

Abstract
Key points
The extent of myosin regulatory light chain phosphorylation (RLC) necessary for smooth muscle contraction depends on the respective activities of Ca2+/calmodulin‐dependent myosin light chain kinase and myosin light chain phosphatase (MLCP), which contains a regulatory subunit MYPT1 bound to the phosphatase catalytic subunit and myosin.
MYPT1 showed significant constitutive T696 and T853 phosphorylation, which is predicted to inhibit MLCP activity in isolated ileal smooth muscle tissues, with additional phosphorylation upon pharmacological treatment with the muscarinic agonist carbachol.
Electrical field stimulation (EFS), which releases ACh from nerves, increased force and RLC phosphorylation but not MYPT1 T696 or T853 phosphorylation.
The conditional knockout of MYPT1 or the knockin mutation T853A in mice had no effect on the frequency‐maximal force responses to EFS in isolated ileal tissues.
Physiological RLC phosphorylation and force development in ileal smooth muscle depend on myosin light chain kinase and MLCP activities without changes in constitutive MYPT1 phosphorylation.
Abstract
Smooth muscle contraction initiated by myosin regulatory light chain (RLC) phosphorylation is dependent on the relative activities of Ca2+/calmodulin‐dependent myosin light chain kinase (MLCK) and myosin light chain phosphatase (MLCP). We have investigated the physiological role of the MLCP regulatory subunit MYPT1 in ileal smooth muscle in adult mice with (1) smooth muscle‐specific deletion of MYPT1; (2) non‐phosphorylatable MYPT1 containing a T853A knockin mutation; and (3) measurements of force and protein phosphorylation responses to cholinergic neurostimulation initiated by electric field stimulation. Isolated MYPT1‐deficient tissues from MYPT1SM−/− mice contracted and relaxed rapidly with moderate differences in sustained responses to KCl and carbachol treatments and washouts, respectively. Similarly, measurements of regulatory proteins responsible for RLC phosphorylation during contractions also revealed moderate changes. There were no differences in contractile or RLC phosphorylation responses to carbachol between tissues from normal mice vs. MYPT1 T853A knockin mice. Quantitatively, there was substantial MYPT1 T696 and T853 phosphorylation in wild‐type tissues under resting conditions, predicting a high extent of MLCP phosphatase inhibition. Reduced PP1cδ activity in MYPT1‐deficient tissues may be similar to attenuated MLCP activity in wild‐type tissues resulting from constitutively phosphorylated MYPT1. Electric field stimulation increased RLC phosphorylation and force development in tissues from wild‐type mice without an increase in MYPT1 phosphorylation. Thus, physiological RLC phosphorylation and force development in ileal smooth muscle appear to be dependent on MLCK and MLCP activities without changes in constitutive MYPT1 phosphorylation.
Key points
The extent of myosin regulatory light chain phosphorylation (RLC) necessary for smooth muscle contraction depends on the respective activities of Ca2+/calmodulin‐dependent myosin light chain kinase and myosin light chain phosphatase (MLCP), which contains a regulatory subunit MYPT1 bound to the phosphatase catalytic subunit and myosin.
MYPT1 showed significant constitutive T696 and T853 phosphorylation, which is predicted to inhibit MLCP activity in isolated ileal smooth muscle tissues, with additional phosphorylation upon pharmacological treatment with the muscarinic agonist carbachol.
Electrical field stimulation (EFS), which releases ACh from nerves, increased force and RLC phosphorylation but not MYPT1 T696 or T853 phosphorylation.
The conditional knockout of MYPT1 or the knockin mutation T853A in mice had no effect on the frequency‐maximal force responses to EFS in isolated ileal tissues.
Physiological RLC phosphorylation and force development in ileal smooth muscle depend on myosin light chain kinase and MLCP activities without changes in constitutive MYPT1 phosphorylation.
Abbreviations
- CPI‐17
protein kinase C‐potentiated protein phosphatase 1 inhibitor protein of 17 kDa
- H&E
haematoxylin & eosin
- MBS85
myosin binding subunit 85
- MLCK
myosin light chain kinase
- MLCP
myosin light chain phosphatase
- MYPT1
myosin phosphatase targeting subunit‐1
- PDBu
phorbol 12, 13‐dibutyrate
- PP1cδ
Protein Phosphatase Type 1 catalyic subunit δ
- PKC
protein kinase C
- PSS
physiological salt solution
- RLC
myosin regulatory light chain
- ROCK
RhoA‐associated protein kinase
- SMMHC
smooth muscle myosin heavy chain
Introduction
Diverse cell signalling pathways increase smooth muscle cell [Ca2+]i, leading to Ca2+ binding to calmodulin. Ca2+/calmodulin then binds and activates myosin light chain kinase (MLCK), which phosphorylates myosin regulatory light chain (RLC) to initiate smooth muscle contraction (Kamm & Stull, 1985 a; Hartshorne, 1987; Kamm & Stull, 2001; Somlyo & Somlyo, 2003). MLCK appears to be the only kinase phosphorylating RLC physiologically in intestinal (He et al. 2008), airway (Zhang et al. 2010), mesenteric arterial (He et al. 2011), aortic (Gao et al. 2013) and urinary bladder smooth muscles (Gao et al. 2013). Additional regulation of activation occurs by the subsequent Ca2+‐dependent phosphorylation of MLCK, which diminishes its activity, thereby fine tuning the RLC phosphorylation response (Stull et al. 1990; Kamm & Stull, 2001; Tsai et al. 2012).
Myosin light chain phosphatase (MLCP) dephosphorylates RLC, and induces relaxation after a decrease in [Ca2+]i returns MLCK to an inactive state. MLCP is a holoenzyme composed of three distinct subunits: a 38 kDa catalytic subunit (PP1cδ), a large 110–130 kDa regulatory subunit (MYPT1) and a small 20 kDa subunit of unknown function (Matsumura & Hartshorne, 2008; Grassie et al. 2011). Biochemically, MYPT1 binds PP1cδ and myosin, and thus functions as a scaffolding protein. Signalling pathways may converge to inhibit MLCP activity by distinct mechanisms, thereby increasing RLC phosphorylation without changing elevated [Ca2+]i (Ca2+ sensitization) (Somlyo & Somlyo, 2003; Hartshorne et al. 2004; Dimopoulos et al. 2007; Matsumura & Hartshorne, 2008; Kitazawa, 2010; Grassie et al. 2011). Studies in many types of smooth muscles, including intestinal smooth muscle, indicate that agonist‐mediated Ca2+ sensitization involving decreased MLCP activity is the result of two major signalling pathways, including phosphorylation of MYPT1 T853 by a RhoA‐associated kinase (ROCK) pathway and phosphorylation of a small inhibitor protein CPI‐17 at T38 by protein kinase C (PKC) (Kitazawa et al. 2000; Hirano et al. 2003; Somlyo & Somlyo, 2003; Murthy, 2006; Mizuno et al. 2008; Eto, 2009). Additionally, constitutive phosphorylation of MYPT1 at T696 as well as T853 inhibits MLCP activity (Khromov et al. 2009; Tsai et al. 2014). Thus, the extent of RLC phosphorylation is determined by the relative Ca2+/calmodulin‐dependent MLCK and MLCP activities, each of which is modulated by phosphorylation involving different cell signalling pathways.
Although pharmacological, biophysical and biochemical approaches have identified generic components of signalling modules that may act on smooth muscle RLC phosphorylation, the physiological relationships among these signalling molecules in many cases are not well understood in different types of smooth muscles. For example, the importance of distinct signalling modules and pathways appears to be different in the vascular bed for large conduit arteries vs. resistance arteries (Cole & Welsh, 2011; Kitazawa & Kitazawa, 2012; Gao et al. 2013). Thus, the relative importance of individual components in the general scheme for the regulation of RLC phosphorylation needs to be understood in the context of various smooth muscles and their physiological contractile responses.
MYPT1, similar to MLCK, was expected to play a central role in signalling to RLC phosphorylation in smooth muscle tissues (Hartshorne et al. 1998; Somlyo & Somlyo, 2000; Grassie et al. 2011). The conditional knockout of MLCK in smooth muscle cells in adult mice is lethal because of contractile failure (He et al. 2008; Zhang et al. 2010; He et al. 2011; Gao et al. 2013). Furthermore, inactivation of one MYLK allele in humans induces aortic dissections as a result of the selective sensitivity of limiting MLCK activity in aortic smooth muscle cells (Wang et al. 2010; Gao et al. 2013). Genetic approaches for investigating the functions of MYPT1 as a scaffolding protein and substrate for ROCK recently provided new insights into our understanding of its physiological role in smooth muscle contraction. Surprisingly, the knockout of MYPT1 specifically in smooth muscle cells (He et al. 2013; Qiao et al. 2014; Tsai et al. 2014) did not result in smooth muscle failure in intact mice, and was associated with modest changes in agonist‐ and KCl‐induced contractile and relaxation responses of isolated ileal (He et al. 2013), mesenteric arterial (Qiao et al. 2014) and bladder (Tsai et al. 2014) tissues. An apparent compensation for MYPT1 loss may be attributed to one of several findings. First, in wild‐type tissues, MLCP activity is diminished by constitutive phosphorylation of MYPT1. Knockout of MYPT1 results in a reduction of PP1cδ protein, rendering the reduced MLCP activity to be similar to the constitutively inhibited MLCP in wild‐type tissues (Tsai et al. 2014). Second, PP1cδ not bound to MYPT1 may also contribute to the dephosphorylation of RLC in smooth muscle tissues from either wild‐type or MYPT1 knockout mice (Tsai et al. 2014). Additionally, the expression of another closely related MYPT family member, MBS85, may provide some compensation because of a greater level of expression in smooth muscle compared to other family members (MYPT2 and MYPT3) (Tsai et al. 2014).
In the present study, we have extended investigations involving genetically modified mice on the physiological regulation of RLC phosphorylation/dephosphorylation in ileal smooth muscle tissues (He et al. 2013). We have also investigated biochemical and contractile responses to the electrical field stimulation that releases neurotransmitter ACh for physiological contractions compared to pharmacological muscarinic stimulation. We used a new method to quantify phosphorylation of MYPT1 T696 and T853 in addition to using established quantitative measurements for CPI‐17 and RLC phosphorylation. The results from these studies led to the conclusion that physiological RLC phosphorylation and force development in ileal smooth muscle appear to be dependent on MLCK and MLCP activities without changes in constitutive MYPT1 phosphorylation.
