive mice were filled with 0.9% saline. Following recovery from anaesthesia, mice were housed in individual cages and allowed free access to food and water. Systolic blood pressure was recorded on days 4, 7, 10, 14, 18 and 21 after implantation of minipumps. The systolic blood pressure in the mice infused with 0.9% saline for 3 weeks was 121 ± 5 mmHg (n = 4), and the systolic blood pressure in the mice infused with Ang II was 180 ± 6 mmHg (n = 4).
Calculation of the number of Kir channels
A linear relationship between channel current (I K) and membrane potential can be explained by the Goldman–Hodgkin–Katz constant field equation (Goldman, 1943; Hodgkin & Katz, 1949), as follows:
where P K is the permeability of the channel to K+ ions (in centimetres per second); [K+]out and [K+]in are the concentrations of K+ in extracellular and intracellular compartments (millimolar), respectively; E is the membrane potential (in volts), [K+] is in moles per millilitre, F is the Faraday constant, R is the gas constant and T is the temperature (in kelvin). Single channel permeability was calculated using a simplified formula when [K+]out = [K+]in (Benham et al. 1986):
where i K is the single channel current. Given that conductance γ = i K/E (with γ in siemens, I K in amperes and E in volts):
The number of Kir2.1 channels per EC was determined from the equation for macroscopic current, I = N × i × P O, where N is the number of channels, i is the single‐channel current, and P O is the open state probability of the channel. Based on the single channel conductance of Kir2.1 channels of 22.7 pS as measured in excised patches and [K+]out and [K+]in of 140 mm (So et al. 2001), the single channel current at −140 mV (−0.14 V) is i = −0.14 × 22.7 pA and = 4.3 × 10−14 cm s−1 at 23°C.
For [K+]out = 60 mm and [K+]in = 140 mm, E = −140 mV, using the Goldman–Hodgkin–Katz equation yielded a value of i of −1.36 pA at 24°C. The average whole‐cell current was −109.8 pA at −140 mV. Based on the whole‐cell current, P O of 0.56 at −140 mV (So et al. 2001), the number of Kir2.1 channels per cell was calculated using the equation:
Reagents
Cyclopiazonic acid was obtained from EMD Chemicals (Billerica, MA, USA). NS309 was a gift from Professor Soren‐Peter Olesen, Neurosearch A/S (Hellerup, Denmark). HC‐067047 was a gift from Hydra Biosciences (Cambridge, MA, USA). Charybdotoxin and apamin were purchased from Peptide Institute (Osaka, Japan) and Enzo Life Sciences (Farmingdale, NY, USA), respectively. Angiotensin II was obtaind from Bachem Americas Inc. (Torrance, CA, USA). All other chemicals were obtained from Sigma‐Aldrich.
Data analysis and statistics
Data are expressed as means ± SEM. Student's paired or unpaired t test wherever appropriate and one‐way ANOVA with post hoc Bonferroni tests were used for comparisons between two groups and among more than two groups, respectively. A value of P < 0.05 was considered significant. The number n represents the number of arteries for diameter and Ca2+ imaging experiments and the number of cells for patch‐clamp experiments. Each treatment was carried out in at least four arteries or five cells from at least three different mice. The n numbers and the statistical tests are indicated in the figure legends along with the resultant P values. Except for the specific treatment to be tested, experimental conditions were not changed throughout the experiments.
Results
Endothelial cells, but not SMCs, display Kir currents in small mesenteric arteries
Kir channels have been well characterized in the SMCs from cerebral arteries and have been shown to require the Kir2.1 isoform (Quayle et al. 1993; Bradley et al. 1999; Zaritsky et al. 2000). In contrast, SMCs in the systemic resistance (mesenteric) arteries from rats do not exhibit Kir channel currents (Crane et al. 2003; Smith et al. 2008). To establish whether functional Kir channels are expressed in the SMCs and/or ECs of resistance‐sized mesenteric arteries from mice, we measured whole‐cell currents in isolated single ECs and SMCs from these arteries using the perforated patch configuration of the patch‐clamp technique. The currents were recorded simultaneously at a physiological membrane potential of −50 mV and with 400 ms ramps from −140 to +50 mV. Barium (100 μm) and ML‐133 (20 μm) were used to inhibit the Kir currents. Barium ions block Kir2 channels potently in a voltage‐dependent manner (Quayle et al. 1993; Longden & Nelson, 2015), and ML‐133 is a selective inhibitor of Kir2 channels (Wu et al. 2010; Wang et al. 2011) that has been shown to inhibit the Kir currents in cerebral ECs (Kochukov et al. 2014). Currents in SMCs exhibited characteristic outward rectification, an indication of large conductance K+ (BK) and voltage‐gated K+ (Kv) channels (Fig. 1 A), without a hint of currents through Kir channels even with 60 mm external K+. We did not detect any Ba2+‐sensitive currents in SMCs from these arteries.
Figure 1. Freshly isolated endothelial cells (ECs), but not smooth muscle cells (SMCs) exhibit functional inward rectifier potassium (Kir) channels in third‐order mesenteric arteries .
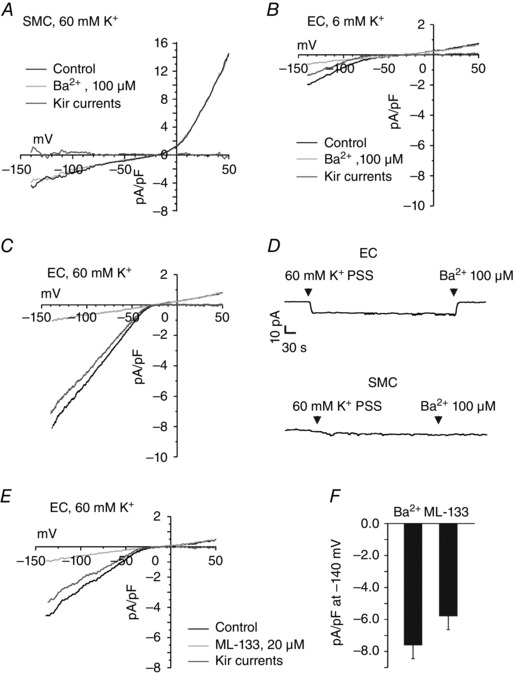
The Kir currents were recorded using the perforated patch configuration of the patch‐clamp technique. A, representative current traces in the SMCs from third‐order mesenteric arteries. There was no difference between the inward currents at −140 mV before and after addition of Ba2+ in the SMCs (n = 7 SMCs; P = 0.2035 using Student's t test). B and C, representative current traces in ECs from third‐order mesenteric arteries in the presence and absence of Ba2+ (100 μm) at extracellular K+ concentrations of 6 (B) and 60 mm (C). The cells were held at −50 mV, and 400 ms ramps from −140 to +50 mV were applied. The [K+]in was 140 mm. Kir current was estimated as the decrease in current following the addition of Ba2+. D, continuous current recordings at physiological membrane potential (−50 mV) in EC and SMCs, starting at 6 mm K+ physiological saline solution (PSS). E, representative traces for ML‐133‐sensitive Kir currents in the ECs. F, a bar graph showing averaged Kir current densities using Ba2+ and ML‐133 in the ECs (n = 10 cells for Ba2+ and n = 5 ECs for ML‐133; P = 0.4931 using Student's unpaired t test).
