

Abstract
Key points
Nitric oxide synthase (NOS) contributes to sweating and cutaneous vasodilatation during exercise in the heat.
Similarly, reports show that Na+/K+‐ATPase activation can modulate sweating and microvascular circulation. In light of the fact that NO can activate Na+/K+‐ATPase, we evaluated whether there is an interaction between Na+/K+‐ATPase and NOS in the regulation of heat loss responses during an exercise‐induced heat stress.
We demonstrate that Na+/K+‐ATPase and NOS do not synergistically influence local forearm sweating during moderate intensity (fixed rate of metabolic heat production of 500 W) exercise in the heat (35°C). Conversely, we show an interactive role between NOS and Na+/K+‐ATPase in the modulation of cutaneous vasodilatation.
These findings provide novel insight regarding the mechanisms underpinning the control of sweating and cutaneous vasodilatation during exercise in the heat. Given that ouabain may be prescribed as a cardiac glycoside in clinical settings, potential heat loss impairments with ouabain administration should be explored.
Abstract
Nitric oxide (NO) synthase (NOS) contributes to the heat loss responses of sweating and cutaneous vasodilatation. Given that NO can activate Na+/K+‐ATPase, which also contributes to sweating and microvasculature regulation, we evaluated the separate and combined influence of Na+/K+‐ATPase and NOS on sweating and cutaneous vasodilatation. Thirteen young (23±3 years) males performed two 30 min semi‐recumbent cycling bouts in the heat (35°C) at a fixed rate of metabolic heat production (500 W) followed by 20 and 40 min recoveries, respectively. Local sweat rate (LSR) and cutaneous vascular conductance (CVC) were measured at four forearm skin sites continuously perfused via intradermal microdialysis with either: (1) lactated Ringer solution (Control); (2) 6 mᴍ ouabain (Ouabain), a Na+/K+‐ATPase inhibitor; (3) 10 mᴍ l‐NG‐nitroarginine methyl ester (l‐NAME), a NOS inhibitor; or (4) 6 mᴍ ouabain and 10 mᴍ l‐NAME (Ouabain+l‐NAME). At the end of both exercise bouts relative to Control, LSR was attenuated with Ouabain (54–60%), l‐NAME (12–13%) and Ouabain+l‐NAME (68–74%; all P < 0.05). Moreover, the sum of attenuations from Control induced by independent administration of Ouabain and l‐NAME was similar to the combined infusion of Ouabain+l‐NAME (both P ≥ 0.74). Compared to Control, CVC at the end of both exercise bouts was similar with Ouabain (both P ≥ 0.30), but attenuated with l‐NAME (%CVCmax reduction from Control, 24–25%). Furthermore, CVC at the Ouabain+l‐NAME site (38–39%; all P < 0.01) was attenuated compared to Control and did not differ from baseline resting values (both P ≥ 0.81). We show that Na+/K+‐ATPase and NOS do not synergistically mediate sweating, whereas they influence cutaneous blood flow in an interactive manner during exercise in the heat.
Key points
Nitric oxide synthase (NOS) contributes to sweating and cutaneous vasodilatation during exercise in the heat.
Similarly, reports show that Na+/K+‐ATPase activation can modulate sweating and microvascular circulation. In light of the fact that NO can activate Na+/K+‐ATPase, we evaluated whether there is an interaction between Na+/K+‐ATPase and NOS in the regulation of heat loss responses during an exercise‐induced heat stress.
We demonstrate that Na+/K+‐ATPase and NOS do not synergistically influence local forearm sweating during moderate intensity (fixed rate of metabolic heat production of 500 W) exercise in the heat (35°C). Conversely, we show an interactive role between NOS and Na+/K+‐ATPase in the modulation of cutaneous vasodilatation.
These findings provide novel insight regarding the mechanisms underpinning the control of sweating and cutaneous vasodilatation during exercise in the heat. Given that ouabain may be prescribed as a cardiac glycoside in clinical settings, potential heat loss impairments with ouabain administration should be explored.
Abbreviations
- CVC
cutaneous vascular conductance
- EDHF
endothelium‐derived hyperpolarizing factor
- l‐NAME
l‐NG‐Nitroarginine methyl ester
- LSR
local sweat rate
- NO
nitric oxide
- NOS
nitric oxide synthase
Introduction
In humans, eccrine sweating and cutaneous vasodilatation are the body's primary heat loss responses for maintaining stable core body temperatures during exercise and/or exposure to a hot environment. The contribution of nitric oxide (NO) synthase (NOS) to local sweating and cutaneous vasodilatation during exercise can be determined by perfusion of a non‐selective NOS inhibitor to the skin, and comparing these responses to those at a non‐treated skin site. Specifically, NOS inhibition has resulted in approximately 13–26% attenuations in local sweat rate (LSR) (Welch et al. 2009; Fujii et al. 2014; McGinn et al. 2014 b; Stapleton et al. 2014) and 25–57% attenuations in cutaneous vasodilatation (Welch et al. 2009; Fujii et al. 2014; McGinn et al. 2014 a,b; McNamara et al. 2014) compared to the control sites during steady‐state exercise. However, although the literature clearly indicates that the inhibition of NOS attenuates the sweating response, the mechanism by which this occurs remains unknown. Interestingly, Fujii et al. (2014) demonstrated that the perfusion of sodium nitroprusside, a widely employed NO donor, did not increase local forearm sweat rate during passive exposure to a warm environment. Thus, the mechanism by which NOS modulates sweating remains to be determined. As postulated by Welch et al. (2009), NOS may influence sweating via the modulation of postsynaptic events associated with cholinergic stimulation of the sweat gland, such as affecting the electrochemical gradient necessary for the production of sweat.