Methods
Ethical approval
Experiments were performed in accordance with the National Institutes of Health and Institutional Animal Care and Use Guidelines. The Institutional Animal Care and Use Committee at the University of Texas Southwestern Medical Centre approved all procedures and protocols in compliance with The Journal of Physiology guidelines. Animals were killed by i.p. administration of a lethal dose of tribromoethanol (250 mg kg−1) for tissue collection.
Generation of genetically modified mice
Mice containing floxed Mypt1 alleles (Mypt1f/f) (He et al. 2013) were crossed with a SMMHC‐CreERT2 transgenic mouse line expressing a fusion protein of the Cre recombinase with the modified oestrogen receptor binding domain (CreERT2) under the control of the smooth muscle myosin heavy chain (SMMHC) promoter (Wirth et al. 2008). Cre‐mediated recombination occurred robustly and exclusively in smooth muscle cells in tissues, although only after tamoxifen treatment (Wirth et al. 2008). Mice were bred and screened as described previously (Wirth et al. 2008; He et al. 2013; Tsai et al. 2014). Male mice (8–10 weeks old) were injected i.p. with tamoxifen or vehicle for five consecutive days each week for 2 weeks at a dose of 1 mg day−1 (Tsai et al. 2014). Ileal tissues were harvested 6–7 weeks after starting tamoxifen treatment from transgenic mice containing Mypt1f/f, Cre+ alleles (denoted MYPT1SM−/− mice) and age‐matched vehicle‐treated male littermate mice (denoted as MYPT1SM+/+ mice). Mice containing the MYPT1 T852A mutation (numbering based on mouse MYPT1 sequence) with alanine substituted for threonine 852 (Chen et al. 2015) were also used for experiments on isolated ileal tissue strips. For consistency with previous biochemical investigations on the phosphorylation properties of MYPT1 phosphorylation by ROCK (Somlyo & Somlyo, 2003; Hartshorne et al. 2004; Grassie et al. 2011), we refer to this mutation as MYPT1T853A with the human isoform numbering sequence, which is also used for the MYPT1 T696 and T853 phosphorylation sites.
Measurements of ileal smooth muscle contraction
Maintained MYPT1SM+/+ or MYPT1SM−/− or MYPT1T853A mutant mice were killed with a lethal dose injection of tribromoethanol. The small intestine was completely removed and immediately immersed in cool physiological salt solution (PSS) (in mm: 118.5 NaCl, 4.75 KCl, 1.2 MgSO4, 1.2 KH2PO4, 24.9 NaHCO3, 1.6 CaCl2 and 10.0 d‐glucose, aerated with 95% O2–5% CO2 to maintain pH 7.4). A 5–6 cm segment from the distal ileum, located ±1 cm away from the ileocecal transition was removed. Segments from ileum (5–6 mm) were mounted with surgical silk to the small hook in a longitudinal orientation on a water‐jacketed 8 ml organ bath in PSS pre‐gassed with 95% O2–5% CO2 at 37°C. After mounting, a resting force of 0.9 g was applied and tissue was allowed to equilibrate for at least 45 min followed by pre‐contraction with alternating treatments with 65 mm KCl twice and 90 mm KCl once every 10 min to establish viability. At the end of the equilibration, 90 mm KCl (pretreated with 1 μm atropine 10 min) or the muscarinic agonist 6 μm carbachol (Sigma, St Louis, MO, USA) were added to initiate contractile responses. Strips were also pretreated with the Rho kinase inhibitor H‐1152 (1 μm) (Calbiochem‐EMD Biosciences, San Diego, CA, USA) or protein kinase C inhibitor GF‐109203X (3 μm (Enzo Life Sciences, Farmingdale, NY, USA). After 20 min of pre‐incubation, 6 μm carbachol was applied to initiate contractile responses.
Nerve‐mediated smooth muscle contractions were elicited by electric field stimulation (EFS) using a pair of electrodes mounted in the tissue bath in parallel to the ileum strip in a similar manner to previous studies (Ding et al. 2009; Tsai et al. 2012). Pulses of 0.5 ms in duration were delivered in trains of increasing frequencies (0.5–80 Hz) by a DC power amplifier (HP 6824A; Hewlett‐Packard, Palo Alto, CA, USA) driven by a Grass S88 stimulator at 20 V (Grass Instrument Co., Quincy, MA, USA). Isometric force development in response to KCl, CCh and EFS was recorded with a Grass FT03 force transducer connected to Powerlab 8/SP data acquisition unit (AD Instruments, Colorado Springs, CO, USA). Force measurements were normalized as grammes of developed force per milligram tissue wet weight. Force responses to EFS were inhibited 75 ± 0.04% and 78 ± 0.04% with 1 μm atropine, a cholinergic muscarinic antagonist. These results show the prominent role of the muscarinic‐mediated contractile response with EFS, similar to the airway (Kamm & Stull, 1985 b) and urinary bladder responses (Tsai et al. 2012).
At the indicated times after specific treatments, tissues were quick frozen by clamps pre‐chilled in liquid nitrogen, and were processed as described previously (Gao et al. 2013). Briefly, frozen tissues were stored at –80°C until they were added to frozen slurry of acetone with 10% trichloroacetic acid and 10 mm dithiothreitol, and slowly thawed at room temperature. Tissues were rinsed with ether (three times for 10 min each), dried (1 h) and suspended in urea sample buffer containing 8 m urea, 18.5 mm Tris (pH 8.6), 20.4 mm glycine, 10 mm dithiothreitol, 4 mm EDTA and 5% sucrose. Proteins were then solubilized in a the Bullet Blender with the addition of urea crystals to saturation (Next Advance, Inc., Averill Park, NY, USA) (with 2 mm zirconium oxide beads, four spins × 3 min each at setting 9). Protein content was determined by a Bradford assay (Bio‐Rad, Hercules, CA, USA) with BSA as the standard. Bromophenol blue was added to 0.004% and the sample was stored at –80°C.
Western blot analysis
Tissue extracts solubilized in urea sample buffer were added to 0.25 volumes of SDS sample buffer containing 250 mm Tris (pH 6.8), 10% SDS, 50 mm dithiothreitol, 40% glycerol and 0.01% bromophenol blue, and boiled for SDS‐PAGE (8–18% polyacrylamide gradient). Proteins were transferred onto a nitrocellulose membrane and visualized by immunoblot staining using antibodies to MYPT1 total (mouse monoclonal; BD Transduction Laboratories, Lexington, KT, USA), pan‐MYPTs (from Dr Masumi Eto, Thomas Jefferson University, Philladelphia, PA, USA), PP1cα (rabbit polyclonal; Millipore, Billerica, MA, USA), PP1cδ (rabbit; Upstate Biotechnology, Lake Placid, NY, USA), PP1cγ (goat; Santa Cruz Biotechnology, Santa Cruz, CA, USA), MLCK (mouse, K36; Sigma, St. Louis, MO, USA), phospho‐MLCK S1760 (rabbit; ProSci, Poway, CA, USA), ROCK1 (rabbit; BD Biosciences, San Jose, CA, USA), CPI‐17 total (Santa Cruz Biotechnology, Santa Cruz, CA, USA), phospho‐MYPT1 T853 (rabbit; Upstate Biotechnology, Lake Placid, NY, USA), phospho‐MYPT1 T696 (rabbit; Upstate Biotechnology, Lake Placid, NY, USA), phospho‐CPI‐17 Thr38(rabbit; Bioworld, Dublin, OH, USA). The pan‐MYPTs antibody recognizes the conserved autoinhibitory sequence in MYPT1, MYPT2 and MBS85. GAPDH was stained with anti‐GAPDH antibody (rabbit polyclonal; Santa Cruz Biotechnology, Santa Cruz, CA, USA) as a loading control. Total protein blots for MYPT1, PP1cα, PP1cδ, PP1cγ, MBS85, ROCK1, MLCK and CPI‐17 were expressed as ratios relative to the immunoblot of GAPDH loading control. The phosphorylation of MYPT1 (T853 or T696) were expressed as a ratio of phospho‐MYPT1 to MYPT1 total and then normalized with the average values response to calyculin A (LC Laboratories, Woburn, MA, USA). The phosphorylation of MYPT1 T696 and T853 in calyculin A‐treated ileal strips was quantitated by comparison with purified GST‐MYPT1 (654‐880) maximally phosphorylated by ROCK1 in vitro (PV3691; Life Technologies, Grand Island, NY, USA) (Khromov et al. 2009; Tsai et al. 2014). Purified GST‐MYPT1 (654‐880) was prepared as described previously (Tsai et al. 2014). The phosphorylation of CPI‐17 was obtained as a ratio of phosphorylated CPI‐17 to CPI‐17 total and was normalized to the phosphorylation response to phorbol 12,13‐dibutyrate (PDBu) (Tsai et al. 2014). MLCK and MBS85 phosphorylation were normalized to the response obtained with calyculin A. Quantification of western blots was determined by quantitative densitometry using the ImageQuant software package (Molecular Dynamics, Sunnyvale, CA, USA).
Measurement of RLC phosphorylation
RLC phosphorylation was measured by urea/glycerol‐PAGE as described previously (Kamm et al. 1989; Gao et al. 2013). Muscle proteins in 8 m urea sample buffer were subjected to urea/glycerol‐PAGE to separate non‐phosphorylated and monophosphorylated RLC. Following electrophoresis, proteins were transferred to nitrocellulose membranes, and probed with mouse monoclonal antibodies against smooth muscle RLC. The ratio of monophosphorylated RLC to total RLC (non‐phosphorylated plus monophosphorylated) was determined by quantitative densitometry and expressed as mol phosphate per mol protein.