In contrast, whole‐cell inward currents from the ECs displayed characteristic properties of Kir2 channels: sharp activation with membrane potential hyperpolarization, shift of activation curve to more positive potentials as the external K+ is changed from 6 to 60 mm, and inhibition by external Ba2+ (100 μm; Fig. 1 B and C). The density of Kir currents at −140 mV was −1.7 ± 0.4 and −7.5 ± 1.0 pA pF−1 at 6 and 60 mm external K+ PSS (n = 5 and 10 cells, respectively), respectively. The reversal potentials of the Kir currents were −70 ± 4 (n = 5) and −22 ± 2 mV (n = 10) at 6 and 60 mm K+, respectively. At physiological membrane potential (−50 mV), switching external solution from 6 to 60 mm K+ PSS increased the inward current in ECs, and this current was completely blocked by Ba2+ (Fig. 1 D). This increase in Ba2+‐sensitive inward current was absent in SMCs, again confirming a lack of functional Kir channels in the SMCs from these arteries. The inward currents at 60 mm external K+ were also inhibited by a selective blocker of Kir2 channels, ML‐133 (20 μm; Fig. 1 E and F). These results confirmed that functional Kir channels are present in the ECs but not the SMCs from mesenteric arteries. In the conditions used, the estimated number of Kir channels per EC was 144 (see Methods for details), which corresponds to approximately one channel per 11 μm2.
Barium does not affect the function of EC IK, SK or TRPV4 channels
We have previously shown that Ba2+ at concentrations of 100 μm or lower does not block other types of ion channels in SMCs (Nelson & Quayle, 1995). Endothelial cells in addition to Kir channels also have functional IK, SK and TRPV4 channels. Therefore, to use Ba2+ as a tool, we evaluated the effect of Ba2+ on IK and SK currents by recording NS309‐induced outward currents in single, freshly isolated ECs in the absence and presence of Ba2+. The currents were recorded simultaneously at a physiological membrane potential of −50 mV (Fig. 2 A) and in response to 250 ms ramps from −140 to +50 mV (Fig. 2 B). At 0 mV, there was no difference in the NS309‐induced outward currents before and after the addition of Ba2+ (n = 4 ECs, P = 0.8016; Fig. 2 C,). The outward currents elicited by NS309 were completely inhibited by a combination of charybdotoxin (ChTx; an IK channel inhibitor) and apamin (Apa; an SK channel inhibitor; Fig. 2 A). Although Ba2+ did not alter NS309‐activated outward currents, it reduced the inward currents, implying a specific inhibition of Kir currents (Fig. 2 B). These results indicate that Ba2+ ions at concentrations that block Kir channels do not affect the activity of IK or SK channels.
Figure 2. Barium does not affect endothelial intermediate and small conductance potassium (IK and SK) channel currents .
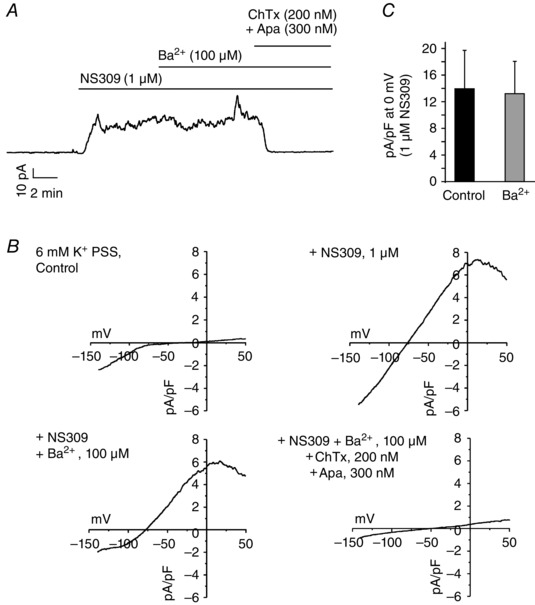
The perforated patch configuration was used to study the effect of Ba2+ on IK and SK currents in freshly isolated ECs. A, a representative trace showing continuous current recording at physiological membrane potential (−50 mV) in a freshly isolated EC. NS309 (1 μm) increased the outward K+ current that was completely inhibited by a combination of charybdotoxin (ChTx; 200 nm) and apamin (Apa; 300 nm; n = 6 ECs). B, representative current recordings in the ECs in response to 250 ms ramps from −140 to +50 mV. C, averaged outward current at 0 mV in response to NS309 before (Control) and after addition of Ba2+ (n = 4 ECs; P = 0.8583 using Student's paired t test).
To evaluate the effect of Ba2+ on TRPV4 channel function, we optically recorded TRPV4 channel activity in the presence or absence of Ba2+ in slit‐open mesenteric arteries from GCaMP2 mice (Tallini et al. 2006, 2007; Sonkusare et al. 2012, 2014 b). The experiments were performed in the presence of cyclopiazonic acid (a SERCA inhibitor) to deplete intracellular stores of Ca2+ and to eliminate IP3R‐mediated Ca2+ release. The TRPV4 channel function was recorded as TRPV4 sparklets (Ca2+ influx through single TRPV4 channels) in physiological conditions of temperature and ionic solutions (Sonkusare et al. 2012, 2014 b). The baseline open state probability of TRPV4 channels is very low in this vascular bed. For accurate quantification of the effect of Ba2+ on TRPV4 sparklet activity, the open probability of TRPV4 channels was increased by the use of 3 nm GSK101. In the presence of 3 nm GSK101, Ba2+ did not affect the average number of open TRPV4 channels per site (Sonkusare et al. 2014 b; P = 0.8861, 77–87 sites from six fields), thus ruling out a direct effect of Ba2+ on TRPV4 channels.
Endothelial cell‐specific knockdown of Kir2.1 channels substantially reduces Kir currents
Based on the inhibition of Kir currents by a selective inhibitor of Kir2.x channels (ML‐133), we postulated that endothelial Kir2.x channels were involved in EDH‐mediated vasodilatations. Kir2.1 channels had been previously identified in the rat mesenteric artery homogenate and were downregulated in the arteries from spontaneously hypertensive rats (SHRs; Weston et al. 2010). Moreover, Kir2.1 was the predominant subtype in the cerebral arteries (Bradley et al. 1999; Zaritsky et al. 2000). Based on these findings, we hypothesized that Kir2.1 is the prominent subunit isoform in the ECs from mesenteric arteries. To verify this hypothesis, we probed for Kir currents in the ECs from mesenteric arteries from the EC‐Kir2.1 −/− mice. The Kir currents in ECs from these mice were significantly lower than those in the ECs from C57BL6 or floxed Kir2.1 mice (Fig. 3 A and B). A continuous current recording at −50 mV showed no Ba2+‐sensitive inward currents (Fig. 3 C), but NS309 was able to activate IK currents sensitive to ChTx in the same cells (n = 6). These results supported the concept that Kir2.1 is the predominant Kir channel isoform in the ECs.
