

Abstract
Key points
The present study investigated the mechanism associated with impaired cardiac mechanosensing that leads to heart failure by examining the factors regulating muscle LIM protein subcellular distribution in myocytes.
In myocytes, muscle LIM protein subcellular distribution is regulated by cell contractility rather than passive stretch via heme oxygenase‐1 and histone deacetylase signalling. The result of the present study provide new insights into mechanotransduction in cardiac myocytes.
Myocyte mechanosensitivity, as indicated by the muscle LIM protein ratio, is also correlated with cardiac function in the transition to failure in a guinea‐pig model of disease. This shows that the loss mechanosensitivity plays an important role during the transition to failure in the heart.
The present study provides the first indication that mechanosensing could be modified pharmacologically during the transition to heart failure.
Abstract
Impaired mechanosensing leads to heart failure and a decreased ratio of cytoplasmic to nuclear CSRP3/muscle LIM protein (MLP ratio) is associated with a loss of mechanosensitivity. In the present study, we tested whether passive or active stress/strain was important in modulating the MLP ratio and determined whether this correlated with heart function during the transition to failure. We exposed cultured neonatal rat myocytes to a 10% cyclic mechanical stretch at 1 Hz, or electrically paced myocytes at 6.8 V (1 Hz) for 48 h. The MLP ratio decreased by 50% (P < 0.05, n = 4) only in response to electrical pacing, suggesting impaired mechanosensitivity. Inhibition of contractility with 10 μm blebbistatin resulted in an ∼3‐fold increase in the MLP ratio (n = 8, P < 0.05), indicating that myocyte contractility regulates nuclear MLP. Inhibition of histone deacetylase (HDAC) signalling with trichostatin A increased nuclear MLP following passive stretch, suggesting that HDACs block MLP nuclear accumulation. Inhibition of heme oxygenase1 (HO‐1) activity with protoporphyrin IX zinc(II) blocked MLP nuclear accumulation. To examine how mechanosensitivity changes during the transition to heart failure, we studied a guinea‐pig model of angiotensin II infusion (400 ng kg–1 min–1) over 12 weeks. Using subcellular fractionation, we showed that the MLP ratio increased by 88% (n = 4, P < 0.01) during compensated hypertrophy but decreased significantly during heart failure (P < 0.001, n = 4). The MLP ratio correlated significantly with the E/A ratio (r = 0.71, P < 0.01, n = 12), a clinical measure of diastolic function. These data indicate for the first time that myocyte mechanosensitivity as indicated by the MLP ratio is regulated primarily by myocyte contractility via HO‐1 and HDAC signalling.
Key points
The present study investigated the mechanism associated with impaired cardiac mechanosensing that leads to heart failure by examining the factors regulating muscle LIM protein subcellular distribution in myocytes.
In myocytes, muscle LIM protein subcellular distribution is regulated by cell contractility rather than passive stretch via heme oxygenase‐1 and histone deacetylase signalling. The result of the present study provide new insights into mechanotransduction in cardiac myocytes.
Myocyte mechanosensitivity, as indicated by the muscle LIM protein ratio, is also correlated with cardiac function in the transition to failure in a guinea‐pig model of disease. This shows that the loss mechanosensitivity plays an important role during the transition to failure in the heart.
The present study provides the first indication that mechanosensing could be modified pharmacologically during the transition to heart failure.
Abbreviations
- αβ‐Crys
αβ crystalline
- Ag II
angiotensin II
- AMPK
AMP kinase
- ATF2
activating transcription factor 2
- DAPI
4′,6‐diamidino‐2‐phenylindole
- GAPDH
glyceraldehyde 3 phosphate
- HDAC
histone deacetylase
- HO‐1
heme oxygenase 1
- HSP60
heat shock protein 60
- LPP
lipoma preferred partner
- MLP
muscle LIM protein
- PPZ II
protoporphyrin IX zinc(II)
- TSA
trichostatin A
Introduction
Myocytes within the heart experience cyclic alterations in passive forces caused by changes in blood volume and pressure during diastole, as well as active forces generated by the contracting sarcomeres. Prolonged increases in haemodynamic load can lead to cardiac hypertrophy and heart failure (Lorell & Carabello, 2000). Cardiac adaptation is mediated through mechanosensing proteins and their impairment leads to heart failure (Buyandelger et al. 2014).
CSRP3 or muscle LIM protein (MLP) forms part of the mechanosensing complex in the sarcomere with telethonin, titin and α‐actinin (Knoll et al. 2002; Buyandelger et al. 2014). Mutations in the gene for MLP lead to cardiomyopathy in humans (Newman et al. 2005; Geier et al. 2008). We have previously shown that there is an increase in nuclear MLP along with a loss of cytoplasmic fraction in human heart failure and also in rodent models of disease (Boateng et al. 2007). This loss impairs the mechanosensitivity of the heart by altering the MLP stoichiometry with the other proteins within the cytoplasmic complex (Boateng et al. 2009). In the nucleus, MLP plays a role in the regulation of muscle‐specific gene expression (Gardiwal et al. 2007) and, in myocytes, it is involved in the regulation of protein synthesis (Boateng et al. 2009). We have reasoned that prolonged haemodynamic stress increases the loss of cytoplasmic MLP as a result of refraction of the mechanosensing elements (Boateng et al. 2009). We propose that MLP nuclear transit is essential for normal function and adaptive physiology, whereas nuclear accumulation is associated with pathology. This contrast between nuclear protein transit and accumulation has been observed for other proteins We showed that lipoma preferred partner (LPP), a focal adhesion protein required for myocyte myofibrillogenesis (Hooper et al. 2013), also transits through the nucleus without accumulating in the absence of CRM1‐dependent inhibition of nuclear export (Hooper et al. 2012). However, the accumulation of LPP in the nucleus is associated with disease in non‐cardiac tissue (Crombez et al. 2005). Similalrly, in myocytes, we suggest that the accumulation of nuclear MLP is associated with cardiac pathology. In myocytes, nuclear accumulation of MLP is also associated with the loss of the cytoplasmic fraction (Boateng et al. 2007). In the present study, we hypothesized that MLP subcellular distribution and mechanosensitivity are regulated by histone deacetylase (HDAC) signalling and heme oxygenase 1 (HO‐1). HDAC4 can directly deacetylate MLP in the sarcomere (Gupta et al. 2008). HO‐1 is upregulated in the failing heart and is anti‐hypertrophic (Hu et al. 2004). The gene for HO‐1 contains regulatory sequences for the binding of transcription factors whose activation is associated with cellular stress and cell/tissue injury (Lavrovsky et al. 1994). Previous work has shown that shear stress upregulates HO‐1 in smooth muscle cells, indicating that the protein may be involved in mechanotransduction (Wagner et al. 1997).
It is unclear how MLP nuclear accumulation is regulated in heart failure, as well as in response to different types of mechanical stimuli. In the present study, we aimed to determine the type of mechanical stresses (passive diastolic or active systolic) that modulate the loss of mechanosensing as measured by the MLP nuclear ratio. We then determined whether HDAC and HO‐1 signalling could influence this process. Our data strongly suggest that it is increased cross‐bridge activity rather than passive stretch that regulates nuclear MLP accumulation. Nuclear accumulation of MLP in response to passive mechanical stretch only occurred in combination with an HDAC inhibitor. By contrast, inhibition of HO‐1 blocked MLP nuclear accumulation in response to electrical pacing. Our data show that the MLP ratio improves during compensated hypertrophy but drops sharply during the transition to failure. The MLP ratio (a measure of cytoplasmic to nuclear MLP) also correlated significantly with the E/A ratio, a clinical marker of diastolic dysfunction. This suggests that there is a direct functional link between the MLP ratio and the mechanosensing capacity of the heart during increased overload and in the transition to heart failure.
Methods
Guinea‐pig model of angiotensin II‐induced heart failure
Guinea‐pigs were divided into two groups: a saline control group and an angiotensin II treatment group. All experiments were performed in accordance with NIH guidelines (Guide for the Care and Use of Laboratory Animals) and the guidelines from Directive 2010/63/EU of the European Parliament on the protection of animals used for scientific purposes. During all invasive surgical procedures, animals were initially anaesthetized by inhalation of 2.0% isoflurane with 100% oxygen in a closed chamber after which the anaesthesia levels were maintained during surgery via a nose cone. The levels of anaesthesia were constantly monitored by toe pinch and eye reflexes. Angiotensin II pumps were surgically implanted (400 ng kg–1 min–1)/saline pumps (0.9%) into female Dunkin‐Hartley guinea‐pigs. Surgery was performed at Loyola University (Maywood, IL, USA). All animal procedures were reviewed and approved by the institutional animal care and use committee. Animals weighed ∼400 g at the time of surgery and were 4–6 weeks old. Angiotensin II‐treated animals developed gradual pressure overload that advanced into diastolic heart failure and were euthanized at 4, 8 and 12 weeks; sham‐operated animals served as age‐matched controls. At 4 weeks, hearts from angiotensin II‐treated animals experienced early stage pressure overload and hypertrophy. At 8 weeks, hearts from angiotensin II‐treated animals had undergone fully compensated hypertrophy. Hearts taken from angiotensin II‐treated animals at 12 weeks had undergone decompensated hypertrophy and failure. For tissue collection, animals were put under deep anaesthesia by an overdose of isoflurane (5%) at the end of the respective study period. Immediately after the beating heart was excised from the chest, it was perfused with ice‐cold phosphate‐buffered saline, weighed for its chamber masses and snap frozen. Tissue samples were later shipped to the UK on dry ice for further analysis.
