

Abstract
Drug nanocrystals comprise unique drug delivery platforms playing a significantly important and distinctive role in drug delivery and as such, the industry and academia are spending a lot of their time and money in developing the nanocrystal products. The current research works in this field depict a vivid shift from lab scale optimization studies to scale up focused studies. In this emerging scenario of nanocrystal technology, a review on some exemplary and progressing research studies with either scalability as their objective or upscaling as their future scope may smoothen the future upscaling attempts in this field. Hence, this paper reviews the efforts of such research works as case studies since an analysis of such research studies may input certain beneficial knowledge to carry out more scale up based research works on nanocrystals.
Keywords: Nanocrystals, Upscaling, Smartcrystals, Experimental design, Bridging, Stabilizers
1. Introduction
Enhanced solubility and dissolution, improved bioavailability and absorption, elimination of food effects, safe dose escalation, enhanced safety, efficacy and tolerability profiles are the inherited advantages of nanoparticles due to their size and surface features. Drug nanocrystals (NCs) are the nanoparticles which offer an additional advantage of 100% drug loading since they are encapsulating-carrier free nanoparticles. An NC formulation contains drug and one/more stabilizers dispersed in aqueous or non-aqueous media. Stabilizers could be one or more generally regarded as safe excipients (surfactants or buffers, salts or sugars). The liquid dispersion NCs could be further post processed into solid or sterile injectable dosage forms (Merisko-Liversidge and Liversidge, 2008). The therapeutic applications of NC products have been identified in oral (Hanafy et al., 2007, Kayser et al., 2003, Mauludin et al., 2009), parenteral (Ganta et al., 2009, Rabinow et al., 2007, Gao et al., 2008a, Gao et al., 2008b), ocular (Kassem et al., 2007, Ali et al., 2011), dermal (Shaal et al., 2011, Mitri et al., 2011, Mishra et al., 2009), pulmonary (El-Gendy et al., 2011, Jacobs and Müller, 2002, Yang et al., 2010) and targeted drug delivery (Kayser, 2001, Muller and Jacobs, 2002). NCs, further offer flexibility of upscaling and downscaling which could be of great value whenever alterations with respect to unit operation functions or formulation are desired during scaling up process (Eerdenbrugh et al., 2009). The formulation simplicity and production scaling flexibility along with their intrinsic small particle size and large surface area make NCs stand a way unique not just among the pharmaceuticals but also among other nanoparticles.
There are already six licensed and regulatory approved NC products launched in the market. Products like Semapimod® (guanylhydrazone), Paxceed® (paclitaxel), Theralux® (thymectacin) and Nucryst® (silver) are currently under clinical phases and there are many more products under preclinical stages (Gasper, 2010). Each of the commercialized NC products represents a rational formulation design. Rapamune® was developed as a tablet dosage form to overcome the unpalatable taste and restricted cold storage conditions of the earlier formulation of sirolimus (rapamycin). Drug absorption being effected by food uptake was the disadvantage encountered by the then existing formulation of aprepitant. Emend® was developed using nanosuspension of aprepitant and was formulated as a spray coated solid capsule dosage form which exhibited enhanced bioavailability due to reduced fast and fed state variations. TriCor® and Triglide® are tablet formulations of fenofibrate designed to improve the bioavailability and to overcome the fast and fed state dependent absorption variations associated with other formulations of this drug. Megace ES® is a liquid dispersion dosage form designed to improve dissolution, and bioavailability of megestrol acetate and thereby provides reduced dosing volume compared to other dosage forms of the drug. Invega Sustenna® was developed as a once monthly extended release sterile injectable liquid dispersion dosage form of paliperidone palmitate (intramuscular suspension) available in prefilled syringes and it stands unique for being available at variable dose strengths with a two year shelf life period. The patient population to whom paliperidone palmitate (antipsychotic) is indicated pose compliance problems to a great extent and the NC product being a once a month administrable medicine scores for its patient friendly therapy (Merisko-Liversidge and Liversidge, 2011, Shen and Wu, 2007, Wu et al., 2004, Eerdenbrugh et al., 2008, Deschamps et al., 2009). On account of such rational formulation development, an NC is considered as a new drug product by Food and Drug Administration (FDA) and not as a “generic” to any other approved product since its pharmacokinetic profile is not bioequivalent to any other solubilized form of the same drug, not even to the drug’s own micronized form, administered at the same dosage. Therefore, an NC could be patented as “new drug” which offers a product line extension for the already existing drug formulations and could serve as a new and beneficial dosage form (Singare et al., 2010).
2. Preparation and characterization of drug nanocrystals
Before delving into the discussions on scale up based research works, a snapshot of typical manufacturing methods available for the production of NCs has been provided. The preparation techniques for NCs could be mainly classified into three categories namely top down, bottom up, and combination methods. A detailed description of the classified methods as well as of the possible sub classifications under each of the classes has already been elucidated by various authors (Patravale et al., 2004, Kocbek et al., 2006, Zhang et al., 2011, Chan and Kwok, 2011). In short, the top down methods are physicomechanical processes mainly involving crushing or attrition principles (fragmentation) while bottom up methods are physicochemical processes involving the principles of atomic or molecular level self organization (amalgamation) as demonstrated in Fig. 1. The top down methods mainly involve milling or homogenization while the bottom up methods are primarily based on the principle of precipitation. The combination approaches involve bottom up plus top down method combinations.
Figure 1.

Production of drug nanocrystals.
Wet ball milling comminutes material loaded into milling chamber with an agitator (milling media). The milling material, the drug to be nanosized is normally provided as a treated (micronized) or untreated solid dispersed in a liquid medium (usually water) with the aid of surfactants as stabilizers. The comminution principle involved is the mechanical attrition and shear that arises due to collision between milling media and drug particles or between two drug particles or also between a drug particle and the walls of the milling chamber. The milling media are small beads or pearls made of ceramic (e.g., yttrium stabilized zirconium dioxide) or highly cross-linked polystyrene resin or stainless steel or glass having different sizes (0.3 mm or higher). However, the first two ensure minimal contamination to the product. The size reduction effectiveness could be further determined by the concentration of drug and surfactant, viscosity of the dispersion medium, temperature conditions and by the initial particle size and hardness of the drug (Niwa et al., 2011, Möschwitzer, 2013, Gao et al., 2008a, Gao et al., 2008b, Merisko-Liversidge and Liversidge, 2011).
HPH is another top down process where in the particle size reduction is brought about by shear forces, cavitation forces and particle collision aided by high pressure conditions. It is of two types namely the microfluidization and piston gap homogenization. Microfluidization is also called as air-jet milling or jet stream homogenization wherein the particles are fragmented in a high pressure air jet induced by collision of two fluid streams. Piston-gap homogenization employs high pressure to force a liquid suspension through a gap or narrow channel inside a pipe. If the medium is aqueous, bubbles are formed inside the gap due to reduced static pressure in the gap region which later collapse upon exiting the narrow gap. The break-up of particles is achieved by the consequently generated cavitation energy. On the other hand if the medium is oil or a non aqueous solvent, the particle comminution is facilitated by the high shear and collision through the gap (Shegokar and Müller, 2010, Müller et al., 2001, Keck et al., 2008, Keck and Müller, 2006).
All bottom up approaches employ two basic principles namely precipitation and evaporation. Accordingly there are numerous variations available which incorporate either of the two principles or a combination of both. ‘Cryogenic solvent evaporation’ is a bottom up method which involves spraying of drug solution into cryogenic liquids using ‘spray freezing into liquid’ technology. Here the drug solution droplets are frozen upon contact with cryogenic liquid (liquid nitrogen) and the organic solvent is removed by lyophilization. Precipitation when performed in conjugation with centrifugation is termed ‘high gravity controlled precipitation’ technique. Performing precipitation at elevated temperatures is called ‘evaporation precipitation into aqueous solution’. A technique such as ‘controlled crystallization during freeze drying’ is also available. There are several methods involving precipitation based on supercritical fluid (SCF) technology. If the drug is soluble in SCF, the method employed is called ‘rapid expansion of supercritical solution’. If SCF is used as antisolvent, there are other variations possible such as ‘gas antisolvent process’, ‘supercritical antisolvent process’ and ‘solution enhanced dispersion of solids’. Each of the different variations was discussed extensively by a few authors. Literature presents the reports of some positive results with the specialized bottom up approaches too but their application is mainly limited due to the requirement of special processing expertise and custom designed equipment as well as the high costs associated with such production equipment. Solvent–antisolvent precipitation is the simplest and single step precipitation process involving low energy, less expense and requires simpler instruments. The process may be designed more efficiently with the incorporation of high speed homogenization or sonication and subsequent solidification. The use of evaporation processes like spray or freeze drying operable at low temperatures (suitable for thermolabile drugs) or fluid bed drying for solidification purpose would still constitute cost-efficient processes when compared to other high energy and sophisticated precipitation processes (Möschwitzer, 2013, Abdelwahed et al., 2006, Chan and Kwok, 2011, Sinha et al., 2013).