Following Na+/K+‐ATPase identification in eccrine sweat gland cells (Adachi & Yamasawa, 1966; Gibbs, 1967), it was determined that Na+/K+‐ATPase was primarily localized on the basolateral membrane (Quinton & Tormey, 1976; Saga & Sato, 1988; Toyomoto et al. 1997; Saga, 2002). Based on the Na–K–2Cl cotransport model, Na+/K+‐ATPase is responsible for maintenance of membrane potential (Sato & Sato, 1987; Sato et al. 1989). The electrochemical gradient established by Na+/K+‐ATPase in conjunction with the other factors of the Na–K–2Cl cotransporter model such as Na–K–2Cl cotransporters, K+ channels and Cl− channels, ultimately result in the transepithelial movement of water from blood to the lumen of the sweat gland (i.e. sweat production) (Sato & Sato, 1987; Sato et al. 1989). It was shown that intradermal injection of ouabain, a potent inhibitor of Na+/K+‐ATPase, markedly attenuated LSR during graded exercise in a hot environment (40–46°C) (Sato & Dobson, 1969; Sato et al. 1969). Na+/K+‐ATPase also contributes to the vasodilatory response in human microcirculation (e.g. coronary circulation) (Miura et al., 2011), although it remains to be determined if this is also true for cutaneous vasodilatation during exercise in the heat. NO has been demonstrated to stimulate Na+/K+‐ATPase in human corpus cavernosum smooth muscle, porcine internal mammary artery and rat aortic rings (Gupta et al. 1994; Palacios et al. 2004; Pagan et al. 2010), but it remains unknown if this interaction also exists in human cutaneous vasculature and eccrine sweat glands and whether it can explain the discrepancy between the attenuation in sweating seen with NOS inhibition and lack of effect from NO donor perfusion.
The purpose of this study was to investigate the underlying mechanisms mediating NOS‐dependent sweating and cutaneous vasodilatation during exercise in the heat (35°C, 20% relative humidity). We hypothesized that Na+/K+‐ATPase and NOS would have an interactive influence in the regulation of sweating and cutaneous vasodilatation during an exercise‐induced heat stress.
Methods
Ethical approval
The current experimental protocol was approved by the University of Ottawa Health Sciences Ethics Board and was in accordance with guidelines set out in the Declaration of Helsinki. Written and informed consent were obtained from all volunteers prior to their involvement in the study.
Participants
Thirteen healthy and habitually active (2–5 days week−1 of structured physical activity; ≥ 30 min day−1) males volunteered for the study. All participants were screened for and free of smoking, prescription and/or over‐the‐counter medications as well as any known cardiovascular, respiratory or metabolic diseases at the time of the study. Participants’ physical characteristics (mean ± SD) were: age, 23 ± 3 years; height, 1.75 ± 0.07 m; body mass, 72 ± 8 kg; body surface area, 1.9 ± 0.1 m2; body fat percentage, 12 ± 4%; and peak rate of oxygen consumption (), 51 ± 6 ml O2 kg−1 min−1.
Experimental procedures
Participants completed one preliminary and one experimental session, abstaining from prescription and over‐the‐counter medications (including vitamin and mineral supplements) for 48 h, from alcohol, caffeine and strenuous exercise for 12 h and from food for 2 h prior to each session. During the preliminary session, anthropometric data were measured including body height determined using a stadiometer (Detecto, model 2391, Webb City, MO, USA) and body mass determined using a digital high‐performance weighing terminal (model CBU150X, Mettler Toledo Inc., Mississauga, ON, Canada). Body surface area was then calculated using body height and mass (Du Bois & Du Bois, 1989). Body density was determined using the hydrostatic weighing technique and was subsequently used to estimate body fat percentage (Siri, 1956). was determined using indirect calorimetry (MCD Medgraphics Ultima Series; MGC Diagnostics, Saint Paul, MN, USA) during an incremental cycling protocol until exhaustion performed on a constant‐load semi‐recumbent cycle ergometer (Corival Recumbent, Lode, Groningen, Netherlands). Participants were instructed to pedal at a cadence of 60–90 revolutions min−1 at a starting external workload of 100 W, which was then increased by 20 W min−1 until they reached volitional fatigue or could not maintain a pedalling cadence of ≥50 revolutions min−1. was taken as the greatest average oxygen uptake measured over 30 s.
The experimental session occurred on a separate day. Participants arrived at the laboratory euhydrated (by consuming 500 ml of water the night prior and 2 h before arrival), which was verified by measuring urine specific gravity (1.010 ± 0.003) (Sawka et al. 2007). A nude body mass measurement was then taken. Thereafter, participants were seated in a semi‐recumbent position in a thermoneutral room (25°C) and were instrumented with four microdialysis fibres (MD2000, Bioanalytical Systems, West Lafayette, IN, USA), placed at least 4 cm apart from one another, in the dermal layer of skin on the left dorsal forearm. To place each of the four microdialysis fibres, a 25‐gauge needle was first inserted into the anaesthetized skin which exited ∼2.5 cm from the point of entry. The microdialysis fibre was then threaded through the lumen of the needle after which the needle was withdrawn leaving the 10 mm semi‐permeable microdialysis membrane in place under the skin. The fibre was then secured using surgical tape. This process was then repeated for each fibre. Participants were then moved to a thermal chamber (Can‐Trol Environmental Systems, Markham, ON, Canada) regulated to an ambient temperature of 35°C and 20% relative humidity and seated on a semi‐recumbent cycle ergometer.
After entering the thermal chamber the skin sites were perfused in a counter‐balanced manner with one of the following pharmacological agents: (1) lactated Ringer solution (Control); (2) 6 mᴍ ouabain (Ouabain; Sigma‐Aldrich, St Louis, MO, USA), a Na+/K+‐ATPase inhibitor; (3) 10 mᴍ l‐NG‐nitroarginine methyl ester (l‐NAME; Sigma‐Aldrich), a non‐selective NOS inhibitor; or (4) a combination of 6 mm ouabain and 10 mm l‐NAME (Ouabain + l‐NAME). Each agent was perfused at a rate of 4 μl min−1 via a microinfusion pump (Model 400, CMA Microdialysis, Solna, Sweden). The simultaneous perfusion of each antagonistic agent (dissolved in lactated Ringer solution) independently as well as in combination allowed for the assessment of the separate and combined contribution of Na+/K+‐ATPase and NOS on the local heat loss responses when compared to the Control site perfused with only lactated Ringer solution (Kellogg et al. 1998; Holowatz et al. 2006 b; Fujii et al. 2014). The concentration of ouabain was selected based on pilot work (n = 4) in which we perfused increasing concentrations of the cholinergic agent methacholine (i.e. 0.0125, 0.25, 5, 100, 2000 mM each for ∼25 min) in combination with three concentrations of ouabain (2, 6 and 12 mm; the last dose being the highest concentration of ouabain dissolvable in lactated Ringer solution) in the forearm skin via intradermal microdialysis. In comparison to the control site that received only methacholine (i.e. no ouabain), 6 and 12 mm ouabain elicited a similar attenuation in LSR, which was greater than that induced at the 2 mm site. Therefore, 6 mm was determined to be the minimal concentration of ouabain that maximally inhibits methacholine‐induced sweating. On the other hand, the concentration used for l‐NAME was chosen based on previous literature using the microdialysis technique in human skin (Holowatz et al. 2003, 2006 a,b; Fujii et al. 2014; McGinn et al. 2014 a,b; Stapleton et al. 2014; Meade et al. 2015). All four skin sites were continuously perfused with their respective agents for at least 60 min to ensure establishment of each blockade. This minimum 75 min period (15 min between fibre placement and drug infusion plus the 60 min habituation period) allowed for the local trauma due to the needle/fibre insertion to subside.
