

The hypothesis that dysregulated lipid metabolism contributes to insulin resistance in muscle is now over a half‐century old (Randle et al. 1963). Although early work implicated reciprocal allosteric regulation of glucose and fat oxidation as a mechanism for lipid‐induced impairments in glucose metabolism (Randle et al. 1963), more recent studies propose that distinct lipid species with signalling properties directly interfere with the insulin signalling cascade to produce insulin resistance (Shulman, 2014). Of the many lipid metabolites proposed to cause insulin resistance, the ceramides have received particular attention.
Early studies demonstrating a relationship between intracellular ceramide levels and insulin resistance were performed in cell culture; the addition of palmitate or cell‐permeant short‐chain ceramides to the culture medium, often at supraphysiological concentrations, was repeatedly shown to impair insulin action (Summers et al. 1998; Zhou et al. 1998; Schmitz‐Peiffer et al. 1999; Chavez & Summers, 2003). Rodent studies in which ceramide levels were modulated pharmacologically or genetically in various models of insulin resistance also demonstrated an inverse relationship between intramyocellular ceramide levels and insulin action, although the interventions often produced pleiotropic physiological effects and non‐specific, potentially confounding effects on the lipidome (Holland et al. 2007; Bruce et al., 2012, 2013). Palmitate is thought to modulate intramyocellular ceramide levels by two major pathways. First, palmitate provides substrate for ceramide biosynthesis, the initial step of which is the condensation of serine and palmitate‐derived palmitoyl‐CoA. Palmitate also activates Toll‐like receptor 4 signalling, which transcriptionally upregulates ceramide biosynthetic enzymes. Because unsaturated fatty acids (USFAs) have neither of these functions, preferential induction of insulin resistance by palmitate rather than USFAs would support a role for ceramides in insulin resistance. Indeed, palmitate is capable of impairing Akt S473 phosphorylation to a greater extent than USFAs. Importantly, however, when insulin action is evaluated functionally (e.g. glycogen synthesis or glucose transport), palmitate and USFAs induce insulin resistance to a similar extent (Schmitz‐Peiffer et al. 1999; Chavez & Summers, 2003; Holland et al. 2007). USFA treatment does not increase ceramide content in myocytes, indicating that ceramides are not necessary for lipid‐induced insulin resistance (Chavez & Summers, 2003).
Another major challenge for the ceramide‐induced insulin resistance hypothesis concerns the molecular mechanism by which ceramides impair insulin signalling. Although there was early disagreement regarding the target(s) of ceramide‐induced insulin resistance, accumulating evidence eventually implicated Akt, downstream of proximal insulin receptor signalling, as the culprit (Summers et al. 1998; Zhou et al. 1998; Schmitz‐Peiffer et al. 1999). The mechanism for ceramide‐induced Akt inhibition is proposed to involve both increased protein phosphatase 2A activity (Salinas et al. 2000; Schubert et al. 2000; Stratford et al. 2004) and impaired insulin‐stimulated Akt translocation secondary to activation of atypical protein kinase Cζ (Powell et al. 2003; Stratford et al. 2004) (Fig. 1, left panel). If ceramides mediate obesity‐associated intramyocellular insulin resistance by this mechanism, it would follow that proximal insulin signalling would be unaffected in insulin resistant muscle: all insulin signalling defects should lie downstream of Akt. Although defective Akt S473 phosphorylation is certainly observed in insulin resistant muscle (Adams et al. 2004), proximal insulin signalling is also impaired (Caro et al. 1987; Cusi et al. 2000). Specifically, skeletal muscle from obese insulin resistant humans displays impairments in insulin receptor tyrosine phosphorylation and activity, insulin receptor substrate‐1 (IRS1) tyrosine phosphorylation, and IRS‐associated phosphatidylinositol‐3‐kinase activity (Caro et al. 1987; Cusi et al. 2000) (Fig. 1, right panel). Defective proximal insulin signalling in insulin resistant muscle challenges the hypothesis that ceramide‐induced Akt inhibition is a central defect in insulin resistance. In fact, a primary impairment of Akt phosphorylation might well enhance proximal insulin signalling owing to the loss of Akt‐mediated negative feedback mechanisms for insulin receptor signalling, such as stabilization of growth factor receptor‐bound protein 10 (Hsu et al. 2011; Yu et al. 2011). Thus, ceramide signalling as currently understood cannot fully account for the commonly observed signalling defects in obesity‐associated muscle insulin resistance.