All the combination approaches are uniquely referred to as smartCrystals® (Keck et al., 2008, Shegokar and Müller, 2010). Examples of smartCrystals® technologies so far explored include Nanoedge™ technology which involves microprecipitation plus high pressure homogenization (HPH) (Kipp, 2004); H42 technology involving non-aqueous spray drying followed by HPH (Salazar et al., 2013); H96 technology which involves freeze-drying followed by HPH (Salazar et al., 2012). There is also one H69 technology which employs the same combination approach as Nanoedge™ but in order to save time between the precipitation and homogenization processes and to yield smaller drug NCs, the precipitation process is carried out directly within the zone of dissipation of homogenizer (Kakrana et al., 2012). Combination Technology (CT) is another smartCrystal technology which involves media milling followed by high pressure homogenization (Shaal et al., 2010). A brief summary of the most commonly employed characterization methods for NC products is presented in Table 1.
Table 1.
Most commonly employed characterization techniques for nanocrystals (Singare et al., 2010, de Waard et al., 2009, Shaal et al., 2010, Hu et al., 2011, Niwa et al., 2011, Shegokar et al., 2011, Quan et al., 2011, Ghosh et al., 2012, Moeschwitzer, 2010a, Moeschwitzer, 2010b).
| Characterization parameter | Examples of analytical methods |
|---|---|
| Structure and morphology | Light microscopy, scanning electron microscopy, transmission electron microscopy, field emission scanning electron microscopy, atomic force microscopy |
| Particle size and particle size distribution | Photon correlation spectroscopy (based on dynamic laser light scattering), laser diffraction (static laser light scattering), microscopic methods |
| Surface charge | Zeta potential |
| Solid state analysis (crystallinity) | Powder X-ray diffraction, differential scanning calorimetry |
| Rheological properties (for liquid nanosuspensions) | Viscometer, rheometer |
3. Scale up
Scale up with respect to pharmaceutical manufacturing process is a translation involving the transformation from microscopic (molecular) lab level to macroscopic (bulk) industrial commercial level production. Operationally scale up ratio is defined as follows:
Scale up ratio = large scale production rate/small scale production rate
However, in literal sense, scale up is a process which could not be detailed by such a simple ratio. The design and development of scale up is emphasized because there is no framed algorithm which can help the formulators predict the large scale performance of a product based on its small scale behavior. Scale up is accomplished by the sameness criterion, the sameness among all the three levels of study, lab, pilot and the production. At each of the three levels, the raw material specifications and controls, in process and finished product specifications and bioequivalence results of any given lot are expected to be in line with the results of previous lots (Levin, 2002). Fig. 2 depicts the similarity concerns to be born on mind while planning a scale up.
Figure 2.
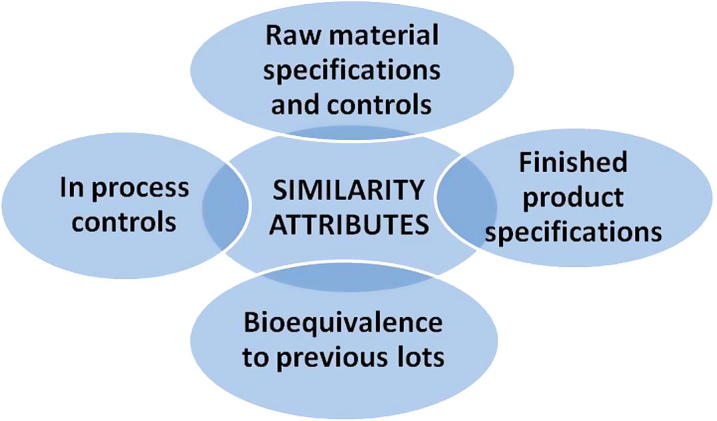
Set of product attributes whose similarity is critical during a scale up process.
The success of any formulation development depends on its transferability to large scale and all the NC products already in the market might have been designed by keeping on mind, the industrial production ever since their lab scale development. Additionally, a scalable formulation/method will remain robust at all the three levels of study, the lab, pilot and industry. Based on the above fact, our interest arose in reviewing the efforts involved in scale up based research works since we believe that the study of success profiles of scalable formulations/methods may increase the early optimization rates of the beginners in the field. Literature shows that there are a few such researches on NCs. Table 2 lists the summary of such works. Our present paper presents a note on all the listed research works as case studies. Each of these works either had scalability as their objective or upscaling as their future scope.
4. Case studies
A survey on the recent publications reveals the vivid shift of the research on drug NCs from the stage of lab level optimization to the stage of upscaling studies. This shift is certainly encouraging on account of the advantages offered by these particular delivery systems and as such, a review of such NC upscaling studies has been incorporated in this paper. The following sections provide a thorough review of the research works with scalability as objective or upscaling as future scope. In order to avoid a monotonous brief or gist of the research works and by bearing reader’s convenience on mind, our understanding of each of the research works was presented in five sections to cover the process (method), equipment, formulation, stability and summary of the works.
4.1. Batch and semi-continuous production of nanocrystals by controlled crystallization using a ‘3-way nozzle equipment’ (de Waard et al., 2009)
The authors of this research work carried out the NC production in batch fashion as well as in semi-continuous fashion using a ‘3-way nozzle equipment’ with large scale production potential. The actual process involved initial mixing of the solution of drug (fenofibrate) in tertiary butyl alcohol (TBA) with the solution of cryoprotectant (mannitol) in water and the subsequent freeze drying at a relatively high temperature (−25 °C). The immediate freezing was employed since the mixture of the above two solutions was thermodynamically unstable by being prone to premature crystallization of the drug which may result in the formation of large drug crystals.
During batch production, just the moment the solutions were mixed manually, the vials were immersed in liquid nitrogen. In order to design a semi-continuous production method for large scale application, the 3-way nozzle model was studied. A nozzle was designed with three separate channels for aqueous solution, TBA-solution, and atomizing air flow in such a way that the atomizing air thoroughly mixes the two solutions as soon as they leave the nozzle. The resulting mixture was then immediately frozen by spraying it directly into liquid nitrogen to prevent premature crystallization. This semi-continuous design provided a more instantaneous freezing since the atomizing air produced small droplets of mixture and resulted in smaller drug crystals with superior drug release profiles which indicates the success of the semi-continuous design for the production of better controlled crystallized dispersions and its suitability to large scale production.
The immediate freezing further to mixing was carried out at a temperature below the glass transition temperature and the subsequent freeze drying was carried above the glass transition temperature but below the eutectic temperature. Such temperature conditions lead to the crystallization of the drug and matrix material in the freeze-concentrated fraction and the crystal size was found to be affected by the freezing rate and water/TBA ratio. The immediate freezing requirement at the large scale was aimed to be achieved by using the 3-way nozzle model designed in this study. The mixing efficiency of the 3-way nozzle model was studied to validate the upscaling suitability of controlled crystallization process from batch to semicontinuous state. Additionally, the crystallinity and dissolution performance of the controlled crystallized dispersions prepared by the batch process and the semi-continuous process were compared.
A method by name Villermaux/Dushman originally developed for microfluidic devices to measure the mixing quality, was used to validate the mixing efficiency of the 3-way nozzle model. The method involved mixing of an acidic and a buffered iodine/iodate solution which can trigger two parallel reactions depending on the mixing quality. A slow mixing leads to the formation of triiodine out of acid and a fast mixing leads to neutralization of acid by the buffer (here no triiodine was formed). Hence the triiodine amount formed was used as a measure for mixing efficiency and a mixing of poor quality could be indicated by the formation of triiodine due to reaction of some amount of acid with iodine and iodate, which may be detected using a spectrophotometer. The Villermaux/Dushman method was applied to the 3-way nozzle model with and without (control) application of atomized air. The spray settings tested were similar to those used for spray freeze drying of controlled crystallized dispersion by setting the rate of atomizing air flow at 500 L/h (i.e. 500 L of air at 1 atm and 0 °C), a total liquid flow as 15 ml/min and the distance to the sprayed surface as 60 mm. A lower spectroscopic absorption indicates formation of less triiodine, in other words indicates faster and better quality mixing. The spectroscopic absorption was much lower (0.222 ± 0.06) for the 3-way nozzle sample produced with the atomizing airflow rate of 500 L/h when compared to the sample (1.163 ± 0.08) produced as control without applying the atomizing airflow (for control, the drug and mannitol solutions were mixed without aid of atomizing air flow by adapting Villermaux/Dushman method). Hence the spectrophotometric results confirmed a proper degree of mixing of two solutions in the presence of atomizing air which validates the use of the 3-way nozzle model for a homogenous mixing of the aqueous-mannitol and TBA-drug solutions to produce a controlled crystallized dispersion. Based on the dissolution results, the authors confirmed that the semi-continuous design was valuable for large scale production and yields a better product.