Histology and immunohistochemistry
The small intestines from MYPT1SM+/+ or MYPT1SM−/− mice were immersed in cool PSS. The ileum was isolated, fixed in 4% (w/v) paraformaldehyde overnight, embedded in paraffin and transversely sectioned at a thickness of 6 μm. After dewaxing and hydration, the sections were stained with standard haematoxylin & eosin (H&E). Quantification of the area of longitudinal and circular smooth muscle layers was determined using ImageJ (BIH, Bethesda, MD, USA). For immunohistochemistry, fresh ileum sections were embedded with OCT and blocked with phosphate‐buffered saline containing 0.1% Triton X‐100, 0.1% Tween 20, 1% BSA and 5% non‐immune goat serum for 1 h at room temperature, and then incubated with primary antibody: anti‐rabbit MYPT1 (BD Transduction Laboratories, Lexington, KY, USA), anti‐mouse smooth muscle α‐actin (Sigma) overnight and incubated with secondary antibodies conjugated with either Alexa Fluor 488 (Life Technologies, Gaithersburg, MD, USA) for smooth muscle α‐actin or 569 (Life Technologies) for MYPT1. Microscopy was performed using an Eclipse 400 fluorescence microscope (Nikon, Tokyo, Japan) or a confocal microscope (Olympus, Tokyo, Japan).
Statistical analysis
All data are presented as the mean ± SEM Statistical comparisons were performed by Student's t test for comparison between the control and treatment groups. For multiple comparisons, one‐way ANOVA followed by Dunnett's post hoc test or two‐way ANOVA with Tukey's post hoc test was used. Data analyses were performed with Prism, version 6.0 (GraphPad Software, San Diego, CA, USA). P < 0.05 was considered statistically significant.
Results
Effect of conditional MYPT1 gene deletion in adult mice on myosin regulatory protein content in ileal tissue
In our initial study, Mypt1 expression was ablated early in development specifically in smooth muscle cells by crossing Mypt1 floxed mice with SMA‐Cre transgenic mice, which express non‐inducible Cre recombinase under the control of the α‐smooth muscle actin promoter, with no change in PP1cδ expression in ileal tissues (Wu et al. 2007; He et al. 2013). However, the conditional knockout of MYPT1 in adult mice resulted in a 70% decrease in PP1cδ in bladder smooth muscle (Tsai et al. 2014). For these reasons, the expression of signalling proteins acting on smooth muscle RLC phosphorylation was examined with the conditional knockout of MYPT1 in ileal smooth muscle of adult mice, including PP1cδ and MBS85. Expression of MYPT1 protein decreased by 90% (Fig. 1 A and B). Using an antibody raised to a common sequence in MYPT1 and MBS85, the amount of MBS85 was 15 ± 1% compared to MYPT1 in wild‐type tissue, which is similar to the results obtained in bladder smooth muscle (Tsai et al. 2014). By contrast, MBS85 expression increased three‐fold in tissues from MYPT1SM−/− mice compared to MYPT1SM+/+ mice. Thus, the amount of MBS85 in MYPT1‐deficient ileal tissues was 45% of the amount of MYPT1 found in ileal tissues from wild‐type mice.
Figure 1. Effect of conditional MYPT1 gene ablation on content of myosin regulatory proteins in ileal tissues .
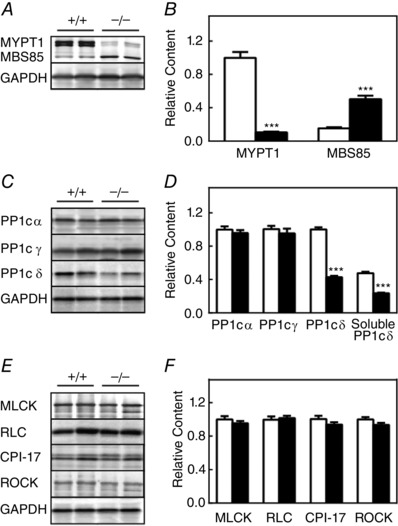
The relative contents of proteins were measured by immunoblotting extracts of ileal smooth muscle tissues from MYPT1SM+/+ and MYPT1SM–/– mice. Representative immunoblots (A, C and E) and quantified results (B, D and F) are shown for MYPT1, MBS85 and PP1C (α, γ,δ), as well as RLC, MLCK, CPI‐17 and ROCK1 for ileal tissues from MYPT1SM+/+ (open bars) and MYPT1SM‐/‐ (solid bars) mice with GAPDH as a loading control. MLCP component proteins MYPT1 and MBS85 were normalized to the MYPT1 content in ileal tissues from MYPT1SM+/+ mice. MYPT1 and PP1cδ contents were similar in ileal tissues with or without mucosa in both MYPT1SM+/+ and MYPT1SM–/– mice (data not shown). Data are presented as the mean ± SEM from ≥10 animals in each group. ***P < 0.001 compared to MYPT1SM+/+.
Expression of PP1 isoforms showed different responses to MYPT1 knockout, with no significant changes in α and γ isoforms, whereas δ decreased by 57% (Fig. 1 C and D). The decrease in PP1cδ in ileal tissues is similar to previous results obtained with bladder smooth muscle, ruling out tissue‐specific responses. In any event, this difference in PP1cδ expression did not result in any apparent changes in expression of other signalling proteins relevant to RLC phosphorylation in isolated ileal tissue contractile properties or in the phenotype of adult mice. Similar to the results obtained with bladder smooth muscle (Tsai et al. 2014), 50% of the total PP1cδ isoform was soluble in ileal tissues from wild‐type animals. Although MYPT1‐deficient tissues showed a decrease in the total amount of PP1cδ, the relative amount that was soluble did not change.
The expression of other relevant proteins in the RLC signalling module, including MLCK, Rho‐kinase (ROCK I), RLC and CPI‐17, was comparable in ileum from MYPT1SM+/+ and MYPT1SM−/− mice (Fig. 1 E and F).
Thus, the conditional ablation of MYPT1 in ileal smooth muscle tissues from adult MYPT1SM−/− mice was associated with a partial reduction of soluble and bound PP1cδ and an increased expression of MBS85.
Mypt1 ablation results in MYPT1 deficiency specifically in ileal smooth muscle cells
At necropsy, the digestive tract appeared normal, including the small intestine (data not shown), which is similar to previously reported results (He et al. 2013). Histological examination revealed a normal ileal structure with some hypertrophy of the smooth muscle layer (Fig. 2 A–C). The hypertrophy response in ileal tissues from MYPT1SM−/− mice may be an adaptive response associated with the loss of the ICC cells, diminution of peristalsis and infiltration of inflammatory cells in the mucosal lamina propria as described previously (He et al. 2013). Immunofluorescence co‐staining for smooth muscle α‐actin and MYPT1 showed deficiency of MYPT1 in smooth muscle cells but not mucosal cells in ileal tissue from Mypt1SM−/− mice (Fig. 2 D). These results are consistent with the smooth muscle‐specific expression of the fusion protein Cre recombinase under the control of the SMMHC promoter (Wirth et al. 2008). Additionally, the results show that residual MYPT1 in ileal tissue from Mypt1SM−/− mice (Fig. 1 A and B) is primarily in non‐smooth muscle cells.
Figure 2. Histological examination of ileal tissues from MYPT1SM+/+ and MYPT1SM–/– mice .
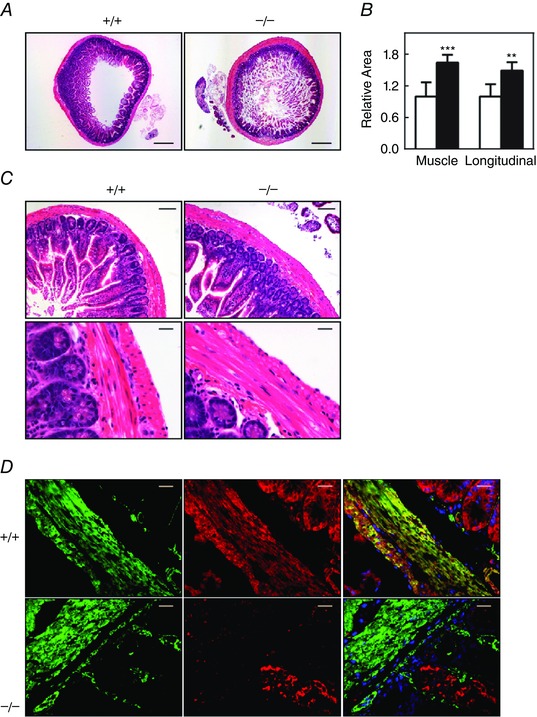
A, H&E stained transverse sections of ileum revealed wall thickening in MYPT1SM−/− mice (right). Scale bars = 400 μm. B, quantified results for total and longitudinal muscle areas are presented as the mean ± SEM (n ≥ 4 mice in each group). **P < 0.01 and ***P < 0.001 compared to MYPT1SM+/+ mice. C, normal histological structure of ileum from MYPT1SM–/– mice with H&E staining. Scale bars = 100 μm (top) and 20 μm (bottom). D, immunofluorescence staining of MYPT1 in ileal smooth muscle tissue from MYPT1SM+/+ and MYPT1SM–/– mice. Green, smooth muscle α‐actin; red, anti‐MYPT1. Scale bars = 20 μm.
MYPT1‐deficient ileum exhibits enhanced RLC phosphorylation and force in the sustained phase of contraction
We found reduced and variable spontaneous force amplitudes from 2.8 ± 0.08 g/mg tissue × 100 for MYPT1SM+/+ mice to 0.8 ± 0.04 g/mg tissue × 100 for MYPT1SM−/− mice without a loss in frequency of contractions. These results were associated with a decrease in immunostaining for c‐Kit and a decreased amount of c‐Kit by western blotting (data not shown). These results are similar to those reported previously (He et al. 2013), indicating that MYPT1 knockout in ileal tissue is associated with impairment of the interstitial cells of Cajal in the myenteric plexus, thereby affecting peristalsis.