Following the 60 min habituation period, a 10 min period of baseline resting data collection occurred. Thereafter, participants performed two successive 30 min bouts of semi‐recumbent cycling, with the first and second bouts followed by 20 and 40 min recovery periods, respectively. Participants exercised at a fixed rate of metabolic heat production of 500 W (equivalent to 50 ± 6% and requiring an external workload of 97 ± 3 W) to ensure similar thermal drive for whole‐body heat loss (Gagnon et al. 2013; Kenny & Jay, 2013). Following the second recovery period, each fibre was perfused with 50 mm sodium nitroprusside (Sigma‐Aldrich) at a rate of 6 μl min−1 for ∼20 min until a stable plateau in cutaneous blood flow was established. Following this plateau, blood pressure was measured for the calculation of maximal cutaneous vascular conductance (CVCmax). Finally, the microdialysis fibres and remaining instrumentation were removed and a nude body mass was measured.
Measurements
Local forearm sweat rate (LSR) was simultaneously measured at each skin site using the ventilated capsule technique. Each sweat capsule was placed directly over the centre of the microdialysis membrane and secured to the skin using adhesive rings and topical glue (Collodion HV, Mavidon Medical Products, Lake Worth, FL, USA). Anhydrous air was passed through each sweat capsule and the water content of the effluent air was measured using capacitance hygrometers (Model HMT333, Vaisala, Helsinki, Finland). Long vinyl tubes connected the gas tanks, located in the thermal chamber, to the sweat capsules as well as the sweat capsules to the hygrometers to ensure that internal gas temperatures were equilibrated to near room temperature (35°C). LSR was calculated every 5 s using the difference in water content between effluent and influent air multiplied by flow rate and normalized for skin surface area beneath the capsule (in mg min−1 cm−2). Due to technical difficulties, the LSR data were not collected for two participants.
Cutaneous red blood cell flux (in perfusion units), an index of cutaneous blood flow, was measured at each of the four skin sites at a sampling rate of 32 Hz with laser Doppler flowmetry (PeriFlux System 5000, Perimed, Stockholm, Sweden). The sweat capsules employed were specially designed to house an integrated laser Doppler flowmetry probe with a seven‐laser array (Model 413, Perimed), positioned directly over the centre of their respective microdialysis membrane to allow for simultaneous measurement of local sweat rate and cutaneous red blood cell flux at each skin site. CVC was calculated as cutaneous red blood cell flux divided by mean arterial pressure and was presented as a percentage of CVCmax as determined during the sodium nitroprusside‐induced maximal cutaneous blood flow protocol (see above). Mean arterial pressure was calculated as diastolic pressure plus one‐third of the difference between systolic and diastolic pressures (i.e. pulse pressure), measured every 5 min throughout the experimental protocol as well as during the plateau phase of CVCmax using manual auscultation with a validated mercury column sphygmomanometer (Baumanometer Standby Model, WA Baum Co., Copiague, NY, USA). Heart rate was recorded at a sampling rate of 15 s using a Polar coded WearLink and transmitter, Polar RS400 interface and Polar Trainer 5 software (Polar Electro, Kempele, Finland).
Oesophageal temperature was continuously measured using a paediatric thermocouple probe, with a diameter of ∼2 mm (Mon‐a‐therm; Mallinckrodt Medical, St Louis, MO, USA), inserted 40 cm past the entrance of the nostril. Mean skin temperature was determined using thermocouples (Concept Engineering, Old Saybrook, CT, USA) at six skin sites weighted to the following regional proportions (Hardy et al., 1938): upper back, 21.0%; chest, 21.0%; biceps, 19.0%; quadriceps, 9.5%; hamstring, 9.5%; and front calf, 20.0%. Both oesophageal and skin temperature data were collected at a sampling rate of 15 s using a data acquisition module (Model 34970A; Agilent Technologies Canada, Mississauga, ON, Canada), which was simultaneously displayed and recorded in a spreadsheet format on a personal computer with LabVIEW software (National Instruments, Austin, TX, USA). From these data, mean body temperature was calculated as 0.9 × core temperature + 0.1 × mean skin temperature (Wilson et al. 2005).
Metabolic heat production during exercise was calculated as the difference between metabolic rate and external workload (Kenny & Jay, 2013). Metabolic rate was determined using indirect calorimetry. Expired air was analysed for concentrations of oxygen and carbon dioxide measured using electrochemical gas analysers (AMETEK model S‐3A/1 and CD3A, Applied Electrochemistry, Pittsburgh, PA, USA). Gas analysers were calibrated ∼20 min prior to the start of the baseline resting period using a gas of known concentrations (∼17% oxygen, ∼4% carbon dioxide, balance nitrogen) and the turbine ventilometer was calibrated using a 3‐litre syringe. Participants wore a full face mask (Model 7600 V2, Hans‐Rudolph, Kansas City, MO, USA) which was attached to a two‐way T‐shape non‐rebreathing valve (Model 2700, Hans‐Rudolph). Metabolic rate was calculated using oxygen uptake and respiratory exchange ratio values obtained at a sampling rate of 30 s.
Data analysis
Baseline resting values were determined by averaging the measurements performed over the last 5 min prior to the start of the first exercise. During the intermittent exercise protocol, LSR and CVC at each forearm skin site, as well as oesophageal temperature, mean skin temperature and heart rate, were determined by averaging data during the final 5 min of each 10 min interval. Blood pressure data were presented by averaging the two measurements taken during each 10 min interval. CVCmax was taken as a 2 min average of CVC data during the plateau phase of the maximal cutaneous blood flow protocol. The difference (∆) in LSR from Control was determined at the Ouabain, l‐NAME and Ouabain + l‐NAME sites for the final 5 min of both exercise bouts (i.e. time points 50 and 80 min).