4.1.1. Process
The aqueous-mannitol and TBA-drug solutions were preheated to 60 °C before subjecting to the batch or semi-continuous process. In case of batch process, 1.2 and 0.8 ml of preheated aqueous-mannitol and TBA-drug solutions were respectively mixed in 20 ml glass vials and the vials were immediately immersed in liquid nitrogen and subsequently freeze dried. In case of semi-continuous process, the preheated aqueous-mannitol and TBA-drug solutions were pumped separately using perfusion pumps through a heated 3-way nozzle at a flow rate of 9 and 6 ml/min. respectively. The total liquid volumes of 55, 70, and 90 ml were mixed to achieve drug loads of 30%, 40%, and 50% (w/w), respectively. The liquid dispersion obtained by the mixing of two solutions was then sprayed directly with the aid of atomized airflow, into liquid nitrogen filled metal tray. Thus frozen material was subsequently freeze dried. During freeze drying, the temperature of samples was initially equilibrated on a pre-cooled shelf (−50 °C) for 1.5 h and the subsequent increase in temperature to −25 °C crystallized the drug and matrix material. Next, the solvents were removed by decreasing the pressure to 0.220 mbar after 3 h. This low pressure was maintained for 10 h after which the temperature was gradually increased to 20 °C. The samples were stored at room temperature for at least 1 day in a dessicator over silica gel before further processing.
4.1.2. Equipment
The semicontinuous process employed a custom designed 3-way nozzle model and the freeze drier used was Christ model Epsilon 2–4 lyophilizer (Salm en Kip, Breukelen, The Netherlands).
4.1.3. Formulation
25 mg/ml of drug in TBA solution was mixed with varying concentrations of mannitol in water so as to produce drug loads of 30%, 40%, and 50% (w/w) at both batch and semi-continuous levels. The scanning electron microscopy (SEM) pictures though showed the presence of aggregates, the particles size (PS) values were less than 1 μm (nanorange). The controlled crystallized dispersions produced by batch and semi-continuous processes were tested for their crystallinity and dissolution characteristics. The differential scanning calorimetric (DSC) analysis showed that the degree of crystallinity for the products produced by batch (86–93%) and semi-continuous (82–86%) processes was similar. Tablets were prepared with physical mixture containing 30% fenofibrate, 30% controlled crystallized product produced by batch process (freeze-dried) and semi-continuous process (spray freeze-dried) and their dissolution profiles were compared. The results indicated that the tablets prepared from the controlled crystallized dispersions irrespective of the batch and semi-continuous process involved, showed identical drug release profiles (almost 100% drug dissolved in 2 h) while the tablets prepared with physical mixtures released lower drug amounts (less than 50% drug dissolved in 2 h). The dissolution rate of the tablets containing controlled crystallized dispersion prepared by semi-continuous process was slightly higher than the batch-wise produced product on account of the higher freezing rate and smaller drug NCs produced during semi-continuous process. Additionally, the effect of drug load on the dissolution rate was studied which showed that the increase in drug loads from 30% to 40% and 50% decreased the dissolution rate for which the authors reasoning was that for a fixed mass of tablet, with the increase in drug concentration, the lipophilicity increased which resulted in decreased wetting and consequently decreased dissolution rate. Besides, with the increasing drug loads, the difference in the release profiles (rate and extent of drug dissolved) of batch process product and semi-continuous process product also increased.
4.1.4. Stability
The controlled crystallized dispersions were prepared by using δ-mannitol which has a reported stability for few years at ambient conditions. So the authors claimed that the shelf life problems need not be expected for the prepared controlled crystallized dispersions.
4.1.5. Summary
A scalable semi-continuous equipment model was designed for the production of drug NCs. The NC production was carried out in batch and semi-continuous fashions and the study designed a new ‘3-way nozzle equipment’ with large scale production potential for the semi-continuous production. The mixing efficiency of the 3-way nozzle model was validated using Villermaux/Dushman method. The NCs were finally formulated as tablet dosage forms and the release studies demonstrated higher dissolution of the tablets prepared out of the 3-way nozzle manufactured NCs as compared to those of batch model.
4.2. Application of design of experiments to optimize the production of a nanosuspension formulation with an industrial perspective (Singare et al., 2010)
Box–Behnken design was applied to study the effect of formulation and processing parameters on the PS, zeta potential (ZP) and scalability of formulation. The model drug chosen for study was meloxicam. The optimization of formulation variables can render a robust and stable formula with ideal characteristics and the processing parameters can affect the production of nanosuspension at large scale.
4.2.1. Process
Top down media milling was the production process chosen. Purified water, yttrium-stabilized zirconium beads and 0.2 mm as bead volume were screened during the initial screening studies as the solvent for nanosuspension production, the media for milling and the milling media volume, respectively. A recirculation mode of milling operation was performed with the pump/ suspension fed rate as 100 ml/min. The product temperature was controlled by circulating cold water through the outer jacket during milling. 250 g batch size, 16 g drug content, the type of polymer and stabilizer were kept constant throughout the study. The polymer used was hydroxyl propylmethyl cellulose (HPMC), 6 cps and the stabilizer was sodium lauryl sulfate (SLS).
Based on the screening studies, the parameters influencing ZP and the mean PS, d(90) of formulation were filtered as the ratio of polymer to drug, ratio of stabilizer to drug, milling time and milling speed. Hence, the process was characterized by studying the effect of the above formulation variables and processing parameters on ZP and PS. A premixed product was subjected to milling and as far as the production process was concerned, both the ZP and PS were found to be influenced by the process parameters, milling time and speed as follows. Lower milling speeds decreased ZP while the effect of milling time was not clarified. Particle size decreased with increasing milling speeds and time. The required ZP in the range of −20 to −25 mV and d(90) values of 350–400 nm were achieved by optimizing the milling time for 3.8 h and milling speed at 2563 rpm.
4.2.2. Equipment
The equipment chosen was bead mill, Model: Lab Star 1, Netzsch Mill, Germany to produce 250 g batches at lab scale.
4.2.3. Formulation
The effect of formulation variables namely polymer to drug ratio and surfactant to drug ratio on ZP and particle size, d(90) were evaluated. The polymer to drug ratio significantly influenced the ZP rather than the surfactant to drug ratio. High polymer concentrations decreased the ZP values independent of the surfactant to drug ratio. The PS was low at lower polymer concentrations. A ZP range from −20 to −25 mV and d(90) of 350–400 nm were achieved by optimizing the polymer to drug and surfactant to drug weight ratios at 0.39 and 0.04, respectively, as understood from the optimized formula. The optimized formula contained 16 g drug, 3.9 g polymer and 0.4 g surfactant dispersed in 250 ml water. The spray dried product was subject to X-ray diffraction (XRD) which confirmed retention of solid state characters.
4.2.4. Stability
The stability study was not performed in the above research work.
4.2.5. Summary
The wet ball milling process for the production of meloxicam nanosuspensions was characterized by studying the effect of the above formulation variables and processing parameters such as ratio of polymer to drug, ratio of stabilizer to drug, milling time and milling speed on ZP and PS of formulation. The authors successfully utilized the Box–Behnken design, ANOVA, perturbation plots, research surface methodology and contour graphs to study the effect of the independent variables on dependent variables and established reliability on design of experiments in the optimization of nanosuspension production. The ratio of polymer to drug and milling speed were found to affect the ZP while the PS was found to be affected by milling time and milling speed and accordingly, the independent variables were optimized. Additionally the authors claimed to have identified important formulation variables and production processing parameters which may affect the nanosuspension production at higher scales. The inevitable industrial scale manufacturing requirements like high polymer concentration and high milling speeds which may be associated with issues such as rise in production temperature and increase in the pressure on the milling equipment were said to have been met by modulating the milling speed and time. Milling at lower speeds for the first 10 min followed by a slow increase in speed was attempted to meet the production level requirements. The authors have not reported the production of any scaled up batches. Hence, no note on the production level equipment had been reported. Singare and his coworkers successfully applied this optimized method of production for the manufacture of NCs of glyburide too (Singh et al., 2011). The applicability of the optimized processing and formulation parameters to another drug signifies the robustness and quality of optimization achieved by these researchers.
4.3. Lab to pilot level upscaling of smartCrystal combination technology for the manufacture of apigenin nanocrystals (Shaal et al., 2010)
Apigenin NCs were produced at lab and pilot levels by applying a scale up factor of 150. 20 g product at lab scale was manufactured by HPH and the production was scaled up to 3 kg at pilot scale using CT (media milling followed by HPH), a smartCrystal technology.
4.3.1. Process
Apigenin nanosuspension at lab scale was manufactured using Micro LAB 40 (discontinuous mode) by applying 30 homogenization cycles at 1500 bar pressure. The process characterization revealed that the homogenization cycles beyond 20 had little effect on PS but decreased the polydispersity index (PDI). The 20 g lab product after 30 cycles showed a mean particle size of 264 ± 5 nm and a PDI of 0.136 ± 0.05. The reproducibility was checked with 3 batches. The production at pilot scale employed CT technology where 3 kg product was manufactured. The product was initially processed for 7 passages using pearl milling (discontinuous mode set up) and was further processed by one cycle of HPH treatment with Avestin C50 at 300 bar pressure. These operating parameters were said to be finalized based on the initial screening studies. At the end of 7 milling cycles, the product attained 440 nm PS and 0.265 PDI. The subsequent homogenization yielded product of PS 413 nm and PDI 0.2.
4.3.2. Equipment
The lab level studies employed Micro LAB 40 for homogenization. At the pilot scale, the CT technology employed use of a discontinuous mode set up of pearl mill and Avestin C50 HPH.