Myocyte cell culture and treatments
Animal experiments were performed in accordance with the Institutional Animal Care of the University of Reading, UK and in accordance with Home Office Guidelines of the Animals (Scientific Procedures) Inspectorate, UK. Myocytes were isolated from the cardiac ventricles harvested from 1‐ to 2‐day‐old Sprague–Dawley rats (which had been killed by cervical dislocation followed by decapitation) by sequential collagenase digestion as described previously (Boateng et al. 2007). Briefly, cells were plated (1 million cells cm–2) on fibronectin (25 μmol ml–1)‐coated Bioflex dishes in PC1 medium (Lonza, Allendale, NJ, USA) for 48 h and transferred to a Dulbecco's modified Eagle's medium: M199 serum free medium. Myocytes were cyclically stretched at 10% maximum strain at 1 Hz for 48 h using the Flexcell 4000 (Flexcell International, Burlington, NC, USA). The cyclic stretch was performed without the posts. For pacing experiments, myocytes were paced at 6.8 V (1 Hz) for 48 h using the IonOptix C‐pace EP system (IonOptix LLC, Westwood, MA, USA). Myocytes were also stretched in combination with electrical pacing. The stimuli were combined by connecting the Flexcell computer signal normally going to the Flexlink to the ‘Advance In’ connection of the IonOptix C‐pace instrument. This allowed the pulse signal from the Flexcell to be synchronized with the electrical pulse delivered by the C‐pace. The electrical pulse was delayed by 0.5 s to ensure that the contraction of the myocytes would coincide with the maximum period of mechanical strain as shown in Fig. 4 A and B. To inhibit myocyte contraction, myocytes were treated with 10 μm blebbistatin for 48 h in combination with stretch and pacing. To study the effect of HDAC inhibition on the nuclear shuttling of MLP, myocytes were treated with 85 nm trichostatin A (TSA) for 48 h in combination with cyclic mechanical stretch. MLP nuclear shuttling was also studied after inhibition of HO‐1 signalling using 10 μm of protoporphyrin IX zinc(II) (PPZII) in combination with electrical pacing for 48 h. The solvent for PPZII was 0.3% DMSO and 0.03% for TSA/blebbistatin treatments. In each case, vehicle‐treated cells were used as a control. We have established that 0.03% does not interfere with MLP nuclear shuttling; however, 0.3% resulted in a slight reduction of nuclear MLP translocation. For inhibition of MLP nuclear translocation, myocytes were treated with synthetic peptides containing the fibroblast growth factor membrane translocating peptide sequence AAVALLPAVLLALLAP with RKYGPK, the nuclear localization sequence of MLP (i.e. AAVALLPAVLLALLAPRKYGPK), as described previously (Boateng et al. 2009). As a control, only AAVALLPAVLLALLAP was used. Peptides were dissolved in ethanol and 20 μm was used to treat the myocytes.
Figure 4. Combined stretch and electrical pacing augments HO‐1 but not nuclear MLP .
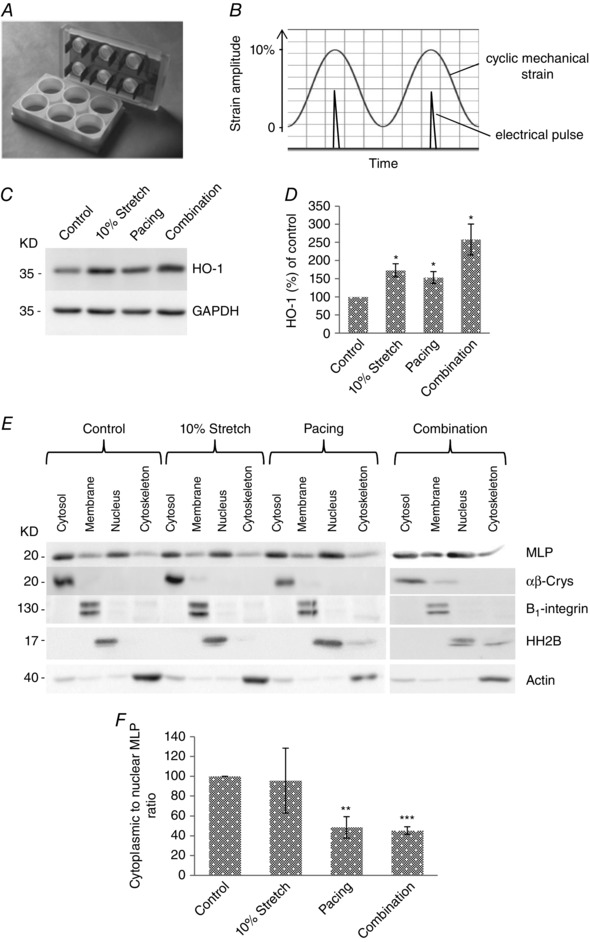
A, electrodes from the IonOptix C‐pace were inserted into the Flexcell six well dishes, allowing myocytes to be simultaneously stretched and paced. B, cells were stretched up to 10% maximal strain and the electrical pulse was applied to coincide with the point of maximal stain. C, western blot of HO‐1 and GAPDH in cultured neonatal myocytes following stretch, pacing and combined stimuli. D, quantification of HO‐1 protein following treatments. E, western blot to assess the subcellular distribution of MLP in cells following 10% stretch/strain, electrical pacing and combination of 10% stretch/strain and electrical pacing. αβ‐Crys, integrin β1, histine H2B and actin have been used as the marker of the cytoplasmic, membrane, nuclear and cytoskeletal pool of protein, respectively, to verify the fractionation process. F, quantification of MLP presented as cytoplasmic to nuclear ratio in cells following treatments. N ≥ 4 cultures.*P < 0.05, **P < 0.01 and ***P < 0.001 compared to control.
Cellular composition and subcellular fractionation
For subcellular fractionation of myocytes, the Proteo Extract Subcellular Proteome Kit from Calbiochem (San Diego, CA, USA) was used in accordance with the manufacturer's instructions and as described previously (Boateng et al. 2009). This method uses a detergent‐based protocol for cell fractionation. Cellular proteins were sequentially extracted into four compartments: cytosolic, membrane/organelles, nuclei and cytoskeleton. After each fraction was removed, cells were observed by microscopy to ensure that they were still attached to the dish. Cell integrity is maintained throughout the fractionation process. For subcellular fractionation of guinea‐pig heart tissue, the NE‐PER Nuclear and Cytoplasmic Extraction Kit from Pierce Biotechnology (Rockford, IL, USA) was used in accordance with the manufacturer's instructions. The fractions were then frozen at –80°C prior to analysis. The accuracy of the fractionation method was verified using antibodies to proteins with a well‐documented subcellular distribution. GAPDH and α‐β crystalline were used to verify the cytosolic fraction; β1 integrin and HO‐1 were used for the membrane fraction; histone H2B and ATF2 were used for the nuclear fraction; and actin was used for the cytoskeletal fraction.
Real‐time PCR gene expression
Guinea‐pig left ventricular tissue was homogenized using TissueRupter (Qiagen) to mechanically break up the tissue. RNA was extracted from the tissue lysate using Qiagen RNeasy Fibrous tissue midi kit (Qiagen) in accordance with the manufacturer's instructions. For each experimental and control group, a minimum of five guinea‐pig left ventricle samples was analysed. The concentration of RNA was determined by absorbance at 260 nm using a Nanodrop ND‐1000 spectrophotometer (Thermo Scientific, Waltham, MA, USA). Then 0.5–2 ug of total RNA was used to synthesize cDNA using a High Capacity cDNA Reverse Transcription Kit (Applied Biosystems, Foster City, CA, USA) in accordance with the manufacturer's instructions. Guinea‐pig specific genes sequences were obtained from Ensemble Genome browser (http://www.ensembl.org) and primers were designed using Primer‐3 software (http://primer3.ut.ee) (ANP: Forward, CCCCATGTACAATGTCGTGT, Reverse, TAAGGGCATCTTCTCCTCCAAACTA1; Forward, TGCTGTCCCTCTATGCCTCT, Reverse, CAGGGCATAGCCCTCATAGA). The concentrations of primers and cDNA were optimized to give a PCR efficiency of between 90% and 100 % and Ct values below 30. For each reaction 1 × SYBR Green PCR master mix (Applied Biosystems) was mixed with 900 nm of reverse and forward primer, 5 ng of cDNA and dH2O to bring the final volume up to 20 μl. PCR reactions were run on 7500 FAST system (Applied Biosystems) using a fast protocol of 95°C for 10 min to activate enzyme, then 40 cycles of denature at 95°C for 1 s and annealing and extension at 60°C for 20 s. The results were analysed using Excel (Microsoft Corp., Redmond, WA, USA).