Figure 3. Kir2.1 channels are the major Kir channel isoform in ECs .
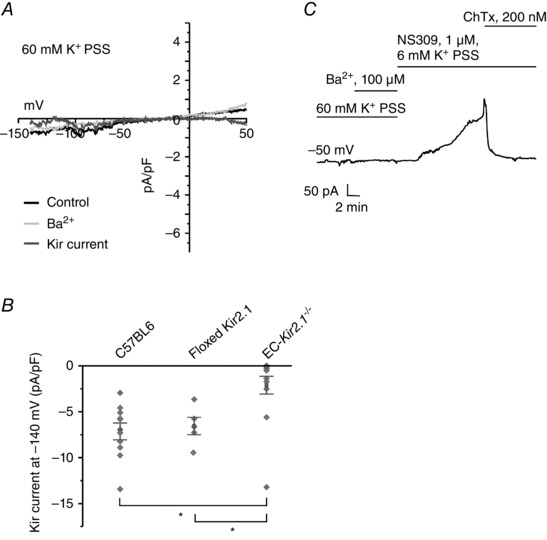
Kir currents were recorded using the perforated patch configuration of the patch‐clamp technique in ECs from third‐order mesenteric arteries. A, representative current traces in an EC from an EC‐Kir2.1 −/− mouse. The Kir currents were calculated as the difference between the traces before and after addition of Ba2+. B, a scatter plot compares the current densities at −140 mV between the ECs from the C57BL6 mice, floxed Kir2.1 mice, and EC‐Kir2.1 −/− mice (n = 5–12 ECs; *P < 0.001 using One‐Way ANOVA). C, NS309 was used to evoke outward IK/SK currents to confirm that the cell being studied was an EC.
Activation of endothelial Kir channels by extracellular K+ dilates third‐order mesenteric arteries
In the cerebral microvasculature, increasing extracellular K+ dilates small arteries by activating Kir currents in the SMCs, thereby hyperpolarizing them (Nelson & Quayle, 1995). Whether activation of endothelial Kir channels by external K+ can relax the SMCs and dilate the arteries is not known. We hypothesized that activation of endothelial Kir channels by external K+ leads to vasodilatation of mesenteric arteries. Intravascular pressure was elevated to 80 mmHg, which causes these arteries to constrict by ∼25% (Sonkusare et al. 2012, 2014 b). Elevation of external K+ from 6 to 14 mm dilated the mesenteric arteries, and this dilatation was inhibited by the Kir channel inhibitors Ba2+ and ML‐133 (Fig. 4 A and B). Removal of the EC layer eliminated the dilatation in response to 14 mm K+ (Fig. 4 C), confirming that endothelial Kir channels mediate the K+‐induced vasodilatation in the mesenteric arteries. K+‐induced dilatations were not affected by inhibitors of TRPV4 (HC‐067047), IK (ChTx) and SK (apamin) channels (Fig. 4 D). Moreover, the dilatations in response to 14 mm K+ were absent in the arteries from EC‐Kir2.1 −/− mice, supporting the idea that endothelial Kir2.1 channels are the main contributors to K+‐induced dilatation in the mesenteric arteries (Fig. 4 E and F). These results indicate that activation of EC Kir channels leads to vasodilatation that is independent of IK, SK and TRPV4 channels.
Figure 4. Endothelial Kir channels dilate mesenteric arteries in response to an increase in extracellular K+ .
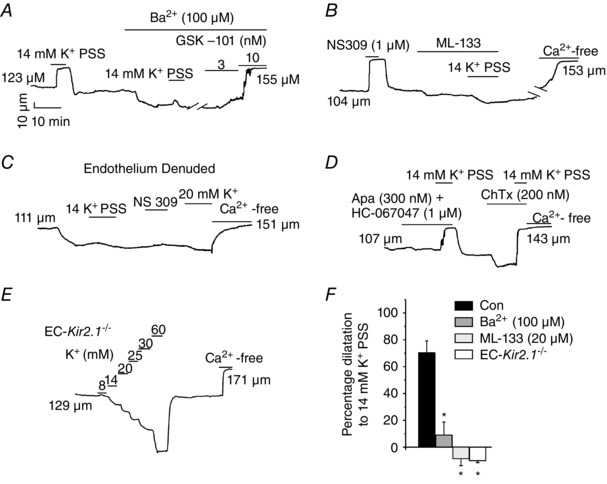
The effect of external K+ on arterial diameter was studied in the small mesenteric arteries constricted by intravascular pressure (80 mmHg). Representative diameter traces show the effect of 14 mm K+ on arterial diameter in control conditions (A), in the presence of Ba2+ (A) and ML‐133 (B), in endothelium‐denuded arteries (C), in the presence of SK/IK and transient receptor potential vanilloid 4 (TRPV4) channel inhibitors (D) and in arteries from EC‐Kir2.1 −/− mice (E). The K+‐induced dilatations were absent in endothelium‐denuded arteries and in the arteries from the EC‐Kir2.1 −/− mice. F, the bar graph shows the averaged data for K+‐induced dilatations in C57BL6 and EC‐Kir2.1 −/− mice (n = 4–9 arteries; *P < 0.05 compared with Control using one‐way ANOVA).
Endothelial Kir channels amplify the vasodilatations in response to muscarinic receptor, TRPV4 and IK/SK channel agonists
Our results demonstrate that activation of EC Kir2.1 channels by external K+ can cause vasodilatation. Kir2.1 channels are also activated by membrane potential hyperpolarization. Therefore, any endothelium‐dependent vasodilatory pathway that leads to activation of IK/SK channels should engage Kir channels through membrane potential hyperpolarization and, possibly, through perivascular K+ accumulation (Edwards et al. 1998). We, therefore, tested the effects of Kir channel inhibitors on dilatations in response to synthetic activators of IK and SK channels, NS309. Vasodilatations in response to NS309 (0.3–1.0 μm) were greatly reduced by Ba2+ (Fig. 5 A and B) or ML‐133 (Fig. 5 C). Interestingly, higher concentrations of NS309 (2 μm) were able to dilate the mesenteric arteries completely in the presence of Ba2+ or ML‐133 (Fig. 5), supporting the concept that EC Kir channels amplify the effects of IK/SK channel activation.
Figure 5. Endothelial cell Kir channels also mediate vasodilatation in response to the IK and SK channel opener NS309 .
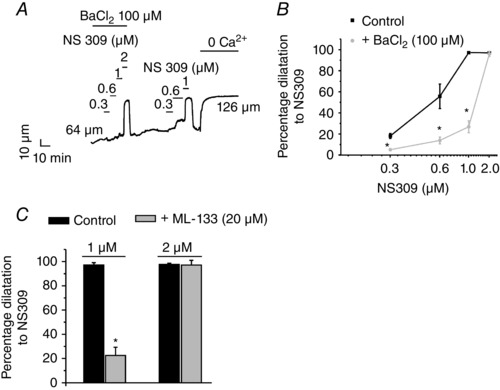
Arterial diameters were recorded in third‐order mesenteric arteries constricted by intravascular pressure (80 mmHg). NS309 was used to activate IK/SK channels. The figure shows a representative diameter trace (A) and averaged data (B) for NS309‐induced vasodilatation of third‐ordered mesenteric arteries in the presence or absence of Ba2+ (n = 4–12 arteries; *P < 0.05 using one‐way ANOVA). C, averaged diameter data for NS309‐induced dilatations in the presence or absence of ML‐133 (n = 3–12 arteries; *P < 0.05 using one‐way ANOVA). A representative trace for the effect of ML‐133 on NS309‐induced dilatation is shown in Fig. 6 D.