4.3.3. Formulation
The optimized formula contained 10% apigenin powder and 1% surfactant (Plantacare 2000) dispersed in purified water as dispersion medium. The formula was maintained constant at lab and pilot scales where 20 g and 3 kg products were manufactured, respectively. However, during the pilot production, the nanosuspension based on the above formula and operated by pearl mill was diluted with 1% surfactant solution prior to homogenization step.
4.3.4. Stability
Irrespective of the batch size and the production method employed, all the samples at lab and pilot scales remained physically stable with respect to ZP, PS and PDI during the 6 month long term study period carried out at 4 °C, room temperature and 40 °C with no signs of Ostwald ripening (OsR). The ZP values at lab level using HPH, LAB 40 were −38 mV in water and −37 mV in the original dispersion medium (water and 1% surfactant solution). The ZPs recorded for pilot batch were −45.0 mV in water and −42.5 mV in the original dispersion medium. The formulation stabilized with Plantacare 2000 at 1% concentration seems to have sufficed the stability (electrostatic and steric) requirement as confirmed by the ZP values studied. The particle size analysis was performed by photon correlation microscopy (PCS), laser diffraction (LD) and light microscopic (LM) studies. The observations of all the three studies on the day of production and at the end of six months of stability study at room temperature indicated no signs of large crystals or visible aggregation and depicted comparable results between lab and pilot batches.
4.3.5. Summary
The authors successfully produced 20 g product at lab scale and 3 kg product at pilot scale with comparable PS, ZP and PDI. It was expressed that the discontinuous arrangement of the CT (media milling followed by HPH), smartCrystal equipment set up may assure the transferability to production level for manufacturing few hundred kg product. The study suggested few rules of thumb and interpretations. The effect of pearl milling on the PS was found to be dependent on the agitator rotation speed, the velocity of pumping of suspension and the number of milling cycles. The PS reduction by HPH was found to be influenced by the number of homogenization cycles and pressure applied which could in turn depend on the initial product hardness, crystalline and amorphous fractions and the crystalline imperfections. It was suggested that a pearl mill could handle higher drug concentration and could process 2–3 times more viscous macrosuspensions in relation to a homogenizer. It was also interpreted that the size of NCs obtained would in general be 1000 times smaller than the employed milling pearls’ volume since the study employed 0.4 mm pearls and yielded 400 nm particles.
4.4. Design of a continuous and scalable process for the development of a water redispersable nanocrystal formulation (Hu et al., 2011)
The aim of this work was to develop a direct, fast, continuous and scalable process for precipitating drug NCs. Fenofibrate was employed as a model low solubility drug for the production of drug NCs. ‘Batch mixing coupled with spray drying’ was developed as proxy for ‘continuous static mixing coupled with spray drying’. The former process was studied in this research work in order to confirm the feasibility of the latter for large scale production of NCs. Based on the characterization results of morphology, PS, crystalline state and drug release, the batch mixing coupled with spray drying was reported successful for the continuous NC production and therefore, the potential of the model, ‘continuous static mixer coupled with spray dryer’ was claimed. The authors previously proposed the use of static mixer for the continuous production of NCs by bottom up precipitation method (Dong et al., 2010). As an extension of the above work, and in order to deal with the OsR problem, the authors examined the effect of the subsequent and immediate spray drying on the precipitated NCs. Hence, the process developed for manufacturing was antisolvent precipitation combined with spray drying followed in a continuous loop as opposed to the typical batch manufacturing.
4.4.1. Process
Though the original aim of authors was to couple the static mixer with spray drier to achieve continuous production, for the sake of lab level study, to avoid the production of large quantities of NCs as obtained with the use of a typical static mixer which would require a costlier spray dryer beyond the lab scale capacity, the authors proposed the batch precipitation process as a proxy for the continuous static mixing. The PS of NCs obtained from the batch and static mixing was compared to investigate the equivalence between the two methods. Hence, the process involved immediate treatment of the batch wise precipitated NCs with a mini spray dryer wherein dried NCs were produced within 1 min.
Scaled up batches were not produced but the aim had been to develop a manufacturing process which could be scalable. Static mixing can maintain the same precipitation conditions at any scale of study and therefore, the stirring and mixing problems could be avoided at the production scale. Hence, a combination of batch wise antisolvent precipitation (proxy for continuous static mixing at production scale) coupled with spray drier was proposed in this study.
4.4.2. Equipment
The authors employed magnetic stirrer (wherein the precipitation was carried out multiple times with a new beaker of antisolvent each time) coupled to a Büchi Mini Spray-dryer B-290 with inlet loop B-295, Germany for the continuous intake of the product precipitated batch wise. The static mixer used was 6-element SMV DN25 from Sulzer Chemtech, Switzerland.
4.4.3. Formulation
For continuous looped batch wise production, 50 mg of fenofibrate was dissolved in 1 ml of ethanol (solvent) which was added at 1000 rpm stirring rate and at room temperature to 10 ml antisolvent (water) containing 10 mg/ml of lactose or mannitol, 0.5 mg/ml of sodium dodecyl sulfate (SDS) and 0.5 mg/ml of HPMC E3. Thus obtained NCs suspension was immediately spray dried.
For static mixing, the drug-solvent and antisolvent solutions were pumped through nozzles of diameters 0.5 and 1.5 mm and at flow rates of 50 and 500 ml/min respectively into the 6-element SMV DN25 static mixer.
Field emission scanning electron microscopy and dynamic laser light scattering studies confirmed the nanosize of freshly precipitated formulation as well as subsequently spray dried and redispersed (in water) formulation. Powder-XRD, DSC studies confirmed the retention of crystallinity of fenofibrate in NCs. The PS results for NCs obtained by batch (magnetic stirring) and static mixing were 318 ± 19 and 328 ± 22 nm, respectively which confirm the usage suitability of the employed batch mixing process as a proxy for static mixing. The freshly prepared NCs from batch mixing, when not subjected to spray drying began to start growing in size to 2.5 μm within 10 min. Hence, an immediate spray drying step was coupled with the precipitation step by batch mixing. The release profile of the developed (spray dried) NCs was compared with sample 1 and sample 2 (described as follows). Sample 1 was a physical mixture consisting of 1 g of micronized fenofibrate, 2 g of lactose, 0.1 g of HPMC E3 and 0.1 g of SDS, and mixed using a mortar and pestle. Sample 2 was a spray dried powder of the above physical mixture in 200 ml water. The dissolution studies showed that 84.2% of drug dissolved from NCs in 5 min as compared to 31.7% drug dissolved from the conventional spray-dried formulation (sample 2) and 9.7% drug dissolved from the physical mixture using micronized fenofibrate (sample 1). The drug from NCs completely dissolved within 15 min while 17% and 44% drug remained undissolved in case of physical mixtures of micronized fenofibrate prepared with (sample 2) and without (sample 1) spray-drying, respectively, even after 60 min. The redispersant used for spray drying was lactose or mannitol, both of which showed similar results.
4.4.4. Stability
Stability was not studied and storage conditions were not reported.
4.4.5. Summary
The static mixing set up mentioned in this work was initially established by these authors for the production of spiranolactone NCs. The experimental set up sufficient to precipitate the drug particles in the submicron range (of about 500 nm) was optimized as SMV DN25 static mixer designed for turbulent flow which contained 6 mixing elements accommodating a total flow rate of 1.0 L/min where the flow rate ratio of solvent to antisolvent was maintained at 1:9. The increase in the number of mixing elements to 12 was found to further decrease the PS and size distribution. The increase in drug concentration led to aggregation and an upper limit concentration of 10 mg/ml could only result in sub micron particles. Further increase in drug concentration did not yield nanosized product. The spiranolactone nanosuspension freeze dried using lactose as redispersant and cryoprotectant exhibited 6.6 and 3.3 times faster dissolution rate than the freeze dried raw drug formulation (containing approximately 33% drug loading, 60% lactose, 3.3% HPMC and 3.3% SDS) in 5 and 10 min, respectively. The optimized formula of NCs was not clearly disclosed. The NCs exhibited a dissolution peak in 10 min in view of the 60 min study period. The XRD and SEM results demonstrated that the freshly precipitated drug NCs formulation subjected to immediate freeze drying was amorphous in nature while the nanosuspensions left alone for several minutes before freeze drying acquired crystalline state on their own and that crystalline state was in accordance to the raw drug (Dong et al., 2010).
In the current work (Hu et al., 2011), the production of fenofibrate NCs was carried out by immediate spray drying of the product precipitated batch wise using a magnetic stirrer (continuously looped to spray drier). The morphology, PS, crystalline state and drug release study results (as discussed in the formulation section) revealed the successful use of this continuous production process (proxy for the continuous static mixing). So, based on the results obtained for the NCs of spiranolactone and fenofibrate, the plausibility of large scale production using a combination of static mixer and spray drier could be endorsed.
