

Abstract
Key points
Cystine is a disulphide amino acid that is normally generated in the lysosomes by the breakdown of cystine‐containing proteins.
Previously, we demonstrated that lysosomal cystine accumulation in kidney proximal tubular epithelial cells (PTECs) dramatically reduced glutathione (GSH) levels, which may result in the disruption of cellular redox balance.
In the present study, we show that lysosomal cystine accumulation following CTNS gene silencing in kidney PTECs resulted in elevated intracellular reactive oxygen species production, reduced antioxidant capacity, induction of redox‐sensitive proteins, altered mitochondrial integrity and augmented cell death.
These alterations may represent different facets of a unique cascade leading to tubular dysfunction initiated by lysosomal cystine accumulation and may present a clear disadvantage for cystinotic PTECs in vivo.
Cystine depletion by cysteamine afforded cytoprotection in CTNS knockdown cells by reducing oxidative stress, normalizing intracellular GSH and ATP content, and preserving cell viability.
Abstract
Cystine is a disulphide amino acid that is normally generated within the lysosomes through lysosomal‐based protein degradation and via extracellular uptake of free cystine. In the autosomal recessive disorder, cystinosis, a defect in the CTNS gene results in excessive lysosomal accumulation of cystine, with early kidney failure a hallmark of the disease. Previously, we demonstrated that silencing of the CTNS gene in kidney proximal tubular epithelial cells (PTECs) resulted in an increase in intracellular cystine concentration coupled with a dramatic reduction in the total GSH content. Because of the crucial role of GSH in maintaining the redox status and viability of kidney PTECs, we assessed the effects of CTNS knockdown‐induced lysosomal cystine accumulation on intracellular reactive oxygen species (ROS) production, activity of classical redox‐sensitive genes, mitochondrial integrity and cell viability. Our results showed that lysosomal cystine accumulation increased ROS production and solicitation to oxidative stress (OS). This was associated with the induction of classical redox‐sensitive proteins, NF‐κB, NRF2, HSP32 and HSP70. Cystine‐loaded PTECs also displayed depolarized mitochondria, reduced ATP content and augmented apoptosis. Treatment of CTNS knockdown PTECs with the cystine‐depleting agent cysteamine resulted in the normalization of OS index, increased GSH and ATP content, and preservation of cell viability. Taken together, the alterations observed in cystinotic cells may represent different facets of a cascade leading to tubular dysfunction and, in combination with cysteamine therapy, may offer a novel link for the attenuation of renal injury and preservation of functions of other organs affected in cystinosis.
Key points
Cystine is a disulphide amino acid that is normally generated in the lysosomes by the breakdown of cystine‐containing proteins.
Previously, we demonstrated that lysosomal cystine accumulation in kidney proximal tubular epithelial cells (PTECs) dramatically reduced glutathione (GSH) levels, which may result in the disruption of cellular redox balance.
In the present study, we show that lysosomal cystine accumulation following CTNS gene silencing in kidney PTECs resulted in elevated intracellular reactive oxygen species production, reduced antioxidant capacity, induction of redox‐sensitive proteins, altered mitochondrial integrity and augmented cell death.
These alterations may represent different facets of a unique cascade leading to tubular dysfunction initiated by lysosomal cystine accumulation and may present a clear disadvantage for cystinotic PTECs in vivo.
Cystine depletion by cysteamine afforded cytoprotection in CTNS knockdown cells by reducing oxidative stress, normalizing intracellular GSH and ATP content, and preserving cell viability.
Abbreviations
- carboxy‐H2DFFDA
5‐(and‐6)‐carboxy‐2′,7′‐difluorodihydrofluorescein diacetate
- DAF‐FMDA
4‐amino‐5‐methylamino‐2′,7′‐difluoroflurescein diacetate
- DHE
dihyroethidine
- ER
endoplasmic reticulum
- GSH
glutathione
- HSE
heat shock element
- HSF‐1
heat shock factor‐1
- HSP
heat shock protein
- iNOS
inducible nitric oxide synthase
- NF‐κB
nuclear factor kappa‐light‐chain‐enhancer of activated B cells
- NOX
NADPH oxidase
- NRF2
nuclear factor erythroid 2‐related factor 2
- OS
oxidative stress
- PI
propidium iodide
- PS
propidium iodide
- PTEC
proximal tubular epithelial cell
- RFU
relative fluorescence units
- ROS
reactive oxygen species
Introduction
Cystine is a disulphide amino acid that is normally generated inside the lysosomes by a cathepsin‐catalysed breakdown of cystine‐containing proteins (Thoene & Lemons, 1980). However, experimental data on cystinotic leukocytes and fibroblasts have shown that part of the lysosomal cystine pool originates from the uptake of extracellular non‐protein cystine (Schulman et al. 1969; Danpure et al. 1986). In renal proximal tubules, most of this non‐protein cystine uptake is mediated by the apical heterodimertic transporter, b0,+AT‐RBAT, encoded by the b0,+AT1/SLC7A9 and rBAT/SLC3A1 genes (Pras et al. 1995; Feliubadaló et al. 1999). Although the extent to which the apical uptake of cystine versus the hydrolysis of reabsorbed proteins contributes to proximal tubular cystine pool has not yet been fully established, the cystine compartmentalized in the lysosome represents > 90% of the intracellular cystine pool (Gahl et al. 1982; Ghal et al., 1982).
In the autosomal recessive disorder cystinosis, cystine accumulates at greatly elevated levels in the lysosomes of the cell (Gahl, 2001; Gahl et al. 2002). Although the widespread lysosomal accumulation of cystine in different tissues leads to multi‐organ dysfunction, the loss of kidney function remains the foremost clinical characteristic of this disease. The cystinosis‐associated kidney dysfunction is clinically manifested by the excessive loss of water, electrolytes, minerals and essential nutrients into the urine, a condition known as Fanconi syndrome (Gahl, 2001; Gahl et al. 2002). The disease is caused by one or more mutations in the CTNS gene, which codes for a lysosomal transmembrane protein, cystinosin (Town et al. 1998; Gahl, 2001; Gahl et al. 2002). Cystinosin is a 367 amino acid cystine/H+ symporter whose only known function, to date, is to facilitate the efflux of cystine from the lysosomal compartment to the cytosol (Kalatzis et al. 2001), where it is subsequently reduced to cysteine by cytosolic reducing systems (Wilmer et al. 2010 a).
It has been reported that the expression of cystinosin is particularly robust in the renal proximal tubules compared with other segments of the nephron (Kalatzis et al. 2004), suggesting that cystinosin is essential for proximal tubular cell function. Theoretically, sequestration of cystine in the lysosomes, a consequence of defective cystinosin, may result in the cytosolic deficit in cysteine, which is the primary limiting factor for the synthesis of the main intracellular antioxidant glutathione (GSH). Because GSH is essential for the tight control and modulation of cellular redox status, it is tempting to speculate that sequestration of cystine within the lysosomal compartment may result in the perturbation of redox status which, in turn, can lead to the activation of redox‐sensitive signalling pathways. These signalling pathways can induce the expression of genes whose protein products can either confer cytoprotection or lead to inappropriate cell death. Furthermore, a deficiency in GSH can result in increased concentration of reactive oxygen species (ROS), which can indiscriminately target mitochondrial proteins, significantly impacting energy generating capacity. In this study, the effects of lysosomal cystine accumulation, associated with CTNS gene inhibition, on intracellular redox status were extensively investigated in kidney proximal tubular epithelial cells (PTECs). The impact of these redox alterations on oxidative damage, ROS generation, mitochondrial integrity and cell viability was assessed. Because loss of cystinosin function can elicit a stress response via redox‐sensitive transcription factors, we attempted to gain further insights into the influence of lysosomal cystine accumulation on the expression of some classical stress‐responsive proteins. The effects of the cystine‐depleting drug cysteamine on rescuing the cellular phenotypes associated with lysosomal cystine accumulation were also investigated.
Methods
Cell culture
HK‐2 cells, a PTEC line derived from a normal adult human kidney, were cultured as described previously (Sumayao et al. 2013). Unless otherwise stated, all reagents were purchased from Sigma‐Aldrich (St Louis, MO, USA).
siRNA transfection and assessment of CTNS knockdown
The selection and production of CTNS‐targeting siRNAs (siCTNS), based on the human CTNS gene sequence, were performed by Dharmacon, Inc. (Lafayette, CO, USA) using the SMARTpool technology, which combines four highly potent siRNAs to mimic the natural silencing pathway. A non‐targeting siRNA (siNT) pool designed to target no known genes in human was supplied by the same vendor as a negative control. HK‐2 cells were cultured in antibiotic‐ and serum‐free medium to obtain ∼40% confluence. siRNA transfection was performed as described previously (Sumayao et al. 2013). The assessment of CTNS gene silencing and intracellular cystine accumulation was also described in Sumayao et al. (2013). Succeeding experiments were performed 72 h following transfection.
Intracellular ROS levels and OS index
Intracellular superoxide (O2 −) and NO levels were determined in live cells using dihyroethidine (DHE) and 4‐amino‐5‐methylamino‐2′,7′‐difluoroflurescein diacetate (DAF‐FMDA), respectively. DHE is a redox‐sensitive probe which is oxidized by O2 − to form a fluorescent product, 2‐hydroxyethidium (2‐OH‐E+) and ethidium (E+) (Zhao et al. 2003). DAF‐FMDA is a cell‐permeant probe which is deacetylated by intracellular esterases/deacetylases to become DAF‐FM. The fluorescence of DAF‐FM increases 160‐fold after reacting with NO (Kojima et al. 1999). The OS index of cells was assessed using the probe 5‐(and‐6)‐carboxy‐2′,7′‐difluorodihydrofluorescein diacetate (carboxy‐H2DFFDA) which is non‐fluorescent until the acetate group is removed by intracellular esterases and oxidation occurs within the cell. The oxidation of carboxy‐H2DFFDA not only depends on the intracellular ROS levels but also on the concentration of intracellular antioxidants such as GSH, and hence can be interpreted as an OS index rather than direct evidence of augmented ROS production (Jakubowski & Bartosz, 2000).