We recently showed that IK and SK channels are downstream mediators of the vasodilatations in response to the TRPV4 channel activator GSK101 and the muscarinic receptor agonist CCh (Sonkusare et al. 2012, 2014 a,b). Inhibiting Kir channels should therefore attenuate the EDH–dilatation via the muscarinic receptor–TRPV4–IK/SK pathway. To examine the possibility that EDH signalling is boosted by Kir channel activation, we tested the effects of Kir channel inhibitors Ba2+ and ML‐133 on vasodilatation in response to muscarinic receptor stimulation (CCh, 0.3–10 μm) and direct TRPV4 channel activation (3–10 nm GSK1016790A or GSK101). Dilatations in response to CCh and GSK101 were inhibited by Ba2+ (Fig. 6 A–C) and ML‐133 (Fig. 6 D), indicating that Kir channels are involved in the CCh‐ and TRPV4‐induced vasodilatations. Higher concentrations of GSK101 (10 nm) or CCh (10 μm) were able to dilate the arteries completely (Fig. 6 A–C), supporting the concept that EC Kir channels act as boosters of EC‐dependent vasodilatators. The vasodilatations in response to CCh, GSK101 and NS309 were also attenuated in the mesenteric arteries from the EC‐Kir2.1 −/− mice (Fig. 6 E), confirming that Kir2.1 channels are required for vasodilatation in response to the muscarinic receptor–TRPV4–IK/SK pathway at lower levels of activation. These results also implied that Kir2.1 channels are downstream from the IK and SK channels in this vasodilatory signalling pathway.
Figure 6. Kir channels mediate vasodilatation in response to muscarinic receptor and TRPV4 channel agonists .
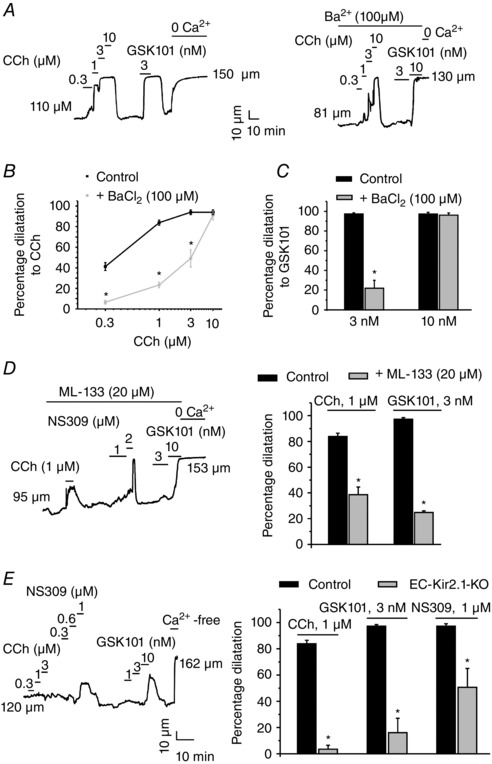
Arterial diameter measurements were performed in third‐order mesenteric arteries constricted by intravascular pressure (80 mmHg). Carbachol (CCh) and GSK101 were used to activate the muscarinic receptors and TRPV4 channels, respectively. A, representative diameter traces show vasodilatation in response to CCh and GSK101 in the presence or absence of Ba2+. Averaged data summarize the effect of Kir channel inhibition with Ba2+ on dilatations in response to CCh (B) and GSK101 (C; n = 6 arteries; *P < 0.05 using one‐way ANOVA). The data are means ± SEM. D, representative diameter trace (left) and averaged diameter data (right) for CCh‐ and GSK101‐induced dilatations in the presence or absence of the selective Kir2.x channel inhibitor ML‐133 (n = 4 arteries; *P < 0.001 using Student's t test). E, a representative diameter trace from an EC‐Kir2.1 −/− mouse showing the dilatations in response to CCh, GSK101 and NS309 in the mesenteric arteries (left) and averaged diameter data for CCh‐, GSK101‐ and NS309‐induced dilatations in the mesenteric arteries from EC‐Kir2.1 −/− mice (right; n = 4 arteries; *P < 0.05 using one‐way ANOVA).
The function of endothelial Kir channels is not altered in angiotensin II‐induced hypertension
Although functional Kir channels have been shown in the ECs from several vascular beds (von Beckerath et al. 1996; Crane et al. 2003; Fang et al. 2005, 2006; Jackson, 2005; Climent et al. 2011), it is unclear whether endothelial dysfunction in hypertension is associated with a reduced function of endothelial Kir channels. Previous studies have shown a reduced expression of Kir2.1 channels (Weston et al. 2010) in mesenteric artery homogenates and an impaired K+‐induced hyperpolarization (Goto et al. 2004) in the mesenteric arteries from SHRs. Given that EC Kir channels are downstream boosters in the TRPV4–IK–Kir vasodilatory pathway, we evaluated the Kir channel function in Ang II‐induced hypertension.
C57BL6 mice were infused with Ang II (1.4 mg kg−1 day−1) for 3 weeks (Kharade et al. 2013; Sonkusare et al. 2014 b), using osmotic minipumps as we have done previously. The Kir currents were studied in freshly isolated ECs from mesenteric arteries from normal (NT) and hypertensive (HT) mice. The Kir current densities at −140 mV were not different between the ECs from NT and HT mice (Fig. 7 A), suggesting that the function of endothelial Kir channels is not affected in this form of hypertension. To confirm that the loss of endothelium‐dependent vasodilatation in hypertension does not involve a dysfunction of endothelial Kir channels, we studied K+‐induced dilatation in the mesenteric arteries from NT and HT mice. The dilatations induced by K+ (14 mm) were also unaffected in the arteries from HT mice (Fig. 7 B and C). The myogenic tone was not different between the arteries from NT and HT mice (27.6 ± 1.2 and 28.5 ± 1.5% in NT and HT mice, respectively). These results suggest that the function of endothelial Kir channels is unaffected in Ang II‐induced hypertension.
Figure 7. The function of endothelial Kir channels is unaffected in angiotensin II‐induced hypertension .