Maximal force development and RLC phosphorylation responses to KCl or carbachol were similar in isolated ileal tissue strips from both Mypt1SM−/− and Mypt1SM+/+ mice (Fig. 3). However, the sustained responses to both treatments were enhanced in tissues from Mypt1SM−/− compared to Mypt1SM+/+ mice. Additionally, the relaxation responses obtained on washout of carbachol from both types of mice were rapid (Fig. 3 A). The half‐time for relaxation was slower for tissues from Mypt1SM−/− (t 1/2 = 11.8 ± 0.7) compared to tissues from Mypt1SM+/+ (t 1/2 = 6.0 ± 0.4) mice (P < 0.001, n = 20). Thus, isolated smooth muscle strips from MYPT1‐deficient mice show modest differences in RLC phosphorylation and contractile responses compared to control tissues. The lack of contractile impairment is consistent with the phenotype of knockout animals that display normal bowel motility, food consumption and defaecation (He et al. 2013).
Figure 3. MYPT1‐deficient ileum exhibits increased force and RLC phosphorylation during the sustained phase of contraction .
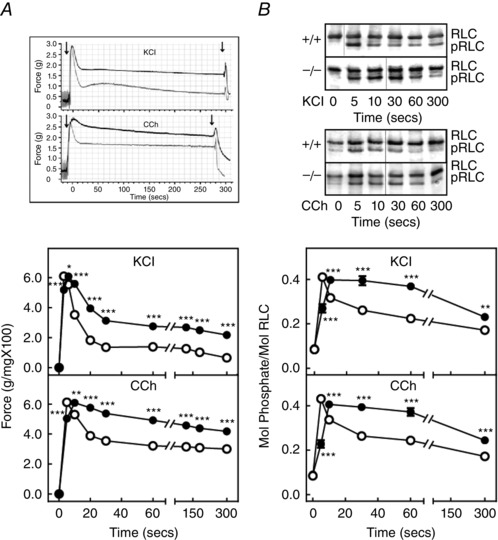
A, representative force tracings (above) of MYPT1SM+/+ (grey trace) and MYPT1SM–/− (black trace) and quantified force responses (below) for ileal strips from MYPT1SM+/+ (open circles) and MYPT1SM–/− (solid circles) mice are shown for 90 mm KCl (top) or 6 μm carbachol (CCh) (bottom) treatments. Data are the mean ± SEM (n ≥ 20). *P < 0.05 and ***P < 0.001 compared to MYPT1SM+/+ mice for the same time. The SEM bars are smaller than the symbols. B, representative immunoblots (above) following glycerol/urea‐PAGE for RLC phosphorylation and quantified RLC phosphorylation (below) responses are shown for 90 mm KCl (top) and 6 μm carbachol (CCh) (bottom) with symbols as in (A). RLC, non‐phosphorylated, pRLC, monophosphorylated. Data are the mean ± SEM from n ≥ 10 measurements each. **P < 0.01 and ***P < 0.001 compared to MYPT1SM+/+ at the same time. The SEM bars may be smaller than the symbols for the means.
Constitutive and agonist‐induced MYPT1 phosphorylation were reduced in MYPT1‐deficient tissues proportional to the reduction in MYPT1 protein
We previously developed a method to quantify the extent of phosphorylation of MYPT1 T696 and T853 (Tsai et al. 2014). This method was applied to isolated ileal tissues as shown in Fig. 4 A, where an expressed GST fragment of MYPT1 (residues 654–880) containing both T696 and T853 was diphosphorylated by purified ROCK. We used this fragment as a standard to quantify the extent of MYPT1 T696 and T853 phosphorylation in ileal tissues treated for 30 min with the MLCP phosphatase inhibitor, calyculin A. The MYPT1 antibody raised to the MYPT1 sequence 723–840 allowed measurement of the total amount of the tissue MYPT1 relative to the GST‐MYPT1 fragment and calculation of the extent of MYPT1 phosphorylation with calyculin A treatment. The results show complete phosphorylation of T696 with calyculin A treatment, whereas T853 was 60% phosphorylated (Fig. 4 A).
Figure 4. Treatment with KCl or carbachol increase phosphorylation of constitutive phosphorylated MYPT1 in ileal tissues from MYPT1SM − /− and MYPT1SM+/+ mice .
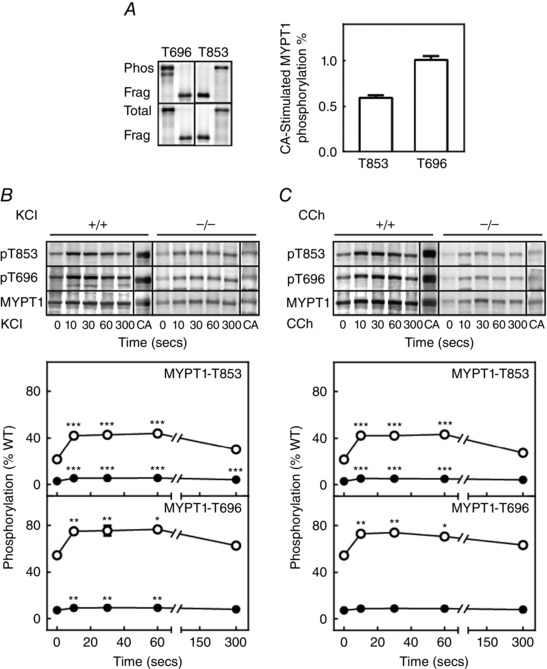
A, quantification of MYPT1 phosphorylation in calyculin A (CA)‐treated ileum strips. Left: representative blots for calyculin A‐treated ileal strips from wild‐type mice and diphosphorylated GST‐MYPT1 fragment are shown for total MYPT1 (bottom), phosphorylated T696 and phosphorylated T853 (top). Purified GST‐ MYPT1 (654‐880) was maximally phosphorylated by ROCK1 in vitro. Right: amounts of calyculin A‐treated tissue MYPT1 phosphorylated at T696 per total T696 and MYPT1 phosphorylated at T853 per total T853 are shown as ratios. B and C, representative MYPT1 immunoblots (top) and quantitative phosphorylation results (bottom) are shown for the response to 90 mm KCl (B) and 6 μm carbachol (CCh) (C) in ileal tissues from MYPT1SM+/+ (open circles) and MYPT1SM–/– (solid circles) mice normalized to the amount of MYPT1 protein in tissues from wild‐type mice. More protein was applied to SDS‐PAGE for tissues from MYPT1SM–/− mice. MYPT1 phosphorylation was calculated relative to values obtained with calyculin A as described in (A). Data are presented as the mean ± SEM (n ≥10 animals in each group). There were no differences in the relative phosphorylation responses for MYPT1 in tissues from MYPT1SM+/+ and MYPT1SM–/– mice. *P < 0.05, **P < 0.01 and ***P < 0.001 compared to values at 0 s. The SEM bars may be smaller than the symbols for the means.
Based on this quantitative procedure, MYPT1 T696 and T853 sites were both found to be significantly phosphorylated under resting conditions in tissues from Mypt1SM+/+ mice (Fig. 4 B and C). Additionally, the extent of MYPT1 T696 phosphorylation was substantially greater (50%) than MYPT1 T853 phosphorylation (22%). Treatment of isolated ileal tissues with KCl or carbachol increased the extent of phosphorylation in T696 and T853 sites by ∼20%, which declined slowly by 300 s. These results show quantitatively that there is significant constitutive MYPT1 phosphorylation predicted to inhibit the activity of PP1cδ bound to MYPT1, and concentrations of KCl and carbachol that elicit maximal contractile responses result in increases in the extent of T696 or T853 phosphorylation.
The residual amount of MYPT1 remaining in tissues from MYPT1SM−/− mice was also phosphorylated constitutively and with KCl and carbachol treatments (Fig. 4 B and C). However, because this residual MYPT1 appears to be present in non‐smooth muscle cells (Fig. 2 D), it probably plays no role in regulating smooth muscle contraction.
MBS85 was phosphorylated in ileal tissues treated with KCl and carbachol
MBS85, the 85 kDa MYPT family member, has a phosphorylation site (T560) corresponding to T696 in MYPT1 but lacks the T853 phosphorylation site (Grassie et al. 2011). Similar to MYPT1 T696, there was significant constitutive phosphorylation of MBS T560 in resting tissues (Fig. 5). MBS85 basal phosphorylation both diminished and stimulated responses to KCl and carbachol in tissues from MYPT1SM−/− mice. This diminution may be a result of the increased expression of MBS85 (Fig. 1) leading to more substrate for an unidentified kinase activity. Although the extent of MBS85 phosphorylation was lower in tissues from MYPT1SM−/− mice, there may be a greater inhibitory effect because of the three‐fold greater expression of MBS85 protein. Thus, MBS phosphorylation (constitutive and agonist‐induced) may inhibit MLCP phosphatase activity in ileal tissues from MYPT1SM−/− and also MYPT1SM+/+ mice.
Figure 5. MBS85 phosphorylation responses to KCl and CCh are enhanced in ileal tissues from MYPT1‐deficient mice .
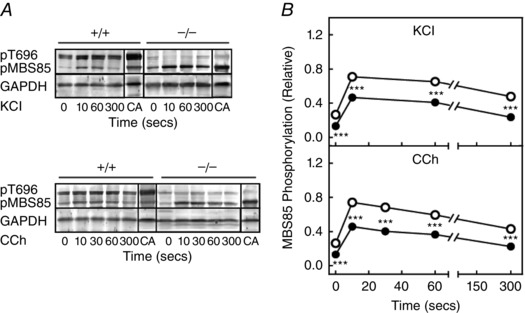
Representative blots for MBS85 phosphorylation (A) and quantification of MBS85 phosphorylation (B) in response to 90 mm KCl and 6 μm carbachol (CCh) in ileal tissues from MYPT1SM+/+ (open circles) and MYPT1SM–/− (solid circles) mice. MYPT1 T696 (pT696) and MBS85 T560 (pMBS85) phosphorylation are shown in the upper panel with both normalized to that obtained with calyculin A (CA), respectively, in MYPT1SM+/+ and MYPT1SM–/− ileal tissue with GAPDH as a loading control. Values are the mean ± SEM (n ≥ 8). **P < 0.0.01 and ***P < 0.001 compared to MYPT1SM+/+ strips at the same time. The SEM bars may be smaller than the symbols for the means.