For live imaging experiments, cells were seeded in 35 mm uncoated glass‐bottom dishes (MatTek Corp., Ashland, MA, USA). The cells were incubated with 5 μm DHE (Invitrogen, Carlsbad, CA, USA) for 30 min, 5 μm DAF‐FMDA (Invitrogen) for 45 min or 20 μm carboxy‐H2DFFDA (Invitrogen) for 45 min at 37°C. The cells were washed three times with PBS to remove excess probe and incubated with serum‐free medium for 30 min to allow complete de‐esterification of intracellular diacetates. The fluorescence emission of the oxidized probes was analysed immediately by fluorescence microscopy (Nikon Instruments, Melville, NY, USA). Images were acquired using a computer‐controlled Zeiss HRc charge‐coupled device (CCD) camera (Carl Zeiss Microscopy GmbH, Jena, Germany) with at least five fields of view collected for each well. Images were analysed using the cell∧A Image Acquisition Software (Olympus, Tokyo, Japan).
For flow cytometry experiments, the cells were seeded and grown to 40–50% confluence in six‐well tissue culture plates. Following siRNA transfection, dye loading was performed following the procedures described previously. The cells were then washed three times with PBS and gently detached with 0.05% trypsin‐EDTA. Trypsin was neutralized by adding culture medium containing 10% fetal calf serum (FCS) to the cell suspension. The cell suspensions were centrifuged at 800 g for 3 min, resuspended in PBS and analysed immediately using a BD Accuri C6 cytometer (BD Biosciences, San Jose, CA, USA). Data were analysed using the CFlow Software (BD Biosciences).
Nitrotyrosine content
After siRNA transfection, cells were washed three times with ice‐cold PBS and lysed with 25 mm Hepes buffer (pH 7.5) which contained 1% NP‐40, 150 mm NaCl, 10 mm MgCl2, 1 mm EDTA and 2% glycerol. The lysates were homogenized using a motorized Teflon pestle for 3 min with vortexing every 30 s. The lysates were centrifuged at 14,000 g for 15 min at 4°C. Nitrotyrosine levels in the supernatants were determined by Western blot using an anti‐nitrotyrosine antibody (Cell Signaling, Danvers, MA, USA) following the procedures described in the Western blot section.
Additionally, the nitrotyrosine content was determined by a competitive enzyme‐linked immunosorbent assay (ELISA) using the OxiSelect Nitrotyrosine ELISA kit (Cell Biolabs, San Diego, CA, USA), according to the instructions of the manufacturer. The nitrotyrosine content was normalized for protein concentration of the supernatant.
Intracellular calcium concentration
Intracellular calcium Ca2+ concentration was assessed using the Ca2+‐sensitive probe Fluo‐4 AM. For live cell imaging experiments, cells were seeded and grown to 40–50% confluence in 35 mm uncoated glass‐bottom dishes (MatTek). Following siRNA transfection, cells were washed three times with PBS and incubated with 1 μm Fluo‐4 AM (Invitrogen) in serum‐free medium at 37°C for 1 h. The cells were washed three times with PBS to remove excess probe and incubated with serum‐free medium for 30 min to allow complete de‐esterification of intracellular AM esters. The fluorescence emission of Ca2+‐bound Fluo‐4 was analysed immediately by fluorescence microscopy (Nikon Instruments) as previously described in the ‘Intracellular ROS and OS index’ section. Images were analysed using the cell^A Image Acquisition Software (Olympus).
For the flow cytometry measurement of intracellular Ca2+ levels, cells were seeded and grown to 40–50% confluence in six‐well tissue culture plates. Following siRNA transfection, the cells were loaded with Fluo‐4 AM as described previously. The cell suspensions were prepared following the procedures described in the ‘Intracellular ROS and OS index’ section. The fluorescence emission of the Ca2+‐bound Fluo‐4 was analysed immediately using a BD Accuri C6 cytometer (BD Biosciences). Data were analysed using the CFlow Software (BD Biosciences).
Mitochondrial transmembrane potential (ΔΨm)
ΔΨm was assessed using the lipophilic cationic dye JC‐1. JC‐1 selectively enters the mitochondria and, at high mitochondrial ΔΨm, it spontaneously forms complexes known as J‐aggregates, which emit an intense red fluorescence. At low mitochondrial ΔΨm, JC‐1 remains in the monomeric form, which shows only green fluorescence.
In brief, cells were washed three times with PBS and incubated with 0.05% trypsin‐EDTA solution to facilitate cell dispersal. Trypsin action was terminated by adding culture medium containing 10% FCS to the cell suspension. The cell suspensions were centrifuged at 800 g for 3 min at room temperature and the cell pellets were resuspended with 2 μm JC‐1 dye (Invitrogen) in serum‐free complete culture medium for 30 min at 37°C. Following centrifugation, the cells were resuspended in PBS and were analysed immediately using a BD Accuri C6 cytometer (BD Biosciences). The ratio of J‐aggregates to monomers was used to assess mitochondrial ΔΨm.
GSH content
Total intracellular GSH content was determined by an enzyme‐recycling method as previously described (Baker et al. 1990). The assay is based on the reaction of GSH with 5,5′‐dithio‐bis‐2‐nitrobenzoic acid (DTNB) which produces the TNB chromophore with a maximal absorbance at 412 nm. In brief, 10 μl of cell lysates was placed into a 96‐well clear‐bottom microplate. Then, 200 μl of freshly prepared reaction mixture [0.1 m potassium phosphate buffer (pH 7.5), 1 mm DTNB, 1 mm NADPH and 1 U ml−1 GSH reductase] was delivered in each sample. The absorbance of the samples was measured at 412 nm, with 30 s read intervals for 5 min using a scanning microplate reader equipped with a kinetic trace (Molecular Devices, Sunnyvale, CA, USA). The GSH content of the samples was calculated using a standard curve prepared from GSH of known concentrations (0–0.28 mm). GSH concentrations were normalized for the protein content of the samples.
ATP content
The preparation of cell suspensions for ATP assay was performed as previously described (Sumayao et al. 2013). Intracellular ATP content was measured using the ATP Bioluminescence Assay Kit HSII (Roche Diagnostics, Mannheim, Germany) following the instructions of the manufacturer. ATP concentrations were normalized for the protein content of cell lysates.
Preparation of protein extracts for Western blot
Cells were washed three times with ice‐cold PBS and lysed in 1× radioimmunoprecipitation assay (RIPA) buffer which contained 5 mm EDTA, 1 mm phenylmethylsulfonylfluoride (PMSF), 5 mm sodium fluoride (NaF), 1 mm sodium orthovanadate (Na3VO4), protease inhibitor cocktail (1:100) and 1× solution of PhosSTOP phosphatase inhibitor cocktail (Millipore, Billerica, MA, USA). The cell lysates were homogenized by frequent vortexing for 15 min and centrifuged at 14,000 g for 15 min at 4°C. The supernatants were collected and stored at −80ºC until analysis.
Western blot
Western blot analysis was performed following the procedures described previously (Sumayao et al. 2013). The following concentrations of primary antibodies in 5% non‐fat milk/Tris‐base saline buffer (pH 7.4) containing 0.1% Tween‐20 (TBST) were used: anti‐nitrotyrosine (1:2000; Cell Signaling), anti‐iNOS (1:1000; Sigma), anti‐p47(phox) (1:1000; Cell Signaling), anti‐NF‐κB (p65) (1:1000; Cell Signaling), Phospho‐NF‐κB (p65) (1:1000; Cell Signaling), anti‐Nrf2 (1:1000; Cell Signaling), anti‐Hsp32 and ‐Hsp70 (1:1000; Cell Signaling), anti‐Beclin (1:1000; Cell Signaling), anti‐LC3 I/II (1:1000; Cell Signaling), β‐actin (1:2000; Thermo Scientific, Waltham, MA, USA) and GAPDH (1:2000; Abnova, Walnut, CA, USA). The membranes were incubated with a 1:2000 dilution of horseradish peroxidase‐conjugated IgG (Thermo Scientific) in 5% non‐fat milk/TBST. The bound secondary antibodies were detected using the Enhanced Chemiluminescence Kit (Thermo Scientific, Rockford, IL, USA) following the instructions of the manufacturer. Bands were developed on autoradiographic films using an automated developer. Densitometry analyses of bands were performed using the ImageJ software (National Institutes of Health, Bethesda, MD, USA).
Apoptosis and necrosis
The levels of apoptosis and necrosis were assessed by Annexin V‐FITC and propidium iodide (PI) co‐staining, respectively. Annexin V is a Ca2+‐dependent phospholipid‐binding protein that has a high affinity for phosphatidylserine (PS) (Koopman et al. 1994; Vermes et al. 1995). In the early stages of apoptosis, the cell loses membrane asymmetry, which results in the translocation of PS to the outer leaflet of the plasma membrane, thereby exposing it to the external cellular environment (Vermes et al. 1995). Therefore, binding of Annexin V to externalized PS can indicate the proportion of cells undergoing apoptosis. PI, on the other hand, is an intercalating agent and a fluorescent molecule that has been widely used for the evaluation of necrosis in different experimental models because of its ability to permeate cells with damaged membranes, a hallmark of late apoptosis or necrosis (Riccardi & Nicoletti, 2006).
The preparation of cell suspensions was performed following the same procedures described in the ‘Intracellular ROS and OS index’ section. Following centrifugation, the cell pellets were resuspended in 500 μl 1× Annexin V binding buffer (BD Biosciences) and dispersed by passing the cell suspension repeatedly through a pipette to produce single cell suspension; 5 μl of Annexin V‐FITC (BD Biosciences) and 5 μl of 1 μg ml−1 PI (Invitrogen) were added to the cell suspensions. The cell suspensions were incubated for 5 min at room temperature and protected from light. The cell suspensions were analysed immediately using a BD Accuri C6 flow cytometer (BD Biosciences).
Cell viability
Cell viability was assessed by a resazurin reduction assay and WST‐1 assay. The resazurin reduction assay uses the metabolic capacity of cells in reducing the indicator dye resazurin to highly fluorescent resorufin. Resazurin is effectively reduced in the mitochondria by NADPH or NADH dehydrogenase, making it useful in assessing mitochondrial metabolic capacity. In brief, 5000 cells per well were plated into a 96‐well black, clear‐bottom tissue culture plate (Corning, New York, USA) and grown to ∼40% confluence at 37°C in a humidified atmosphere with 5% CO2. The cells were incubated with 0.005% resazurin at 37°C for 90 min. The fluorescence of resorufin was measured using a scanning fluorescence microplate reader (Molecular Devices, Sunnyvale, CA, USA) with excitation and emission wavelengths set at 530 and 590 nm, respectively.