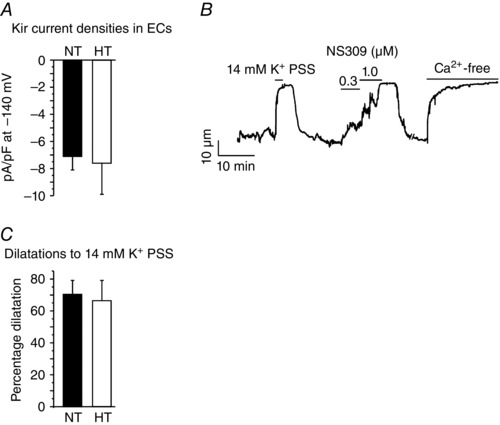
Kir currents were recorded using the perforated patch configuration of the patch‐clamp technique in ECs from third‐order mesenteric arteries. Arterial diameter measurements were performed in third‐order mesenteric arteries constricted by intravascular pressure (80 mmHg). A, Ba2+‐sensitive Kir currents in the ECs from normal (NT) and hypertensive (HT) mice. The bar graph summarizes Kir current densities at −140 mV (n = 8–10 cells; P = 0.8403 using Student's t test). B, a representative diameter trace in a third‐order mesenteric artery from a HT mouse. The bar graph (C) shows summarized diameter data in the arteries from NT and HT mice (n = 4–9 arteries; P = 0.5895 using Student's unpaired t test).
Discussion
Our results support the concept that any endothelium‐dependent vasodilator (e.g. acetycholine) that elevates intracellular Ca2+ and hyperpolarizes the endothelium through the activation of IK and SK channels will engage EC Kir channels to boost their signal strength (Fig. 8). The hyperpolarization (charge) spreads through myoendothelial gap junctions to hyperpolarize and relax the adjacent smooth muscle through deactivation of voltage‐dependent calcium channels (Sandow et al. 2006; Dora, 2010). The hyperpolarization may also spread through gap junctions to adjacent ECs to cause a regenerating electrical signal to promote a propagating vasodilatation (‘vasoconduction’; Welsh & Segal, 1998; Emerson & Segal, 2000).
Figure 8. Endothelial Kir channels boost the dilatations to endothelial vasodilators .
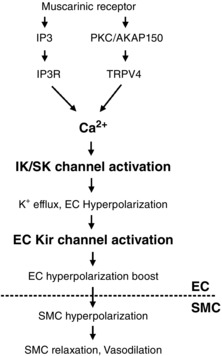
Activation of inositol trisphosphate receptors (IP3Rs) or TRPV4 channels increases endothelial Ca2+, which activates IK/SK channels. The ensuing K+ efflux from the ECs and membrane hyperpolarization can activate endothelial Kir channels, which provide a hyperpolarization boost and amplify the vasodilatory signal. Abbreviations: AKAP150, A‐kinase anchoring protein 150; PKC, protein kinase C.
Strong inward rectifier K+ (Kir2) in ECs
Kir channels have been well characterized in smooth muscle cells in different vascular beds, including cerebral and coronary arteries (Quayle et al. 1997). Based on biophysical properties and genetic knockout studies, the Kir2.1 subtype appears to be crucial for Kir channel function in the SMCs from cerebral arteries (Bradley et al. 1999; Zaritsky et al. 2000). However, Kir channels are not uniformly expressed in vascular smooth muscle; SMCs of mesenteric arteries do not express Kir channels (Crane et al. 2003; Smith et al. 2008; Fig. 1).
Although Kir channel currents have been measured in cultured ECs (Olesen et al. 1988; Zhang et al. 1994; Jacobs et al. 1995; Jow & Numann, 1998; Romanenko et al. 2002; Fang et al. 2005), there have been few measurements of Kir channel properties in native ECs. Barium‐sensitive Kir currents were shown in the ECs from rat mesenteric arteries (Crane et al. 2003; Smith et al. 2008) and hamster cremaster arterioles (Jackson, 2005). Endothelial cells from rat pulmonary arteries (Hogg et al. 2002) and descending vasa recta (Cao et al. 2007) also displayed Kir currents. It was suggested that Kir channels maintain the resting membrane potential in the endothelium from rat mesenteric arteries (Climent et al. 2011). The biophysical properties (strong rectification and shift in activation curve with external K+; Fig. 1) and pharmacology (block by low Ba2+ concentrations and ML‐133; Fig. 1) of Kir currents strongly support the idea that Kir2 channels underlie the membrane currents reported in the present study. In the rat mesenteric artery homogenates, messenger RNAs for Kir2.1, Kir2.2 and Kir2.4 were detected (Goto et al. 2004). Another study confirmed the expression of the Kir2.1 channel at the protein level in rat mesenteric arteries, but did not probe for other channel isoforms (Weston et al. 2010). Our results demonstrating that the inward rectifier currents are substantially reduced in the EC‐Kir2.1 −/− mice (Fig. 3) suggest that Kir2.1 is the crucial isoform for functional Kir channels in the ECs. Out of the 11 ECs from four EC‐Kir2.1 −/− mice, two ECs showed Kir currents in the normal range (Fig. 3 B). This could be explained by the following factors: (i) less than 100% efficiency of Cre excision on the lox pair; and (ii) a compensatory upregulation of another Kir2.x channel isoform in these cells. In sharp contrast to the cells from the knockdown mice, 100% of the ECs from C57BL6 mice (12 ECs from eight mice) showed robust Kir currents. It is surprising that the Kir currents were substantially reduced with elimination of only one Kir isoform, mainly because the expression of at least three different Kir isoforms has been detected previously. This result brings about two possibilities: (i) only the Kir2.1 isoform forms functional channels in the ECs; and (ii) different Kir isoforms form heteromultimers, but the presence of Kir2.1 is crucial for forming heteromultimers that make a functional channel. Indeed, heteromultimeric interactions have been demonstrated between many different Kir channels (Glowatzki et al. 1995; Fakler et al. 1996; Pessia et al. 1996; Schram et al. 2002).
Mechanism of activation of EC Kir channels
Our previous results indicate that the muscarinic receptor agonists, such as carbachol, only activate TRPV4 channels at the myoendothelial projections in mesenteric arteries (Sonkusare et al. 2014 b) and that Ca2+ influx through TRPV4 channels at MEPs primarily activates co‐localized IK channels. Membrane potential hyperpolarization through activation of IK and SK channels would engage all Kir channels in the cell membrane. It has also been proposed that Kir channels are activated by local K+ accumulation in restricted spaces between the endothelial and smooth muscle membranes (Edwards et al. 1998). However, endothelium‐dependent vasodilators only activate the TRPV4–IK pathway at MEPs; therefore, if K+ accumulated it would only happen near MEPs. Our estimation of the number of channels indicates that each EC expresses ∼144 functional Kir channels (see Methods for the calculation of the number of channels). With a cell surface area of ∼1600 μm2, this corresponds to an extremely low channel density of one Kir channel per 11 μm2. Considering the low density of Kir channels, it is likely that the primary activator of EC Kir channels is membrane potential hyperpolarization. Membrane hyperpolarization through IK/SK channels will activate Kir channels throughout the EC membrane to boost vasodilatory signals. Local K+ accumulation at MEPs could contribute to this amplification; however, there is no evidence to support this conjecture.