Signalling mechanisms leading to phosphorylation of MLCK and CPI‐17 appear intact in MYPT1‐deficient smooth muscles
Under resting conditions, the extent of MLCK and CPI‐17 phosphorylation was low and both increased in response to treatment with KCl or carbachol in ileal tissues from both MYPT1SM−/− and MYPT1SM+/+ mice (Fig. 6). Phosphorylation of MLCK was enhanced, whereas CPI‐17 phosphorylation decreased in tissues from MYPT1SM−/− mice. The enhanced MLCK phosphorylation may be related to the decrease in PP1cδ that dephosphorylates phosphorylated MLCK (Nomura et al. 1992). Additionally, the phosphorylation of MLCK diminishes its ability to be activated by Ca2+/calmodulin and thus acts to dampen RLC phosphorylation (Tsai et al. 2012).
Figure 6. MLCK and CPI‐17 are phosphorylated in MYPT1‐deficient ileal tissues .
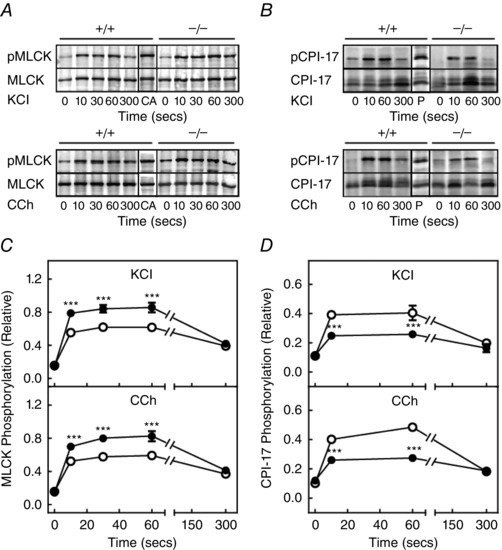
A, representative blots for MLCK phosphorylation and (C) its quantification are shown in response to 90 mm KCl (top) and 6 μm carbachol (CCh) (bottom) in ileal tissues from MYPT1SM+/+ (open circles) and MYPT1SM–/− (solid circles) mice. MLCK phosphorylation was normalized to the response obtained with calyculin A (CA) with GAPDH as a loading control. Values are the mean ± SEM (n ≥ 10). ***P < 0.001 compared to MYPT1SM+/+ strips at the same time. B, representative blots for CPI‐17 phosphorylation and (D) its quantification are shown in response to 90 mm KCl (top) and 6 μm CCh (bottom) ileum tissues from MYPT1SM+/+ (open circle) and MYPT1SM–/− (solid circle) mice. CPI‐17 phosphorylation normalized to the response obtained with PDBu. Values are the mean ± SEM (n ≥ 10). ***P < 0.001 compared to MYPT1SM+/+ strips at the same time. The SEM bars may be smaller than the circles for the means.
Effects of ROCK and PKC inhibitors on RLC phosphorylation and developed force responses to carbachol are attenuated in MYPT1‐deficient mice
Pre‐treatment of ileal smooth muscles from MYPT1SM+/+ mice with ROCK inhibitor H‐1152 or PKC inhibitor GF109203X decreased force responses, as well as RLC phosphorylation to carbachol (Fig. 7 A and B). However, there were no significant effects to either inhibitor with tissues from MYPT1SM−/− mice. Constitutive phosphorylation of MYPT1 T853 (but not T696) was inhibited with H‐1152 in tissues from both MYPT1SM+/+ and MYPT1SM−/− mice (Fig. 7 C). H‐1152 also inhibited MYPT1 T853 phosphorylation induced by carbachol (Fig. 7 D). The phosphorylation of residual MYPT1 in ileal tissues from MYPT1SM−/− mice would be predicted to have no effect on MLCP activity in smooth muscle cells as a result of its presence primarily in non‐smooth muscles cells in the tissue (Fig. 2). Hence, RLC phosphorylation and force responses were not inhibited in tissues from MYPT1SM−/− mice. Interestingly, the PKC inhibitor GF109203X attenuated CPI‐17 phosphorylation in smooth muscle tissues from both MYPT1SM+/+ and MYPT1SM−/− mice (Fig. 7 B), whereas it had no effect on force development (Fig. 7 A) or RLC phosphorylation (Fig. 7 B) in smooth muscle tissue from MYPT1SM−/− mice. Phosphorylated CPI‐17 may not be able to inhibit PP1cδ not bound to MYPT1 (Eto, 2009).
Figure 7. ROCK and PKC inhibitors affect force as well as RLC and CPI‐17 phosphorylation responses differently in ileal tissues from MYPT1SM+/+ and MYPT1SM − /− mice .
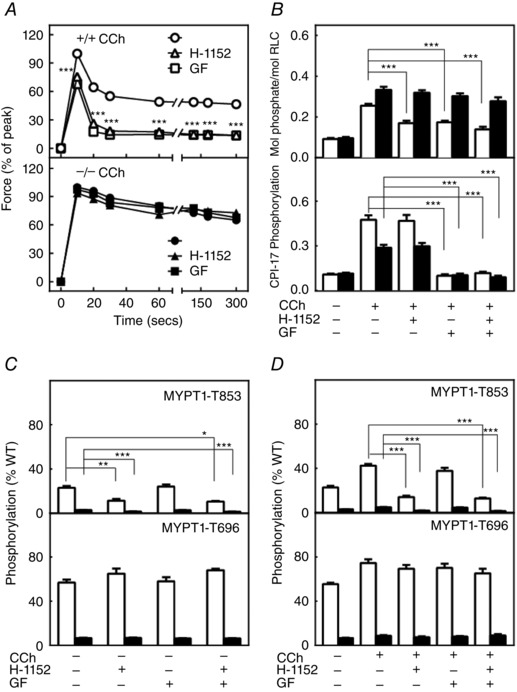
A, carbachol (CCh)‐induced force development responses are shown in the absence (circles) or presence of ROCK inhibitor H‐1152 (triangles) or PKC inhibitor GF109203X (squares) in ileal tissues from MYPT1SM+/+ (+/+, top) or MYPT1SM−/− (–/–, bottom) mice. Force values are expressed relative to the peak forces in the absence of inhibitors. Data are the mean ± SEM (n ≥ 10 measurements from different animals). ***P < 0.001 compared to no treatment. The SEM bars may be smaller than the circles for the means. B, CCh‐induced RLC (top) and CPI‐17 (bottom) phosphorylation at 30 s are shown following H‐1152 or GF109203X in ileal tissues from MYPT1SM+/+ (open bars) or MYPT1SM–/− (solid bars) mice. CPI‐17 phosphorylation was quantified relative to the results obtained with PDBu. Data are presented as the mean ± SEM (n ≥ 6 animals in each group). ***P < 0.001 compared to CCh responses at 30 s. C, resting and CCh‐induced (D) phosphorylation of MYPT1 T853 (upper) and T696 (lower) were analysed following ROCK H‐1152 and PKC GF109203X inhibitor treatments, respectively, for ileal tissues from MYPT1SM+/+ (open bars) and MYPT1SM–/− (solid bars) mice. MYPT1 phosphorylation was quantified relative to the results obtained with calyculin A as shown in Fig. 4 A and normalized to the amount of MYPT1 protein of wild‐type mice. Data are presented as the mean ± SEM (n ≥10 animals in each group). *P < 0.05 and ***P < 0.001 compared to resting values (C) or CCh simulation at 30 s (D) in the absence of inhibitors.
RLC phosphorylation and contractile responses to KCl and carbachol were not different in ileal smooth muscle from MYPT1 T853A knockin mice
The non‐phosphorylatable knockin mutation T853A in MYPT1 did not affect the amount of MYPT1 expression in adult ileal smooth muscle tissues in MYPT1T853A mice compared to tissues from MYPT1SM+/+ mice (Fig. 8 A). MYPT1 T853 was phosphorylated in MYPT1SM+/+ but not MYPT1T853A mice with carbachol treatment, demonstrating the expression of the T853A mutation. Similar amounts of expression were also found with RLC and CPI‐17 (data not shown). There were no differences in force development, RLC phosphorylation or CPI‐17 phosphorylation responses to KCl and carbachol between tissues from MYPT1SM+/+ and MYPT1T853A mice (Fig. 8 B and C). Neither the maximal, nor sustained force responses were affected by the knockin mutation. Additionally, we saw no changes in spontaneous force amplitudes in tissues from MYPT1T853A mice compared to tissues from MYPT1SM+/+ mice (data not shown). Thus, the knockin mutation T853A had no discernable effects on agonist‐induced RLC phosphorylation or contractile responses.
Figure 8. The knockin mutation MYPT1 T853A has no effect on force development and RLC phosphorylation in ileal tissues from MYPT1T853A knockin mice .
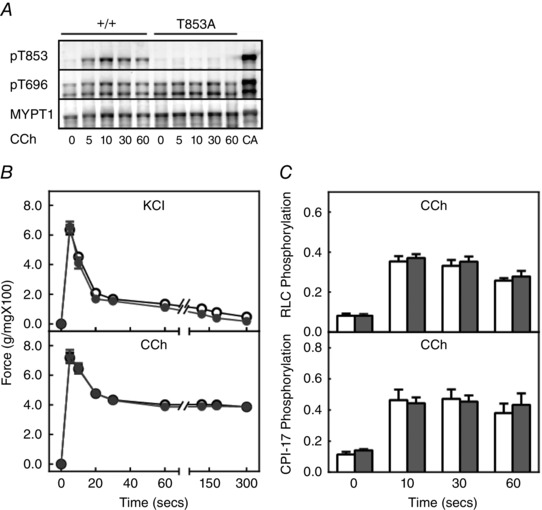
A, representative blots for MYPT1 phosphorylation in response to 6 μm carbachol (CCh) showing no MYPT1 T853 phosphorylation in tissues from homozygous MYPT1T853A knockin mice. B, force development responses in ileal strips from MYPT1SM+/+ (open circles) and MYPT1T853A (grey circles) are shown for 90 mm KCl (top) or 6 μm carbachol (CCh) (bottom) treatments. Data are the mean ± SEM (n ≥ 20). The SEM bars may be smaller than the symbols for the means. C, quantified RLC (top) and CPI‐17 phosphorylation (bottom) responses are shown for ileal strips from MYPT1SM+/+ (open bars) and MYPT1T853A (grey bars) mice to treatment with 6 μm CCh. CPI‐17 phosphorylation normalized to the response obtained with PDBu. Data are the mean ± SEM (n ≥10 animals).