The WST‐1 assay uses the stable tetrazolium salt WST‐1, which is cleaved to soluble formazan by a mechanism largely dependent on the glycolytic production of NADPH by viable cells. The amount of formazan formed in the culture medium directly correlates with the number of metabolically active cells in the culture. The same procedures as in the resazurin reduction assay were followed for plating of cells. Ten microlitres of WST‐1 reagent (Roche Diagnostics) was added to each well and incubated at 37°C for 60 min. The absorbance of formazan was measured at 450 and 630 nm (test and reference wavelengths, respectively) using a scanning microplate reader (Molecular Devices). The net absorbance was obtained from the difference in absorbance measured at 630 and 450 nm.
Protein concentration
Determination of protein concentration was performed using the BCA Protein Assay Kit (Thermo Scientific) following the manufacturer's instructions.
Statistical analysis
Statistical analysis was performed using GraphPad Prism 6.0 (GraphPad, San Diego, CA, USA). Results were expressed as mean ± standard deviation (SD) of at least three independent experiments. A standard t‐test was used to test for statistical significance between siCTNS‐ and siNT‐transfected cells. Differences between multiple group means were analysed using ANOVA followed by post hoc analysis using Tukey's multiple comparisons test. For the data expressed as percentage or as fraction/fold, the data were first log10‐transformed followed by an appropriate parametric test. A P of 0.05% or less was deemed statistically significant.
Results
Assessment of CTNS gene silencing and intracellular cystine accumulation
The gene silencing efficiency of siCTNS transfection into HK‐2 cells was reported previously by our group (Sumayao et al. 2013). The transfection of CTNS‐targeting siRNAs into HK‐2 cells resulted in ∼60 and ∼42% knockdown of CTNS mRNA and cystinosin, respectively, as assessed by Western blot and RT‐PCR, respectively. The siRNA‐mediated knockdown of the CTNS gene was also associated with a ∼70% increase in intracellular cystine content. Furthermore, the cystine levels observed in CTNS knockdown cells were comparable to the levels reported by Bellomo et al. (2010), indicating that the cystine storage defect is preserved in our in vitro model (Sumayao et al. 2013).
Lysosomal cystine accumulation increases intracellular O2 − and NO production but does not alter basal OS index
To assess if lysosomal cystine accumulation caused alterations in intracellular ROS production and OS index, cells were probed with ROS‐specific and redox‐sensitive dyes following CTNS gene knockdown. Live cell fluorescence imaging showed a more pronounced DHE and DAF‐FM oxidation in CTNS knockdown cells compared with siNT transfected cells (Fig. 1 A and C, respectively), indicating increased intracellular O2 − and NO production, respectively. The levels of O2 − and NO were further analysed using flow cytometry, which provided us with relative quantities of these ROS. Flow cytometry analysis of intracellular O2 − and NO levels showed an ∼30% increase (P < 0.05) (Fig. 1 B) and ∼35% increase (P < 0.01) (Fig. 1 D) in CTNS knockdown cells, respectively, compared with controls. Interestingly, despite the increased intracellular O2 − and NO levels, basal OS index was not significantly increased in CTNS knockdown cells, as assessed by fluorescence microscopy (Fig. 1 E) and flow cytometry (siNT vs. siCTNS, P = 0.1949, Fig. 1 F). Interestingly, under oxidizing conditions (200 μm hydrogen peroxide (H2O2), CTNS knockdown cells displayed a more pronounced increase in OS index than siNT‐transfected cells, as assessed by live fluorescence imaging (Fig. 1 E) and flow cytometry (siNT vs. siCTNS, P < 0.05, Fig. 1 F).
Figure 1. Effect of CTNS knockdown‐induced lysosomal cystine accumulation on intracellular ROS level and OS index .
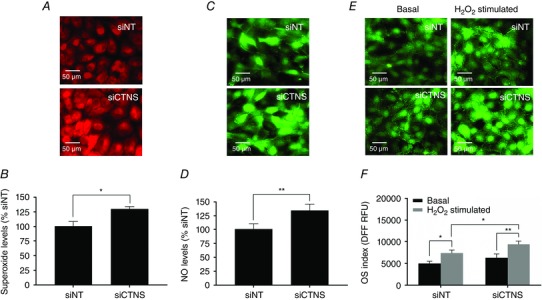
HK‐2 cells were transfected with CTNS siRNA (siCTNS) pool (50 nM) for 24 h. A separate batch of cells was transfected with a non‐targeting siRNA (siNT) pool (50 nm) as a negative control. After 72 h, the intracellular ROS levels and OS index were assessed. Intracellular O2 − production was monitored in live cells by fluorescence microscopy using a O2 −‐sensitive probe DHE. Representative fluorescence images are shown (A). Using the same probe, intracellular O2 − levels were assessed by flow cytometry. O2 − levels are expressed as mean % siNT ± SD of three independent experiments (B). Intracellular NO production was monitored in live cells by fluorescence microscopy using an NO‐specific fluorogenic probe, DAF‐FMDA. Representative fluorescence images are shown (C). Intracellular NO levels were also assessed by flow cytometry using DAF‐FMDA. NO levels are expressed as mean % siNT ± SD of three independent experiments (D). The general OS index of the cells was assessed by fluorescence microscopy using the redox‐sensitive dye DFFDA under basal conditions and in the presence of H2O2 (200 μm) for 1 h. Representative fluorescence images are shown (E). In a separate experiment, the intracellular oxidation of DFFDA was assessed by flow cytometry. The OS indices are expressed as mean DFF fluorescence (relative fluorescence units, RFU) ± SD of three independent experiments (F). Asterisks indicate statistically significant differences at *P < 0.01 and **P < 0.001.
Lysosomal cystine accumulation promotes nitrosative damage under oxidizing conditions
Because increased intracellular levels of O2 − and NO can lead to the generation of highly oxidizing peroxynitrite (ONOO−) which, in turn, can induce nitrosative damage by nitrating tyrosine residues in proteins, we determined the effect of CTNS knockdown‐induced lysosomal cystine accumulation on nitrotyrosine levels. Using Western blot analysis, CTNS knockdown cells displayed an increase in nitrotyrosine levels compared with siNT‐transfected cells (1.08 vs. 1.67 fold change, respectively, P < 0.05, Fig. 2 A and B). However, no statistical difference between siNT‐ and siCTNS‐transfected cells was observed when ELISA was used to measure nitrotyrosine levels (P = 0.3642, Fig. 2 C). Under oxidizing conditions, CTNS knockdown cells showed a more pronounced increase in nitrotyrosine levels compared with siNT‐transfected cells, as assessed by either Western blot (1.69 vs. 2.35 fold change, respectively, P < 0.05, Fig. 2 A and B) or ELISA (6.4 vs. 8.9 μmol nitrotyrosine mg protein–1, respectively, P < 0.05, Fig. 2 B). The discrepancy observed between Western blot and ELISA may be attributed to the sensitivity of detecting nitrotyrosines associated with these techniques. While ELISA is generally considered more sensitive than Western blot, the prerequisite for most ELISAs is a huge amount of highly purified antigen for optimal detection. The ELISA described in the present study utilized cell lysates which, arguably, might present a limitation for nitrotyrosine detection under basal conditions.
Figure 2. Effect of CTNS knockdown‐induced lysosomal cystine accumulation on nitrotyrosine levels .
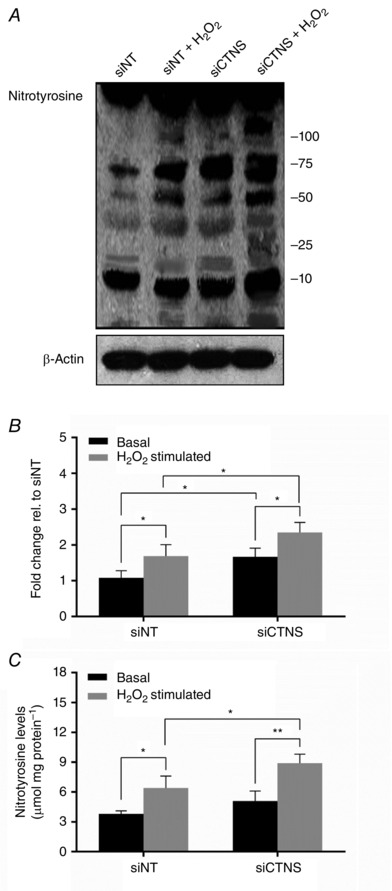
HK‐2 cells were transfected with CTNS siRNA pool (siCTNS) for 24 h. A separate batch of cells was transfected with a non‐targeting siRNA (siNT) pool (50 nm) as a negative control. Nitrotyrosine levels were assessed 72 h after transfection. Nitrotyrosine levels were determined by Western blot using an anti‐nitrotyrosine antibody. A representative blot for nitrotyrosine is shown (A). β‐Actin was used as a loading control. Densitometry analysis of nitrotyrosine expression was performed by normalizing the band densities of nitrotyrosine for β‐actin. Nitrotyrosine levels are expressed as fold change relative (rel) to siNT‐transfected cells +/‐ SD of three independent experiments (B). In a separate experiment, nitrotyrosine content was determined by competitive ELISA using an OxiSelect Nitrotyrosine ELISA Kit (Cell Biolabs). Nitrotyrosine levels are expressed as nmol nitrotyrosine mg protein–1 ± SD of three independent experiments (C). Asterisks indicate statistically significant differences at *P < 0.01 and **P < 0.001.
Lysosomal cystine accumulation promotes induction of ROS‐generating enzymes
Because iNOS and NOX represent two major sources of cellular ROS in kidney PTECs (Gwinner et al. 1998; Beltowski et al. 2004; Liu et al. 2012), we examined the effect of lysosomal cystine accumulation on the expression of these ROS‐generating enzymes by Western blot. The expression of iNOS increased significantly in CTNS knockdown cells by 1.8 ± 0.4‐fold (P < 0.05) when compared with siNT‐transfected cells (Fig. 3 A and B). The expression of p47phox, the cystosolic subunit of NOX, also increased significantly in CTNS knockdown cells by 2.3 ± 0.3‐fold (P < 0.05) when compared with siNT‐transfected cells (Fig. 3 A and B).
Figure 3. Effect of CTNS knockdown‐induced lysosomal cystine accumulation on iNOS and NOX expression .