Potassium‐induced vasodilatation in rat mesenteric arteries preconstricted with phenylephrine has previously been attributed to activation of Na+,K+‐ATPase (Crane et al. 2003; Smith et al. 2008). In rat femoral arteries constricted with noradrenaline, both Kir channels and Na+,K+‐ATPase were required for the vasodilatation in response to K+ (Savage et al. 2003). However, our results demonstrating that the Kir channel inhibitors almost completely inhibit K+‐induced vasodilatation in the arteries with myogenic tone and the K+‐induced dilatation is absent in the arteries from EC‐Kir2.1 −/− mice (Fig. 4) suggest that endothelial Kir channels mediate the vasodilatation in response to external K+ in the mouse small mesenteric arteries. Whether these differences in the results could be explained by the method used to constrict the arteries (phenylephrine/noradrenaline vs. intravascular pressure) is not known. In the rat mesenteric arteries, Dora & Garland (2001) reported that external K+ induces vasodilatation via Kir channels in the presence of endothelium; results that are consistent with our findings.
Endothelial cell Kir channels and hypertension
We have previously shown that muscarinic receptor stimulation activates TRPV4 channels through protein kinase C anchored to A‐kinase anchoring protein 150 (AKAP150) and that Ang II‐induced hypertension (3 weeks) leads to the loss of AKAP150, which is concentrated at the MEPs. This loss of AKAP150 uncouples TRPV4 channels from receptor activation, and thereby cripples endothelium‐dependent vasodilatation (Sonkusare et al. 2014 b). Our results indicate that Kir channel function and K+‐induced vasodilatation are unaltered in Ang II‐induced hypertension (Fig. 7), which further supports the concept that the signalling elements upstream of IK/SK channels in the TRPV4–IK/SK–Kir pathway are involved in the endothelial dysfunction in hypertension. This is different from what has been reported for mesenteric arteries in SHRs. Weston et al. (2010) indicated that expression of the Kir2.1 channel is decreased at the protein level in whole‐artery homogenates from mesenteric arteries of SHRs. Another study demonstrated a key role for EC Kir channels in conducted vasodilatation in mesenteric arteries from normotensive control rats but not from SHRs. Moreover, the K+‐induced, endothelium‐dependent hyperpolarization of SMCs was absent in the arteries from SHRs (Goto et al. 2004). These studies indicate impairment in the expression and function of endothelial Kir channels in the arteries from SHRs. There is no evidence on impairment of endothelial Kir channel function in a non‐genetic model of hypertension, and our results reveal unaltered Kir channel function in Ang II‐induced hypertension. Tajada et al. (2012) indicated small Kir currents in the SMCs from the mesenteric arteries from BPN mice (Jackson Laboratories). In that study, Tajada et al. (2012) also report that the function of Kir channels is impaired in the SMCs from hypertensive mice. We were unable to detect any Ba2+‐sensitive currents in the SMCs from small mesenteric arteries from the C57BL6 mice. The strain differences in Kir channel function could be one possible reason for the divergence between the two studies.
Our results imply that endothelial Kir2.1 channels are crucial downstream amplifiers for the vasodilatory signals in response to increase in endothelial Ca2+ and activation of IK/SK channels. Circulating factors and mechanical stimuli that increase endothelial Ca2+ may recruit Kir2.1 channels to regulate the blood flow. Although the function of endothelial Kir2.1 channels is not altered in Ang II‐induced hypertension, it remains to be seen whether these channels are affected in other vascular disorders and other forms of hypertension. Indeed, it has been shown that hypercholesterolaemia suppresses Kir channel function in the aortic endothelium (Fang et al. 2006). Endothelial Kir2.1 channels could therefore be targeted to treat endothelial dysfunction and impairment of tissue perfusion in vascular disorders.
Additional information
Competing interests
None declared.
Author contributions
S.K.S. and M.T.N. conceptualized and designed the study; S.K.S., T.D. and A.D.B. acquired and analysed the data in the laboratory of M.T.N.; S.K.S. and M.T.N. interpreted the data; and S.K.S. and T.D. drafted the manuscript. All authors contributed to editing and revising the manuscript. All authors have approved the final version of the manuscript and agree to be accountable for all aspects of the work. All persons designated as authors qualify for authorship, and all those who qualify for authorship are listed.
Funding
We would like to acknowledge the grants from National Institutes of Health to S.K.S. (R00HL121484‐02), a postdoctoral fellowship from the Lundbeck Foundation to T.D., and M.T.N. (National Institutes of Health, HL044455, 1P01HL095488, R37DK053832, R01HL098243, 1R01HL131181‐01 and R01HL121706‐01; Totman Medical Research Trust, Horizon 2020 and Fondation Leducq).
References
- Benham CD, Bolton TB, Lang RJ & Takewaki T (1986). Calcium‐activated potassium channels in single smooth muscle cells of rabbit jejunum and guinea‐pig mesenteric artery. J Physiol 371, 45–67. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bogatcheva NV, Sergeeva MG, Dudek SM & Verin AD (2005). Arachidonic acid cascade in endothelial pathobiology. Microvasc Res 69, 107–127. [DOI] [PubMed] [Google Scholar]
- Bradley KK, Jaggar JH, Bonev AD, Heppner TJ, Flynn ER, Nelson MT & Horowitz B (1999). Kir2.1 encodes the inward rectifier potassium channel in rat arterial smooth muscle cells. J Physiol 515, 639–651. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Busse R, Edwards G, Feletou M, Fleming I, Vanhoutte PM & Weston AH (2002). EDHF: bringing the concepts together. Trends Pharmacol Sci 23, 374–380. [DOI] [PubMed] [Google Scholar]
- Cao C, Lee‐Kwon W, Payne K, Edwards A & Pallone TL (2007). Descending vasa recta endothelia express inward rectifier potassium channels. Am J Physiol Renal Physiol 293, F1248–F1255. [DOI] [PubMed] [Google Scholar]
- Climent B, Zsiros E, Stankevicius E, de la Villa P, Panyi G, Simonsen U, García‐Sacristán A & Rivera L (2011). Intact rat superior mesenteric artery endothelium is an electrical syncytium and expresses strong inward rectifier K+ conductance. Biochem Biophys Res Commun 410, 501–507. [DOI] [PubMed] [Google Scholar]
- Crane GJ, Walker SD, Dora KA & Garland CJ (2003). Evidence for a differential cellular distribution of inward rectifier K channels in the rat isolated mesenteric artery. J Vasc Res 40, 159–168. [DOI] [PubMed] [Google Scholar]
- Dora KA (2010). Coordination of vasomotor responses by the endothelium. Circ J 74, 226–232. [DOI] [PubMed] [Google Scholar]