Statistical analysis
LSR and CVC were analysed using a two‐way repeated measures ANOVA with the factors of time (13 levels: baseline resting, 10, 20, 30, 40, 50, 60, 70, 80, 90, 100, 110, 120 min) and treatment site (four levels: Control, Ouabain, l‐NAME, Ouabain + l‐NAME). Core and skin temperatures as well as cardiovascular variables were analysed using a one‐way repeated measures ANOVA with the factor of time (six levels: baseline resting, Ex 1, Rec 1, Ex 2, Rec 2 at 20 min and Rec 2 at 40 min). Local forearm absolute maximal CVC (expressed in perfusion units mmHg−1 multiplied by 100) that was obtained during the sodium nitroprusside infusion protocol was analysed with a one‐way repeated measures ANOVA with the factor of treatment site (four levels). When a significant main effect was observed, post hoc analyses were carried out using two‐tailed Student's paired samples t tests adjusted for multiple comparisons using the Holm–Bonferroni procedure. Two‐tailed Student's paired samples t tests were used (1) to assess whether the sum of attenuations in LSR from Control with the lone perfusions of Ouabain and l‐NAME differed from the attenuation with co‐perfused Ouabain + l‐NAME; and (2) to compare the magnitude of attenuation in LSR from Control induced by independent perfusion of Ouabain versus l‐NAME. For all analyses, P ≤ 0.05 was considered statistically significant. All values are reported as the mean ± 95% confidence interval (i.e. 1.96 × SEM).
Results
Cardiovascular responses
Relative to baseline resting, mean arterial pressure was elevated during both exercise bouts (both P < 0.01) and lower during the second recovery period (both P ≤ 0.05). Heart rate was elevated throughout all exercise and recovery periods compared to baseline resting values (all P < 0.01), and was greater in the second exercise and recovery periods compared to the first (all P ≤ 0.01).
Body temperatures
Oesophageal, mean skin and mean body temperatures were elevated during both exercise and recovery periods in comparison to baseline values (all P < 0.01; Table 1). Moreover, mean skin temperature was greater in the second exercise bout (P < 0.05) and lower at the 40 min time point in the second recovery (P < 0.01) compared to the first exercise and recovery period, respectively. In parallel, oesophageal and mean body temperatures were greater at the end of the second exercise bout as well as the 20 and 40 min time points of the second recovery in comparison to the first exercise and recovery period, respectively (all P < 0.01).
Table 1.
Body temperatures and cardiovascular responses at rest and during exercise and recovery periods
| Exercise 1 | Recovery 1 | Exercise 2 | Recovery 2 | |||
|---|---|---|---|---|---|---|
| BL | 30 min | 20 min | 30 min | 20 min | 40 min | |
| Oesophageal temperature (°C) | 37.1 ± 0.1 | 37.7 ± 0.2* | 37.4 ± 0.1* | 38.0 ± 0.2*† | 37.6 ± 0.2*† | 37.5 ± 0.2*† |
| Mean skin temperature (°C) | 35.0 ± 0.2 | 35.9 ± 0.2* | 35.6 ± 0.2* | 36.0 ± 0.3*† | 35.6 ± 0.3* | 35.3 ± 0.2*† |
| Mean body temperature (°C) | 36.9 ± 0.1 | 37.5 ± 0.2* | 37.2 ± 0.1* | 37.8 ± 0.2*† | 37.4 ± 0.2*† | 37.3 ± 0.2*† |
| Mean arterial pressure (mmHg) | 93 ± 3 | 102 ± 3* | 90 ± 3 | 99 ± 4* | 90 ± 2 | 89 ± 3 |
| Heart rate (b.p.m.) | 75 ± 8 | 131 ± 12* | 83 ± 9* | 139 ± 13*† | 88 ± 8*† | 88 ± 10*† |
Presented values are mean ± 95% confidence interval. Oesophageal, mean skin and mean body temperatures, as well as heart rate values represent an average of the final 5 min for the corresponding time period. Mean arterial pressure values represent an average of two measurements from the final 10 min for the corresponding time period. Baseline resting represents the first 5 min of the first 10 min interval. BL, baseline resting; b.p.m., beats per minute. *P < 0.05 vs. BL, † P < 0.05 Exercise 1 vs. Exercise 2 or Recovery 1 vs. Recovery 2.
Sweating response
During baseline resting, LSR was attenuated at all treatment sites compared to the Control site (all P < 0.05) (Fig. 1). Throughout both exercise bouts, LSR was attenuated relative to Control at all three treatment sites (P < 0.05) with the exception of the l‐NAME site at the 10 min point in Exercise 1 (P = 0.12). Compared to Control, LSR was lower at the Ouabain (all P < 0.05) and Ouabain + l‐NAME (all P ≤ 0.05) sites during the first 10 min of the first recovery and 20 to 40 min time points of the second recovery, whereas LSR at the l‐NAME site was attenuated throughout the last 10 min of both recovery periods (both P ≤ 0.05). Additionally, LSR was increased from baseline resting values at all treatment sites at the end of each exercise bout (all P < 0.05). Moreover, at the end of both exercise bouts, the sum of attenuations in LSR from Control with the independent administrations of Ouabain and l‐NAME was similar to the attenuation with co‐perfused Ouabain + l‐NAME (both P ≥ 0.74). Lastly, the attenuation in LSR from Control at the Ouabain site was greater than that of the l‐NAME site at the end of both exercise bouts (both P < 0.01).
Figure 1. Time‐course changes in local sweat rate during intermittent exercise at a fixed rate of metabolic heat production (500 W) in the heat (35°C; 20% relative humidity) (n = 11) .