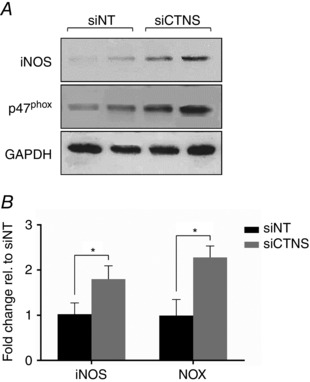
HK‐2 cells were transfected with CTNS siRNA pool (siCTNS) for 24 h. A separate batch of cells was transfected with a non‐targeting siRNA (siNT) pool (50 nm) as a negative control. iNOS and NOX expression were determined by Western blot 72 h after transfection. Representative blots for iNOS and NOX are shown (A). GAPDH was used as a loading control. Densitometry data for iNOS and NOX were normalized for GAPDH expression and are expressed as fold change relative (rel.) to siNT ± SD of three independent experiments (B). *Statistically significant difference at P < 0.05.
Lysosomal cystine accumulation increases intracellular Ca2+ flux
Increased intracellular Ca2+ flux has been shown to be essential in the activation of NOX via activation of Ca2+‐dependent stress kinases, such as protein kinase C (Jiang et al. 2011). To determine whether the observed NOX activation in cystine‐loaded cells was associated with mobilization intracellular Ca2+ stores, the levels of Ca2+ in CTNS knockdown cells were assessed using the Ca2+‐sensitive fluorogenic probe Fluo‐4‐AM. Using fluorescence microscopy, CTNS knockdown cells exhibited increased Fluo‐4 fluorescence compared with siNT‐transfected cells (Fig. 4 A), indicating increased intracellular Ca2+ flux. To validate this observation, intracellular Ca2+ levels were further measured by flow cytometry. Consistent with fluorescence microscopy, flow cytometry analysis of Fluo‐4 fluorescence showed that CTNS knockdown cells had increased intracellular Ca2+ levels compared with siNT‐transfected cells [711.6 ± 156.5 vs. 1035.0 ± 81.39 relative fluorescence units (RFU), respectively, P < 0.01] (Fig. 4 B and C).
Figure 4. Effect of CTNS knockdown‐induced lysosomal cystine accumulation on intracellular Ca2+ levels .
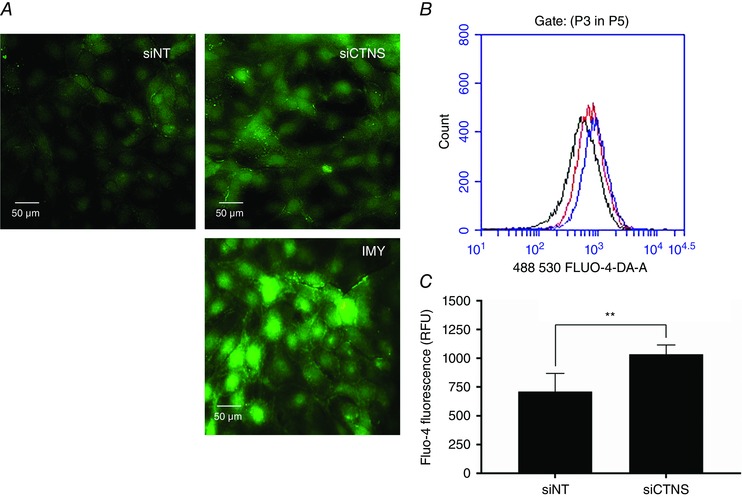
HK‐2 cells were transfected with CTNS siRNA (siCTNS) pool (50 nm) for 24 h. A separate batch of cells was transfected with a non‐targeting siRNA (siNT) pool (50 nm) as a negative control. After 72 h, the intracellular Ca2+ levels were assessed using the Ca2+‐sensitive probe Fluo‐4 AM. In one experiment, the fluorescence emission Ca2+‐bound Fluo‐4 was monitored in live adherent cells by fluorescence microscopy. Representative images are shown (A). Using the same probe, intracellular Ca2+ levels were quantified in cell suspensions using flow cytometry. A representative single‐parameter overlay histogram is shown (B) [black curve, siNT; red curve, siCTNS; blue curve, ionomycin (IMY)]. The relative concentration of intracellular Ca2+ is expressed as mean Fluo‐4 fluorescence (relative fluorescence units, RFU) ± SD of four independent experiments (C). IMY (1 mg ml−1), an ionophore that can raise intracellular Ca2+ levels, was used as a positive control in both experiments. **Statistically significant difference at P < 0.01.
Lysosomal cystine accumulation promotes induction redox‐sensitive transcription factors and cytoprotective proteins
To gain further insight into the stress responses associated with lysosomal cystine accumulation, we examined the effect of CTNS gene inhibition on the expression of various stress‐responsive transcription factors and cytoprotective proteins. One of these transcription factors is NF‐κB, which can be activated as a cellular defence mechanism in response to oxidative insult (Flohé et al. 1997; Kabe et al. 2005). Total NF‐κB levels were not significantly different between CTNS‐ and siNT‐transfected cells (P = 0.19, Fig. 5 A and B). Interestingly, however, the levels of phosphorylated NF‐κB(p65) increased significantly in CTNS knockdown cells compared with siNT‐transfected cells (1.6 ± 0.1‐fold, P < 0.05, Fig. 5 A and B), indicating an augmentation of NF‐κB activity.
Figure 5. Effect of CTNS knockdown‐induced lysosomal cystine accumulation on the expression of stress‐responsive proteins .
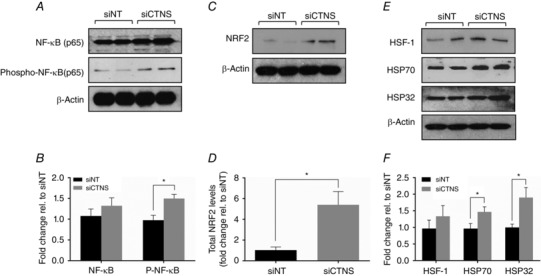
HK‐2 cells were transfected with CTNS siRNA (siCTNS) pool (50 nm) for 24 h. A separate batch of cells was transfected with a non‐targeting siRNA (siNT) pool (50 nm) as a negative control. The expression of various stress‐responsive proteins was determined 72 h after transfection. Representative blots are shown for NF‐κB(p65) and phospho‐NF‐κB(p65) (A), NRF2 (C), and HSF‐1, HSP70 and HSP32 (E). β‐Actin was used as a loading control in all Western blot experiments. Densitometry analyses of NF‐κB(p65) and phospho‐NF‐κB(p65) (B), NRF2 (D), and HSF‐1, HSP70 and HSP32 (F) were performed after normalization for β‐actin expression and are expressed as fold‐change relative to siNT ± SD of three independent experiments. *Statistically significant difference at P < 0.05.
NRF2 represents another important stress‐responsive transcription factor especially during OS (Nguyen et al. 2009). Interestingly, despite the lack of generalized OS, total NRF2 levels increased markedly by ∼5‐fold (P < 0.05) under increased lysosomal cystine conditions (Fig. 5 C and D).
Heat shock proteins (HSP) are key proteins that play a cytoprotective role during cellular stress and redox imbalance (Kregel, 2002; Kalmar & Greensmith, 2009). The induction of HSP expression is regulated by the interaction of heat shock factors (HSF) with regulatory elements in DNA termed heat shock elements (HSE) (Kregel, 2002). To assess whether lysosomal cystine accumulation induced heat shock response, we determined the effect of CTNS gene inhibition on the expression of HSF1, HSP70 and HSP32. Total HSF1 levels remained unchanged in CTNS knockdown cells when compared with siNT‐transfected cells (P = 0.3643, Fig. 5 E and F). While total HSF1 levels were unaffected, both HSP70 and HSP32 increased significantly in CTNS knockdown cells [1.5 ± 0.2‐fold (P < 0.05) and 1.9 ± 0.4‐fold (P < 0.05), respectively] (Fig. 5 E and F).
Lysosomal cystine accumulation results in the depolarization of ΔΨm and in the reduction of intracellular ATP content
Recognizing the crucial role of GSH in preserving mitochondrial integrity, we determined the effects of lysosomal cystine accumulation on ΔΨm. CTNS knockdown cells displayed a significantly increased J‐aggregates/J‐monomer ratio compared with siNT‐transfected cells (2.6 ± 0.3 vs. 1.3 ± 0.1, P < 0.01, Fig. 6 A and B), indicating depolarization in ΔΨm.
Figure 6. Effect of CTNS knockdown‐induced lysosomal cystine accumulation on ΔΨm and intracellular ATP content .
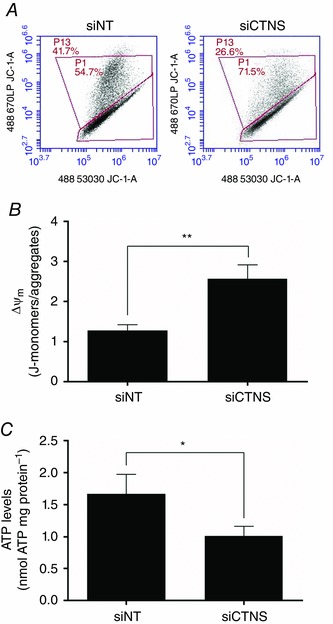
HK‐2 cells were transfected with CTNS siRNA (siCTNS) pool (50 nm) for 24 h. A non‐targeting siRNA (siNT) pool (50 nm) was used as a negative control. ΔΨm and intracellular ATP levels were determined 72 h after transfection. ΔΨm was assessed by flow cytometry using the JC‐1 dye. Representative two‐dimensional density plots of J‐monomers vs. J‐aggregates are shown (A). Flow cytometry data are expressed as the mean ratio of the fluorescence of J‐monomers to J‐aggregates ± SD of three independent experiments (B). Intracellular ATP concentrations were determined using the ATP Bioluminescence Assay Kit HSII (Roche). ATP levels are expressed as mean nmol ATP mg protein–1 ± SD of three independent experiments (C). Asterisks indicate statistically significant differences at *P < 0.05 and **P < 0.01.
Because the cellular energy status is tightly coupled to mitochondrial integrity, the ATP levels were then measured following CTNS gene silencing. As expected, the ATP levels in CTNS knockdown cells were reduced significantly compared with siNT‐transfected cells (1.7 ± 0.3 vs. 1.0 ± 0.1 nmol ATP mg protein–1, P < 0.05) (Fig. 6 C).