Figure 1. Hypothesized changes in intramyocellular ceramide content and insulin signalling do not reflect observed changes in either of these parameters .
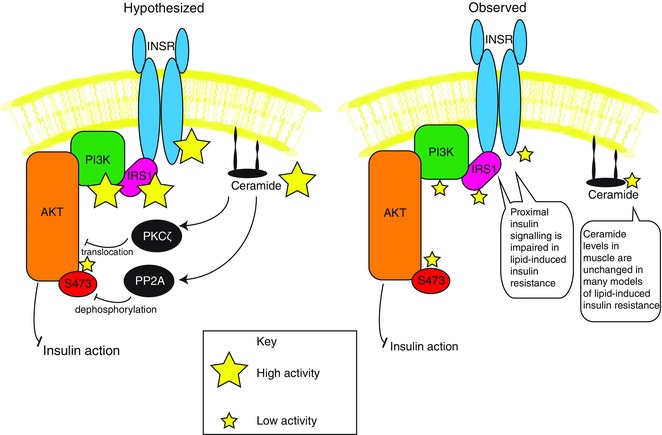
Left, the hypothesized increase in myocellular ceramide levels is proposed to inhibit Akt phosphorylation downstream of proximal insulin signalling. Right, changes in myocellular ceramide levels are frequently unchanged in insulin resistant muscle and proximal defects in insulin signalling are readily detected in lipid‐induced insulin resistance. Abbreviations: INSR, insulin receptor; IRS1, insulin receptor substrate‐1; PI3K, phosphoinositide 3‐kinase.
As ceramides gained prominence as putative mediators of muscle insulin resistance, an increasing number of human studies determined skeletal muscle ceramide content in parallel with measures of whole‐body glucose disposal or insulin sensitivity. Though not entirely unequivocal, available data suggest that changes in skeletal muscle insulin sensitivity and ceramide content can be dissociated under multiple conditions and in diverse patient populations. One well‐cited study of the relationship between muscle ceramide levels and insulin sensitivity in humans compared lean and obese subjects using a hyperinsulinaemic–euglycaemic clamp, with muscle biopsies taken at baseline and during the infusion (Adams et al. 2004). As expected, obese subjects displayed reduced whole‐body insulin sensitivity and, interestingly, elevated muscle ceramide levels at baseline (though not during the clamp). However, there was no significant association between muscle ceramide content and muscle insulin sensitivity (Adams et al. 2004). Several similar comparative studies examining the relationship between intramyocellular ceramide content and insulin sensitivity in humans sometimes report a negative relationship (Straczkowski et al. 2007; Coen et al. 2010; Amati et al. 2011), but just as frequently report no relationship at all (Serlie et al. 2007; Skovbro et al. 2008; Vistisen et al. 2008; Amati et al. 2011). Comparing these studies is difficult due to differences in study groups and the methods used for measuring insulin sensitivity and ceramide content, highlighting the need for interventional approaches or studies with randomized crossover design.
In one such study, older obese subjects were studied by hyperinsulinaemic–euglycaemic clamp and muscle biopsy before and after a 16‐week dietary intervention that produced a 10% weight loss (Dubé et al. 2011). Despite marked improvements in insulin‐stimulated glucose uptake, there was no effect of weight loss on intramyocellular ceramide levels (Dubé et al. 2011). Another study used a randomized crossover approach in which men and women ate diets that were either palmitate‐rich or oleate‐rich (Kien et al. 2013). After 3 weeks on each diet, muscle biopsies were collected for lipid profiling and frequently sampled intravenous glucose tolerance tests were performed to assess insulin sensitivity. In men, the palmitate‐rich diet increased muscle ceramide levels by > 20%, but did not affect insulin sensitivity (Kien et al. 2013). Interestingly, although insulin sensitivity was impaired in women fed the palmitate‐rich diet compared with the oleate‐rich diet, and a non‐significant increase in muscle ceramide content during palmitate consumption in women was reported, there was no overall association between insulin sensitivity and intramyocellular ceramides (Kien et al. 2013). These data argue against a role for ceramide‐induced insulin resistance in response to short‐term changes in dietary lipids in man.