Western blotting for analysis of protein expression
Guinea‐pig whole heart tissue and neonatal rat ventricular myocytes were prepared for western blotting as described previously (Boateng et al. 1998; Hooper et al. 2013). First 10–15 μg of protein was loaded onto each lane. Blots were probed for anti‐desmin (rabbit, dilution 1:1000; AB15200; Abcam), MLP (mouse, dilution 1:500; custom made; Invitrogen, Carlsbad, CA, USA), HSP60 (rabbit, dilution 1:1000; GTX61350; Gentex, Zeeland, MI, USA), AMPK (rabbit, dilution 1:1000; 57C12; Cell Signaling Technology, Beverly, MA, USA), GAPDH (mouse, dilution 1:5000; GTX627408; Gentex), β‐actin (mouse, dilution 1:1000; ab13496; Abcam), ATF2 (rabbit, dilution 1:1000; 20F1; Cell Signaling Technology), CREB (rabbit, dilution 1:1000; 9197S; Cell Signaling Technology), heme oxygenase 1 (rabbit, dilution 1:1000; Ab13243; Abcam), integrin β1 (rabbit, dilution 1:1000; AB30394; Abcam), α‐β crystalline (mouse, dilution 1:1000; ab13496; Abcam) and histone H2B (rabbit, dilution 1:500; GTX87385; Gentex). Horseradish peroxidase‐conjugated goat anti‐mouse or goat anti‐rabbit secondary antibodies (Invitrogen) were used to visualize proteins by enhanced chemiluminescence (ECL; Bio‐Rad, Hercules, CA, USA) using ImageQuant (GE Healthcare, Little Chalfont, UK). The bands corresponding to the various proteins were quantified by laser densitometry using Gel‐Pro Analyser, version 6.0 (Media Cybernetics, Inc., Bethesda, MD, USA). Protein bands were further standardized to total protein loading on the same membranes using the Amido black‐stained nitrocellulose membrane as described previously (Boateng et al. 1998). After membranes were probed for the desired antibodies, they were incubated in stripping buffer [Tris, pH 6.8, 1 m, 2% (w/v) SDS and purified water] and 2‐mercaptoethanol at 50°C for 30 min. Membranes were rinsed in water and stained for total protein using Amido black stain (Sigma, St Louis, MO, USA) for 10 min. The membranes were then destained in destain solution [45% (v/v) methanol, 45% ddH20 and 10% (v/v) acetic acid] for 1 h. Optical densities for individual protein bands were normalized against the total protein within the lane as determined by Amido black staining.
Immunocytochemistry
After the various experimental protocols, myocytes for immunocytochemical staining were fixed in 4% paraformaldehyde in PBS plus 0.2% Triton X 100 for 10 min and then 70% ethanol for storage at −20°C. Cells were rehydrated in PBS and then immunostained with antibodies as described previously (Hooper et al. 2013). Cells were stained for α‐actinin (rabbit, dilution 1:1000 dilution; Ab11007; Abcam), MLP (rabbit, 1:1000 dilution; AB42504; Abcam) and desmin (rabbit, 1:1000 dilution; AB15200; Abcam). Alexa Fluor‐conjugated secondary antibodies (Molecular Probes, Carlsbad, CA, USA) were used to visualize the specific proteins. Fluorescence‐labelled cells were then viewed using an AxioCam HRc camera attached to an AxioPhot 2 microscope (Carl Zeiss, Oberkochen, Germany).
Myocyte cell size measurements
Cells were immunostained with desmin (as described above) to reveal the area of the cell. The same cells were co‐stained with 4′,6‐diamidino‐2‐phenylindole (DAPI) to reveal the nucleus. The area of the nucleus and the whole cell was measured using ImageJ (NIH, Bethesda, MD, USA). The value obtained for the cell area was divided by the value obtained for the nuclear area to obtain the cell to nuclear size ratio.
Statistical analysis
At least three separate primary cultures were averaged. Each culture used ∼30 neonatal hearts. For the analysis of guinea‐pig hearts, samples of four to six hearts were used from each group. All values are the mean ± SEM. All values of significance were calculated using the appropriate comparisons: one‐way ANOVA when comparing three of more groups. However, the coefficient of determination was used for regression analysis shown in Fig. 8. P < 0.05 was considered statistically significant for differences among means. Data were analysed using Minitab (Minitab Inc., State College, PA, USA).
Figure 8. Nuclear MLP ratio correlates with cardiac function .
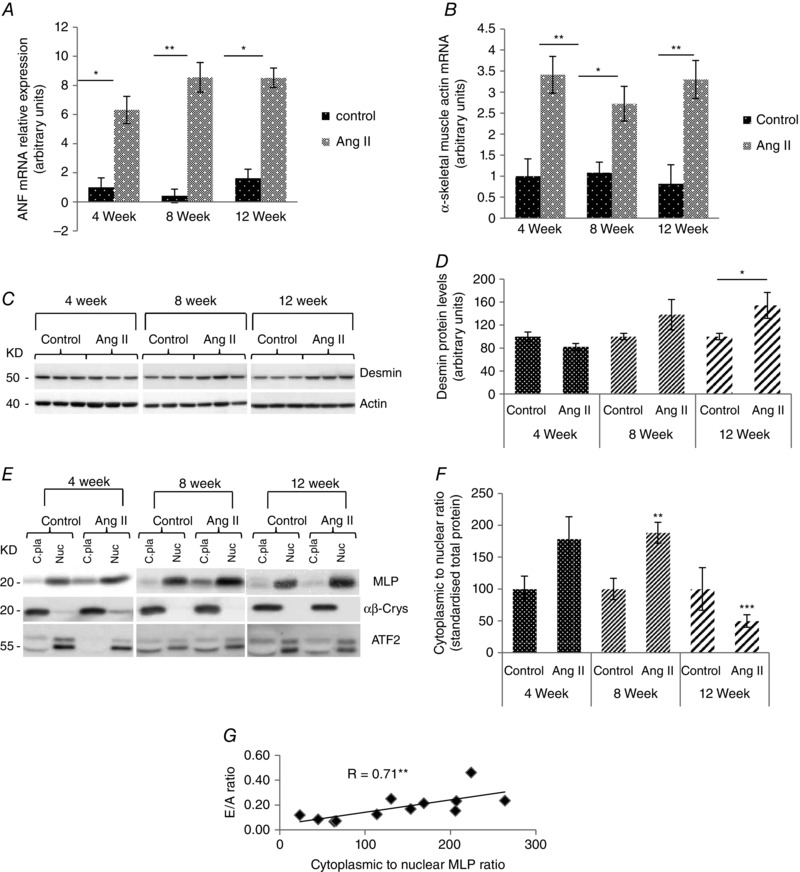
A, relative expression of ANP and (B) α skeletal muscle actin 1 (Acta1) by quantitative PCR in left ventricles of guinea‐pig animals treated with angtiotensin II for 4, 8 and 12 weeks compared to age‐matched controls. The data are normalized to hypoxanthine‐guanine phosphoribosyltransferase (HGPRT). C, western blot of desmin in response to angiotensin II treatment in guinea‐pig left ventricles. Actin was used as a loading control. D, quantified desmin from guinea‐pig left ventricles treated with angiotensin II for 4, 8 and 12 weeks, against age‐matched controls. E, western blot of αβ‐crystallin and activating transcription factor 2 (Atf2) in the cytoplasmic and nuclear fraction of guinea‐pigs left ventricles following angiotensin II treatment. αβ‐crystallin was used as cytoplasmic marker and Aft2 was used as a nuclear marker. F, quantification of MLP presented as the cytoplasmic to nuclear ratio following subcellular fractionation in the six treatment groups. G, The MLP ratio correlated significantly with the E/A ratio in the guinea‐pig hearts (r = 0.71, P < 0.01, n = 12), a clinical measure of diastolic function. N ≥ 3. *P < 0.05, **P < 0.01 and ***P < 0.001 compared to control.
Results
Passive stretch and electrical pacing of cardiac myocytes upregulates HO‐1
Myocytes within the heart experience mechanical stress as a result of contractile activity (active systolic), as well as cyclic changes in blood pressure and volume (passive diastolic stress/strain). To determine how myocytes respond to these two types of mechanical stress, we either electrically stimulated cardiac myocytes for 48 h (6.8 V at 1 Hz) or mechanically stretched the cells (10% maximum cyclic mechanical stretch/strain) for 48 h at 1 Hz. As a positive marker for cellular stress (Lavrovsky et al. 1994), we measured HO‐1. Levels of HO‐1 protein significantly increased following both mechanical stretch and electrical pacing (Fig. 1 A and B). As a positive control for passive stretch/strain based on our previous work (Hooper et al. 2013), we also quantified desmin protein levels, which increased significantly only in response to cyclic stretch/strain (Fig. 1 A and C). By contrast, there was no significant change in the level of MLP protein with either of the two treatments compared to controls (Fig. 1 A and 1 D).
Figure 1. Active contractility induces MLP nuclear accumulation .