Lysosomal cystine accumulation does not alter autophagic activity
Mitochondrial autophagy has previously been documented in cystinotic PTECs and has been hypothesized to be an important event leading to ATP depletion (Sansanwal et al. 2010 b). To determine whether similar perturbation in autophagy could be observed in PTECs with reduced CTNS levels, we examined the expression of key autophagy markers in PTECs following CTNS gene inhibition. Interestingly, despite elevated lysosomal cystine load, no alteration in the expression of autophagy markers, Beclin (P = 0.54), LC3‐I (P = 0.51) and LC3‐II (P = 0.07), was observed in CTNS knockdown cells (Fig. 7 A and B). These data suggest that lysosomal cystine accumulation alone may not be the sole requirement for the induction of autophagy in PTECs and may not be the predominant underlying mechanism leading to ATP depletion observed in CTNS knockdown cells.
Figure 7. Effect of CTNS knockdown‐induced lysosomal cystine accumulation on autophagic activity .
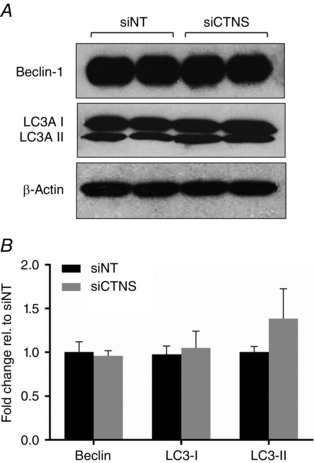
HK‐2 cells were transfected with CTNS siRNA (siCTNS) pool (50 nm) for 24 h. A separate batch of cells was transfected with a non‐targeting siRNA (siNT) pool (50 nm) as a negative control. The expression of various autophagy markers was determined by Western blot 72 h after transfection. Representative blots for Beclin‐1, LC3A I and LC3A II are shown (A). β‐Actin was used as a loading control. Densitometry analyses of Beclin 1, LC3A I and LC3A II expression were performed after normalization for β‐actin expression and are expressed as fold‐change relative to siNT ± SD of three independent experiments (B).
Lysosomal cystine accumulation augments apoptosis and reduces cell viability
To assess whether the observed alterations in redox and energy status impacted cell survival, the effects of CTNS knockdown‐induced lysosomal cystine accumulation on the levels of apoptosis and necrosis were determined. Since the test described in the present study was an endpoint analysis, it does not distinguish cells that have undergone apoptotic death versus those that have died as a result of a necrotic pathway. In general, cells that are in early apoptosis are FITC Annexin V positive and PI negative; cells that are in late apoptosis are both FITC Annexin V and PI positive; cells that are FITC Annexin V negative and PI positive are necrotic cells (Vermes et al. 1995). As shown in Fig. 8 A and B, most of the siNT‐ and siCTNS‐transfected cells were in the early apoptotic phase. However, the percentage of early apoptotic cells remained unchanged following CTNS gene knockdown compared with siNT‐transfected cells (P = 0.54) (Fig. 8 A and B). On the other hand, the percentage of late apoptotic cells increased significantly in CTNS knockdown cells compared with siNT‐transfected cells (12.4 ± 1.6 vs. 3.1 ± 1.2%, P < 0.05) (Fig. 8 A and B). CTNS knockdown cells also displayed increased necrosis compared with siNT‐transfected cells (3.1 ± 0.9 vs. 0.96 ± 0.2%, P < 0.05) (Fig. 8 A and B).
Figure 8. Effect of CTNS knockdown‐induced lysosomal cystine accumulation on the levels of apoptosis and necrosis and on cell viability .
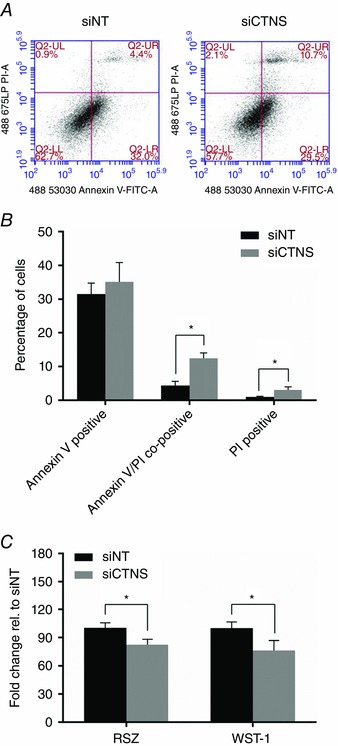
HK‐2 cells were transfected with CTNS siRNA (siCTNS) pool (50 nm) for 24 h. A separate batch of cells was transfected with a non‐targeting siRNA (siNT) pool (50 nm) as a negative control. The proportion of apoptotic and necrotic cells were determined by flow cytometry 72 h after transfection using Annexin V‐FITC and PI, respectively. Representative two‐dimensional plots of Annexin V‐FITC (ex: 488 nm; em: 530 ± 30 nm) vs. PI (ex: 488 nm; em: 675 nm) are shown (A). The levels of early apoptosis, late apoptosis and necrosis are expressed as the percentage of Annexin V‐positive, Annexin V/PI‐co‐positive and PI‐positive cells ± SD of three independent experiments, respectively (B). Cell viability was determined by assessing the metabolic capacity of cells in reducing the indicator dyes resazurin and WST‐1. The resazurin and WST‐1 reduction capacity of cells are both expressed as fold‐change relative to siNT ± SD of three independent experiments (C). *Statistically significant difference at P < 0.05.
To further assess the viability of cells under increased lysosomal cystine conditions, we determined the metabolic capacity of the cells in reducing the indicator dyes resazurin and WST‐1 to resorufin and formazan, respectively, following CTNS gene knockdown. As shown in Fig. 8 C, CTNS knockdown cells showed a ∼17% reduction (P < 0.05) in metabolic capacity in reducing resazurin. Consistent with this observation, CTNS knockdown cells showed a 24% reduction (P < 0.01) in metabolic capacity in reducing WST‐1 (Fig. 8 C). These results indicate that the biochemical and metabolic insults initiated by lysosomal cystine accumulation impacted on the viability of kidney PTECs.
Cysteamine treatment reverses some effects associated with lysosomal cystine accumulation
The main treatment for cystinosis is based on lysosomal cystine depletion using the aminothiol, cysteamine. To assess whether cystine depletion can afford protection in cystine‐loaded cells, cells were treated with cysteamine following CTNS gene silencing. The optimal cysteamine concentration was first determined by resazurin reduction and WST‐1 assay. These experiments established 1000 μm as the optimal cysteamine concentration which consistently resulted to > 90% viability in HK‐2 cells following 24 h incubation (results not shown).
We first examined whether cysteamine alone will alter the redox status of cells and, as shown in Fig. 9 A, cysteamine treatment had no effect on the DFF fluorescence in both siNT‐ (P = 0.9999) and siCTNS‐transfected cells (P = 0.9914), indicating that cysteamine does not alter the intracellular OS index. To determine the effect of cysteamine on the OS index, cells were incubated with 200 μm H2O2 for 1 h to induce OS. As expected, exposure of cells to H2O2 increased the OS index in both siNT‐ (7339.8 ± 858.2 vs. 4699.07± 592.6 RFU at basal, P < 0.01) and siCTNS‐transfected cells (9089.8 ± 816.8 vs. 5220.3 ± 792.4 RFU at basal, P < 0.001) (Fig. 9 A). However, in the presence of cysteamine, the oxidizing effect of H2O2 was blunted in both siNT‐ (5404.8 ± 629.8, P < 0.05) and siCTNS‐transfected cells (6275.8 ± 893.7 RFU, P < 0.01), suggesting that cysteamine can afford antioxidant protection in kidney PTECs subjected to OS.
Figure 9. Effect of cysteamine on OS index and intracellular GSH levels .
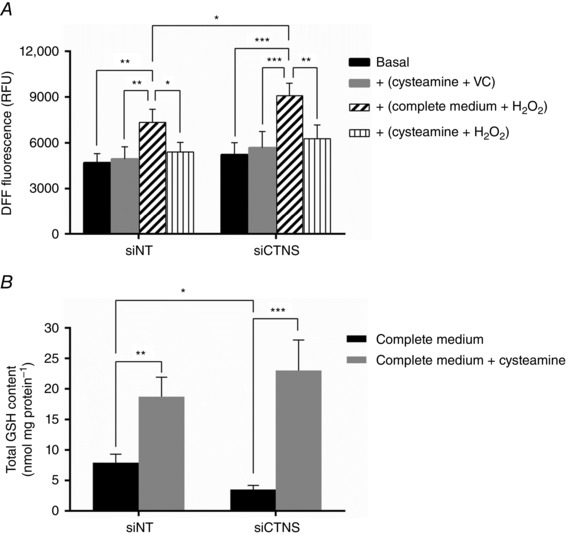
HK‐2 cells were transfected with CTNS siRNA (siCTNS) pool (50 nm) for 24 h. A separate batch of cells was transfected with a non‐targeting siRNA (siNT) pool (50 nm) as a negative control. After 48 h, the cells were incubated with 1000 μm cysteamine or with complete culture medium for 24 h. Subsequently, 200 μm H2O2 or vehicle control was added to the cell culture for 1 h. The OS index was assessed by measuring the fluorescence of the oxidized DFF‐DA (i.e. DFF) by flow cytometry. The OS index is expressed as mean DFF fluorescence (relative fluorescence units, RFU) ± SD of four independent experiments (A). The effect of cysteamine on the intracellular GSH content was measured by an enzyme recycling assay following incubation of cells with 1000 μm cysteamine or with complete culture medium for 24 h, 48 h after transfection. Total GSH levels are expressed as mean nmol GSH mg protein–1 ± SD of four independent experiments (B). Asterisks indicate statistically significant differences at *P < 0.05, **P < 0.01 and ***P < 0.001.
We have previously observed that GSH depletion was one of the biochemical consequences of CTNS knockdown‐induced lysosomal cystine accumulation in kidney PTECs (Sumayao et al. 2013). Because of the potential of cysteamine as a precursor for cysteine, we examined the effect of cysteamine on GSH levels following CTNS gene knockdown. Not only did cysteamine result in the repletion of intracellular GSH levels, cysteamine treatment markedly increased GSH levels in both siNT‐ (18.7 ± 3.2 vs. 7.9 ± 1.4 nmol GSH mg protein–1 at basal, P < 0.01) and siCTNS‐tranfected cells (23.0 ± 5.0 vs. 3.5 ± 0.8 nmol GSH mg protein–1 at basal, P < 0.001) (Fig. 9 B). These data indicate that cysteamine, aside from its lysosomal cystine depleting capacity, can promote GSH synthesis in kidney PTECs.