4.5. Particle sizing using a universal wet milling designs applicable since discovery stage to the preclinical studies (Niwa et al., 2011)
Miniature, middle and large scale wet milling designs were developed which could be operable for drug loadings in the range of 50 mg to 30 g. The manufacturing level scale up was not reported. This drug loading range would cover the early screening studies’ requirement (10–100 mg) as well as the late safety studies’ requirement (10–100 g). The designs were basically developed to facilitate the economic use of time, compound and investment. The solid state characterization studies indicated that the crystal form and crystallinity of the drug were retained after milling process. The milling designs proposed in this research work may be applied by the scientists as simple and easy techniques during the discovery phase since the desired sub micron range particles could be produced in a run time as less as 10 min. The milling designs differed only in the nature of collision impact applied. At the middle scale, oscillating beads-milling apparatus was used for which the process operational conditions and the formula (type and composition of dispersing agents) were optimized so as to achieve finer drug particles. The mode of mixing employed at miniature and large scales was stirring and turbulence using a laboratory magnetic stirrer and a turbulent rotating shaking mixer (turbulent mixer) respectively. The developed designs may find use in evaluating the poorly water soluble drug candidates at discovery stage from the pharmacological, toxicological and pharmacokinetic perspectives.
4.5.1. Process
At the middle scale, oscillating beads mill was operated using a 50 ml conical tube containing 0.6 g drug and 60 g zirconia beads (making up around 16 ml volume of the tube) suspended in an aqueous dispersion medium of 15 ml (40 mg/ml drug concentration). The tube was placed into a holder and oscillated at 2700 rpm for 12 min (optimized driving or running time independent of drug loading) and the holder was cooled by circulating a refrigerant to maintain a temperature under 15 °C. A 12 min run time could produce particles in a nano range for a drug concentration up to 100 mg/ml (1.5 g total drug). The authors reported that the increase in run time produced nano sized particles with ease up to as high as 160 mg/ml drug concentrations (data not presented). The milling process was found to be influenced by the milling media (zirconium beads) size and 0.3 mm beads were set as optimum after trying the effect of different sizes (0.1–1 mm).
For miniature scale production using magnetic stirrer, the same quantity ratio of the drug, beads and dispersion medium (0.1 or 0.6 g drug was milled in 2.5 ml or 15 ml medium using 10 g or 60 g beads) as used in the middle scale was filled into glass vials of 10 and 50 ml capacities. This suspension was milled using 0.3 mm beads on the magnetic stirrer at 700 rpm for 24 h. 0.05–0.5 g and 0.6–3 g range drug loadings were studied using 10 and 50 ml vials respectively.
For large scale manufacture, the turbulent mixer was employed. A turbulent mixer mixes samples by rotating the container with samples in planetary and twisting motion. The drug loads in the range of 7.2–28.8 g of phenytoin were studied. 570 ml steel bottle was filled with a 180 ml aqueous medium containing drug and 720 g of 0.3 mm zirconia beads. The bottle was rotated using the turbulent mixer at 96 rpm for 90 min.
4.5.2. Equipment
Laboratory magnetic stirrer (HS4-SP, AsOne, Osaka, Japan), oscillating bead mill (Multi-Beads Shocker, Yasui Kikai, Osaka, Japan), and turbulent rotating shaking mixer (Turbula Mixer T2F, Willy Bachofen, Basel, Switzerland) were used at the miniature scale, middle scale and large scale respectively. For morphology studies, all the samples were dried using freeze-dryer, model PFR-1000/UT-2000, Tokyo Rikakiki, Tokyo, Japan. For crystallinity studies, the samples from oscillating bead mill were dried using a tray drier (DRA-630DA, Advantec Toyo Ltd., Tokyo, Japan) as well as freeze drier (RLE-52, Kyowa Vacuum Engineering Co., Ltd., Tokyo).
4.5.3. Formulation
Phenytoin was used as a model bcs class 2 drug. Additionally, nifedipine and pranlukast hydrate were also formulated using the middle scale design to generalize the significance of the approach, confirm its robustness independent of the physicochemical properties of drugs and to expand its application. The preparations were subjected to particle size analysis, zeta potential, morphology and crystallinity studies (tray dried and freeze dried products were subjected to XRD and DSC). The SEM photographs gave visual confirmation of milling in the nano range. The XRD and DSC studies confirmed the retention of crystallinity in all the processed samples.
The milling process was found to be influenced by the choice of dispersing agents and as such their composition was finalized as 0.5% polyvinylpyrrolidone (PVP) as steric stabilizer and 0.1% SLS as electrostatic stabilizer which resulted in zeta potential of −30 mV required to stabilize the colloidal particles. Use of different surfactants and stabilizers and trials of milling without addition of stabilizers filtered the above mentioned combination as optimized dispersing agent composition. The results also indicated that the size reduction through wet milling could be attributed not just to the mechanical stress but also to the drug molecular interactions with the dispersing agents.
Milling performance using three model drugs (phenytoin, nifedipine and pranlukast) was studied only at the middle scale using oscillating beads-milling apparatus. The particle size of the starting material of nifedipine was larger compared to that of phenytoin while that of pranlukast was smaller. The study indicated that irrespective of the compounds and their original particle sizes employed, the milled particles reported the particle size distribution (PSD) at around 0.3 μm and 180 nm when analyzed with laser diffraction and dynamic light scattering methods, respectively. Not only that the nanometric range production was confirmed but the PSD was also found to be quite narrow and nearly monodipersed.
The formulations manufactured using the lab magnetic stirrer and with drug loads in the range of 50–400 mg (in 10 ml vials with 2.5 ml aqueous media) showed PSD results in submicron range equivalent to those produced by the middle scale apparatus. The studies in 50 ml vials yielded similar results for 0.6–2.4 g of phenytoin.
Regardless of the milling mechanism applied at different scales, 160 mg/ml drug load is the maximum concentration of drug that could be nano-milled with reproducible and robust PSD results. Authors reported with practical results that 28.8 g of phenytoin could be effectively treated as a maximum amount using the employed turbulence mixer while further scale up experiments were yet to be done.
Nanosized particles in the range of 200–400 nm were successfully prepared with variable drug loads in the range of 10 mg to 10 g independent of the equipment used. 95% of drug was recovered and the drug loss was accounted to the adhesion of particles to the container walls and beads. Independent of the drug loads and manufacturing equipment, 0.5% PVP + 0.1% SLS combination in the aqueous solution was found to serve as efficient dispersing medium. The redispersion of formulations upon drying and the dissolution properties were not studied.
4.5.4. Stability
Stability studies were not reported in the publication under discussion.
4.5.5. Summary
The authors successfully designed universally applicable wet ball mill designs for evaluating the poorly water soluble drug candidates ever since the discovery stage (early screening studies) till preclinical stage (late safety studies) from the pharmacological, toxicological and pharmacokinetic perspectives. Laboratory magnetic stirrer, oscillating bead mill and turbulent rotating shaking mixer were used at the miniature scale, middle scale and large scale respectively. The proposed ball mill designs would be operable covering drug loadings in the range of 50 mg to 30 g. The study indicated that irrespective of the compounds and their original particle sizes employed, the milled particles reported nanometric PSD results which signifies the universal applicability of the approach and confirms its robustness independent of the physicochemical properties of drugs. Additionally, the results also indicated that the size reduction through wet milling could be attributed not just to the mechanical stress but also to the drug molecular interactions with the dispersing agents which emphasizes the coordinated role of process, equipment and formula in the success of a pharmaceutical design.
4.6. Preparation of nevirapine nanosuspensions at lab and pilot scale levels by high pressure homogenization and media milling – a comparative study (Shegokar et al., 2011)
This work produced redispersible white colored homogenous nanosuspensions of pH between 6.87 and 7.02 at three different scales. The PSD analysis confirmed that the manufactured particles were below 5 μm which makes the formulation suitable for intravenous (IV) administration.
4.6.1. Process
Three batches at lab (40 ml), medium (2 kg) and pilot scales (150 ml) were produced using piston gap homogenizers at lab and medium scales and using a bead mill at pilot scale. A coarse suspension was premixed and subsequently processed using HPH or bead mill.
At lab scale, 40 ml nanosuspension was processed using Micro Lab APV 40. Premilling was carried out for 2 cycles at each of the 200, 500, and 1000 bar low pressures followed by actual homogenization for 20 cycles at 1500 bar. The homogenization tower was maintained at 4 °C all through the processing period and the formulation was cooled after each 5 cycles. The relation between the decrease in PS and the increase in homogenization cycles remained linear till 15 cycles after which the further increase in the number of homogenization cycles had not favored any further reduction in PS. However 20 cycles were run to obtain a homogeneous white nanosuspension with a mean PS of 481 ± 10 nm and PDI of 0.212 ± 0.085.
At medium scale, 2 kg coarse suspension was homogenized for 30 min in continuous mode using EmulsiFlex C50 or Avestin C50. The homogenization tower was pre cooled at 4 °C by passing ice cold distilled water and during the processing period, the nanosuspension temperature was controlled using water bath. 30 min processing using Avestin C50 at 1500 bar pressure (without any premilling) yielded nanosuspension of PS 429 ± 16 nm and PDI of 0.158.
At pilot scale, 120 ml coarse suspension of nevirapine was milled using Bühler PML-2 bead mill at 1000 rpm and 4 °C. The mill was run in continuous mode for 3 h and the medium employed for milling was 0.2–0.4 mm yttria stabilized zirconium oxide beads. At the end of 3 h, the mean PS obtained was 202 ± 12 nm and PDI of 0.182 ± 0.093.