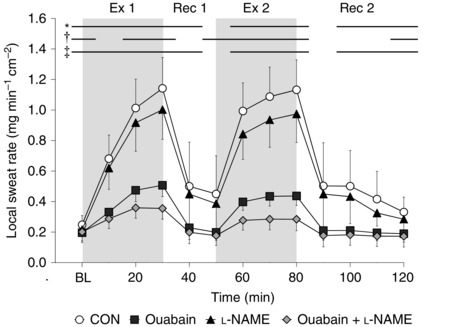
Four forearm skin sites were continuously perfused with (1) lactated Ringer solution (Control, open circles); (2) 6 mm ouabain (Ouabain; squares), a Na+/K+‐ATPase inhibitor; (3) 10 mm l‐NAME (triangles), a non‐selective NOS inhibitor; or (4) a combination of 10 mm l‐NAME and 6 mm ouabain (Ouabain + l‐NAME, diamonds). Values are presented as mean ± 95% confidence interval. Baseline resting represents the first 5 min of the first 10 min interval, and each data point thereafter represents an average of the final 5 min of each 10 min interval. BL, baseline resting; Ex 1, first exercise bout; Rec 1, first recovery period; Ex 2, second exercise bout; Rec 2, second recovery period. *Control significantly different from Ouabain. †Control significantly different from l‐NAME. ‡Control significantly different from Ouabain + l‐NAME (all P ≤ 0.05).
Cutaneous vascular response
There were no differences in CVCmax between treatment sites (P = 0.29). At baseline resting, CVC was greater at the Ouabain‐treated site (P < 0.01), as well as attenuated at the l‐NAME (P < 0.01) and Ouabain + l‐NAME sites (P < 0.01) compared to Control (Fig. 2). During both exercise bouts, CVC was similar at the Ouabain site (all P ≥ 0.16) and attenuated at the l‐NAME (all P < 0.01) and Ouabain + l‐NAME sites (all P < 0.01) in comparison to Control, whereas it was greater at the Ouabain site (all P ≤ 0.01) and attenuated at the l‐NAME (all P < 0.01) and Ouabain + l‐NAME (all P < 0.05) sites during both recovery periods. Relative to baseline resting values, CVC at the Ouabain (both P ≥ 0.09) and Ouabain + l‐NAME (both P ≥ 0.81) sites was similar at the end of both exercise bouts, whereas it was increased with l‐NAME (both P < 0.01).
Figure 2. Time‐course changes in cutaneous vascular conductance (CVC) during intermittent exercise at a fixed rate of metabolic heat production (500 W) in the heat (35°C; 20% relative humidity) (n = 13) .
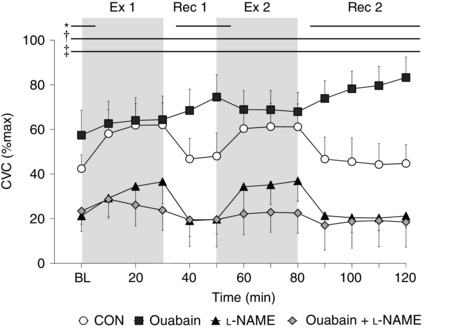
Four forearm skin sites were continuously perfused with (1) lactated Ringer solution (Control, open circles); (2) 6 mm ouabain (Ouabain; squares), a Na+/K+‐ATPase inhibitor; (3) 10 mm l‐NAME (triangles), a non‐selective NOS inhibitor; or (4) a combination of 10 mm l‐NAME and 6 mm ouabain (Ouabain + l‐NAME, diamonds). Values are presented as mean ± 95% confidence interval. Baseline resting represents the first 5 min of the first 10 min interval, and each data point thereafter represents an average of the final 5 min of each 10 min interval. BL, baseline resting; Ex 1, first exercise bout; Rec 1, first recovery period; Ex 2, second exercise bout; Rec 2, second recovery period. *Control significantly different from Ouabain. †Control significantly different from l‐NAME. ‡Control significantly different from Ouabain + l‐NAME (all P ≤ 0.05).
Discussion
The current study showed that the sweating response was attenuated with inhibition of Na+/K+‐ATPase and NOS, albeit the magnitude of attenuation was greater during inhibition of the former. Furthermore, we demonstrate that Na+/K+‐ATPase and NOS do not synergistically influence the sweating response. This is supported by the observation that the sum of reductions in local sweat rate with independent Na+/K+‐ATPase and NOS inhibition was similar to the magnitude of effect when both were inhibited simultaneously. During baseline resting and recovery from exercise, CVC was increased with Na+/K+‐ATPase inhibition, but given this response was reversed in the presence of l‐NAME, it seems this vasodilatation was attributable to increases in NO. On the other hand, Na+/K+‐ATPase inhibition had no effect on CVC during exercise, although the combined blockade of Na+/K+‐ATPase and NOS abolished the cutaneous vasodilatory response to intermittent exercise in the heat.
Sweating
The ∼54–60% reduction in sweat rate from the Control site seen with ouabain perfusion in the current study at the end of exercise (Fig. 1) suggests that Na+/K+‐ATPase may play a role in the sweating response (Sato & Dobson, 1969; Sato et al. 1969). Consistent with previous findings (Welch et al. 2009; Fujii et al. 2014; McGinn et al. 2014 b; Stapleton et al. 2014), we confirm the important influence that NOS plays in the modulation of sweating during exercise given that NOS inhibition caused a ∼12–13% attenuation in sweat rate from Control. We demonstrated that the simultaneous inhibition of Na+/K+‐ATPase and NOS reduced local forearm sweating by 69 and 75% at the end of the first and second exercise bouts, respectively. An additive effect was seen with the attenuations in sweating from Control induced by sole inhibitions of Na+/K+‐ATPase and NOS (i.e. Ouabain only and l‐NAME only sites), such that the sum of these attenuations was similar to that seen with simultaneous administration of both agents (i.e. Ouabain + l‐NAME). This finding would suggest that NOS and Na+/K+‐ATPase do not synergistically mediate sweating. NO may therefore modulate sweating through one or more of the remaining channels of the Na–K–2Cl cotransport model including the Na–K–2Cl cotransporter, K+ channel and Cl− channel (Sato et al. 1989; Saga, 2002). Indeed, NO has been shown to activate K+ (Bolotina et al. 1994; Tanaka et al. 2006) and Cl− (Kamosinska et al., 1997) channels in various tissues. Clearly, further examination in humans in vivo is required to evaluate these possibilities.