A common model of acute muscle insulin resistance employs intravenous lipid infusion to raise plasma fatty acids during a hyperinsulinaemic–euglycaemic clamp. Using this approach, several groups observed impaired insulin‐stimulated glucose disposal with unchanged muscle ceramide levels in lean, overweight and obese men and women (Itani et al. 2002; Vistisen et al. 2008; Hoeks et al. 2012; Nowotny et al. 2013). Only one study, performed in lean male subjects, reported increased muscle ceramide content under similar conditions (Straczkowski et al. 2004). Additional acute interventions that reduced insulin‐stimulated glucose uptake, including oral lipid challenge or intravenous lipopolysaccharide infusion, likewise produced no effect on muscle ceramide content (Nowotny et al. 2013). Finally, increasing and decreasing plasma fatty acid levels over 6 h in lean, overweight and obese subjects with a Intralipid infusion or a hypolipidaemic agent did not affect muscle ceramide levels (Serlie et al. 2007). Taken together, these studies demonstrate that acute lipid‐induced insulin resistance occurs without concomitant changes in intramyocellular ceramide, dissociating the two phenomena.
Conclusion
Pharmacological doses of non‐physiological, short‐chain ceramides are sufficient to induce insulin resistance in cultured myocytes, and various genetic and pharmacological perturbations of ceramide metabolism, often accompanied by broad changes in lipid metabolism, can alter insulin sensitivity in rodents. However, the data implicating ceramide signalling in typical obesity‐associated muscle insulin resistance in humans are weak. Some of the strongest evidence against a role for ceramides in mediating insulin resistance comes from human studies, in which changes in skeletal muscle ceramide levels are frequently dissociated from changes in insulin sensitivity. Further, the proposed mechanism for ceramide‐induced insulin resistance – Akt inhibition – is inconsistent with the signalling abnormalities observed in insulin‐resistant skeletal muscle. Overall, available data do not support a model in which increased myocellular ceramides are either necessary or sufficient for skeletal muscle insulin resistance.
Call for comments
Readers are invited to give their views on this and the accompanying CrossTalk articles in this issue by submitting a brief (250 word) comment. Comments may be submitted up to 6 weeks after publication of the article, at which point the discussion will close and the CrossTalk authors will be invited to submit a ‘Last Word’. Please email your comment, including a title and a declaration of interest, to jphysiol@physoc.org. Comments will be moderated and accepted comments will be published online only as ‘supporting information’ to the original debate articles once discussion has closed.
Additional information
Competing interests
None.
Author contributions
Both authors have approved the final version of the manuscript and agree to be accountable for all aspects of the work. All persons designated as authors qualify for authorship, and all those who qualify for authorship are listed.
Funding
This work was supported by NIH/NIDDK grant K01DK099402 (to M.J.J.). and F30DK104596 (to M.C.P.)
Supporting information
Comments.
Last words by Summers and Goodpaster.
Last words by Petersen and Jurczak.
Biographies
Michael J. Jurczak, PhD, is an Assistant Professor of Medicine in the Division of Endocrinology and Metabolism at the University of Pittsburgh and member of the Center for Metabolism and Mitochondrial Medicine. He completed his graduate training at the University of Chicago and was a postdoctoral fellow at Yale University before joining the research faculty and serving as Co‐Director of the NIH‐funded Yale Mouse Metabolic Phenotyping Center In Vivo Metabolism Core. His lab is primarily interested in the relationship between nutrient excess, mitochondrial overload and the pathogenesis of metabolic diseases, such as fatty liver, insulin resistance and type 2 diabetes, and is supported by funding from the NIH.

Max C. Petersen is an MD/PhD student at the Yale University School of Medicine. He is performing his dissertation research in the lab of Gerald I. Shulman. His research interests concern cellular control of insulin action and mechanisms for insulin resistance.
Linked articles This article is part of a CrossTalk debate. Click the links to read the other articles in this debate: http://dx.doi.org/10.1113/JP271676, http://dx.doi.org/10.1113/JP272136, http://dx.doi.org/10.1113/JP272137.