4.6.2. Equipment
The equipment used for lab scale, medium scale and pilot production were Micro Lab APV 40, Avestin C50 (piston gap high pressure homogenizers) and Bühler PML-2 bead mill respectively.
4.6.3. Formulation
2% w/w coarse nevirapine powder was dispersed in 2.8% w/w aqueous surfactant solution. The 2.8% surfactant solution was made up using 1% tween 80, 0.9% volpol4, 0.5% poloxamer, 0.3% PVP and 0.1% plasdone. A premixed coarse suspension was processed using HPH and bead mill. The drug content as low as 2% was said to have been used in order to achieve maximum possible PS reduction to suite IV administration. The formulation was characterized for mean PS, PDI and ZP. PS and PDI were observed using PCS, LD and LM. ZP was measured in water and in original surfactant solution. All the ZP values were around −15 mV. The XRD studies confirmed retention of crystalline character.
4.6.4. Stability
12 month long term stability was performed at 4 °C, room temperature and 40 °C. The particle size was analyzed at the end of one year which suggested formation of aggregates and hence physical instability, particularly for the HPH processes. The WBM processed product as well showed an increase in mean PS from 210 to 669 nm at the end of one year.
4.6.5. Summary
The authors conveyed that the −15 mV ZP values could suffice the stabilizing requirement of nanosuspension since they claim that the general rule of ±30 mV ZP requirement would be applicable only in case of electrostatic stabilization and that the inclusion of steric stabilizers may stabilize the particle surface even at ZP values as low as −15 mV. However, the physical instability reported at the end of one year confirms the instability of formulation. The stabilizer system and composition may require alteration and optimization. Further, the authors opted air drying of nanosuspension with no data reported about the method. Freeze/spray/fluid bed drying, if applied, may have enhanced the stability of formulations. Though the product was scaled up by a factor 50 using HPHs (homogenization yielded products with comparable PS, PDI, and ZP values), the stability profile was not acceptable. The authors, however, finally confirmed the scalability of milling process (for IV administration purpose) by also referring to one of their previous works on apigenin which involved application of a scale up factor of 150. They conveyed that the use of bigger milling chamber, transfer from continuous mode to discontinuous mode of milling and application of 6–7 milling cycles could process up to 20% solid content and render a robust product (with only a few changes in the nanometer size of particles).
4.7. In-vitro and in vivo evaluation of nitrendipine nanocrystals – a miniature scale up study (Quan et al., 2011)
Nitrendipine NCs for oral delivery were prepared by precipitation-homogenization and by subsequent spray drying. The production was successfully scaled up from 20 to 300 ml.
4.7.1. Process
The nanonization process employed was precipitation followed by homogenization. 100 mg/ml solution of nitrendipine in acetone was dispersed in water containing 1 mg/ml polyvinyl alcohol at 10 °C. The drug solution concentration and temperature were the parameters optimized for precipitation process. The increase in nitrendipine concentration in acetone from 100 to 120 mg/ml was reported to increase the PS and span values. The temperature for precipitation process had been optimized at 10 °C since the increase in temperature leads to crystal growth. The parameters, pressure and number of homogenization cycles of HPH process were optimized as 1000 bar and 20 cycles respectively. While the particle size decreased with increasing HPH pressure, it increased with increasing cycles. The processing conditions, 20 homogenization cycles and 1000 bar pressure yielded smaller PS with a lower span value. The homogenization of presuspension yielded NCs of 175 ± 13 nm mean PS with a span value of 0.9766 ± 0.1658. The NCs were dried by spray drying process and the production was successfully scaled up from 20 to 300 ml.
4.7.2. Equipment
The equipment used for carrying out precipitation was not described. The homogenization was performed using piston-gap homogenizer, ATS AH100D model and spray drying was done using SD-1000 spray-drier, Eyela, Tokyo, Japan.
4.7.3. Formulation
100 mg/ml of nitrendipine in acetone was dispersed in water containing 1 mg/ml polyvinyl alcohol. Acetone was used as solvent, water as antisolvent and polyvinyl alcohol was employed to inhibit the unwanted crystal growth during the HPH step. Only 300 ml batches were spray dried for which carriers like lactose or mannitol were added by dissolving in stabilizer solution before precipitation. The carriers were reported to have a great influence on the dried powder flowability rather than on the PS. The carrier concentration at 1.7% ensured a reproducibly redispersable formulation although there were slight differences between the non-dried product and redispersed spray dried product. The DSC, XRD studies of initial unprocessed coarse powder and dried NCs confirmed the retention of crystalline character with no change in glass transition temperature of nitrendipine. Lyophilized sample (without carrier) was used as control for XRD studies for testing the spray dried samples in which, mannitol’s diffraction pattern interfered with that of the drug. The comparison of in vitro dissolution profiles of the NC formulation, physical mixture and commercial tablet showed that NCs released 100% drug within 1 min while physical mixture and commercial tablet released only up to 9% and 55% of drug in 1 min. Even after 10 min, the physical mixture and commercial tablet released only 40% and 90% drug respectively.
The in vivo bioavailability was studied in rats by HPLC which concluded that NCs showed 15- and 10-fold greater Cmax and 41- and 10-fold greater AUC0→24 than the physical mixture and commercial tablet, respectively. While the difference in the elimination rate constant values observed for NCs, physical mixture and commercial tablet remained statistically insignificant, the relative bioavailability values of NCs and physical mixture in relation to the commercial tablet were 1018.98% and 24.57%, respectively.
4.7.4. Stability
The stability studies were not reported. However it has been mentioned as authors’ observation that the NCs retained their PS constantly and exhibited good redispersibility over a storage period of 1 year under ambient conditions.
4.7.5. Summary
The current research work employs a combination technology to produce NCs wherein the bottom up precipitation was followed by top down HPH. Nanoedge™ is an example of technology which involves microprecipitation plus HPH for the production of drug NCs. The authors successfully produced up to 300 ml volume of nitrendipine NCs which presented superior in vitro dissolution and in vivo pharmacokinetic profiles compared to the physical mixture and commercial tablet.
4.8. Nanosuspension production by wet ball milling: optimization of process parameters and formulation variables (Ghosh et al., 2012)
In this particular work, the production parameters for scale up were optimized with respect to two equipments (a screening purpose planetary mill and a lab scale stirred media mill). The scale up factor for the volume of nanosuspension manufactured was however not reported.
4.8.1. Process
The wet ball milling process employed for the production of nanosuspension was optimized for process parameters as follows. A 23 factorial design of experiments and the knowledge and experience from the previous studies were applied to study the effect of variations in drug content (2%, 5%), milling media size (0.1 mm, 0.5 mm of yttrium stabilized zirconium beads) and mill rotation frequency (150 rpm, 400 rpm). The milling beads size and rotation frequency of mill were optimized as 0.2 mm beads (this number, though was not studied as variable, was optimized since the experimental design and outcomes of previous experiments suggested that at higher rpm, the bead size would be the least significant parameter) and 400 rpm respectively for scale up studies. The total milling time was fixed as 4 h for all the trials.
4.8.2. Equipment
The production scale up was attempted with respect to the equipment design optimization (similarity). The agitation rates were correlated for two equipments by conducting the bridging studies. Screening experiments were performed in planetary mill (early development phase). After optimizing the critical process parameters and formulation variables, the final variant was produced in a lab scale stirred media mill, Lab Star with zeta agitator in recirculation mode (lab scale development phase). The principle of agitation was different for the equipments. The agitator was a rotating jar in the planetary mill while it was an impeller in the Lab Star. The bridging studies concluded that 2500 rpm run in Lab Star would equal a 400 rpm run in the planetary mill to achieve similar particle sizes. All the formulations designed for the in vitro and in vivo studies were manufactured using Lab Star with the processing temperature maintained at 35 °C (temperature maintained by circulating cooled water through the outer jacket in a controlled fashion). The agitating speed was 2500 rpm and the pump speed was 250 rpm. Also once the bridging was done, the grinding efficiency was evaluated (based on PS results) by comparing the performance of the two mills by analyzing the samples collected at different time intervals. The results showed insignificant difference with respect to the PS values obtained with the two mills.
4.8.3. Formulation
The batch size was 25 ml for the preliminary phase of research carried out using planetary mill. The formulation variables like the drug content, ratio of drug:surfactant and type and concentration of additional surfactants were optimized. Compound NVS-102 was used as the model drug. d-α-tocopheryl polyethylene glycol 1000 succinate (TPGS) was used as surfactant. 23 factorial design was applied to optimize the drug content. 5% drug content in suspension, 2:1 drug:TPGS ratio along with 1% HPMC (3 cps) were finalized as efficient to design a robust and stable formula. However the same formula manufactured by Lab Star yielded PS < 300 nm for one batch, F2 for which the sample was collected 1 h after milling and PS < 750 nm for another batch, F3 for which the sample was collected at the end of 4 h of milling and these batches were studied for stability and in vivo performance. Additionally, F2 presented narrower PSD and the PSD for F3 was broader. The dissolution studies showed that F2 and F3 presented superior dissolution profiles in relation to the unmicronised samples while F2 and F3 had similar release profiles.