Cutaneous vascular conductance
Interestingly, during the baseline resting and post‐exercise recovery periods, CVC was greater with Na+/K+‐ATPase inhibition (i.e. Ouabain site) in comparison to Control (Fig. 2). Given that this ouabain‐induced vasodilatory response during baseline resting and recovery was not seen during the co‐inhibition of Na+/K+‐ATPase and NOS (i.e. Ouabain + l‐NAME site), it seems that this response was attributable to NO. It has been previously shown that in the presence of ouabain, there is a greater translocation of NOS in endothelial cells secondary to activation of phosphatidylinositol 3‐kinase (PI3K) and protein kinase B (PKB), resulting in a concomitant increase in NO production (Eva et al. 2006). Additionally, the increase in NO production following Na+/K+‐ATPase blockade may have been due to a rise in intracellular Na+, which would induce an influx of Ca2+ into the endothelial cell via the Na+/Ca2+‐exchanger, thereby activating NOS (Dong et al. 2004). Interestingly, vasoconstriction was not observed which would have presumably resulted from elevated levels of intracellular Ca2+ in vascular smooth muscle cell secondary to Na+/K+‐ATPase inhibition. Furthermore, it is important to consider that the apparent differences in CVC between the Ouabain and Control sites during baseline resting and post‐exercise recovery can in part be explained by the reduction in CVC at the Control site following the cessation of exercise, whereas the elevated CVC with ouabain perfusion was sustained.
During exercise, the CVC response seen at the combination site (i.e. Ouabain + l‐NAME) indicates that ouabain can also modulate a NO‐independent mediator of CVC. This NO‐independent mediator is unlikely to be cyclooxygenase, given that Fujii et al. (2014) demonstrated no role for cyclooxygenase during exercise in the heat involving a protocol similar to that employed in the current study. Given that NOS was simultaneously inhibited at the Ouabain + l‐NAME site, the abolished vasodilatory response may have been attributable to endothelium‐derived hyperpolarizing factors (EDHFs). This may also explain the similar CVC responses between the Ouabain and Control site in each exercise bout. Specifically, one of the pathways by which EDHFs initiate their effects is associated with the increase in intracellular Ca2+ in the endothelial cell, which stimulates calcium‐activated potassium channels (KCa). The resultant K+ efflux into the interstitial space activates inwardly rectifying potassium (KIR) channels and Na+/K+‐ATPase on the vascular smooth muscle cell (Edwards et al. 2010), ultimately leading to hyperpolarization of the vascular smooth muscle and therefore cutaneous vasodilatation. It is plausible that inhibition of Na+/K+‐ATPase may have blunted EDHF‐mediated vasodilatation as Na+/K+‐ATPase is a contributor to this pathway. This may in part explain the lack of increase in CVC at the Ouabain site during exercise.
The combined blockade of Na+/K+‐ATPase and NOS (i.e. Ouabain + l‐NAME site) attenuated the increase in CVC from baseline resting values during exercise. This suppression in CVC may also be explained by the previously described diminished EDHF‐dependent vasodilatation associated with Na+/K+‐ATPase. The CVC responses demonstrated in the current study should be regarded as the combined influences of vascular smooth muscle‐ and endothelium‐derived factors. Taken together, it can be postulated that endothelial Na+/K+‐ATPase may indirectly regulate the production of NO, in the sense that Na+/K+‐ATPase moderates levels of endothelial Ca2+ secondary to maintenance of the electrochemical gradient via Na+ and K+ transport across the endothelial cell membrane. In this regard, an interaction between Na+/K+‐ATPase and NOS in the regulation of cutaneous blood flow appears to exist.
Woolfson & Poston (1991) demonstrated that in the presence of ouabain, there were attenuations in the acetylcholine‐induced endothelium‐mediated vasodilatation of human subcutaneous resistance arteries in situ, which is in contrast to the findings of the current study. The inconsistent findings regarding ouabain may be explained by the involvement of several factors in cutaneous vascular control during exercise in the heat in humans in vivo (as in the current study), which do not occur in the acetylcholine‐dependent in situ model (Woolfson & Poston, 1991). These factors may include the influence of skin temperature (Kellogg, 2006), oxidative stress (Meade et al. 2015), activation of heat shock protein 90 (HSP90) (Shastry & Joyner, 2002), vasoactive intestinal peptide (VIP), pituitary adenylate cyclase activating peptide (PACAP) (Kellogg et al. 2010), neuropeptide substance P (Wong et al. 2005) and other yet undetermined factors.
Limitations
Although ouabain generally has a high affinity for Na+/K+‐ATPase, it is possible that ouabain‐resistant Na+/K+‐ATPase was unaffected and continued to function uninhibited (Kaplan, 2002). Nonetheless, the sweating response was considerably attenuated with blockade of Na+/K+‐ATPase, consistent with previous findings (Sato & Dobson, 1969; Sato et al., 1969). Another consideration is that Na+/K+‐ATPase is located on both endothelial and vascular smooth muscle cells. Hence, the findings of the current study are probably influenced by both the vascular endothelial and smooth muscle cells concomitantly. Moreover, using the intradermal microdialysis technique employed in the present study, it is not possible to selectively target Na+/K+‐ATPase on either the cutaneous vasculature or the sweat gland in order to study their isolated response.
Perspectives
Clinically, ouabain and other cardiac glycosides such as digoxin, due to the increase in intracellular Ca2+ with Na+/K+‐ATPase inhibition, may be used as medications to treat congestive heart failure and arrhythmias by increasing contractility strength and an increased vagal activity‐mediated reduction in heart rate, respectively (Fleckenstein, 1977). We determined that, although local administration of ouabain had no effect on cutaneous vasodilatation, the sweating response was reduced during exercise in the heat. Evaporation of sweat is the primary avenue of heat loss during exercise, especially when performed in hot environments (Kenny & Jay, 2013). Taken together, administration of cardiac glycosides may attenuate an individual's ability to dissipate heat during exercise and/or exposure to hot environments; however, the systemic effects of cardiac glycosides have not been studied from a thermoregulatory standpoint. Furthermore, given our observation of Na+/K+‐ATPase and NOS not synergistically modulating sweating, there may be greater risk associated with the reduction in sweating with cardiac glycosides in populations with diminished NO‐dependent sweating such as older males (Stapleton et al. 2014).
Conclusion
The current study demonstrates that Na+/K+‐ATPase and NOS do not modulate local forearm sweating synergistically during exercise in the heat. Furthermore, our data suggest that there are interactive roles of Na+/K+‐ATPase and NOS in the regulation of cutaneous vasodilatation during exercise in the heat.