Because GSH is essential for mitochondrial function and integrity to maintain the intracellular energy status, we then investigated whether the GSH boosting effect of cysteamine could also rescue ATP depletion in kidney PTECs with high cystine load. As shown in Fig. 10 A, cysteamine normalized the ATP levels in CTNS knockdown cells (2.24 ± 0.4 vs. 1.30 ± 0.5 nmol ATP mg protein–1 at basal, P < 0.05), approaching the levels observed in siNT‐transfected cells (2.4 ± 0.3 nmol ATP mg protein–1). ATP levels remained unchanged in siNT‐transfectd cells following cysteamine treatment (P = 0.5268), suggesting that ATP reduction in CTNS knockdown cells may be, at least in part, a consequence of intracellular GSH depletion.
Figure 10. Effect of cysteamine on intracellular ATP content and cell viability .
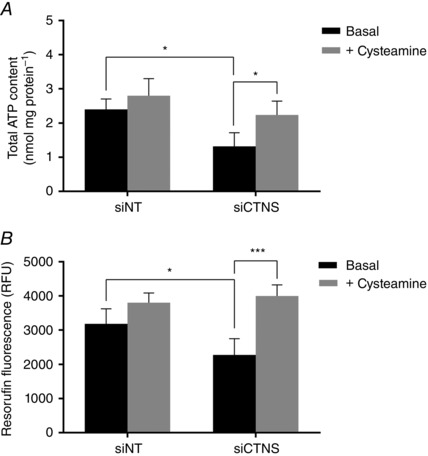
HK‐2 cells were transfected with CTNS siRNA (siCTNS) pool (50 nm) for 24 h. As a negative control, a separate batch of cells was transfected with a non‐targeting siRNA (siNT) pool (50 nm). After 48 h, the cells were incubated with 1000 μm cysteamine or with complete culture medium for 24 h. Intracellular ATP content was measured using the ATP Bioluminescence Assay Kit HSII (Roche). ATP levels are expressed as mean nmol ATP mg protein–1 ± SD of four independent experiments (A). The effect of cysteamine on cell viability was assessed by the metabolic capacity of cells in reducing resazurin to resorufin following incubation of cells with 1000 μm cysteamine or with complete culture medium for 24 h, 48 h after transfection. Cell viability is expressed as mean resorufin fluorescence (relative fluorescence units, RFU) ± SD of four independent experiments (B). Asterisks indicate statistically significant differences at *P < 0.05 and ***P < 0.001.
To determine whether the observed cytoprotective effects mediated by cysteamine could preserve cell viability in kidney PTECs with high cystine load, we determined the effect of cysteamine on the metabolic capacity of cells in reducing resazurin to fluorescent resorufin following CTNS gene knockdown. CTNS knockdown cells displayed increased resorufin fluorescence compared with its matched basal control (3998.8 ± 324.7 vs. 2278.6 ± 469.4 RFU, respectively, P < 0.001) (Fig. 10 B) and was comparable to the levels observed in siNT‐transfected cells (3181.0 ± 440.7). Interestingly, cysteamine did not further increase the resorufin fluorescence in siNT transfected cells when compared with its matched basal control (P = 0.1261). These results indicate that cysteamine treatment promotes preservation of the viability of kidney PTECs under elevated lysosomal cystine conditions, which further substantiates the cytoprotective effect of cysteamine.
Discussion
Early cystinosis research identified lysosomes as the main cystine storage site in cells (Schneider et al. 1967 a; Schulman et al. 1969). The physiological and pathological roles of lysosomal cystine accumulation remain poorly understood partly because cystine should not be toxic especially when isolated within the lysosomal compartment, suggesting no obvious disadvantage for cystinotic cells (Park et al. 2002). However, it was proposed that lysosomal cystine reserves may possibly be a limiting factor for the synthesis of GSH in the cytosol (Chol et al. 2004; Laube et al. 2006; Bellomo et al. 2010). GSH serves as an important cofactor for the GSH peroxidase family of enzymes, which metabolize H2O2 and lipid peroxides, preventing damage to cellular components. Therefore, GSH depletion has become a compelling candidate responsible for the cellular dysfunction associated with cystinosis (Chol et al. 2004; Laube et al. 2006). Indeed, GSH depletion has been shown to result in mitochondrial dysfunction in several cell and animal models (Han et al. 2003). However, GSH deficiency is unlikely to be the sole factor responsible for the complexity of cell stress in cystinosis and this raises the possibility that cells may also respond to other stimuli, which merits further investigation.
We have demonstrated in the present study that CTNS gene inhibition in kidney PTECs was associated with a marked increase in intracellular O2 − and NO production but did not seem to affect the cells’ general OS index. Interestingly, under oxidative challenge induced by H2O2, cells with deficient cystinosin displayed a more pronounced increase in OS index, indicating compromised antioxidant capacity. Consistent with our findings, Wilmer et al. (2011) observed that exposure of conditionally immortalized PTECs obtained from cystinosis patients to high levels of H2O2 resulted in more pronounced ROS production compared with healthy cells. Therefore, lysosomal cystine accumulation, as a consequence of lack of cystinosin activity, reduces the capacity of the cell to deal with OS.
Note, however, that the levels of O2 − and NO represent only one aspect of oxidative stress because these reactive molecules may give rise to a more oxidizing ROS. For example, simultaneous formation of O2 − and NO produces peroxynitrite (ONOO−), which can induce a more aggressive oxidation, nitrosation, and nitration of biological molecules (Kohen & Nyska, 2002). Uncontrolled production of ONOO− can indiscriminately target proteins by nitrating tyrosine residues which can adversely affect the function or activity of the protein (Kohen & Nyska, 2002), potentially leading to apoptotic or necrotic cell death. Interestingly, basal levels of nitrotyrosines in CTNS knockdown cells remained normal, suggesting that the antioxidant capacity of these cells may still be sufficient to prevent oxidative damage at basal conditions. However, under oxidizing conditions, CTNS knockdown cells exhibited a more pronounced nitrotyrosination, suggesting increased susceptibility of these cells to oxidative insult.
NOX and iNOS represent major sources of intracellular O2 − and NO in kidney PTECs, respectively (Beltowski et al. 2004; Liu et al. 2012). To our knowledge, the present study has provided the first evidence of increased expression of NOX and iNOS in cystinotic‐like cells, which may have influenced the observed augmented production of O2 − and NO in these cells. Although the NOX system may be activated by various cellular stresses, the sequence of events from lysosomal cystine accumulation leading to NOX induction is largely unknown. One possibility is that altered mitochondrial integrity and subsequent Ca2+ release to the cytoplasm may activate Ca2+‐dependent stress kinases, such as protein kinase C, which has been shown to be essential in the activation of NOX (Jiang et al. 2011). Indeed, we observed a significant increase in the intracellular Ca2+ levels in kidney PTECs following CTNS knockdown, a physiological event that may be an upstream signal for the activation of NOX.
NO, if generated at high levels in mitochondria, may result in the auto‐oxidation of ubiquinol with concomitant production of O2 −, H2O2, and ONOO− (Turrens, 2003). These reactive molecules may induce irreversible damage to mitochondrial respiratory complexes and inhibition of ATP synthesis (Turrens, 2003), potentially leading to apoptotic or necrotic cell death. Indeed, we have observed an extensive depolarization of mitochondria and a significant reduction in ATP content following CTNS knockdown, suggesting that mitochondrial integrity and function are compromised under elevated lysosomal cystine conditions. These alterations present a clear disadvantage for cystinotic PTECs in vivo as the mass transport of most solutes across the proximal tubular epithelium is largely influenced by the cellular energy (Wilmer et al. 2010 a), which concomitantly supplies some of the essential metabolites to generate energy and preserve redox status. Both ATP and GSH are critical for the survival of kidney PTECs, which are highly metabolically active cells and therefore are constantly exposed to high levels of ROS generated by oxidative mitochondrial metabolism.
Apart from mitochondria, the lysosomes are an important site for ROS production (Nohl & Gille, 2005). It is possible that the elevated ROS and augmented cell death observed in the present study were caused by ROS leakage from the lysosomes. Indeed, lysosomal permeabilization has been proposed to be one of the mechanisms leading to proximal tubular cell death associated with cystinosis (Park et al. 2002). Lysosomal permeabilization may be an upstream event leading to ROS leakage, which could then alter the redox status of cells and may lead to inappropriate cell death.
Despite the compendium of studies addressing the role of ATP depletion in the pathogenesis of cystinosis, measurement of ATP levels in cystinotic cells has yielded inconsistent results (Wilmer et al. 2005, 2011; Levtchenko et al. 2006; Sansanwal et al. 2010 b). It appears that the hypothesis implicating reduced ATP generation as the keystone in the pathogenesis of cystinosis understates the complexity of the problem. Most recently, a striking degree of mitochondrial autophagy and a reduced mitochondrial ATP generation have been observed in cystinotic PTECs, which may be an important event leading to ATP depletion (Sansanwal et al. 2010 b). However, in the present study, the expression of various markers of autophagy was unaltered in CTNS knockdown cells despite increased cystine levels. This observation suggests that cystine accumulation, although regarded as the biochemical hallmark of cystinosis, may not be obligatory for the dysregulation of autophagy.
While small fluctuations in the steady‐state concentrations of ROS may play a role in intracellular signalling (Cadenas, 2004; Valko et al. 2007), uncontrolled production of ROS can lead to the expression of genes primarily controlled by redox‐sensitive transcription factors. In the present study, CTNS knockdown cells had increased levels of the phosphorylated NF‐κB(p65) subunit. It was previously reported that overexpressing the transcription factor NF‐κB in renal proximal tubular cells increased the levels of antioxidative enzymes (George et al. 2012), indicating that NF‐κB plays a direct role in cellular antioxidant homeostasis. Although the activity of NF‐κB is principally regulated by the IκB proteins (also known as the canonical pathway) (Gilmore, 2006), NF‐κB may be activated by the phosphorylation of its p65 subunit which augments its transcriptional activity (Janssen‐Heininger et al. 2000; Gilmore, 2006), an event that occurs independently of IκB. Therefore, the phosphorylation of p65 may represent a non‐canonical pathway for the activation of NF‐κB. Although there is no direct evidence herein demonstrating that increased levels of ROS augmented the phosphorylation of NF‐κB, it has been reported that mechanisms exist for direct and indirect activation of NF‐κB by redox‐sensitive enzymes (Flohé et al. 1997; Kabe et al. 2005; Pantano et al. 2006). Furthermore, ROS‐mediated NF‐κB activation is likely to depend on the type of ROS involved and as well as the cell type under investigation (Janssen‐Heininger et al. 2000).