Electrical field stimulation elicits similar responses in MYPT1‐deficient, MYPT1 T853A mutant and control tissues
Based on the modest changes in contractile and biochemical properties for MYPT1‐deficient tissues, and no changes in tissues containing the knockin mutation MYPT1 T853A to agonist‐induced contractions, we investigated the responses to EFS used to release ACh from parasympathetic nerves intrinsic in isolated tissues (Mang et al. 2002). The maximal force responses at different frequencies were similar in isolated ileal tissues from all three mouse lines, including MYPT1SM+/+, MYPT1SM−/− and MYPT1T853A mice (Fig. 9 B and D). The maximal forces [60 mg (mg wet weight tissue)–1] obtained with EFS are similar to the maximal forces elicited with KCl and carbachol (Fig. 3). The rate of decline in the sustained force was less in MYPT1SM−/− mice. However, there was no attenuation of the force decline in the sustained phase in tissues from MYPT1 T853A mice.
Figure 9. Contractile responses to EFS in MYPT1‐deficient ileum .
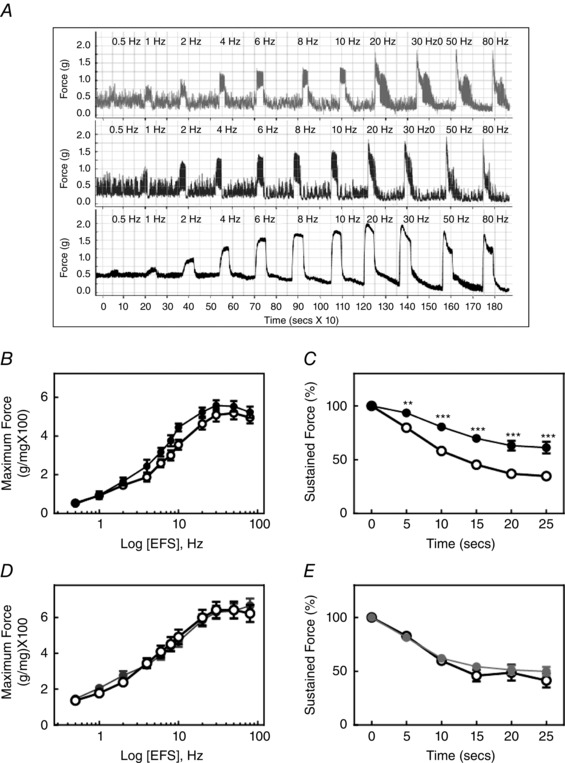
A, representative force responses are shown for indicated EFS frequencies for ileal tissues from MYPT1SM+/+ (upper trace), MYPT1T853A (middle trace) and MYPT1SM–/− (lower trace) mice. B, quantified maximum force‐frequency responses are shown for ileum from MYPT1SM+/+ (open circles) and MYPT1SM–/− (solid circles) mice. C, quantified forces following maximal responses with continuous 30 Hz EFS in ileal strips from MYPT1SM+/+ (open circles) and MYPT1SM–/− (solid circles) mice. D, quantified maximum force‐frequency responses in ileal strips from MYPT1SM+/+ (open circles) and MYPT1T853A (solid circles) mice. E, quantified forces following maximal responses with continuous 30 Hz EFS in ileal strips from MYPT1SM+/+ (open circles) and MYPT1T853A (solid circles) mice. Force measurements were normalized to the maximal developed forces marked as 0 s in (C) and (E). Data are the mean ± SEM (n ≥12 animals). **P < 0.01 and ***P < 0.001 compared to MYPT1SM+/+ at the same time. The SEM bars may be smaller than the symbols for the means.
EFS also elicited similar phosphorylation responses in RLC and MLCK in ileal tissues from MYPT1SM+/+ and MYPT1SM−/− mice (Fig. 10 A). Interestingly, there was no induced phosphorylation of MYPT1 T696 and T853 with EFS (Fig. 10 B), although robust responses were obtained with force development (Fig. 9) and RLC phosphorylation (Fig 10 A). These results with EFS are similar to those obtained in the urinary bladder (Tsai et al. 2012) and gastric fundus (Bhetwal et al. 2013) smooth muscles. Thus, physiological smooth muscle contraction responses do not appear to recruit Ca2+‐sensitization mechanisms by increasing phosphorylation of the constitutively phosphorylated MYPT1 T696 or T853.
Figure 10. MYPT1 T853 and T696 are not phosphorylated in response to EFS in ileal tissues from MYPT1SM+/+ or MYPT1SM − /− mice .
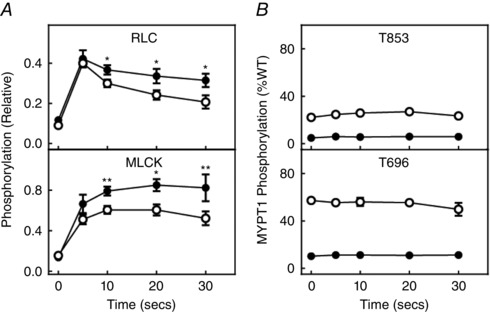
A, RLC and MLCK phosphorylation responses to 30 Hz EFS. B, MYPT1 T853 and T696 phosphorylation responses to 30 Hz EFS. Ileal tissues were from MYPT1SM+/+ (open circles) and MYPT1SM–/− (solid circles) mice. Data are the mean ± SEM (n ≥ 4 from different ileum). **P < 0.0.01 compared to MYPT1SM+/+ at the same time. All RLC and MLCK phosphorylation values were significantly greater than the values under resting conditions. Phosphorylation values for MYPT1 T696 or T853 were not significantly changed with EFS. The SEM bars may be smaller than the symbols for the means.
Discussion
Defining the potential signalling pathways involved in the regulation of the actomyosin system necessary for smooth muscle contraction has involved a variety of experimental approaches, including biochemical, biophysical, cell biological, pharmacological and physiological approaches, leading to consensus on a general scheme involving the potential regulation of MLCK and MLCP activities (Kamm & Stull, 1985 a; Hartshorne, 1987; Hai & Murphy, 1989; Kamm & Stull, 2001; Somlyo & Somlyo, 2003; Kim et al. 2008; Puetz et al. 2009; Cole & Welsh, 2011). A significant number of proposed physiological signalling modules have been simplified with subsequent genetic approaches for specific proteins, as well as with genetically modified mice. For example, conditional knockout of MLCK specifically in smooth muscle cells of adult mice showed that it was the physiologically important kinase for RLC phosphorylation in several different types of smooth muscles (He et al. 2008; Zhang et al. 2010; He et al. 2011; Gao et al. 2013).
Recently, genetic approaches have also been used to study MLCP with conditional knockouts of MYPT1 in smooth muscle cells of mice. The original study reported that MYPT1 knockout mice were viable, had a normal body size and reached adulthood (He et al. 2013), and similar results were found in the present study. Curiously, there was also a decrease in c‐Kit staining, indicating a disruption of the interstitial cells of Cajal in the myenteric plexus region, as well as less rhythmic spontaneous contractile activity. We also found decreased c‐Kit staining in ileal tissue from MYPT1SM−/− mice (data not shown), as well as more asynchronous spontaneous contractions. At necropsy, the whole digestive tract appeared normal, including the small intestine. These studies raise questions about why MYPT1 knockout does not cause significant impairment of ileal smooth muscle contraction. PP1cδ bound to MYPT1 (and presumably MBS85) may be inhibited substantially by an intramolecular phosphorylation mechanism (Khromov et al. 2009) with 77% constitutive phosphorylation of MYPT at T696 and 853, and 28% constitutive phosphorylation of MBS85, which is 15% of that of MYPT1 (present study). Thus, most of the bound PP1cδ may be inhibited via phosphorylation, representing a constitutive Ca2+‐sensitization mechanism. With the knockout of MYPT1, the amount of PP1cδ decreased by 60% in ileal tissues in the present study. However, there was an increase in MBS85 expression to 50% of the amount of MYPT1 in wild‐type tissues in ileal smooth muscle but not bladder smooth muscle (Tsai et al. 2014). In MYPT‐deficient ileal smooth muscle cells, some of the PP1cδ could be bound to MBS85. However, we found that half of the PP1cδ was not bound to the contractile protein system with myosin or MBS85 in wild‐type or MYPT1 deficient tissues (Tsai et al. 2014). Furthermore, soluble PP1cδ was capable of dephosphorylating heavy meromyosin (Tsai et al. 2014), a physiological substrate for MLCP (Hartshorne, 1987; Hartshorne et al. 1998; Somlyo & Somlyo, 2003). Thus, in wild‐type tissues, the constitutive phosphorylation of MYPT1 and MBS85 in ileal tissues may inhibit most (∼75–80%) of the bound PP1cδ, with half of the MLCP phosphatase activity contributed by soluble PP1cδ. With MYPT1 ablation, the reduction in PP1cδ protein may occur because it is not in a stable complex with MYPT1 (Scotto‐Lavino et al. 2010; Tsai et al. 2014). Most of the remaining PP1cδ would appear to be active because (1) the half bound to MBS85 would be less inhibited with constitutive phosphorylation of MBS85 and (2) the remaining soluble half of PP1cδ is active towards phosphorylated RLC. Although these considerations describe compensatory mechanisms associated with MYPT1 knockout, a key perspective is the potential importance of the soluble PP1cδ as part of the MLCP activity in smooth muscle cells, including ileal smooth muscle cells. This possibility needs investigation.