- Dora KA & Garland CJ (2001). Properties of smooth muscle hyperpolarization and relaxation to K+ in the rat isolated mesenteric artery. Am J Physiol Heart Circ Physiol 280, H2424–H2429. [DOI] [PubMed] [Google Scholar]
- Edwards G, Dora KA, Gardener MJ, Garland CJ & Weston AH (1998). K+ is an endothelium‐derived hyperpolarizing factor in rat arteries. Nature 396, 269–272. [DOI] [PubMed] [Google Scholar]
- Emerson GG & Segal SS (2000). Endothelial cell pathway for conduction of hyperpolarization and vasodilation along hamster feed artery. Circ Res 86, 94–100. [DOI] [PubMed] [Google Scholar]
- Fakler B, Bond CT, Adelman JP & Ruppersberg JP (1996). Heterooligomeric assembly of inward‐rectifier K+ channels from subunits of different subfamilies: Kir2.1 (IRK1) and Kir4.1 (BIR10). Pflugers Arch 433, 77–83. [DOI] [PubMed] [Google Scholar]
- Fang Y, Mohler ER 3rd, Hsieh E, Osman H, Hashemi SM, Davies PF, Rothblat GH, Wilensky RL & Levitan I (2006). Hypercholesterolemia suppresses inwardly rectifying K+ channels in aortic endothelium in vitro and in vivo. Circ Res 98, 1064–1071. [DOI] [PubMed] [Google Scholar]
- Fang Y, Schram G, Romanenko VG, Shi C, Conti L, Vandenberg CA, Davies PF, Nattel S & Levitan I (2005). Functional expression of Kir2.x in human aortic endothelial cells: the dominant role of Kir2.2. Am J Physiol Cell Physiol 289, C1134–C1144. [DOI] [PubMed] [Google Scholar]
- Filosa JA, Bonev AD, Straub SV, Meredith AL, Wilkerson MK, Aldrich RW & Nelson MT (2006). Local potassium signaling couples neuronal activity to vasodilation in the brain. Nat Neurosci 9, 1397–1403. [DOI] [PubMed] [Google Scholar]
- Fleming I & Busse R (1999). NO: the primary EDRF. J Mol Cell Cardiol 31, 5–14. [DOI] [PubMed] [Google Scholar]
- Garland CJ, Hiley CR & Dora KA (2011). EDHF: spreading the influence of the endothelium. Br J Pharmacol 164, 839–852. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Glowatzki E, Fakler G, Brändle U, Rexhausen U, Zenner HP, Ruppersberg JP & Fakler B (1995). Subunit‐dependent assembly of inward‐rectifier K+ channels. Proc Biol Sci 261, 251–261. [DOI] [PubMed] [Google Scholar]
- Goldman DE (1943). Potential, impedance, and rectification in membranes. J Gen Physiol 27, 37–60. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Goto K, Rummery NM, Grayson TH & Hill CE (2004). Attenuation of conducted vasodilatation in rat mesenteric arteries during hypertension: role of inwardly rectifying potassium channels. J Physiol 561, 215–231. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hodgkin AL & Katz B (1949). The effect of sodium ions on the electrical activity of giant axon of the squid. J Physiol 108, 37–77. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hogg DS, McMurray G & Kozlowski RZ (2002). Endothelial cells freshly isolated from small pulmonary arteries of the rat possess multiple distinct K+ current profiles. Lung 180, 203–214. [DOI] [PubMed] [Google Scholar]
- Isakson BE & Duling BR (2005). Heterocellular contact at the myoendothelial junction influences gap junction organization. Circ Res 97, 44–51. [DOI] [PubMed] [Google Scholar]
- Jackson WF (2005). Potassium channels in the peripheral microcirculation. Microcirculation 12, 113–127. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jacobs ER, Cheliakine C, Gebremedhin D, Birks EK, Davies PF & Harder DR (1995). Shear activated channels in cell‐attached patches of cultured bovine aortic endothelial cells. Pflugers Arch 431, 129–131. [DOI] [PubMed] [Google Scholar]
- Jow F & Numann R (1998). Divalent ion block of inward rectifier current in human capillary endothelial cells and effects on resting membrane potential. J Physiol 512, 119–128. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kharade SV, Sonkusare SK, Srivastava AK, Thakali KM, Fletcher TW, Rhee SW & Rusch NJ (2013). The β3 subunit contributes to vascular calcium channel upregulation and hypertension in angiotensin II‐infused C57BL/6 mice. Hypertension 61, 137–142. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kochukov MY, Balasubramanian A, Abramowitz J, Birnbaumer L & Marrelli SP (2014). Activation of endothelial transient receptor potential C3 channel is required for small conductance calcium‐activated potassium channel activation and sustained endothelial hyperpolarization and vasodilation of cerebral artery. J Am Heart Assoc 3, e000913. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Köhler R, Heyken WT, Heinau P, Schubert R, Si H, Kacik M, Busch C, Grgic I, Maier T & Hoyer J (2006). Evidence for a functional role of endothelial transient receptor potential V4 in shear stress‐induced vasodilatation. Arterioscler Thromb Vasc Biol 26, 1495–1502. [DOI] [PubMed] [Google Scholar]
- Ledoux J, Taylor MS, Bonev AD, Hannah RM, Solodushko V, Shui B, Tallini Y, Kotlikoff MI & Nelson MT (2008). Functional architecture of inositol 1,4,5‐trisphosphate signaling in restricted spaces of myoendothelial projections. Proc Natl Acad Sci USA 105, 9627–9632. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Leech CA & Stanfield PR (1981). Inward rectification in frog skeletal muscle fibres and its dependence on membrane potential and external potassium. J Physiol 319, 295–309. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Longden TA & Nelson MT (2015). Vascular inward rectifier K+ channels as external K+ sensors in the control of cerebral blood flow. Microcirculation 22, 183–196. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Marrelli SP, O'Neil R G, Brown RC & Bryan RM Jr (2007). PLA2 and TRPV4 channels regulate endothelial calcium in cerebral arteries. Am J Physiol Heart Circ Physiol 292, H1390–H1397. [DOI] [PubMed] [Google Scholar]
- Mather S, Dora KA, Sandow SL, Winter P & Garland CJ (2005). Rapid endothelial cell‐selective loading of connexin 40 antibody blocks endothelium‐derived hyperpolarizing factor dilation in rat small mesenteric arteries. Circ Res 97, 399–407. [DOI] [PubMed] [Google Scholar]
- Nelson MT & Quayle JM (1995). Physiological roles and properties of potassium channels in arterial smooth muscle. Am J Physiol Cell Physiol 268, C799–C822. [DOI] [PubMed] [Google Scholar]
- Olesen SP, Davies PF & Clapham DE (1988). Muscarinic‐activated K+ current in bovine aortic endothelial cells. Circ Res 62, 1059–1064. [DOI] [PubMed] [Google Scholar]
- Pessia M, Tucker SJ, Lee K, Bond CT & Adelman JP (1996). Subunit positional effects revealed by novel heteromeric inwardly rectifying K+ channels. EMBO J 15, 2980–2987. [PMC free article] [PubMed] [Google Scholar]
- Quayle JM, McCarron JG, Brayden JE & Nelson MT (1993). Inward rectifier K+ currents in smooth muscle cells from rat resistance‐sized cerebral arteries. Am J Physiol Cell Physiol 265, C1363–C1370. [DOI] [PubMed] [Google Scholar]