The unmicronised control batches as well as unmicronised batches with similar formula as F2 and F3 showed inferior (statistically significant) AUC and Cmax data compared to F2 and F3 emphasizing that the improvement in bioavailability could be attributed notably to nanonization alone. Additionally, the individual plasma concentrations conveyed that NCs of F2 with smaller PS and narrow or homogeneous PSD produced less variability compared to F3 possibly due to minimal instances of OsR with F2 (though the dissolution profiles of F2 and F3 were similar). Though statistically insignificant, the AUC and Cmax of F2 increased by 16% and 28%, respectively in comparison to F3 indicating the importance of narrow PSD as well.
4.8.4. Stability
The stability studies were performed at 2–8 °C for 6/12 weeks storage time. PS analysis was performed at the end of 6/12 weeks which indicated that the crystal growth was minimum during the storage period (though there was a slight increase in the mean PS values, the statistical significance was not reported). F2 showed superior stability compared to F3 with most minimal instance of OsR.
4.8.5. Summary
During the early development phase, nanosuspension was manufactured using planetary mill for screening purpose. Experimental design was applied to optimize the formulation aspects. Later, a bridging study was conducted and product performance similarity was successfully achieved between the planetary mill and lab scale stirred media mill by optimizing the equipment operation conditions. All the formulations for in vitro and in vivo testing were produced using media mill with zeta agitator in recirculation mode. Finally, based on the in vitro drug dissolution and in vivo pharmacokinetic studies, it was concluded that the prepared nanosuspension presented enhanced dissolution and bioavailability profiles.
5. Upscaling of NCs: challenges and handling
Gradual movement from small to larger scales may generate unanticipated new problems at any stage of development. However, the success of scale up lies in reproductive yielding of a robust product confirming to its quality specifications, irrespective of the scale of study. The experience at the lab and pilot plant scale levels could be an additional asset at production or commercial scale. Understanding of the process parameters becomes mandatory and scale up issues further demand the joint attention of pharmaceutical formulators and engineers. Such an understanding is, however, a challenging task since the list of influential parameters to be scrutinized at the lab level does not always remain the same when it comes to dealing with a production plant process. The plan for scale up commences ever since making the choice of technology which may depend on the indications of the final formulation, dose and route of administration. Each manufacturing technology has its own unique impact on the pharmacokinetic profile of the therapeutic agent and a slight manipulation of any given technology yields a significant difference in the pharmacokinetics of the finished product. The difference in pharmacokinetic profiles may be largely due to the differences in the size of the NCs obtained with different methods. Hence, the decision making at the early stages of formulation development always demands the involvement of scientists equipped with tools of drug delivery, fundamental understanding and past knowledge. The choice of technology (Fig. 3) should consider the target drug profile, optimal patient benefit, technical issues (differences in bioavailability and pharmacokinetic studies), economical aspects (frontloading, manufacturability and marketing) and the intellectual property rights (IPR) wisdom (non-infringement of patents).
Figure 3.

Various considerations associated with the choice of technology.
Many of the challenges during upscaling studies could be met by emphasizing on four basic parameters namely process characterization, choice of equipment, development of robust formulation and stability studies, ever since the formulation development stage (Fig. 4) (Levin, 2002).
Figure 4.
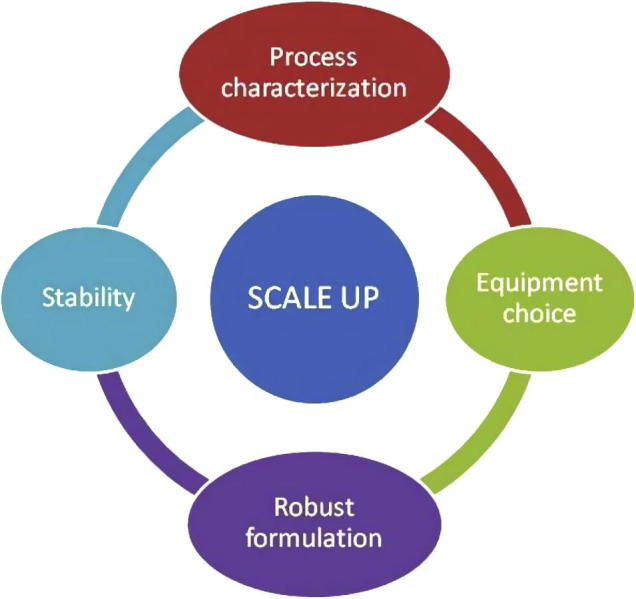
Basic quadrupole prerequisites for successful scale up of nanocrystals.
5.1. Process characterization and choice of equipment
The variations applied and observed in equipment (scales), process (modifications), and product (quantities) during the process of scale up complicate the task of scale up. The parameters to be controlled and monitored differ starting from the material handling (viscosities encountered and heat dissipation capacities of the systems), flow rates and shear stress to the storage and transfer levels. The effects of all these variations adopted in the process of scale up may in combination and/or with potential synergy yield the outcome of scale up beyond any prior expectations and experiences. The formation of NCs is governed by various rates such as the rate of addition of solvents, rate of shearing, rate of mixing, rate of subdividing and the rates of solvent removal and pumping. The knowledge of individual unit operations of a manufacturing process, the physical and physicochemical aspects of each of the operation, and the interactions among the components of process becomes mandatory. If the unit operation mixing is considered for instance (say, the precipitation method of preparation), the lab and industry offer a bewildering array of choices for mixing equipment. The dynamic components of mixing devices may be stirrers at lab scale which transform into impellers of different forms such as blades, paddles, propellers at higher scales. In addition, the mixing efficiency still could be varied by varying the number of impellers, impeller location, number of blades per impeller etc. For a given formulation, since the phenomenon of vortex formation is common with large tanks than the small tanks, the full scale production is likely to require baffles while lab level does not. Additionally, since the time required for executing a unit operation increases with upscaling, the temporal effects need to be studied by and large. It is to be noted that the lab scale equipment presents higher surface area to volume ratios while the production level equipments presents a higher volume to surface ratio. This difference is one of the considerable factors which slows down the processing during scale up. The finite time required for an operation at lab gets redefined while scaling. For the given extent of heat dissipation (heating and cooling), large scale equipment consumes more time when compared to lab equipment. These long processing times are likely to lead to adverse conditions such as adsorption, precipitation, viscosity alterations and may affect the mass and momentum transfer rates, the shear rate and stress factors. Hence, the temporal effects on the product’s physiochemical stability also require a guardianship. The basic idea of study plan should always be to use a scalable ‘smart experimental set up’ or the standard equipment similar to that at production so that the processes and equipment could later be scaled to larger batch sizes and also to identify since the earlier stages, the critical process parameters and dimensions.
Equipment upscaling concerns are fundamentally based on preserving similarities. Geometric similarity requires a three dimensional point to point correspondence between two systems. It is concerned with the linear dimensions of the two systems in question, the previous small scale (lab/ pilot) system and the current scaled up system. The consistency in the ratio of their linear dimensions assures geometric similarity. The mechanical similarity describes the status of application of force to a stationary or moving object and is defined in terms of the static, kinematic and dynamic similarity of the processing equipment. Static similarity correlates the deformation under constant stress of one system to that of another. If the two systems of different sizes in addition to being geometrically similar exhibit an equal ratio of velocities between corresponding points, they are said to have kinematic similarity. Dynamic similarity is attested when the two systems, small scale and scaled up besides passing geometric and kinematic similarities additionally present the same ratio of forces (gravitational, pressure, centrifugal) between corresponding points. Thermal similarity is established by comparing the thermal ratios of heat fluxes by convection, conduction, radiation and bulk transport. Thermal ratio is assured when the quantity of heat transferred per unit time is constant for the two systems under study. For systems in motion, thermal similarity requires attainment of kinematic similarity. Chemical similarity refers to the degree of the point to point variation in chemical composition of the systems as a function of time and it presumes the attainment of kinematic and thermal similarities. Two systems under investigation are said to be chemically similar if they have comparable concentration gradients as a function of time. The choice of equipment should consider the final dosage form requirements of administration. In general, the PS obtained from HPH process could be more suitable for oral application and that obtained from milling could suit parenteral application because of the smaller particle sizes achievable with milling (Lieberman et al., 1998, Shegokar et al., 2011, Moeschwitzer, 2010a).
5.2. Robust formulation and stability
Pharmaceutical formulators, not surprisingly tend to initiate the formulation development with a trial-and-error method since serendipity has a huge role to play in the pursuit of a successful scale up. However, in order to understand the statistics of the literature or the compiled experimental data and to understand the physics behind the problem, the application of trial and error experimental methods becomes a necessity. Experimental designs are in great demand in the current scenario to handle the formulation development process and to arrive at a rational formulation. The trial and error experimental approach before the start up and the design based experimentation in the subsequent phase equips the formulator with certain practical experience which is a prerequisite to the further creative proceedings. Design of a robust formulation at lab and pilot levels is vital to proceed with scale up study. Development of a robust formulation for the production scale is a further tedious process since the formula robust at the lab scale may or may not serve the purpose at the higher scales. This is because the mere application of the scale up volume ratio concept does not result in the same quality product at production level. Regeneration or alteration of the model developed at lab scale tends to be inevitable unless in cases of luck and serendipity. The whole experimental work may require a repetition at every scale of study and accordingly new optimums need to be derived for the variables. Response surface methodologies and the like could be used to compare different models and to optimize the variables according to the required results (Faure et al., 2001).