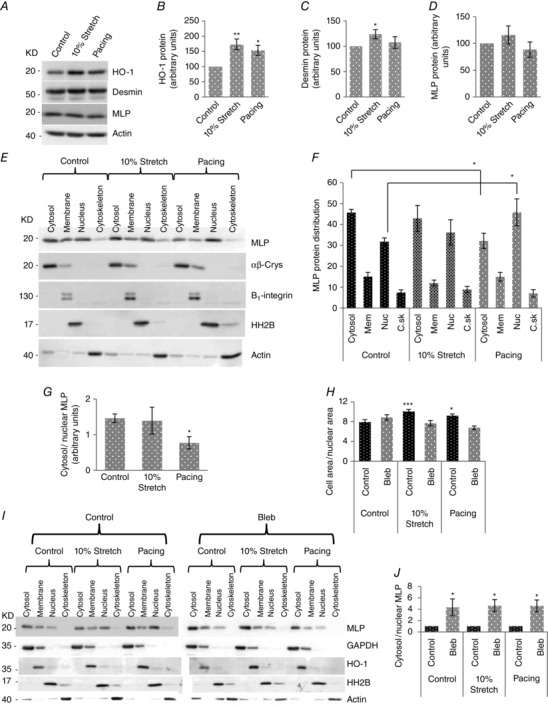
A, western blot of HO‐1, desmin, MLP and actin following either 10% cyclic stretch/strain (10% maximum strain at 1 Hz for 48 h) or electrical pacing (6.8 V at 1 Hz for 48 h). B, quantification of protein levels of HO‐1), (C) desmin and (D) MLP in myocytes following either cyclic stretch/strain or electrical pacing. E, western blot to assess the subcellular distribution of MLP in myocytes following stretch/strain and electrical pacing. α‐β crystalline (αβ‐Crys), integrin β1, histone H2B and actin have been used as the marker of the cytoplasmic, membrane, nuclear and cytoskeletal pool of protein, respectively, to verify the fractionation process. F, Quantification of subcellular distribution of MLP. Data are presented as the percentage of total MLP in each of four fractions in control cells and following stretch and electrical pacing. G, quantification of MLP presented as the cytoplasmic to nuclear ratio. H, quantification of cell area/nuclear area for the six experimental groups. In total, 100–150 cells were counted in each treatment group. I, western blot of subcellular distribution of MLP in control and blebbistatin‐treated cells, in combination with stretch or electrical pacing. J, quantification of MLP presented as the cytoplasmic to nuclear ratio. N ≥ 4. *P < 0.05, **P < 0.01 and ***P < 0.001 compared to control.
Nuclear accumulation of MLP following electrical pacing but not passive stretch
We have shown previously that nuclear MLP increases and cytoplasmic levels decrease in the failing heart (Boateng et al. 2007). Therefore, we aimed to determine what type of mechanical stimulus (passive stretch/strain or active contractility) modulates MLP nuclear translocation. Using subcellular fractionation, we isolated protein from myocyte cytosolic, membrane, nucleus and cytoskeletal compartments following the two stimuli. The methodology was verified using proteins with known subcellular locations (Fig. 1). In control cells, ∼45% of MLP is found in the cytoplasm and ∼30% is found in the nucleus. The remainder is distributed between the membrane and the cytoskeleton. Upon 10% cyclic mechanical stretch/strain, there was no change in the distribution of MLP after 48 h of treatment. However, MLP accumulated in the nucleus from the cytoplasm in response to electrical pacing (Fig. 1). When the data were expressed as cytoplasmic to nuclear ratio, there was a significant decrease only after prolonged electrical pacing (Fig. 1).
Myocyte contractility is required for MLP nuclear accumulation and cellular hypertrophy
The above data suggest that myocyte contractility regulates nuclear MLP accumulation. To examine this further, we inhibited myocyte contractility by treating cells with blebbistatin, a potent inhibitor of myosin II (Kovacs et al. 2004). The treatment was also performed in the presence of stretch/strain or electrical pacing. Following these treatments, we measured myocyte cell area/nuclear area as a measure of cellular hypertrophy. Myocyte cell area/nuclear area increased significantly in response to both stretch and pacing and this was abolished by blebbistatin treatment (Fig. 1). Next, we measured the cytoplasmic to nuclear MLP ratio, which increased by 3‐fold in the presence of blebbistatin, regardless of whether this was combined with cyclic stretch/strain or pacing (Figs 1 I and J). HO‐1 was also used as a membrane marker and remained predominantly within that fraction, with no significant nuclear translocation (Fig. 1). These data were confirmed by immunocytochemistry on myocytes by staining for MLP following blebbistatin, cyclic stretch/strain and electrical pacing. Figure 2 A–L shows that blebbistatin treatment significantly reduced nuclear MLP in cardiac myocytes. Subcellular fractionation of cardiac myocytes shows that in all treatments, MLP is required for the passive stretch response in cardiac myocytes.
Figure 2. Immunostaining of cultured neonatal myocytes showing MLP distribution following cyclic mechanical strain, electrical pacing and MLP NLS peptide treatment to block nuclear MLP .
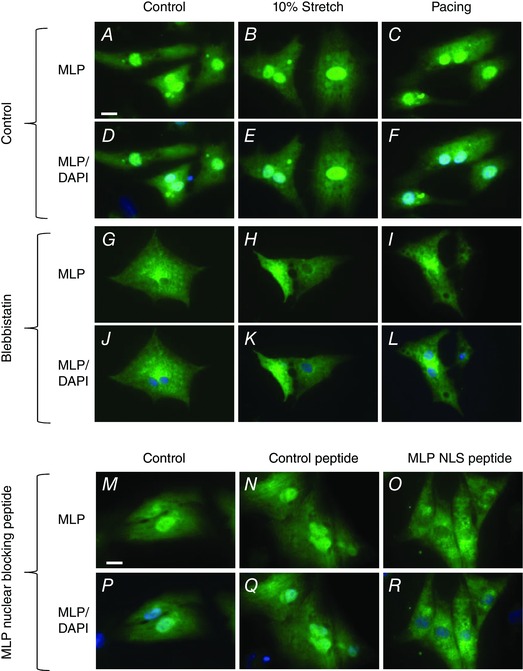
A–C, G–I and M–O are stained for MLP in green. D–F, J–L and P–R are stained for MLP in green and DAPI for nuclei in blue. Scale bar = 10 μm.
We have previously shown that desmin levels increase in response to passive cyclic stretch/strain (Hooper et al. 2013). This makes desmin protein expression a good marker of the cellular response to passive stretch/strain. In the present study, we aimed to determine whether the passive stretch/strain response also required nuclear MLP (despite the fact that nuclear MLP accumulation did not occur following cyclic stretch/strain; Fig. 1 E and F). To examine this, we treated cultured myocytes with a synthetic peptide containing the fibroblast growth factor membrane translocating peptide sequence AAVALLPAVLLALLAP attached to the nuclear localization signal of MLP (RKYGPK) as described previously (Boateng et al. 2009). As a control, cells were treated with AAVALLPAVLLALLAP only without the MLP NLS. We have previously shown that this peptide selectively blocks MLP nuclear entry without significantly altering the nuclear shuttling of other proteins (Boateng et al. 2009). Following MLP‐NLS peptide treatment, nuclear MLP decreased significantly compared to controls and control peptide‐treated cells (Figs 2 M–R and 3A). MLP protein levels were also significantly reduced (by 88%) following MLP‐NLS peptide treatment and this could not be reversed by stretch/strain or electrical pacing (Fig. 3 B and C). Next, we measured desmin protein levels following peptide treatment. The MLP‐NLS peptide significantly reduced desmin levels (by 66%) and remained depressed following either stretch/strain or electrical pacing (Fig. 3 B and D). Therefore desmin upregulation in response to stretch/strain was lost in cells where total and nuclear MLP is significantly reduced. Finally, we measured the levels of actin protein following the various treatments and this remained unchanged in all the treatment groups (Fig. 3 B and E).
Figure 3. Reduced MLP expression inhibits the passive stretch response .