NRF2 represents an important stress‐responsive transcription factor which interacts with the cis‐acting elements in DNA termed antioxidant response elements (ARE) (Nguyen et al. 2009). These elements control the expression of genes involved in the detoxification and elimination of ROS (Nguyen et al. 2009). Interestingly, in the present study, CTNS knockdown cells displayed an increase in NRF2 levels. However, the unaltered generalized OS index in CTNS knockdown cells suggests an alternative mechanism for the activation of NRF2. Activation of NRF2 can occur at the level of endosplasmic reticulum (ER) through a redox‐independent pathway (Cullinan et al. 2003; Ho et al. 2005). Interestingly, microarray and ultrastructural analyses of cystinotic specimens have suggested a strong link between NRF2 upregulation and ER stress in cystinosis (Sansanwal et al. 2010 a). Most recently, Johnson et al. (2013) showed a marked ER expansion and an increase in unfolded protein response‐induced chaperones Grp78 and Grp94 in Ctns –/– PTECs, a hallmark of ER stress. The authors also suggested that ER stress may influence the intracellular Ca2+ levels in cystinotic cells. The increased Ca2+ influx following CTNS knockdown observed in the present study may be a consequence of ER stress. Therefore, it can be speculated that NRF2 activation in cystinosis may be mediated by perturbations in ER homeostasis in an attempt to rescue the cells from inappropriate apoptosis.
HSPs play a role as molecular chaperones to prevent protein misfolding during and after exposure to oxidative insults (Kalmar & Greensmith, 2009). The induction of HSP32 and HSP70 in the absence of OS may suggest an involvement of a signalling pathway independent of redox perturbation. It has been suggested that HSP32 is highly specialized to sense stress conditions even at low levels, in which protein folding is compromised (Kalmar & Greensmith, 2009). Arguably, it is possible that in the present study, the redox perturbations resulting from lysosomal cystine accumulation were transient and/or below detection limits that may be sufficient to trigger HSP induction. Furthermore, the compromised antioxidant capacity in CTNS knockdown cells suggests the presence of early‐phase redox perturbations in these cells.
Cysteamine has revolutionized the treatment and management of cystinosis patients by retarding both renal and non‐renal complications (Gahl et al. 2002). However, the mechanisms of protection by cysteamine have not been fully elucidated. In the present study, cysteamine reversed the various biochemical alterations observed in CTNS knockdown cells such as reduced intracellular GSH and ATP content, decreased antioxidant capacity and augmented apoptosis. It is possible that the cysteine‐boosting effect of cysteamine, being an aminothiol, may have enhanced the antioxidant capacity of kidney PTECs by promoting GSH synthesis. Indeed, we observed a marked increase in the cysteine levels in HK‐2 cells following CTNS knockdown (results not shown). In agreement with our findings, Bellomo et al. (2010) showed that GSH levels and GSH redox status of cystinotic ciPTECs normalized following cysteamine treatment. Therefore, cytoprotective effects that cysteamine affords may be attributed to its antioxidant capacity, which can rescue the cells from the damaging effects of reactive molecules such as ROS and reactive nitrogen species.
How do these findings reconcile with the current understanding of the pathogenesis of cystinosis? The homeostatic modulation of ROS is a highly efficient mechanism allowing cells to tightly control their redox status within a very narrow range (Finkel, 2003; Cadenas, 2004; Valko et al. 2007). While small fluctuations in the steady‐state levels of ROS may play a role in intracellular signalling (Cadenas, 2004), an uncontrolled increase in these oxidants, especially under conditions of high metabolic activity, can lead to induction of protective genes for subverting ‘danger signals’ and may determine cell fate. Cystinotic cells may be disadvantaged because they are more vulnerable to oxidative insults than healthy cells. Indeed, Vaisbich et al. (2011) reported increased levels of serum thiobarbituric acid reactive substances (TBAR) in patients with nephropathic cystinosis, which is a direct evidence of OS in cystinosis. Therefore, cystinotic cells and tissues may be more vulnerable to oxidative insult than estimated in vitro and may warrant the need for antioxidant therapy to retard organ demise.
Overall, our results have demonstrated that lysosomal cystine accumulation in kidney PTECs, as a consequence of CTNS gene inhibition, promotes perturbation of cellular redox status, compromised mitochondrial function and augmented cell death. The apparent upregulation of classical stress‐responsive and cytoprotective proteins and the reduction in antioxidant capacity suggest that significant disruption in redox balance initiated by lysosomal sequestration of cystine may be involved in the kidney proximal tubular dysfunction associated with cystinosis. Further studies of stress response proteins and their associated signalling pathways may offer critical insights into the important events leading to renal injury and cell death in cystinosis. This may offer novel links for targeted therapy of the disease, in combination with the cystine‐depleting therapies, which can potentially lead to the attenuation of renal injury and preservation of the functions of other tissues and organs affected in cystinosis. However, it is not clear whether ‘acute’ in vitro models of cystine accumulation reflect the molecular alterations associated with the chronic cystine accumulation in cystinosis in vivo. To add to this complexity, there is accumulating evidence suggesting that cystine accumulation alone is not obligatory for the manifestation of molecular and physiological alterations in cystinosis (Wilmer et al. 2010 a). The use of animal models that can reproduce the clinical phenotype in cystinosis, such as the Ctns –/– mouse (Nevo et al. 2010), will be hugely beneficial in shedding light into the molecular events that ultimately lead to renal injury in cystinosis.
Additional information
Competing Interests
None declared.
Author contributions
R.S., with contribution from B.M., performed all experiments. R.S., T.M. and P.N. designed the study and all authors participated in the interpretation of the results. All authors contributed to writing the manuscript and to subsequent revisions.
Funding
This study was supported by the Cystinosis Foundation Ireland and the Cystinosis Research Foundation.
Acknowledgements
We gratefully acknowledge Dr Craig Slattery and the members of the UCD Renal Disease Research Group for their technical assistance and scientific discussions.
References
- Baker MA, Cerniglia GJ & Zaman A (1990). Microtiter plate assay for the measurement of glutathione and glutathione disulfide in large numbers of biological samples. Anal Biochem 190, 360–365. [DOI] [PubMed] [Google Scholar]
- Bellomo F, Corallini S, Pastore A, Palma A, Laurenzi C, Emma F & Taranta A (2010). Modulation of CTNS gene expression by intracellular thiols. Free Radic Biol Med 48, 865–872. [DOI] [PubMed] [Google Scholar]
- Beltowski J, Marciniak A, Jamroz‐Wiśniewska A & Borkowska E (2004). Nitric oxide–superoxide cooperation in the regulation of renal Na+K+ATPase. Acta Biochim Pol 51, 933–942. [PubMed] [Google Scholar]
- Cadenas E (2004). Mitochondrial free radical production and cell signaling. Mol Aspects Med 25, 17–26. [DOI] [PubMed] [Google Scholar]
- Chol M, Nevo N, Cherqui S, Antignac C & Rustin P (2004). Glutathione precursors replenish decreased glutathione pool in cystinotic cell lines. Biochem Biophys Res Commun 324, 231–235. [DOI] [PubMed] [Google Scholar]
- Cullinan SB, Zhang D, Hannink M, Arvisais E, Kaufman RJ & Diehl JA (2003). Nrf2 is a direct PERK substrate and effector of PERK‐dependent cell survival. Mol Cell Biol 23, 7198–7209. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Danpure C, Jennings P & Fyfe D (1986). Further studies on the effect of chloroquine on the uptake, metabolism and intracellular translocation of [35S] cystine in cystinotic fibroblasts. Biochim Biophys Acta 885, 256–265. [DOI] [PubMed] [Google Scholar]
- Feliubadaló L, Font M, Purroy J, Rousaud F, Estivill X, Nunes V, Golomb E, Centola M, Aksentijevich I & Kreiss Y (1999). Non‐type I cystinuria caused by mutations in SLC7A9, encoding a subunit (b0,+AT) of rBAT. Nat Genet 23, 52–57. [DOI] [PubMed] [Google Scholar]
- Finkel T (2003). Oxidant signals and oxidative stress. Curr Opin Cell Biol 15, 247–254. [DOI] [PubMed] [Google Scholar]
- Flohé L, Brigelius‐Flohé R, Saliou C, Traber MG & Packer L (1997). Redox regulation of NF‐κB activation. Free Radic Biol Med 22, 1115–1126. [DOI] [PubMed] [Google Scholar]
- Gahl W, Bashan N, Tietze F, Bernardini I & Schulman J (1982). Cystine transport is defective in isolated leukocyte lysosomes from patients with cystinosis. Science 217, 1263–1265. [DOI] [PubMed] [Google Scholar]
- Gahl W, Thoene JG, Schneider JA (2001). Cystinosis: a disorder of lysosomal membrane transport In The Metabolic and Molecular Bases of Inherited Disease, 8 edn, ed. Scriver CRBA, Sly WS, Valle D, Vogelstein B, pp. 5085–5108. McGraw‐Hill, New York. [Google Scholar]
- Gahl WA, Thoene JG & Schneider JA (2002). Cystinosis. N Engl J Med 347, 111–121. [DOI] [PubMed] [Google Scholar]
- Gahl WA, Tietze F, Bashan N, Steinherz R & Schulman J (1982). Defective cystine exodus from isolated lysosome‐rich fractions of cystinotic leucocytes. J Biol Chem 257, 9570–9575. [PubMed] [Google Scholar]