References
- Adams JM, Pratipanawatr T, Berria R, Wang E, DeFronzo RA, Sullards MC & Mandarino LJ (2004). Ceramide content is increased in skeletal muscle from obese insulin‐resistant humans. Diabetes 53, 25–31. [DOI] [PubMed] [Google Scholar]
- Amati F, Dubé JJ, Alvarez‐Carnero E, Edreira MM, Chomentowski P, Coen PM, Switzer GE, Bickel PE, Stefanovic‐Racic M, Toledo FGS & Goodpaster BH (2011). Skeletal muscle triglycerides, diacylglycerols, and ceramides in insulin resistance: another paradox in endurance‐trained athletes? Diabetes 60, 2588–2597. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bruce CR, Risis S, Babb JR, Yang C, Kowalski GM, Selathurai A, Lee‐Young RS, Weir JM, Yoshioka K, Takuwa Y, Meikle PJ, Pitson SM & Febbraio MA (2012). Overexpression of sphingosine kinase 1 prevents ceramide accumulation and ameliorates muscle insulin resistance in high‐fat diet‐fed mice. Diabetes 61, 3148–3155. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bruce CR, Risis S, Babb JR, Yang C, Lee‐Young RS, Henstridge DC & Febbraio MA (2013). The sphingosine‐1‐phosphate analog FTY720 reduces muscle ceramide content and improves glucose tolerance in high fat‐fed male mice. Endocrinology 154, 65–76. [DOI] [PubMed] [Google Scholar]
- Caro JF, Sinha MK, Raju SM, Ittoop O, Pories WJ, Flickinger EG, Meelheim D & Dohm GL (1987). Insulin receptor kinase in human skeletal muscle from obese subjects with and without noninsulin dependent diabetes. J Clin Invest 79, 1330–1337. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chavez JA & Summers SA (2003). Characterizing the effects of saturated fatty acids on insulin signaling and ceramide and diacylglycerol accumulation in 3T3‐L1 adipocytes and C2C12 myotubes. Arch Biochem Biophys 419, 101–109. [DOI] [PubMed] [Google Scholar]
- Coen PM, Dubé JJ, Amati F, Stefanovic‐Racic M, Ferrell RE, Toledo FGS & Goodpaster BH (2010). Insulin resistance is associated with higher intramyocellular triglycerides in type I but not type II myocytes concomitant with higher ceramide content. Diabetes 59, 80–88. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cusi K, Maezono K, Osman A, Pendergrass M, Patti ME, Pratipanawatr T, DeFronzo RA, Kahn CR & Mandarino LJ (2000). Insulin resistance differentially affects the PI 3‐kinase‐ and MAP kinase‐mediated signaling in human muscle. J Clin Invest 105, 311–320. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dubé JJ, Amati F, Toledo FGS, Stefanovic‐Racic M, Rossi A, Coen P & Goodpaster BH (2011). Effects of weight loss and exercise on insulin resistance, and intramyocellular triacylglycerol, diacylglycerol and ceramide. Diabetologia 54, 1147–1156. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hoeks J, Mensink M, Hesselink MKC, Ekroos K & Schrauwen P (2012). Long‐ and medium‐chain fatty acids induce insulin resistance to a similar extent in humans despite marked differences in muscle fat accumulation. J Clin Endocrinol Metab 97, 208–216. [DOI] [PubMed] [Google Scholar]
- Holland WL, Brozinick JT, Wang L‐P, Hawkins ED, Sargent KM, Liu Y, Narra K, Hoehn KL, Knotts TA, Siesky A, Nelson DH, Karathanasis SK, Fontenot GK, Birnbaum MJ & Summers SA (2007). Inhibition of ceramide synthesis ameliorates glucocorticoid‐, saturated‐fat‐, and obesity‐induced insulin resistance. Cell Metab 5, 167–179. [DOI] [PubMed] [Google Scholar]
- Hsu PP, Kang SA, Rameseder J, Zhang Y, Ottina KA, Lim D, Peterson TR, Choi Y, Gray NS, Yaffe MB, Marto JA & Sabatini DM (2011). The mTOR‐regulated phosphoproteome reveals a mechanism of mTORC1‐mediated inhibition of growth factor signaling. Science 332, 1317–1322. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Itani SI, Ruderman NB, Schmieder F & Boden G (2002). Lipid‐induced insulin resistance in human muscle is associated with changes in diacylglycerol, protein kinase C, and IκB‐α. Diabetes 51, 2005–2011. [DOI] [PubMed] [Google Scholar]