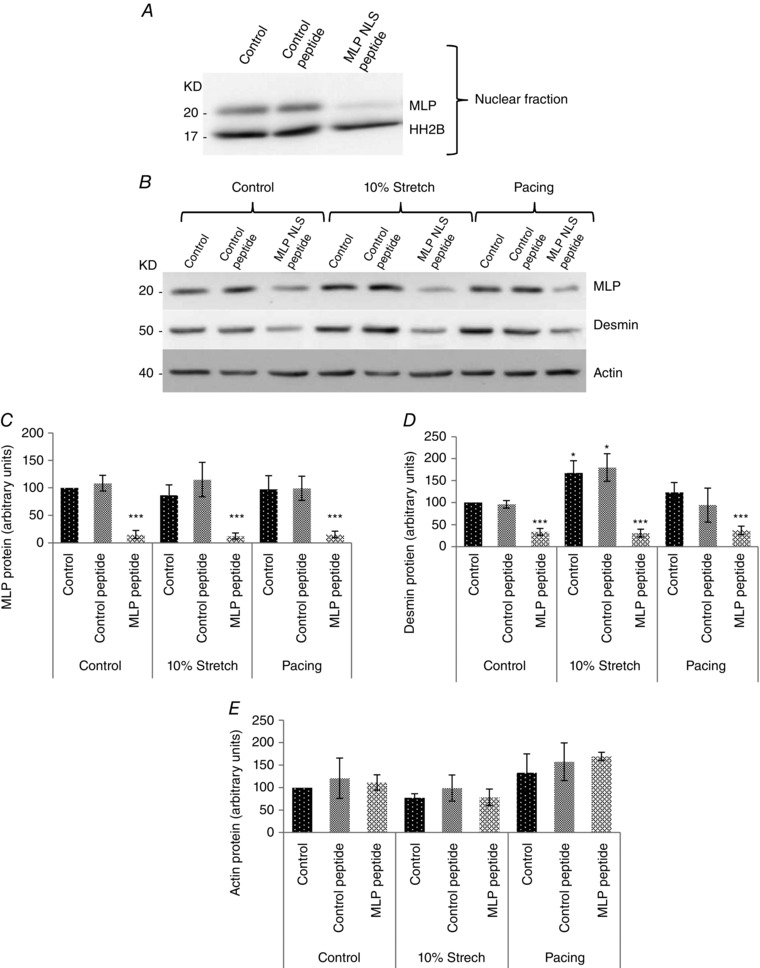
A, western blot of MLP and histone 2B from cultured neonatal myocyte nuclear fractions. Cells were treated with either 20 μM control peptide or MLP‐NLS containing peptide. B, western blot of MLP, desmin and actin from cultured myocytes from controls, treated with either 20 μM control peptide or MLP‐NLS containing peptide. C, quantified MLP from the three treatment groups. D, quantified desmin from the three treatment groups. E, quantified actin from the same treatment groups. N ≥ 6. *P < 0.05 and ***P < 0.001 compared to control.
Mechanical strain when combined with electrical pacing augments HO‐1 but not nuclear MLP
In the present study, we have separated the two main sources of mechanical strain on myocytes aiming to determine their effect on MLP nuclear shuttling. Here, we have combined the two stimuli to simulate what happens in vivo by synchronizing our Flexcell and C‐pace instruments using a common TTL (transistor‐to‐transistor logic) signal such that the electrical stimulus was delivered at the peak of the mechanical strain (Fig. 4).
This ensures that the myocytes contracted when ‘loaded’ mechanically once stimulated to contract (Fig. 4). Stretch and electrical pacing significantly increased HO‐1 expression by 50–60%, although the combination of the two stimuli increased it by more than 100% (Fig. 4 C and D). As shown in Fig. 1, stretch did not alter MLP nuclear accumulation, although stretch might modulate nuclear MLP in response to pacing. When the two stimuli were combined, the ratio of cytoplasmic to nuclear MLP decreased significantly, although this was similar to what occurred with pacing alone (Fig. 4 E and F).
HO‐1 regulates MLP nuclear shuttling in response to myocyte contractility
Nuclear accumulation of MLP is influenced primarily by myocyte contractility rather than stretch; however, the mechanisms by which the subcellular localization of the protein is regulated in myocytes are unknown. Fig. 1 shows that HO‐1 is upregulated by both stretch and electrical pacing. Therefore, we tested the hypothesis that HO‐1 signalling is upstream of MLP activity using the MLP nuclear blocking peptide that decreases both nuclear and total MLP. When MLP levels were significantly reduced by the blocking peptide, HO‐1 levels still increased in response to electrical pacing, (Fig. 5 A and B). This strongly suggests that MLP is downstream of HO‐1 signalling. We then inhibited HO‐1 enzymatic activity with PPZII, which resulted in a significant reduction of nuclear MLP, even when combined with electrical pacing (Fig. 5 C and D). In this experiment, the normal increase in nuclear MLP following electrical pacing alone was not observed because of the presence of 0.3% DMSO used as a solvent for PPZII. However, this does not detract from the finding that inhibition of HO‐1 almost completely removes nuclear MLP.
Figure 5. Regulation of nuclear MLP by HO‐1 and HDAC signalling .
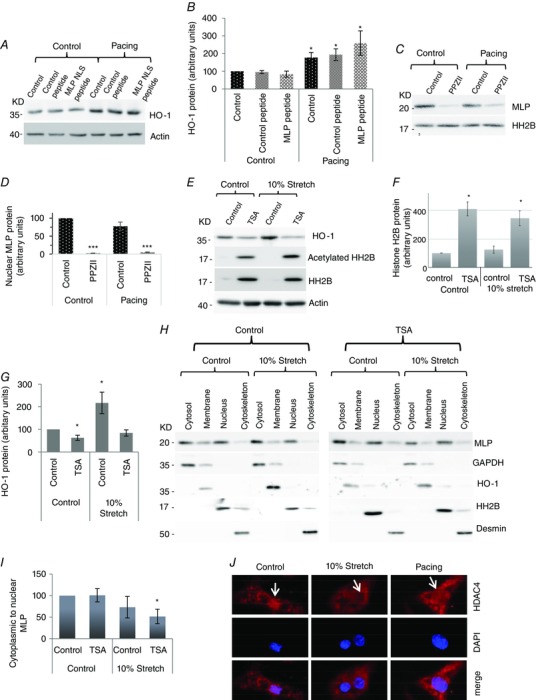
(A) Western blot showing H0‐1 and actin following pacing, with and without 20 μM control/MLP NLS peptide in cardiac myocytes. (B) Quantification of HO‐1 protein following treatments (C) Western blot showing nuclear MLP protein following PPZII treatment to inhibit HO‐1 with and without electrical pacing. (D) Quantitation of nuclear MLP following treatment. (E) Western blot of total histone H2B, acetylated histone and actin following treatment of cardiac myocytes with TSA to inhibit HDAC activity, with and without 10% cyclic stretch. (F) Quantification of total histone H2B following the various treatments (G) Quantification of HO‐1 following TSA and stretch treatments. (H) Western blot to assess the subcellular distribution of MLP in cells following treatment with TSA with and without 10% stretch/strain. GAPDH, HO‐1, histone H2B and desmin have been used as markers for cytoplasmic, membrane, nuclear and cytoskeletal pool of protein respectively. (I) Quantification of MLP presented as cytoplasmic to nuclear ratio in myocytes following treatments. (J) Immunostaining of cultured neonatal myocytes showing HDAC4 distribution (in red) and dapi (in blue) following cyclic mechanical strain and electrical pacing. White arrows point to the cell nucleus. N ≥ 4. *donates P < 0.05 and ***P < 0.001 compared to control.
Inhibition of HDAC activity blocks the HO‐1 stress response to stretch
We hypothesized that the HO‐1 stress response to mechanical stretch might be influenced by HDAC signalling because both are known to influence the hypertrophic response in myocytes. Inhibition of HDAC activity by TSA resulted in a significant increase in both the expression and acetylation of histone 2B (Fig. 5 E and F). Examination of HO‐1 protein expression following TSA treatment showed a significantly reduced expression (Fig. 5). Moreover, the normal increase of HO‐1 expression following stretch was completely blocked by inhibition of HDAC signalling by TSA. This suggests that HDAC signalling is upstream of HO‐1 activity.
HDAC activity inhibits MLP nuclear accumulation in response to passive stretch
The present study shows that mechanical strain does not result in nuclear accumulation of MLP compared to electrical pacing. We also aimed to determine whether HDAC activity influenced MLP nuclear shuttling considering that HDAC4 can deacetylate the protein (Gupta et al. 2008). Cyclic mechanical stretch did not result in nuclear accumulation of MLP unless it was combined with HDAC inhibition using TSA (Fig. 5 H and I). This data suggests that HDAC activity inhibits nuclear accumulation of MLP in response to stretch.
Stretch but not pacing activates nuclear export of HDAC4 in cardiac myocytes
Because the data indicate that HDAC signalling is upstream of HO‐1 activity, we aimed to determine whether the observed difference between stretch and pacing on MLP nuclear accumulation might be a result of differential effects on HDAC4. HDAC4 has been shown to deacetylate MLP (Gupta et al. 2008). To examine this, we investigated HDAC4 subcellular localization by immunostaining following both 10% stretch and electrical pacing in cardiac myocytes. HDAC4 protein showed both a cytoplasmic and nuclear distribution in control cells (Fig. 5). However, following mechanical stretch, there was significant nuclear export of the protein such that very little remained in the nucleus (Fig. 5). Electrical pacing did not result in a significant nuclear export of HDAC4 (Fig. 5).
Changes in cardiac function during disease progression following angiotensin II infusion in guinea‐pigs
Having established that the reduction in the MLP ratio (and loss of mechanosensing) is induced primarily by increased contractile activity, we aimed to determine how this index changed during the progression of myocardial disease. To investigate this, we established a model of cardiac hypertrophy and failure in guinea‐pigs in which angiotensin II was administered at 400 ng kg–1 min–1. Left ventricular tissue samples were taken at 4, 8 and 12 weeks after initiation of the angiotensin II treatment along with age‐matched controls. Various cardiac parameters were measured to correlate this with the protein changes investigated in our in vitro work to help explain some of the mechanisms of disease. Left ventricular mass to body weight ratio increased significantly at 4, 8 and 12 weeks in angiotensin II hearts compared to age‐matched controls (Fig. 6), providing evidence for the presence of left ventricular hypertrophy in these animals. To determine cardiac function at each of these three stages various parameters were measured. To determine systolic function, we measured dP max and end‐systolic pressure. dP max and end‐systolic pressure were significantly reduced in the failing group at 12 weeks, suggesting impaired systolic function (Fig. 6 B and C respectively). Stroke volume increased during the compensated phase at 8 weeks but decreased significantly at 12 weeks (Fig. 6). End‐diastolic volume decreased at 8 weeks and increased again at 12 weeks (Fig. 6). To determine diastolic function, we measured dP min, Tau Weiss and the E/A ratio. dP min decreased significantly at 12 weeks (Fig. 6), Tau Weiss increased at 12 weeks (Fig. 6) and the E/A ratio (a clinical marker of diastolic function) decreased significantly at 12 weeks (Fig. 6).