Interestingly, MYPT1 T696 and MBS85 T560 showed significant constitutive phosphorylation, which increased with carbachol and KCl treatments in ileal smooth muscle. High KCl concentrations cause membrane depolarization with activation of voltage‐gated Ca2+ channels that increase [Ca2+]i for MLCK activation by Ca2+/calmodulin. In addition, KCl also increases Ca2+ sensitization by activation of RhoA and ROCK from the generation of arachidonic acid and lysophospholipids by phospholipase A2 leading to phosphorylation of MYPT1 (Sakurada et al. 2003; Ratz et al. 2005; Urena & Lopez‐Barneo, 2012). The contribution of high K+‐inducing RhoA and ROCK activation to RLC phosphorylation and contraction responses varies in different types of smooth muscles, including arteries (coronary, femoral and renal), aorta, urinary bladder and intestinal smooth muscles (Sakurada et al. 2003; Ratz et al. 2005; Mori et al. 2011; Urena & Lopez‐Barneo, 2012; Gao et al. 2013; Tsai et al. 2014).
In previous work, bladder smooth muscle from a mouse line with alanine substitution for MYPT1 T696 showed a significant reduction of force with reduced RLC phosphorylation (Chen et al. 2015). The contractile responses of T696A mutant smooth muscle were also independent of ROCK activation. These results highlight the importance of constitutive and agonist‐induced MYPT1 T696 phosphorylation as a primary mechanism contributing to inhibition of MLCP activity and enhancement of RLC phosphorylation and contractile force. However, there was no induced phosphorylation with EFS in bladder smooth muscle (Tsai et al. 2012). Remarkably, it is not yet clear what protein kinase accounts for the constitutive phosphorylation in different types of smooth muscle cells. There is a consensus that ROCK and PKC are not the responsible kinases based on pharmacological results (Wang et al. 2009; Grassie et al. 2011; Tsai et al. 2014; Chen et al. 2015). Considering the apparent importance of MYPT1 T696 (and presumably MBS85 T560 phosphorylation), additional investigations are needed to identify the physiologically involved protein kinase(s).
The role of MYPT1 T853 phosphorylation has proved controversial. MYPT1 T853 phosphorylation may not inhibit MLCP activity (Feng et al. 1999), although other studies concluded that MYPT1 T853 phosphorylation inhibited MLCP activity equivalently to T696 (Muranyi et al. 2005). Instead of direct inhibition of PP1cδ activity through MYPT1 T853 phosphorylation, its phosphorylation may trigger dissociation of PP1 from MYPT1 (Guillermo et al. 2002). Khasnis et al. (2014) recently co‐expressed PP1cδ and MYPT1 in mammalian cells to reconstitute MLCP retaining characteristics of the native enzyme. MYPT1 T696 phosphorylation inhibited MLCP activity, whereas T853 phosphorylation did not. Our recent results showed no changes in RLC phosphorylation and force responses in bladder smooth muscle tissues containing the knockin MYPT1 T853A mutation (Chen et al. 2015). We found similar results in ileal smooth muscle tissues. Thus, agonist‐induced MYPT1 T853 phosphorylation does not appear to play a role in agonist‐induced Ca2+‐sensitization in bladder and ileal smooth muscles.
We extended these pharmacological studies on isolated tissues with EFS aiming to investigate the signalling modules involved with activation of smooth muscle cells by the release of neurotransmitter. Studies using EFS to initiate cholinergic neurotransmission in mouse bladder smooth muscle tissues found that phosphorylation of constitutively phosphorylated MYPT1 T696 and T853 was not increased, in contrast to cholinergic agonist treatment (Tsai et al. 2012). Similar results were subsequently reported for gastric fundus (Bhetwal et al. 2013) where the data suggested that the accessibility of nerve‐released ACh may be limited to a restricted population of receptors, possibly expressed by intramuscular interstitial cells of Cajal. ACh from neurons does not appear to be available to smooth muscle receptors that are linked to ROCK activation and MYPT1 phosphorylation, perhaps as a result of the unique anatomical organization of the neuroeffector junction in gastrointestinal smooth muscles, which involves information transfer between motor neurons, intramuscular interstitial cells of Cajal and smooth muscle cells. We obtained similar results with EFS in ileal tissues showing that EFS did not induce additional phosphorylation over constitutive phosphorylation of MYPT1. It is important to note that the amount of force development with EFS in these studies was similar to that obtained with cholinergic agonist‐induced force development. Thus, bath application of a high concentration of contractile agonists to urinary bladder, gastric fundus and ileal smooth muscles does not mimic the physiological responses to cholinergic neurotransmission. It is not yet clear whether these neurophysiological responses are restricted to phasic smooth muscles vs. tonic smooth muscles. However, it should be noted that the myogenic response in cerebral and skeletal muscle resistance arteries appears to include changes in MYTP1 T853 phosphorylation as part of the physiological signalling module affecting RLC phosphorylation (Cole & Welsh, 2011). It would be interesting to investigate these responses in MYPT1 knockout and MYPT1 T853A mice, which do not show any apparent vascular derangements.
In summary, the results of the present study show that the conditional knockout of MYPT1 in smooth muscle tissues of adult mice results in a reduced amount of PP1cδ without markedly affecting RLC dephosphorylation and relaxation of pre‐contracted ileal smooth muscle strips. The knockin mutation MYPT1 T853A had no significant effect on RLC phosphorylation and force development/relaxation responses. Finally, EFS increased RLC phosphorylation and force development in ileal tissues from wild‐type mice without an increase in MYPT1 phosphorylation. Thus, physiological RLC phosphorylation and force development in ileal smooth muscle appear to be dependent on MLCK and MLCP activities without changes in MYPT1 phosphorylation.
Additional information
Competing interests
The authors declare that they have no competing interests.
Author contributions
NG, ANC, WH, C‐PC, MZ, KEK and JTS conceived the experimental approaches. NG, ANC, KEK and JTS designed the experiments. NG and ANC performed the experiments. NG, ANC, MZ, KEK and JTS analysed data and wrote the manuscript. All authors have approved the final version of the manuscript and agree to be accountable for all aspects of the work. All persons designated as authors qualify for authorship, and all those who qualify for authorship are listed.
Funding
This work was supported by National Institutes of Health Grants R01 HL112778 and P01 HL110869, Moss Heart Fund, and the Fouad A. and Val Imm Bashour Distinguished Chair in Physiology, National Basic Research Program of China Grant 2009CB942602 and National Natural Science Funding of China Grant 31272311.
Acknowledgements
We appreciate receipt of the SMMHC‐CreERT2 transgenic mice from Stefan Offermanns, Goethe University, Frankfurt, Germany, as well as biochemical reagents for quantifying MYPT T696 and T853 phosphorylation from Masumi Eto (Thomas Jefferson University, Philadelphia, PA, USA).
References
- Bhetwal BP, Sanders KM, An C, Trappanese DM, Moreland RS & Perrino BA (2013). Ca2+ sensitization pathways accessed by cholinergic neurotransmission in the murine gastric fundus. J Physiol 591, 2971–2986. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chen CP, Chen X, Qiao YN, Wang P, He WQ, Zhang CH, Zhao W, Gao YQ, Chen C, Tao T, Sun J, Wang Y, Gao N, Kamm KE, Stull JT & Zhu MS (2015). In vivo roles for myosin phosphatase targeting subunit‐1 phosphorylation sites T694 and T852 in bladder smooth muscle contraction. J Physiol 593, 681–700. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cole WC & Welsh DG (2011). Role of myosin light chain kinase and myosin light chain phosphatase in the resistance arterial myogenic response to intravascular pressure. Arch Biochem Biophys 510, 160–173. [DOI] [PubMed] [Google Scholar]
- Dimopoulos GJ, Semba S, Kitazawa K, Eto M & Kitazawa T (2007). Ca2+‐dependent rapid Ca2+ sensitization of contraction in arterial smooth muscle. Circ Res 100, 121–129. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ding HL, Ryder JW, Stull JT & Kamm KE (2009). Signalling processes for initiating smooth muscle contraction upon neural stimulation. J Biol Chem 284, 15541–15548. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Eto M (2009). Regulation of cellular protein phosphatase‐1 (PP1) by phosphorylation of the CPI‐17 family, C‐kinase‐activated PP1 inhibitors. J Biol Chem 284, 35273–35277. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Feng J, Ito M, Ichikawa K, Isaka N, Nishikawa M, Hartshorne DJ & Nakano T (1999). Inhibitory phosphorylation site for Rho‐associated kinase on smooth muscle myosin phosphatase. J Biol Chem 274, 37385–37390. [DOI] [PubMed] [Google Scholar]
- Gao N, Huang J, He W, Zhu M, Kamm KE & Stull JT (2013). Signalling through myosin light chain kinase in smooth muscles. J Biol Chem 288, 7596–7605. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Grassie ME, Moffat LD, Walsh MP & MacDonald JA (2011). The myosin phosphatase targeting protein (MYPT) family: a regulated mechanism for achieving substrate specificity of the catalytic subunit of protein phosphatase type 1delta. Arch Biochem Biophys 510, 147–159. [DOI] [PubMed] [Google Scholar]
- Guillermo V, Chris A, Nick M, Sheelagh F & Philip C (2002). Phosphorylation of the regulatory subunit of smooth muscle protein phosphatase 1 M at Thr850 induces its dissociation from myosin. FEBS Lett 527, 101–104. [DOI] [PubMed] [Google Scholar]