- Kien CL, Bunn JY, Poynter ME, Stevens R, Bain J, Ikayeva O, Fukagawa NK, Champagne CM, Crain KI, Koves TR & Muoio DM (2013). A lipidomics analysis of the relationship between dietary fatty acid composition and insulin sensitivity in young adults. Diabetes 62, 1054–1063. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nowotny B, Zahiragic L, Krog D, Nowotny PJ, Herder C, Carstensen M, Yoshimura T, Szendroedi J, Phielix E, Schadewaldt P, Schloot NC, Shulman GI & Roden M (2013). Mechanisms underlying the onset of oral lipid‐induced skeletal muscle insulin resistance in humans. Diabetes 62, 2240–2248. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Powell DJ, Hajduch E, Kular G & Hundal HS (2003). Ceramide disables 3‐phosphoinositide binding to the pleckstrin homology domain of protein kinase B (PKB)/Akt by a PKCzeta‐dependent mechanism. Mol Cell Biol 23, 7794–7808. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Randle PJ, Garland PB, Hales CN & Newsholme EA (1963). The glucose fatty‐acid cycle. Its role in insulin sensitivity and the metabolic disturbances of diabetes mellitus. Lancet 1, 785–789. [DOI] [PubMed] [Google Scholar]
- Salinas M, López‐Valdaliso R, Martín D, Alvarez A & Cuadrado A (2000). Inhibition of PKB/Akt1 by C2‐ceramide involves activation of ceramide‐activated protein phosphatase in PC12 cells. Mol Cell Neurosci 15, 156–169. [DOI] [PubMed] [Google Scholar]
- Schmitz‐Peiffer C, Craig DL & Biden TJ (1999). Ceramide generation is sufficient to account for the inhibition of the insulin‐stimulated PKB pathway in C2C12 skeletal muscle cells pretreated with palmitate. J Biol Chem 274, 24202–24210. [DOI] [PubMed] [Google Scholar]
- Schubert KM, Scheid MP & Duronio V (2000). Ceramide inhibits protein kinase B/Akt by promoting dephosphorylation of serine 473. J Biol Chem 275, 13330–13335. [DOI] [PubMed] [Google Scholar]
- Serlie MJ, Meijer AJ, Groener JE, Duran M, Endert E, Fliers E, Aerts JM & Sauerwein HP (2007). Short‐term manipulation of plasma free fatty acids does not change skeletal muscle concentrations of ceramide and glucosylceramide in lean and overweight subjects. J Clin Endocrinol Metab 92, 1524–1529. [DOI] [PubMed] [Google Scholar]
- Shulman GI (2014). Ectopic fat in insulin resistance, dyslipidemia, and cardiometabolic disease. N Engl J Med 371, 1131–1141. [DOI] [PubMed] [Google Scholar]
- Skovbro M, Baranowski M, Skov‐Jensen C, Flint A, Dela F, Gorski J & Helge JW (2008). Human skeletal muscle ceramide content is not a major factor in muscle insulin sensitivity. Diabetologia 51, 1253–1260. [DOI] [PubMed] [Google Scholar]
- Straczkowski M, Kowalska I, Baranowski M, Nikolajuk A, Otziomek E, Zabielski P, Adamska A, Blachnio A, Gorski J & Gorska M (2007). Increased skeletal muscle ceramide level in men at risk of developing type 2 diabetes. Diabetologia 50, 2366–2373. [DOI] [PubMed] [Google Scholar]
- Straczkowski M, Kowalska I, Nikolajuk A, Dzienis‐Straczkowska S, Kinalska I, Baranowski M, Zendzian‐Piotrowska M, Brzezinska Z & Gorski J (2004). Relationship between insulin sensitivity and sphingomyelin signaling pathway in human skeletal muscle. Diabetes 53, 1215–1221. [DOI] [PubMed] [Google Scholar]
- Stratford S, Hoehn KL, Liu F & Summers SA (2004). Regulation of insulin action by ceramide: dual mechanisms linking ceramide accumulation to the inhibition of Akt/protein kinase B. J Biol Chem 279, 36608–36615. [DOI] [PubMed] [Google Scholar]
- Summers SA, Garza LA, Zhou H & Birnbaum MJ (1998). Regulation of insulin‐stimulated glucose transporter GLUT4 translocation and Akt kinase activity by ceramide. Mol Cell Biol 18, 5457–5464. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Vistisen B, Hellgren LI, Vadset T, Scheede‐Bergdahl C, Helge JW, Dela F & Stallknecht B (2008). Effect of gender on lipid‐induced insulin resistance in obese subjects. Eur J Endocrinol Eur Fed Endocr Soc 158, 61–68. [DOI] [PubMed] [Google Scholar]
- Yu Y, Yoon S‐O, Poulogiannis G, Yang Q, Ma XM, Villén J, Kubica N, Hoffman GR, Cantley LC, Gygi SP & Blenis J (2011). Phosphoproteomic analysis identifies Grb10 as an mTORC1 substrate that negatively regulates insulin signaling. Science 332, 1322–1326. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhou H, Summers SA, Birnbaum MJ & Pittman RN (1998). Inhibition of Akt kinase by cell‐permeable ceramide and its implications for ceramide‐induced apoptosis. J Biol Chem 273, 16568–16575. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Comments.
Last words by Summers and Goodpaster.
Last words by Petersen and Jurczak.