Figure 6. Angiotensin infusion induces heart failure in guinea‐pigs .
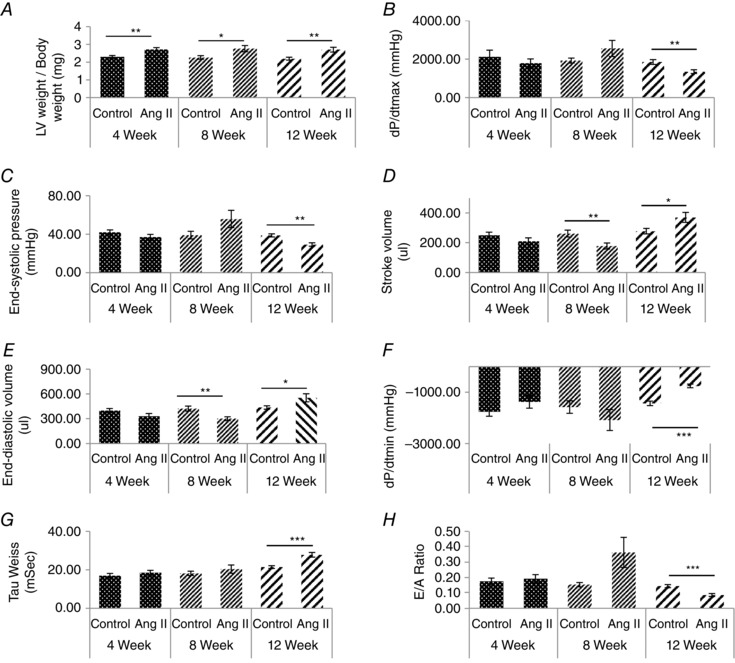
A, measurement of left ventricle weight to body weight ratio. B, dP/dT max. C, end‐systolic pressure. D, stroke volume. E, end‐diastolic volume (F) dP/dT min. G, Tau Weiss and (H) E/A ratio in guinea‐pig animals treated with angiotensin II for 4, 8 and 12 weeks, with age‐matched controls. N ≥ 7. *P < 0.05, **P < 0.01 and ***P < 0.001 compared to control.
Hypertrophic and metabolic markers are altered in the progression to heart failure
During progression to heart failure, there are significant changes to metabolism and, to investigate some of these, we measured levels of AMPK, GAPDH and HSP60 proteins by western blotting. The results show that the level of AMPK was significantly increased in response to angiotensin II treatment for 4 weeks compared to age‐matched control (Fig. 7 A and B). However, this difference disappeared during the progression to failure. By contrast, GAPDH was found to be significantly increased in response to angiotensin II treatment at 12 weeks compared to age‐matched controls (Fig. 7 A and C). HSP60 is a mitochondrial chaperon that functions to aid the correct folding of the proteins during stress. We examined the level of HSP60 protein in the guinea‐pig hearts treated with angiotensin II for 4, 8 and 12 weeks. Levels of HSP60 protein increased significantly only at 12 weeks (Fig. 7 A and D). Total MLP protein expression remained unchanged in all guinea‐pig groups as shown in Fig. 7 E and F. However, in control animals, there was a steady age‐dependent increase in MLP expression (Fig. 7).
Figure 7. Metabolic genes and MLP expression following angiotensin II induced heart failure in guinea‐pigs .
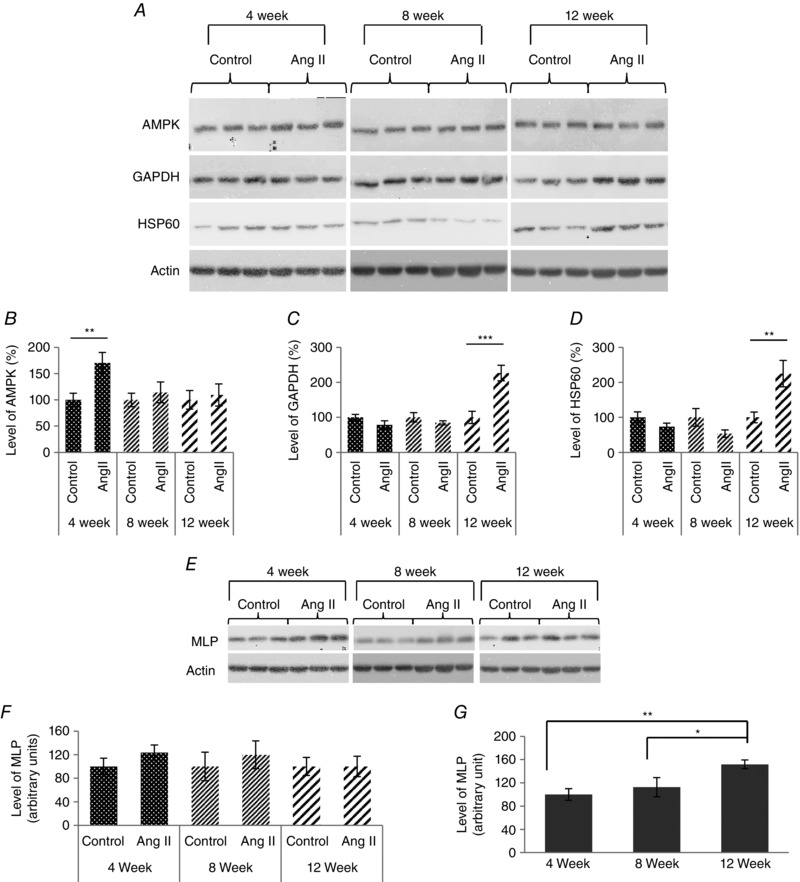
A, western blot of AMP‐activated protein kinase (AMPK), glyceraldehyde 3‐phosphate dehydrogenase (GAPDH), heat shock protein 60 (HSP60) and MLP following angiotensin II treatment in guinea‐pigs undergoing adaptive cardiac hypertrophy and failure. Actin was used as a visual loading control. B, quantification of AMPK (B), GAPDH (C) and (D) HSP60 in guinea‐pig left ventricles treated with angiotensin II for 4, 8 and 12 weeks, with age‐matched controls, normalized to total protein. N = 7. *P < 0.05 compared to age‐matched control animals. E, western blot of MLP following angiotensin II treatment in guinea‐pigs undergoing adaptive cardiac hypertrophy and failure. F, quantification of MLP in left ventricles of guinea‐pig animals treated with angiotensin II for 4, 8 and 12 weeks, with age‐matched controls, normalized to actin. G, quantification of MLP in response to the age of control guinea‐pig animals at 4, 8 and 12 weeks.
The mRNA expression level of genes involved in the hypertrophic response was determined during disease progression in the guinea‐pig hearts using quantitative RT‐PCR. The mRNA expression profiles for each gene were normalized to hypoxanthine phophoribosyltransferease 1. ANP mRNA expression was increased in the angiotensin II hearts at 4, 8 and 12 weeks compared to age‐matched controls (P < 0.05, n = 7) (Fig. 8). α‐skeletal muscle actin is a cytoskeletal protein and its expression is normally augmented during hypertrophy (Schwartz et al. 1986; Bishopric et al. 1987). α‐Skeletal muscle actin mRNA expression also increased in response to angiotensin II treatment at all three time points compared to age‐matched controls (P < 0.05, n = 7) (Fig. 8).
Mechanosensitivity improves during compensated hypertrophy but decreases during heart failure
To determine how changes in passive forces within the heart might alter during the progression of disease, desmin protein levels were measured in the guinea‐pig hearts. Desmin protein increased by ∼50% in response to angiotensin II treatment after 12 weeks (P < 0.05, n = 7) (Fig. 8 C and D).
To determine how the subcellular localization of MLP altered during the progression of cardiac disease, guinea‐pig heart tissue samples were fractionated into cytoplasmic and nuclear compartments for further analysis (Fig. 8 E and F). At 4 weeks, the MLP ratio increased but was not statistically significant (P > 0.05, n = 4). However, at 8 weeks, the MLP ratio increased significantly by ∼90% (P < 0.01, n = 4). At 12 weeks, there was a significant decrease in the MLP ratio (P < 0.01, n = 4). α‐β crystallin was used as a cytoplasmic marker, whereas ATF2 was used for the nucleus. Unfortunately, the histone H2B antibodies did not work for guinea‐pig samples.