Additional information
Competing interests
None.
Author contributions
J.C.L., N.F., R.D.M and G.P.K. conceived and designed experiments. J.C.L., N.F. and R.D.M. contributed to data collection. J.C.L. performed data analysis. J.C.L., N.F., R.D.M. and G.P.K. interpreted the experimental results. J.C.L. prepared the figures. J.C.L. drafted the manuscript. J.C.L., N.F., R.D.M. and G.P.K. edited and revised the manuscript. All authors approved the final version of the manuscript. All experiments took place at the Human and Environmental Physiology Research Unit located at the University of Ottawa.
Funding
This study was supported by grants from the Natural Sciences and Engineering Research Council of Canada [Discovery Grant (RGPIN‐06313–2014) and Discovery Grants Program ‐ Accelerator Supplements (RGPAS‐462252–2014); funds held by G.P.K.]. J.C.L. is supported by a Queen Elizabeth II Graduate Scholarship in Science and Technology. N.F. is supported by the Human and Environmental Physiology Research Unit. R.D.M. is supported by a Natural Sciences and Engineering Research Council Alexander Graham Bell Graduate Scholarship. G.P.K. is supported by a University Research Chair in Environmental Physiology.
Acknowledgements
We greatly appreciate all of the volunteers for taking their time to participate in this study and Martin Poirier for his technical assistance. We thank Mr Michael Sabino of Can‐Trol Environmental Systems Limited (Markham, ON, Canada) for his support as well as Ms Sarah Zhang and Ms Mercy Danquah for their assistance in the study.
References
- Adachi K & Yamasawa S (1966). Enzymatic basis for active transport of Na+ in the sweat gland unit. J Invest Dermatol 46, 510–511. [DOI] [PubMed] [Google Scholar]
- Bolotina VM, Najibi S, Palacino JJ, Pagano PJ & Cohen RA (1994). Nitric oxide directly activates calcium‐dependent potassium channels in vascular smooth muscle. Nature 368, 850–853. [DOI] [PubMed] [Google Scholar]
- Dong XH, Komiyama Y, Nishimura N, Masuda M & Takahashi H (2004). Nanomolar level of ouabain increases intracellular calcium to produce nitric oxide in rat aortic endothelial cells. Clin Exp Pharmacol Physiol 31, 276–283. [DOI] [PubMed] [Google Scholar]
- Du Bois D & Du Bois EF (1989). A formula to estimate the approximate surface area if height and weight be known. 1916. Nutrition 5, 303–311; discussion 312–303. [PubMed] [Google Scholar]
- Edwards G, Feletou M & Weston AH (2010). Endothelium‐derived hyperpolarising factors and associated pathways: a synopsis. Pflugers Arch 459, 863–879. [DOI] [PubMed] [Google Scholar]
- Eva A, Kirch U & Scheiner‐Bobis G (2006). Signaling pathways involving the sodium pump stimulate NO production in endothelial cells. Biochim Biophys Acta 1758, 1809–1814. [DOI] [PubMed] [Google Scholar]
- Fleckenstein A (1977). Specific pharmacology of calcium in myocardium, cardiac pacemakers, and vascular smooth muscle. Annu Rev Pharmacol Toxicol 17, 149–166. [DOI] [PubMed] [Google Scholar]
- Fujii N, McGinn R, Stapleton JM, Paull G, Meade RD & Kenny GP (2014). Evidence for cyclooxygenase‐dependent sweating in young males during intermittent exercise in the heat. J Physiol 592, 5327–5339. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gagnon D, Jay O & Kenny GP (2013). The evaporative requirement for heat balance determines whole‐body sweat rate during exercise under conditions permitting full evaporation. J Physiol 591, 2925–2935. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gibbs GE (1967). Quantitative microdetermination of enzymes in sweat glands in cystic fibrosis. Bibl Paediatr 86, 95–99. [PubMed] [Google Scholar]
- Gupta S, McArthur C, Grady C & Ruderman NB (1994). Role of endothelium‐derived nitric oxide in stimulation of Na+–K+‐ATPase activity by endothelin in rabbit aorta. Am J Physiol 266, H577–582. [DOI] [PubMed] [Google Scholar]
- Hardy JD, Du Bois EF & Soderstrom GF (1938). The technic of measuring radiation and convection one figure. J Nutrition 15, 461–475. [Google Scholar]
- Holowatz LA, Houghton BL, Wong BJ, Wilkins BW, Harding AW, Kenney WL & Minson CT (2003). Nitric oxide and attenuated reflex cutaneous vasodilation in aged skin. Am J Physiol Heart Circ Physiol 284, H1662–1667. [DOI] [PubMed] [Google Scholar]
- Holowatz LA, Thompson CS & Kenney WL (2006. a). Acute ascorbate supplementation alone or combined with arginase inhibition augments reflex cutaneous vasodilation in aged human skin. Am J Physiol Heart Circ Physiol 291, H2965–2970. [DOI] [PubMed] [Google Scholar]
- Holowatz LA, Thompson CS & Kenney WL (2006. b). l‐Arginine supplementation or arginase inhibition augments reflex cutaneous vasodilatation in aged human skin. J Physiol 574, 573–581. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kamosinska B, Radomski MW, Duszyk M, Radomski A & Man SF (1997). Nitric oxide activates chloride currents in human lung epithelial cells. Am J Physiol 272, L1098–1104. [DOI] [PubMed] [Google Scholar]
- Kaplan JH (2002). Biochemistry of Na,K‐ATPase. Annu Rev Biochem 71, 511–535. [DOI] [PubMed] [Google Scholar]
- Kellogg DL, Jr (2006). In vivo mechanisms of cutaneous vasodilation and vasoconstriction in humans during thermoregulatory challenges. J Appl Physiol (1985) 100, 1709–1718. [DOI] [PubMed] [Google Scholar]
- Kellogg DL, Jr , Crandall CG, Liu Y, Charkoudian N & Johnson JM (1998). Nitric oxide and cutaneous active vasodilation during heat stress in humans. J Appl Physiol (1985) 85, 824–829. [DOI] [PubMed] [Google Scholar]