- Hai CM & Murphy RA (1989). Ca2+, crossbridge phosphorylation, and contraction. Annu Rev Physiol 51, 285–298. [DOI] [PubMed] [Google Scholar]
- Hartshorne DA (1987). Biochemistry of the contractile process in smooth muscle In Physiology of the Gastrointestinal Tract, 2nd edn, pp. 423–482. Raven Press, New York, NY. [Google Scholar]
- Hartshorne DJ, Ito M & Erdodi F (1998). Myosin light chain phosphatase: subunit coMopsition, interactions and regulation. J Muscle Res Cell Motil 19, 325–341. [DOI] [PubMed] [Google Scholar]
- Hartshorne DJ, Ito M & Erdodi F (2004). Role of protein phosphatase type 1 in contractile functions: myosin phosphatase. J Biol Chem 279, 37211–37214. [DOI] [PubMed] [Google Scholar]
- He WQ, Peng YJ, Zhang WC, Lv N, Tang J, Chen C, Zhang CH, Gao S, Chen HQ, Zhi G, Feil R, Kamm KE, Stull JT, Gao X & Zhu MS (2008). Myosin light chain kinase is central to smooth muscle contraction and required for gastrointestinal motility in mice. Gastroenterology 135, 610–620. [DOI] [PMC free article] [PubMed] [Google Scholar]
- He WQ, Qiao YN, Peng YJ, Zha JM, Zhang CH, Chen C, Chen CP, Wang P, Yang X, Li CJ, Kamm KE, Stull JT & Zhu MS (2013). Altered contractile phenotypes of intestinal smooth muscle in mice deficient in myosin phosphatase target subunit 1. Gastroenterology 144, 1456–1465, 1465 e1451‐1455. [DOI] [PMC free article] [PubMed] [Google Scholar]
- He WQ, Qiao YN, Zhang CH, Peng YJ, Chen C, Wang P, Gao YQ, Chen X, Tao T, Su XH, Li CJ, Kamm KE, Stull JT & Zhu MS (2011). Role of myosin light chain kinase in regulation of basal blood pressure and maintenance of salt‐induced hypertension. Am J Physiol Heart Circ Physiol 301, H584–H591. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hirano K, Derkach DN, Hirano M, Nishimura J & Kanaide H (2003). Protein kinase network in the regulation of phosphorylation and dephosphorylation of smooth muscle myosin light chain. Mol Cell Biochem 248, 105–114. [DOI] [PubMed] [Google Scholar]
- Kamm KE, Hsu LC, Kubota Y & Stull JT (1989). Phosphorylation of smooth muscle myosin heavy and light chains. Effects of phorbol dibutyrate and agonists. J Biol Chem 264, 21223–21229. [PubMed] [Google Scholar]
- Kamm KE & Stull JT (1985. a). The function of myosin and myosin light chain kinase phosphorylation in smooth muscle. Annu Rev Pharmacol Toxicol 25, 593–620. [DOI] [PubMed] [Google Scholar]
- Kamm KE & Stull JT (1985. b). Myosin phosphorylation, force, and maximal shortening velocity in neurally stimulated tracheal smooth muscle. Am J Physiol Cell Physiol, C238–C247. [DOI] [PubMed] [Google Scholar]
- Kamm KE & Stull JT (2001). Dedicated myosin light chain kinases with diverse cellular functions. J Biol Chem 276, 4527–4530. [DOI] [PubMed] [Google Scholar]
- Khasnis M, Nakatomi A, Gumpper K & Eto M (2014). Reconstituted human myosin light chain phosphatase reveals distinct roles of two inhibitory phosphorylation sites of the regulatory subunit, MYPT1. Biochemistry 53, 2701–2709. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Khromov A, Choudhury N, Stevenson AS, Somlyo AV & Eto M (2009). Phosphorylation‐dependent autoinhibition of myosin light chain phosphatase accounts for Ca2+ sensitization force of smooth muscle contraction. J Biol Chem 284, 21569–21579. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kim HR, Appel S, Vetterkind S, Gangopadhyay SS & Morgan KG (2008). Smooth muscle signalling pathways in health and disease. J Cell Mol Med 12, 2165–2180. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kitazawa T (2010). G protein‐mediated Ca2+‐sensitization of CPI‐17 phosphorylation in arterial smooth muscle. Biochem Biophys Res Comm 401, 75–78. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kitazawa T, Eto M, Woodsome TP & Brautigan DL (2000). Agonists trigger G protein‐mediated activation of the CPI‐17 inhibitor phosphoprotein of myosin light chain phosphatase to enhance vascular smooth muscle contractility. J Biol Chem 275, 9897–9900. [DOI] [PubMed] [Google Scholar]
- Kitazawa T & Kitazawa K (2012). Size‐dependent heterogeneity of contractile Ca2+ sensitization in rat arterial smooth muscle. J Physiol 590, 5401–5423. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mang CF, Truempler S, Erbelding D & Kilbinger H (2002). Modulation by NO of acetylcholine release in the ileum of wild‐type and NOS gene knockout mice. Am J Physiol Gastrointest Liver Physiol 283, G1132–G1138. [DOI] [PubMed] [Google Scholar]
- Matsumura F & Hartshorne DJ (2008). Myosin phosphatase target subunit: many roles in cell function. Biochem Biophys Res Commun 369, 149–156. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mizuno Y, Isotani E, Huang J, Ding H, Stull JT & Kamm KE (2008). Myosin light chain kinase activation and calcium sensitization in smooth muscle in vivo . Am J Physiol Cell Physiol 295, C358–C364. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mori D, Hori M, Murata T, Ohama T, Kishi H, Kobayashi S & Ozaki H (2011). Synchronous phosphorylation of CPI‐17 and MYPT1 is essential for inducing Ca(2+) sensitization in intestinal smooth muscle. Neurogastroenterol Motil 23, 1111–1122. [DOI] [PubMed] [Google Scholar]
- Muranyi A, Derkach D, Erdodi F, Kiss A, Ito M & Hartshorne DJ (2005). Phosphorylation of Thr695 and Thr850 on the myosin phosphatase target subunit: inhibitory effects and occurrence in A7r5 cells. FEBS Lett 579, 6611–6615. [DOI] [PubMed] [Google Scholar]
- Murthy KS (2006). Signalling for contraction and relaxation in smooth muscle of the gut. Ann Rev Physiol 68, 345–374. [DOI] [PubMed] [Google Scholar]
- Nomura M, Stull JT, Kamm KE & Mumby MC (1992). Site‐specific dephosphorylation of smooth muscle myosin light chain kinase by protein phosphatases 1 and 2A. Biochemistry 31, 11915–11920. [DOI] [PubMed] [Google Scholar]
- Puetz S, Lubomirov LT & Pfitzer G (2009). Regulation of smooth muscle contraction by small GTPases. Physiology 24, 342–356. [DOI] [PubMed] [Google Scholar]
- Qiao YN, He WQ, Chen CP, Zhang CH, Zhao W, Wang P, Zhang L, Wu YZ, Yang X, Peng YJ, Gao JM, Kamm KE, Stull JT & Zhu MS (2014). Myosin phosphatase target subunit 1 (MYPT1) regulates the contraction and relaxation of vascular smooth muscle and maintains blood pressure. J Biol Chem 289, 22512–22523. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ratz PH, Berg KM, Urban NH & Miner AS (2005). Regulation of smooth muscle calcium sensitivity: KCl as a calcium‐sensitizing stimulus. Am J Physiol Cell Physiol 288, C769–C783. [DOI] [PubMed] [Google Scholar]
- Sakurada S, Takuwa N, Sugimoto N, Wang Y, Seto M, Sasaki Y & Takuwa Y (2003). Ca2+‐dependent activation of Rho and Rho kinase in membrane depolarization‐induced and receptor stimulation‐induced vascular smooth muscle contraction. Circ Res 93, 548–556. [DOI] [PubMed] [Google Scholar]
- Scotto‐Lavino E, Garcia‐Diaz M, Du G & Frohman MA (2010). Basis for the isoform‐specific interaction of myosin phosphatase subunits protein phosphatase 1c beta and myosin phosphatase targeting subunit 1. J Biol Chem 285, 6419–6424. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Somlyo AP & Somlyo AV (2000). Signal transduction by G‐proteins, rho‐kinase and protein phosphatase to smooth muscle and non‐muscle myosin II. J Physiol 522, 177–185. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Somlyo AP & Somlyo AV (2003). Ca2+‐sensitivity of smooth and non‐muscle myosin II: modulation by G proteins, kinases and myosin phosphatase. Physiol Rev 83, 1325–1358. [DOI] [PubMed] [Google Scholar]
- Stull JT, Hsu LC, Tansey MG & Kamm KE (1990). Myosin light chain kinase phosphorylation in tracheal smooth muscle. J Biol Chem 265, 16683–16690. [PubMed] [Google Scholar]
- Tsai MH, Chang AN, Huang J, He W, Sweeney HL, Zhu M, Kamm KE & Stull JT (2014). Constitutive phosphorylation of myosin phosphatase targeting subunit‐1 in smooth muscle. J Physiol 592, 3031–3051. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tsai MH, Kamm KE & Stull JT (2012). Signalling to contractile proteins by muscarinic and purinergic pathways in neurally stimulated bladder smooth muscle. J Physiol 590, 5107–5121. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Urena J & Lopez‐Barneo J (2012). Metabotropic regulation of RhoA/Rho‐associated kinase by L‐type Ca2+ channels. Trends Cardiovas Med 22, 155–160. [DOI] [PubMed] [Google Scholar]
- Wang L, Guo DC, Cao J, Gong L, Kamm KE, Regalado E, Li L, Shete S, He WQ, Zhu MS, Offermanns S, Gilchrist D, Elefteriades J, Stull JT & Milewicz DM (2010). Mutations in myosin light chain kinase cause familial aortic dissections. Am J Hum Genet 87, 701–707. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wang T, Kendig DM, Smolock EM & Moreland RS (2009). Carbachol‐induced rabbit bladder smooth muscle contraction: roles of protein kinase C and Rho kinase. Am J Physiol Renal Physiol 297, F1534–F1542. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wirth A, Benyo Z, Lukasova M, Leutgeb B, Wettschureck N, Gorbey S, Orsy P, Horvath B, Maser‐Gluth C, Greiner E, Lemmer B, Schutz G, Gutkind S & Offermanns S (2008). G12‐G13‐LARG‐mediated signalling in vascular smooth muscle is required for salt‐induced hypertension. Nat Med 14, 64–68. [DOI] [PubMed] [Google Scholar]
- Wu Z, Yang L, Cai L, Zhang M, Cheng X, Yang X & Xu J (2007). Detection of epithelial to mesenchymal transition in airways of a bleomycin induced pulmonary fibrosis model derived from an alpha‐smooth muscle actin‐Cre transgenic mouse. Respir Res 8, 1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhang WC, Peng YJ, Zhang GS, He WQ, Qiao YN, Dong YY, Gao YQ, Chen C, Zhang CH, Li W, Shen HH, Ning W, Kamm KE, Stull JT, Gao X & Zhu MS (2010). Myosin light chain kinase is necessary for tonic airway smooth muscle contraction. J Biol Chem 285, 5522–5531. [DOI] [PMC free article] [PubMed] [Google Scholar]