Cytoplasmic to nuclear MLP ratio correlates with the diastolic function of the heart during progression to the failure
To investigate how the molecular changes that take place during the progression to heart failure correlate with heart function, we compared various functional parameters with the MLP ratio (following correction for age). There was a significant positive correlation between the MLP ratio and the E/A ratio (P < 0.01, n = 12), a clinical marker of diastolic function (Fig. 8).
Discussion
We have previously shown that the MLP ratio can be used as an index of mechanosensitivity in the heart because the presence of the protein in the cytoplasm and sensing complex is essential for adaptation (Boateng et al. 2007). In the present study, we show for the first time that this index improves during compensated adaptation but decreases significantly upon the progression to heart failure. Furthermore, there was a significant correlation between this ratio and cardiac function, the E/A ratio, which is a clinical measure of diastolic function. This is significant because the MLP ratio provides a much better correlation with cardiac function compared to other established markers, such as ANF or α‐skeletal actin, which also increase in response to mechanical stress. This is unsurprising because mechanosensitivity provides an index of myocyte/heart adaptability to mechanical stress. Impaired mechanosensitivity will eventually lead to impaired cardiac function. A number of previous studies have focused on the molecular events associated with heart failure, whereas, in the present study, our focus was to determine the events leading up to impaired cardiac function. Angiotension II induces cardiac hypertrophy and failure through neurohumoral activation and pressure overload, whereas the guinea‐pig heart provides functional parameters that are more similar to those of man (Sadoshima et al. 1996). Markers such as ANP and α‐skeletal muscle actin were both elevated in the early stages of hypertrophy and remained elevated beyond the transition to failure, whereas the MLP ratio changed in synchrony with cardiac function.
One remaining consideration was the determination the type of mechanical stimuli resulting in the nuclear accumulation of MLP associated with the changes observed in the failing heart. There are two main sources of mechanical stress experienced by myocytes within the heart: (i) the passive stress/strain arising from the cyclic change in the volume and the filling pressure and (ii) the intracellular forces arising from cross‐bridge cycling during systole. We have previously established that elevated desmin protein expression is a good marker for passive mechanical stress/strain, and cross‐bridge activity appears to have little effect on this protein (Hooper et al. 2013). However, the loss of MLP results in a decreased desmin expression and inhibition of the normal response to passive stretch. This indicates that MLP may directly regulate desmin expression and is required for the passive stretch response despite the absence of nuclear accumulation. We have previously shown that proteins such as LPP function via rapid nuclear transit without ever accumulating in the nucleus (Hooper et al. 2012). Nuclear accumulation of LPP only occurs when CRM1‐dependent nuclear export is blocked by leptomycin. Similar to LPP, whose nuclear accumulation is associated with pathology of adipose tissue (Crombez et al. 2005), the accumulation of MLP in the myocyte nucleus appears to be associated with cardiac pathology.
In the present study, we compared passive stretch/strain with active stress/stain to determine which was more important in modulating MLP nuclear accumulation. Surprisingly, passive stress/strain alone did not affect the subcellular levels of MLP in the absence of HDAC inhibition, whereas electrical pacing alone increased the MLP ratio similar to the failing heart. This may imply that the altered MLP ratio and mechanosensitivity become impaired in the failing heart as a result of prolonged mechanical stress within the contracting myofilaments. Previous work has shown that, in MLP null hearts, there is an impaired interaction between the cytoskeleton and mitochondria, suggesting that MLP may link the sensing of cross‐bridge activity and ATP production (van den Bosch et al. 2005). This is consistent with our previous finding that nuclear MLP also increases in response to phenylephrine treatment that increases contractility (Boateng et al. 2009). These findings suggest that MLP has a more complex role in myocytes beyond being a mechanosensor.
We show for the first time that MLP nuclear accumulation requires HO‐1 enzymatic activity, although the process can be blocked by HDAC signalling. Inhibition of HDACs by TSA increased MLP nuclear accumulation in the presence of passive stretch, whereas TSA alone had no effect. This suggests that stretch may activate HDAC‐induced inhibition of MLP nuclear accumulation. We previously observed a decrease in nuclear MLP following longitudinal passive stretch in myocytes (Boateng et al. 2009). A previous study also showed that the acetylation of MLP could be modified by HDAC4 and PCAF in the myofilaments (Gupta et al. 2008). These data, when combined with our own, indicate that HDAC deacetylation of MLP inhibits nuclear accumulation without necessarily inhibiting nuclear transit.
Our data show for the first time that the activity of the stress sensor HO‐1 lies upstream of MLP function and regulates its nuclear accumulation, suggesting that this protein could potentially influence the progression of myocyte adaptation to haemodynamic load. The mechanism by which HO‐1 influences MLP nuclear accumulation is unclear, although it is known to catalyze the breakdown of heme to CO, biliverdin and iron. Biliverdin is then converted to bilirubin and the iron is taken up by ferritin. Many of the effects of HO‐1 activity can be mimicked by the administration of exogenous CO. There is growing interest in the biological activity of CO as an endogenous signalling molecule, including effects on mitochondrial function and calcium signalling, as well as alteration of the activity of protein kinases (Bauer & Pannen, 2009). Calcium fluxes appear to play a major role in the activation and regulation of HO‐1 (Kim et al. 2010) and this would increase in response to either stretch or electrical pacing of myocytes. This is consistent with the finding that stretch combined with electrical pacing (as would occur in vivo) augments HO‐1 expression. However, MLP nuclear accumulation in response to the combined stimulus was similar to pacing alone. These data strongly suggest that cross‐bridge activity has a bigger influence over MLP subcellular localization compared to passive stretch alone. However, in vivo, the situation may be more complex and the effect on MLP subcellular localization could depend on the interaction between these two main mechanical stimuli and neurohumeral factors.
We show that inhibition of myocyte contractility with blebbistatin significantly decreased nuclear MLP regardless of whether this was combined with stretch or electrical pacing. Unlike BDM, which has some effects on calcium cycling (Jayawant et al. 1999), blebbistatin inhibits myosin II at the same time as preserving intracellular calcium dynamics (Kovacs et al. 2004). These data taken together suggest that MLP nuclear shuttling is regulated primarily by myocyte contractility. A recent study showed that MLP in postnatal neuronal rat retinal cells only localizes to the cytoplasm (Levin et al. 2014). That study supports the idea that the cellular contractility is required for the nuclear cycling of MLP.
By combining our in vitro myocyte data with the guinea‐pig model of heart failure, we can deduce that the change in MLP ratio, and thus the subsequent loss of mechanosensing, may be the result of an increased workload within the myofilaments as the disease progresses. A hypothetical model of mechanosensing in cardiac myocytes is shown in Fig. 9. In chronic human hypertension and aortic stenosis, there is a gradual increase in afterload during the progression of the disease over many years, which eventually results in heart failure (Gaasch, 1982). A study in a sheep model of heart failure found that left ventricular pressure continued to rise during the transition from compensated to non‐compensated hypertrophy, resulting in increased myocardial stress (Aoyagi et al. 1993). This progressive increase in myocardial stress is further augmented by apoptosis and necrosis of the myocytes, resulting in a loss of contractile function (Dorn, 2009). These events would further impair myocyte mechanosensitivity.
Figure 9. Hypothetical model of mechanosensing in cardiomyocytes .
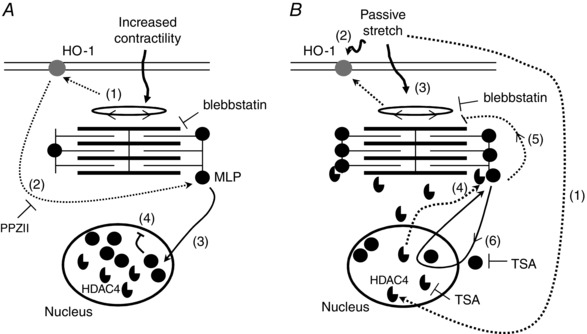
A, effects of myocyte cross‐bridge activity on MLP subcellular location. 1, increased cross‐bridge activity activates HO‐1. 2, activation of HO‐1 enables the translocation of MLP to the nucleus in response to myocyte contraction. Blebbstatin may be used to block cell contractility. MLP nuclear translocation can be blocked by PPZII, an inhibitor of HO‐1. 3, MLP translocates to the nucleus. 4, protein accumulates in the nucleus, resulting in decreased levels of the cytoplasmic protein and decreased mechanosensitivity. B, effects of passive stretch on MLP subcellular location. 1, passive stretch activates HDAC4 activity and its translocation out of the nucleus. 2, stretch also directly activates HO‐1 and indirectly through increased contractility (3). 4, HDAC4 in the cytoplasm interacts with and deacetylates MLP in the myofilaments. 5, deacetylated MLP alters myocyte contractility and calcium sensitivity. 6, deacetylated MLP is able to enter and exit the nucleus in response to cross‐bridge activity, meaning that the protein does not accumulate there.
In conclusion, we show for the first time that there is an improved index of mechanosensing in the compensated heart as measured by the MLP ratio, whereas the transition to failure results in a significant decline. The MLP ratio also correlated significantly with the E/A ratio, a clinical index of cardiac diastolic function. In vitro work that compared passive strain with active contractility in myocytes showed that the index of mechanosensitivity and subsequent heart failure may be the result of increased stress within the contracting myofilaments. MLP nuclear accumulation is regulated by both HO‐1 and HDACs, thereby providing possible therapeutic approaches for the manipulation of mechanosensing during the transition to heart failure.