- Kellogg DL, Jr , Zhao JL, Wu Y & Johnson JM (2010). VIP/PACAP receptor mediation of cutaneous active vasodilation during heat stress in humans. J Appl Physiol (1985) 109, 95–100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kenny GP & Jay O (2013). Thermometry, calorimetry, and mean body temperature during heat stress. Compr Physiol 3, 1689–1719. [DOI] [PubMed] [Google Scholar]
- McGinn R, Fujii N, Swift B, Lamarche DT & Kenny GP (2014. a). Adenosine receptor inhibition attenuates the suppression of postexercise cutaneous blood flow. J Physiol 592, 2667–2678. [DOI] [PMC free article] [PubMed] [Google Scholar]
- McGinn R, Paull G, Meade RD, Fujii N & Kenny GP (2014. b). Mechanisms underlying the postexercise baroreceptor‐mediated suppression of heat loss. Physiol Rep 2, e12168. [DOI] [PMC free article] [PubMed] [Google Scholar]
- McNamara TC, Keen JT, Simmons GH, Alexander LM & Wong BJ (2014). Endothelial nitric oxide synthase mediates the nitric oxide component of reflex cutaneous vasodilatation during dynamic exercise in humans. J Physiol 592, 5317–5326. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Meade RD, Fujii N, Alexander LM, Paull G, Louie JC, Flouris AD & Kenny GP (2015). Local infusion of ascorbate augments NO‐dependent cutaneous vasodilatation during intense exercise in the heat. J Physiol 593, 4055–4065. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Miura H, Toyama K, Pratt PF & Gutterman DD (2011). Cigarette smoking impairs Na+–K+‐ATPase activity in the human coronary microcirculation. Am J Physiol Heart Circ Physiol 300, H109–117. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pagan RM, Prieto D, Hernandez M, Correa C, Garcia‐Sacristan A, Benedito S & Martinez AC (2010). Regulation of NO‐dependent acetylcholine relaxation by K+ channels and the Na+–K+ ATPase pump in porcine internal mammary artery. Eur J Pharmacol 641, 61–66. [DOI] [PubMed] [Google Scholar]
- Palacios J, Marusic ET, Lopez NC, Gonzalez M & Michea L (2004). Estradiol‐induced expression of N+–K+‐ATPase catalytic isoforms in rat arteries: gender differences in activity mediated by nitric oxide donors. Am J Physiol Heart Circ Physiol 286, H1793–1800. [DOI] [PubMed] [Google Scholar]
- Quinton PM & Tormey JM (1976). Localization of Na/K‐ATPase sites in the secretory and reabsorptive epithelia of perfused eccrine sweat glands: a question to the role of the enzyme in secretion. J Membr Biol 29, 383–399. [DOI] [PubMed] [Google Scholar]
- Saga K (2002). Structure and function of human sweat glands studied with histochemistry and cytochemistry. Prog Histochem Cytochem 37, 323–386. [DOI] [PubMed] [Google Scholar]
- Saga K & Sato K (1988). Ultrastructural localization of ouabain‐sensitive, K‐dependent p‐nitrophenyl phosphatase activity in monkey eccrine sweat gland. J Histochem Cytochem 36, 1023–1030. [DOI] [PubMed] [Google Scholar]
- Sato K & Dobson RL (1969). Reversal of ouabain‐induced inhibition of human eccrine sweat gland function by diphenylhydantoin. J Invest Dermatol 53, 283–288. [DOI] [PubMed] [Google Scholar]
- Sato K, Kang WH, Saga K & Sato KT (1989). Biology of sweat glands and their disorders. I. Normal sweat gland function. J Am Acad Dermatol 20, 537–563. [DOI] [PubMed] [Google Scholar]
- Sato K & Sato F (1987). Nonisotonicity of simian eccrine primary sweat induced in vitro . Am J Physiol 252, R1099–1105. [DOI] [PubMed] [Google Scholar]
- Sato K, Taylor JR & Dobson RL (1969). The effect of oubain on eccrine sweat gland function. J Invest Dermatol 53, 275–282. [DOI] [PubMed] [Google Scholar]
- Sawka MN, Burke LM, Eichner ER, Maughan RJ, Montain SJ & Stachenfeld NS (2007). American College of Sports Medicine position stand. Exercise and fluid replacement. Med Sci Sports Exerc 39, 377–390. [DOI] [PubMed] [Google Scholar]
- Shastry S & Joyner MJ (2002). Geldanamycin attenuates NO‐mediated dilation in human skin. Am J Physiol Heart Circ Physiol 282, H232–236. [DOI] [PubMed] [Google Scholar]
- Siri WE (1956). The gross composition of the body. Adv Biol Med Phys 4, 239–280. [DOI] [PubMed] [Google Scholar]
- Stapleton JM, Fujii N, Carter M & Kenny GP (2014). Diminished nitric oxide‐dependent sweating in older males during intermittent exercise in the heat. Exp Physiol 99, 921–932. [DOI] [PubMed] [Google Scholar]
- Tanaka Y, Tang G, Takizawa K, Otsuka K, Eghbali M, Song M, Nishimaru K, Shigenobu K, Koike K, Stefani E & Toro L (2006). Kv channels contribute to nitric oxide‐ and atrial natriuretic peptide‐induced relaxation of a rat conduit artery. J Pharmacol Exp Ther 317, 341–354. [DOI] [PubMed] [Google Scholar]
- Toyomoto T, Knutsen D, Soos G & Sato K (1997). Na–K–2Cl cotransporters are present and regulated in simian eccrine clear cells. Am J Physiol 273, R270–277. [DOI] [PubMed] [Google Scholar]
- Welch G, Foote KM, Hansen C & Mack GW (2009). Nonselective NOS inhibition blunts the sweat response to exercise in a warm environment. J Appl Physiol (1985) 106, 796–803. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wilson TE, Cui J & Crandall CG (2005). Mean body temperature does not modulate eccrine sweat rate during upright tilt. J Appl Physiol (1985) 98, 1207–1212. [DOI] [PubMed] [Google Scholar]
- Wong BJ, Tublitz NJ & Minson CT (2005). Neurokinin‐1 receptor desensitization to consecutive microdialysis infusions of substance P in human skin. J Physiol 568, 1047–1056. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Woolfson RG & Poston L (1991). Effect of ouabain on endothelium‐dependent relaxation of human resistance arteries. Hypertension 17, 619–625. [DOI] [PubMed] [Google Scholar]