- Quayle JM, Nelson MT & Standen NB (1997). ATP‐sensitive and inwardly rectifying potassium channels in smooth muscle. Physiol Rev 77, 1165–1232. [DOI] [PubMed] [Google Scholar]
- Romanenko VG, Rothblat GH & Levitan I (2002). Modulation of endothelial inward‐rectifier K+ current by optical isomers of cholesterol. Biophys J 83, 3211–3222. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sandow SL, Neylon CB, Chen MX & Garland CJ (2006). Spatial separation of endothelial small‐ and intermediate‐conductance calcium‐activated potassium channels (KCa) and connexins: possible relationship to vasodilator function? J Anat 209, 689–698. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sandow SL, Senadheera S, Bertrand PP, Murphy TV & Tare M (2012). Myoendothelial contacts, gap junctions, and microdomains: anatomical links to function? Microcirculation 19, 403–415. [DOI] [PubMed] [Google Scholar]
- Savage D, Perkins J, Hong Lim C & Bund SJ (2003). Functional evidence that K+ is the non‐nitric oxide, non‐prostanoid endothelium‐derived relaxing factor in rat femoral arteries. Vascul Pharmacol 40, 23–28. [DOI] [PubMed] [Google Scholar]
- Schram G, Melnyk P, Pourrier M, Wang Z & Nattel S (2002). Kir2.4 and Kir2.1 K+ channel subunits co‐assemble: a potential new contributor to inward rectifier current heterogeneity. J Physiol 544, 337–349. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Smith PD, Brett SE, Luykenaar KD, Sandow SL, Marrelli SP, Vigmond EJ & Welsh DG (2008). KIR channels function as electrical amplifiers in rat vascular smooth muscle. J Physiol 586, 1147–1160. [DOI] [PMC free article] [PubMed] [Google Scholar]
- So I, Ashmole I, Davies NW, Sutcliffe MJ & Stanfield PR (2001). The K+ channel signature sequence of murine Kir2.1: mutations that affect microscopic gating but not ionic selectivity. J Physiol 531, 37–50. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sonkusare S, Dalsgaard T, Bonev A, Hill‐Eubanks D, Kotlikoff M, Santana L, Scott J & Nelson M (2014. a). AKAP150‐dependent cooperative TRPV4 channel gating is central to endothelium‐dependent vasodilation and is disrupted in hypertension (678.10). FASEB J 28, http://www.fasebj.org/content/28/1_Supplement/678.10.abstract?sid=806ac414‐eca6‐4c30‐ae52‐039a72d7dd76. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sonkusare SK, Bonev AD, Ledoux J, Liedtke W, Kotlikoff MI, Heppner TJ, Hill‐Eubanks DC & Nelson MT (2012). Elementary Ca2+ signals through endothelial TRPV4 channels regulate vascular function. Science 336, 597–601. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sonkusare SK, Dalsgaard T, Bonev AD, Hill‐Eubanks DC, Kotlikoff MI, Scott JD, Santana LF & Nelson MT (2014. b). AKAP150‐dependent cooperative TRPV4 channel gating is central to endothelium‐dependent vasodilation and is disrupted in hypertension. Sci Signal 7, ra66. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tajada S, Cidad P, Moreno‐Domínguez A, Pérez‐García MT & López‐López JR (2012). High blood pressure associates with the remodelling of inward rectifier K+ channels in mice mesenteric vascular smooth muscle cells. J Physiol 590, 6075–6091. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tallini YN, Brekke JF, Shui B, Doran R, Hwang SM, Nakai J, Salama G, Segal SS & Kotlikoff MI (2007). Propagated endothelial Ca2+ waves and arteriolar dilation in vivo: measurements in Cx40BAC–GCaMP2 transgenic mice. Circ Res 101, 1300–1309. [DOI] [PubMed] [Google Scholar]
- Tallini YN, Ohkura M, Choi BR, Ji G, Imoto K, Doran R, Lee J, Plan P, Wilson J, Xin HB, Sanbe A, Gulick J, Mathai J, Robbins J, Salama G, Nakai J & Kotlikoff MI (2006). Imaging cellular signals in the heart in vivo: cardiac expression of the high‐signal Ca2+ indicator GCaMP2. Proc Natl Acad Sci USA 103, 4753–4758. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Taylor MS, Bonev AD, Gross TP, Eckman DM, Brayden JE, Bond CT, Adelman JP & Nelson MT (2003). Altered expression of small‐conductance Ca2+‐activated K+ (SK3) channels modulates arterial tone and blood pressure. Circ Res 93, 124–131. [DOI] [PubMed] [Google Scholar]
- von Beckerath N, Dittrich M, Klieber HG & Daut J (1996). Inwardly rectifying K+ channels in freshly dissociated coronary endothelial cells from guinea‐pig heart. J Physiol 491, 357–365. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wang HR, Wu M, Yu H, Long S, Stevens A, Engers DW, Sackin H, Daniels JS, Dawson ES, Hopkins CR, Lindsley CW, Li M & McManus OB (2011). Selective inhibition of the Kir2 family of inward rectifier potassium channels by a small molecule probe: the discovery, SAR, and pharmacological characterization of ML133. ACS Chem Biol 6, 845–856. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Welsh DG & Segal SS (1998). Endothelial and smooth muscle cell conduction in arterioles controlling blood flow. Am J Physiol Heart Circ Physiol 274, H178–H186. [DOI] [PubMed] [Google Scholar]
- Weston AH, Porter EL, Harno E & Edwards G (2010). Impairment of endothelial SKCa channels and of downstream hyperpolarizing pathways in mesenteric arteries from spontaneously hypertensive rats. Br J Pharmacol 160, 836–843. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wu M, Wang H, Yu H, Makhina E, Xu J, Dawson ES, Hopkins CR, Lindsley CW, McManus OB & Li M (2010). A potent and selective small molecule Kir2.1 inhibitor In Probe Reports from the NIH Molecular Libraries Program. Bethesda, MD, USA: Available from: http://www.ncbi.nlm.nih.gov/books/NBK50692 [PubMed] [Google Scholar]
- Yamamoto Y, Imaeda K & Suzuki H (1999). Endothelium‐dependent hyperpolarization and intercellular electrical coupling in guinea‐pig mesenteric arterioles. J Physiol 514, 505–513. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yang D, MacCallum DK, Ernst SA & Hughes BA (2003). Expression of the inwardly rectifying K+ channel Kir2.1 in native bovine corneal endothelial cells. Invest Ophthalmol Vis Sci 44, 3511–3519. [DOI] [PubMed] [Google Scholar]
- Zaritsky JJ, Eckman DM, Wellman GC, Nelson MT & Schwarz TL (2000). Targeted disruption of Kir2.1 and Kir2.2 genes reveals the essential role of the inwardly rectifying K+ current in K+‐mediated vasodilation. Circ Res 87, 160–166. [DOI] [PubMed] [Google Scholar]
- Zhang DX, Mendoza SA, Bubolz AH, Mizuno A, Ge ZD, Li R, Warltier DC, Suzuki M & Gutterman DD (2009). Transient receptor potential vanilloid type 4‐deficient mice exhibit impaired endothelium‐dependent relaxation induced by acetylcholine in vitro and in vivo. Hypertension 53, 532–538. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhang H, Inazu M, Weir B & Daniel E (1994). Endothelin‐1 inhibits inward rectifier potassium channels and activates nonspecific cation channels in cultured endothelial cells. Pharmacology 49, 11–22. [DOI] [PubMed] [Google Scholar]