Additional information
Competing interests
The authors declare that they have no competing interests.
Author contributions
AP, SD, PPdT and SYB drafted the article and revised it critically for important intellectual content. AP, SD, CI and SYB collected, assembled, analysed and interpreted data. BJW, PPdT and SYB conceived and designed the experiments. All authors have approved the final version of the manuscript and agree to be accountable for all aspects of the work. All persons designated as authors qualify for authorship, and all those who qualify for authorship are listed.
Funding
This work was supported by the British Heart Foundation project grant PG/10/64/28520 and NIH grant NIH HL62426.
Acknowledgements
The authors would like to thank Ryan Lupton and Jenny Aktins for their assistance with some of the Western blotting and Liam Kelly for immunochemistry and microscopy.
References
- Aoyagi T, Fujii AM, Flanagan MF, Arnold LW, Brathwaite KW, Colan SD & Mirsky I (1993). Transition from compensated hypertrophy to intrinsic myocardial dysfunction during development of left ventricular pressure‐overload hypertrophy in conscious sheep. Systolic dysfunction precedes diastolic dysfunction. Circulation 88, 2415–2425. [DOI] [PubMed] [Google Scholar]
- Bauer I & Pannen BH (2009). Bench‐to‐bedside review: carbon monoxide – from mitochondrial poisoning to therapeutic use. Crit Care 13, 220. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bishopric NH, Simpson PC & Ordahi CP (1987). Induction of the skeletal α‐actin gene in α1‐adrenoceptor‐mediated hypertrophy of rat cardiac myocytes. J Clin Invest 80, 1194–1199. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Boateng SY, Belin RJ, Geenen DL, Margulies KB, Martin JL, Hoshijima M, de Tombe PP & Russell B (2007). Cardiac dysfunction and heart failure are associated with abnormalities in the subcellular distribution and amounts of oligomeric muscle LIM protein. Am J Physiol Heart Circ Physiol 292, H259–H269. [DOI] [PubMed] [Google Scholar]
- Boateng SY, Senyo SE, Qi L, Goldspink PH & Russell B (2009). Myocyte remodeling in response to hypertrophic stimuli requires nucleocytoplasmic shuttling of muscle LIM protein. J Mol Cell Cardiol 47, 426–435. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Boateng SY, Seymour AM, Bhutta NS, Dunn MJ, Yacoub MH & Boheler KR (1998). Sub‐antihypertensive doses of ramipril normalize sarcoplasmic reticulum calcium ATPase expression and function following cardiac hypertrophy in rats. J Mol Cell Cardiol 30, 2683–2694. [DOI] [PubMed] [Google Scholar]
- Buyandelger B, Mansfield C & Knoll R (2014). Mechano‐signaling in heart failure. Pflügers Arch 466, 1093–1099. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Crombez KR, Vanoirbeek EM, Van de Ven WJ & Petit MM (2005). Transactivation functions of the tumor‐specific HMGA2/LPP fusion protein are augmented by wild‐type HMGA2. Mol Cancer Res 3, 63–70. [DOI] [PubMed] [Google Scholar]
- Dorn GW, 2nd (2009). Apoptotic and non‐apoptotic programmed cardiomyocyte death in ventricular remodelling. Cardiovasc Res 81, 465–473. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gaasch WH (1982). Management of aortic valve disease. Hosp Pract 17, 133–138. [DOI] [PubMed] [Google Scholar]
- Gardiwal A, Klein G, Kraemer K, Durgac T, Koenig T, Niehaus M, Heineke J, Mohammadi B, Krampfl K, Schaefer A, Wollert KC & Korte T (2007). Reduced delayed rectifier K+ current, altered electrophysiology, and increased ventricular vulnerability in MLP‐deficient mice. J Card Fail 13, 687–693. [DOI] [PubMed] [Google Scholar]
- Geier C, Gehmlich K, Ehler E, Hassfeld S, Perrot A, Hayess K, Cardim N, Wenzel K, Erdmann B, Krackhardt F, Posch MG, Osterziel KJ, Bublak A, Nagele H, Scheffold T, Dietz R, Chien KR, Spuler S, Furst DO, Nurnberg P & Ozcelik C (2008). Beyond the sarcomere: CSRP3 mutations cause hypertrophic cardiomyopathy. Hum Mol Genet 17, 2753–2765. [DOI] [PubMed] [Google Scholar]
- Gupta MP, Samant SA, Smith SH & Shroff SG (2008). HDAC4 and PCAF bind to cardiac sarcomeres and play a role in regulating myofilament contractile activity. J Biol Chem 283, 10135–10146. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hooper CL, Dash PR & Boateng SY (2012). Lipoma preferred partner is a mechanosensitive protein regulated by nitric oxide in the heart. FEBS open bio 2, 135–144. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hooper CL, Paudyal A, Dash PR & Boateng SY (2013). Modulation of stretch‐induced myocyte remodeling and gene expression by nitric oxide: a novel role for lipoma preferred partner in myofibrillogenesis. Am J Physiol Heart Circ Physiol 304, H1302–H1313. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hu CM, Chen YH, Chiang MT & Chau LY (2004). Heme oxygenase‐1 inhibits angiotensin II‐induced cardiac hypertrophy in vitro and in vivo. Circulation 110, 309–316. [DOI] [PubMed] [Google Scholar]
- Jayawant AM, Stephenson ER, Jr. & Damiano RJ, Jr (1999). 2,3‐Butanedione monoxime cardioplegia: advantages over hyperkalemia in blood‐perfused isolated hearts. Ann Thorac Surg 67, 618–623. [DOI] [PubMed] [Google Scholar]
- Kim AN, Jeon WK, Lee JJ & Kim BC (2010). Up‐regulation of heme oxygenase‐1 expression through CaMKII‐ERK1/2‐Nrf2 signaling mediates the anti‐inflammatory effect of bisdemethoxycurcumin in LPS‐stimulated macrophages. Free Rad Biol Med 49, 323–331. [DOI] [PubMed] [Google Scholar]
- Knoll R, Hoshijima M, Hoffman HM, Person V, Lorenzen‐Schmidt I, Bang ML, Hayashi T, Shiga N, Yasukawa H, Schaper W, McKenna W, Yokoyama M, Schork NJ, Omens JH, McCulloch AD, Kimura A, Gregorio CC, Poller W, Schaper J, Schultheiss HP & Chien KR (2002). The cardiac mechanical stretch sensor machinery involves a Z disc complex that is defective in a subset of human dilated cardiomyopathy. Cell 111, 943–955. [DOI] [PubMed] [Google Scholar]
- Kovacs M, Toth J, Hetenyi C, Malnasi‐Csizmadia A & Sellers JR (2004). Mechanism of blebbistatin inhibition of myosin II. J Biol Chem 279, 35557–35563. [DOI] [PubMed] [Google Scholar]
- Lavrovsky Y, Schwartzman ML, Levere RD, Kappas A & Abraham NG (1994). Identification of binding sites for transcription factors NF‐kappa B and AP‐2 in the promoter region of the human heme oxygenase 1 gene. Proc Natl Acad Sci USA 91, 5987–5991. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Levin E, Leibinger M, Andreadaki A & Fischer D (2014). Neuronal expression of muscle LIM protein in postnatal retinae of rodents. PLoS ONE 9, e100756. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lorell BH & Carabello BA (2000). Left ventricular hypertrophy: pathogenesis, detection, and prognosis. Circulation 102, 470–479. [DOI] [PubMed] [Google Scholar]
- Newman B, Cescon D, Woo A, Rakowski H, Erikkson MJ, Sole M, Wigle ED & Siminovitch KA (2005). W4R variant in CSRP3 encoding muscle LIM protein in a patient with hypertrophic cardiomyopathy. Mol Genet Metab 84, 374–375. [DOI] [PubMed] [Google Scholar]
- Sadoshima J, Malhotra R & Izumo S (1996). The role of the cardiac renin‐angiotensin system in load‐induced cardiac hypertrophy. J Card Fail 2, S1–S6. [DOI] [PubMed] [Google Scholar]
- Schwartz K, de la Bastie D, Bouveret P, Oliviero P, Alonso S & Buckingham M (1986). Alpha‐skeletal muscle actin mRNA's accumulate in hypertrophied adult rat hearts. Circ Res 59, 551–555. [DOI] [PubMed] [Google Scholar]
- van den Bosch BJ, van den Burg CM, Schoonderwoerd K, Lindsey PJ, Scholte HR, de Coo RF, van Rooij E, Rockman HA, Doevendans PA & Smeets HJ (2005). Regional absence of mitochondria causing energy depletion in the myocardium of muscle LIM protein knockout mice. Cardiovasc Res 65, 411–418. [DOI] [PubMed] [Google Scholar]
- Wagner CT, Durante W, Christodoulides N, Hellums JD & Schafer AI (1997). Hemodynamic forces induce the expression of heme oxygenase in cultured vascular smooth muscle cells. J Clin Invest 100, 589–596. [DOI] [PMC free article] [PubMed] [Google Scholar]