- George LE, Lokhandwala MF & Asghar M (2012). Novel role of NF‐κB‐p65 in antioxidant homeostasis in human kidney‐2 cells. Am J Physiol Renal Physiol 302, F1440–F1446. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gilmore T (2006). Introduction to NF‐κB: players, pathways, perspectives. Oncogene 25, 6680–6684. [DOI] [PubMed] [Google Scholar]
- Gwinner W, Deters‐Evers U, Brandes RP, Kubat B, Koch KM, Pape M & Olbricht CJ (1998). Antioxidant–oxidant balance in the glomerulus and proximal tubule of the rat kidney. J Physiol 509, 599–606. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Han D, Canali R, Rettori D & Kaplowitz N (2003). Effect of glutathione depletion on sites and topology of superoxide and hydrogen peroxide production in mitochondria. Mol Pharmacol 64, 1136–1144. [DOI] [PubMed] [Google Scholar]
- Ho HK, White CC, Fernandez C, Fausto N, Kavanagh TJ, Nelson SD & Bruschi SA (2005). Nrf2 activation involves an oxidative‐stress independent pathway in tetrafluoroethylcysteine‐induced cytotoxicity. Toxicol Sci 86, 354–364. [DOI] [PubMed] [Google Scholar]
- Jakubowski W & Bartosz G (2000). 2, 7‐dichlorofluorescin oxidation and reactive oxygen species: what does it measure? Cell Biol Int 24, 757–760. [DOI] [PubMed] [Google Scholar]
- Janssen‐Heininger Y, Poynter ME & Baeuerle PA (2000). Recent advances towards understanding redox mechanisms in the activation of nuclear factor κB. Free Radic Biol Med 28, 1317. [DOI] [PubMed] [Google Scholar]
- Jiang F, Zhang Y & Dusting GJ (2011). NADPH oxidase‐mediated redox signaling: roles in cellular stress response, stress tolerance, and tissue repair. Pharmacol Rev 63, 218–242. [DOI] [PubMed] [Google Scholar]
- Johnson JL, Napolitano G, Monfregola J, Rocca CJ, Cherqui S & Catz SD (2013). Upregulation of the rab27a‐dependent trafficking and secretory mechanisms improves lysosomal transport, alleviates endoplasmic reticulum stress, and reduces lysosome overload in cystinosis. Mol Cell Biol 33, 2950–2962. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kabe Y, Ando K, Hirao S, Yoshida M & Handa H (2005). Redox regulation of NF‐κB activation: distinct redox regulation between the cytoplasm and the nucleus. Antioxid Redox Signal 7, 395–403. [DOI] [PubMed] [Google Scholar]
- Kalatzis V, Cherqui S, Antignac C & Gasnier B (2001). Cystinosin, the protein defective in cystinosis, is a H+‐driven lysosomal cystine transporter. EMBO J 20, 5940–5949. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kalatzis V, Nevo N, Cherqui S, Gasnier B & Antignac C (2004). Molecular pathogenesis of cystinosis: effect of CTNS mutations on the transport activity and subcellular localization of cystinosin. Hum Mol Genet 13, 1361–1371. [DOI] [PubMed] [Google Scholar]
- Kalmar B & Greensmith L (2009). Induction of heat shock proteins for protection against oxidative stress. Adv Drug Deliv Rev 61, 310–318. [DOI] [PubMed] [Google Scholar]
- Kohen R & Nyska A (2002). Invited review: Oxidation of biological systems: oxidative stress phenomena, antioxidants, redox reactions, and methods for their quantification. Toxicol Pathol 30, 620–650. [DOI] [PubMed] [Google Scholar]
- Kojima H, Urano Y, Kikuchi K, Higuchi T, Hirata Y & Nagano T (1999). Fluorescent indicators for imaging nitric oxide production. Angew Chem Int Ed Engl 38, 3209–3212. [DOI] [PubMed] [Google Scholar]
- Koopman G, Reutelingsperger C, Kuijten G, Keehnen R, Pals S & Van Oers M (1994). Annexin V for flow cytometric detection of phosphatidylserine expression on B cells undergoing apoptosis. Blood 84, 1415–1420. [PubMed] [Google Scholar]
- Kregel KC (2002). Invited review: heat shock proteins: modifying factors in physiological stress responses and acquired thermotolerance. J Appl Physiol 92, 2177–2186. [DOI] [PubMed] [Google Scholar]
- Laube GF, Shah V, Stewart VC, Hargreaves IP, Haq MR, Heales SJR & van't Hoff WG (2006). Glutathione depletion and increased apoptosis rate in human cystinotic proximal tubular cells. Pediatr Nephrol 21, 503–509. [DOI] [PubMed] [Google Scholar]
- Levtchenko EN, Wilmer MJG, Janssen AJM, Koenderink JB, Visch HJ, Willems PHGM, de Graaf‐Hess A, Blom HJ, van Den Heuvel LP & Monnens LA (2006). Decreased intracellular ATP content and intact mitochondrial energy generating capacity in human cystinotic fibroblasts. Pediatr Res 59, 287–292. [DOI] [PubMed] [Google Scholar]
- Liu J, Kennedy DJ, Yan Y & Shapiro JI (2012). Reactive oxygen species modulation of Na/K‐ATPase regulates fibrosis and renal proximal tubular sodium handling. Int J Nephrol 2012, 1–14. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nevo N, Chol M, Bailleux A, Kalatzis V, Morisset L, Devuyst O, Gubler MC & Antignac C (2010). Renal phenotype of the cystinosis mouse model is dependent upon genetic background. Nephrol Dial Transplant 25, 1059–1066. [DOI] [PubMed] [Google Scholar]
- Nguyen T, Nioi P & Pickett CB (2009). The Nrf2‐antioxidant response element signaling pathway and its activation by oxidative stress. J Biol Chem 284, 13291–13295. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nohl H & Gille L (2005). Lysosomal ROS formation. Redox Report 10, 199–205. [DOI] [PubMed] [Google Scholar]
- Pantano C, Reynaert NL, Vliet AVD & Janssen‐Heininger YMW (2006). Redox‐sensitive kinases of the nuclear factor‐κB signaling pathway. Antioxid Redox Signal 8, 1791–1806. [DOI] [PubMed] [Google Scholar]
- Park M, Helip‐Wooley A & Thoene J (2002). Lysosomal cystine storage augments apoptosis in cultured human fibroblasts and renal tubular epithelial cells. J Am Soc Nephrol 13, 2878–2887. [DOI] [PubMed] [Google Scholar]
- Pras E, Raben N, Golomb E, Arber N, Aksentijevich I, Schapiro JM, Harel D, Katz G, Liberman U & Pras M (1995). Mutations in the SLC3A1 transporter gene in cystinuria. Am J Hum Genet 56, 1297–1303. [PMC free article] [PubMed] [Google Scholar]
- Riccardi C & Nicoletti I (2006). Analysis of apoptosis by propidium iodide staining and flow cytometry. Nat Protoc 1, 1458–1461. [DOI] [PubMed] [Google Scholar]
- Sansanwal P, Li L, Hsieh SC & Sarwal MM (2010. a). Insights into novel cellular injury mechanisms by gene expression profiling in nephropathic cystinosis. J Inherit Metab Dis 33, 775–786. [DOI] [PubMed] [Google Scholar]
- Sansanwal P, Yen B, Gahl WA, Ma Y, Ying L, Wong LJC & Sarwal MM (2010. b). Mitochondrial autophagy promotes cellular injury in nephropathic cystinosis. J Am Soc Nephrol 21, 272–283. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schneider JA, Bradley K & Seegmiller J (1967). Increased cystine in leukocytes from individuals homozygous and heterozygous for cystinosis. Science 157, 1321–1322. [DOI] [PubMed] [Google Scholar]
- Schulman J, Bradley K & Seegmiller J (1969). Cystine: compartmentalization within lysosomes in cystinotic leukocytes. Science 166, 1152. [DOI] [PubMed] [Google Scholar]
- Sumayao R, McEvoy B, Martin Martin N, McMorrow T & Newsholme P (2013). Cystine dimethylester loading promotes oxidative stress and a reduction in ATP independent of lysosomal cystine accumulation in a human proximal tubular epithelial cell line. Exp Physiol 98, 1505–1517. [DOI] [PubMed] [Google Scholar]
- Thoene JG & Lemons R (1980). Modulation of the intracellular cystine content of cystinotic fibroblasts by extracellular albumin. Pediatr Res 14, 785–787. [DOI] [PubMed] [Google Scholar]
- Town M, Jean G, Cherqui S, Attard M, Forestier L, Whitmore SA, Callen DF, Gribouval O, Broyer M & Bates GP (1998). A novel gene encoding an integral membrane protein is mutated in nephropathic cystinosis. Nat Genet 18, 319–324. [DOI] [PubMed] [Google Scholar]
- Turrens JF (2003). Mitochondrial formation of reactive oxygen species. J Physiol 552, 335–344. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Vaisbich M, Guimaraes LP, Shimizu MM & Seguro AC (2011). Oxidative stress in cystinosis patients. Nephron Extra 1, 73–77. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Valko M, Leibfritz D, Moncol J, Cronin MTD, Mazur M & Telser J (2007). Free radicals and antioxidants in normal physiological functions and human disease. Int J Biochem Cell Biol 39, 44–84. [DOI] [PubMed] [Google Scholar]
- Vermes I, Haanen C, Steffens‐Nakken H & Reutellingsperger C (1995). A novel assay for apoptosis flow cytometric detection of phosphatidylserine expression on early apoptotic cells using fluorescein labelled annexin V. J Immunol Methods 184, 39–51. [DOI] [PubMed] [Google Scholar]
- Wilmer MJ, Emma F & Levtchenko EN (2010). The pathogenesis of cystinosis: mechanisms beyond cystine accumulation. Am J Physiol Renal Physiol 299, F905–F916. [DOI] [PubMed] [Google Scholar]
- Wilmer MJ, Kluijtmans LA, van der Velden TJ, Willems PH, Scheffer PG, Masereeuw R, Monnens LA, van den Heuvel LP & Levtchenko EN (2011). Cysteamine restores glutathione redox status in cultured cystinotic proximal tubular epithelial cells. Biochim Biophys Acta 1812, 643–651. [DOI] [PubMed] [Google Scholar]
- Wilmer MJG, de Graaf‐Hess A, Blom HJ, Dijkman HBPM, Monnens LA, van den Heuvel LP & Levtchenko EN (2005). Elevated oxidized glutathione in cystinotic proximal tubular epithelial cells. Biochem Biophys Res Commun 337, 610–614. [DOI] [PubMed] [Google Scholar]
- Zhao H, Kalivendi S, Zhang H, Joseph J, Nithipatikom K, Vásquez‐Vivar J & Kalyanaraman B (2003). Superoxide reacts with hydroethidine but forms a fluorescent product that is distinctly different from ethidium: potential implications in intracellular fluorescence detection of superoxide. Free Radic Biol Med 34, 1359–1368. [DOI] [PubMed] [Google Scholar]