Shelf stability is crucial to reap maximum biological benefits of the nanoproduct. Performing short term stability studies at 5 °C or 25 °C and ensuring minimal changes in the particle size, particle size distribution and zeta potential (ZP) for at least one month will suffice to prepare a nanosuspension formulation for preclinical testing. Drug nanoparticulate formulations could be studied for long term stability at 40 °C, room temperature and refrigeration and analyzed for their typical properties. They could be characterized for single NC properties like particle size, zeta potential and drug loading abilities as well as bulk properties like viscosity and redispersability (wherever applicable) (Mishra et al., 2009, Cerdeira et al., 2010). At every stage of study, the noted list of observed changes needs to be evaluated to identify the cause for instability. The drug expulsion, crystal formation, aggregates or crystal growth investigation and the related study needs to be performed to account for the causes like OsR. Stability of NCs is affected by a variety of factors such as nature of drug (small molecule or large biomolecule), delivery route (IV, oral, inhalation or others), dosage form (liquid formulation or dry solid), production technique (top down, bottom up or combination), manufacturing conditions (pressure and temperature), dispersion medium (aqueous or non aqueous), storage and shipping conditions. However, one unanimous consideration is that dry powder formulations have limited stability issues (Kipp, 2004).
5.3. Miscellaneous parameters
Nanoparticulate system is a kind of formulation where the size has it all to do with its properties and unique applications and scale up is a process which as such alters the performance of an operation. So the maintenance of particle size, size distribution and polymorphism poses a particularly big challenge. Whenever spray, freeze or fluid bed drying is involved in drying the nano sized product for producing a finished solid dosage form, the drying effects are to be paid sufficient attention to retain the benefits of nano sizing. The final processed formulation should not only ensure the complete release of the stable nanoproduct in biorelevant media but also that the particle size profile is retained. Variations in the particle size, size distribution of the formulation and the polymorphism of the API alter finished drug product’s in vitro and biological performance. When polymorphism is an issue associated with the API in question, scientists need to produce NPs of consistent size and polymorphic form since polymorphs of an API are compounds with differences in their crystal packing structures as well as bioavailability profiles. So it has to be ensured at the lab level itself that you have a good control over the manufacturing operation in practice and the form of polymorph attained. The final target is always to produce an ideal nano sized formulation with narrow particle size distribution and an ideal and reproducible polymorph wherever applicable.
Literature conveys that polymorphic transitions have not been prominently reported with WBM since most often, it is the heat generated during processing which induces the crystal modifications and in wet media milling, the process uses water which effectively dissipates the generated heat. Further the milling process is carried out under controlled temperature conditions. However performing basic analytical tests before and after the processing will ensure the chemical integrity of the compounds (Merisko-Liversidge et al., 2003). The licensed and regulatory approved products already launched in the market attest the capability of WBM process in maintaining the appropriate polymorphic form of the processed API’s. All the commercialized products contained actives with melting points ranging from 80 °C to >200 °C but as the WBM is performed under controlled conditions, the polymorphic transitions could be eliminated and/ or controlled. Thus the formulation could be optimized with the desired polymorphic form using WBM (Merisko-Liversidge and Liversidge, 2011).
The solid–solid morphology and crystal habit transitions are the phenomena that are widely observed with precipitation technique. When bottom up precipitation is used for manufacturing, the polymorphic form is influenced by processing parameters, type of solvent-antisolvent-stabilizer combination and super saturation degree. Hence the form could be altered by changing the solvent types and modulating the degree of super saturation. But a reproducible and desirable form of polymorphic state is crucial for regulatory approval. Additionally the content of residual solvent during precipitation process should conform with the limit of residual solvents as per ICH guideline Q3C. When a compound is subjected to transition in polymorphic state, it becomes critical to identify the processing conditions which can maintain the compound in the preferred polymorphic state. The solid state characterization to study the amorphous or crystalline nature and the crystalline forms of nanonized compounds could be carried out by analytical testing methods such as FTIR, DSC, XRD and NMR at every stage of processing and formulation development. The general notion is that the solid state forms with highest energy and lowest melting point exhibit the best solubility. But any system at high energy state will tend to lose energy and acquire a lower and stable energy state. Therefore amorphous state though increases saturation solubility, it is still a metastable state with higher energy and may transform to a lower energy stable state during shelf life. Solvent removal and formation of dried solid powder can solve this problem. Amorphousness during the process of precipitation is observed because the molecules get less time to organize themselves in order. Sometimes the solvent molecules may get entrapped into the drug molecules and induce solvomorphism. The analytical testing methods employed should also characterize the prevalence of polyamorphism (existence of the same chemical substance in different amorphous forms) associated with the nanoproducts produced by precipitation techniques. However, the crystal lattice arrangement is an intrinsic property of an API. Thermodynamically, albeit crystalline form is at the lowest, free and stable energy level, some molecules (ibuprofen) always precipitate as crystalline forms irrespective of the solvents used whereas other molecules (cefuroxime axetil) always tend to precipitate as amorphous forms (Sinha et al., 2013, Sharma et al., 2011).
It has also been noticed that the morphology of nanoparticles has its own distinguished effects on the drug bioavailability (Venkataraman et al., 2011). The crystal morphology and shape could be more qualitative aspects of nanocrystals rather than polymeric and lipid NPs. The shape and size of the nanocrystals could be determined by the morphology of the initial crystal material, the plane of fracture of crystals and the drug-stabilizer interactions. If the shape of the initial crystal material is spherical, the drug nanoparticles could be nanosized from 10 microns to 200 nm range with a 50-fold increase in surface to volume ratio (Merisko-Liversidge et al., 2003). Strategic control of crystal morphology and shape is by and large an expertise of precipitation techniques (bottom up approaches) and is a face of research which is yet to be explored effectively. Investigations have commenced in this area to identify the benefits of controlling the crystal morphology and shape of drug nanoparticles. If positive effects on nanoproduct stability and biological performance could be unearthed, it would be really interesting. The molecular transport of solutes induced by the change in process temperature and viscosity of the medium (high drug concentrations) can affect the morphology of final nanocrystal product. High temperatures and drug solution concentrations are known to yield fragile and spherical particles. Literature reports that morphology could be altered by changing the stabilizer type too. For example, PVP (polyvinyl pyrrolidone) with SDS (sodium dodecyl sulfate) yielded rod shaped particles and PEG (poly ethylene glycol) with SDS yielded irregular flake shaped particles (Sinha et al., 2013).
6. Conclusion
The ease of their manufacturing makes NCs the choicest nanoparticles if one needs to check the existence of any correlation between the particle size and drug candidate bioavailability at the discovery phase and initial screening stages. Such feasibility makes an NC formulation a rationale development. The drug NCs being unique nanoformulation options with notable advantages, the researches encompassing scalable formulations/methods for the production of drug NCs open a new vista and pave a new platform for the design of these nanopharmaceuticals. An analysis of the NC research works which either had scalability as their objective or upscaling as their future scope leads to our understanding that few authors applied the same approach for the production of NCs at different scales of study and claimed the significance of similarity preserving equipment. Few others employed different approaches at different scales and therefore used different equipment for producing different scales of products. Few authors uniquely attempted the design of custom made apparatus like a 3-way nozzle model for the production of NCs in a continuous and scalable fashion. While the importance of quality by design in the scale up studies was realized by some authors, prolonged shelf life of finished product aided by the incorporation of stable excipients was proposed by some others. Still few other researchers included the trend of studying the in vivo pharmacokinetics of their scaled up formulations. It was understood that a successful scale up demands an adequate process characterization, proper choice of equipment, development of a robust formula and satisfactory stability study results.
7. Future scope
Irrespective of the scale up targets achieved by the discussed research works, each of such work had been a fruitful attempt to gain beneficial knowledge to carry out future upscaling attempts in the field of NCs. In a nut shell, it could be proposed that a successful scale up demands an adequate process characterization, proper choice of equipment, development of a robust formula and satisfactory stability study results (Srivalli and Mishra, 2014). It was understood that process characterization is required for filtering the processing parameters influencing the product quality; the design of equipment at different scales of study should ensure the consistency in product quality; a robust formulation incorporating proper choice of stabilizers and redispersants is a must to retain the stability of the product all through its shelf life; and stability studies are imperative to determine the stability of the product and to predict its shelf life. More studies based on the scale up of NCs are expected to be taken up by the researchers so as to take an advantage of the product line extension offered by FDA for these products. Future upscaling studies on NCs may incorporate the in vivo studies on animals as a definitive part of their formulation characterization.
Declaration of interest
The authors declare no conflicts of interest.
Table 2.
Examples of scale up based research works on nanocrystals.
| Active/actives | Method | Media | Stabilizer/stabilizers | Refs. |
|---|---|---|---|---|
| Fenofibrate | Controlled crystallization during freeze-drying (made semi-continuous process by applying a 3-way nozzle) | Active in tertiary butyl alcohol and excipients in water | Matrix material, mannitol | de Waard et al. (2009) |
| Meloxicam | Media milling (and subsequent spray drying for characterization) | Water | Hydroxylpropyl methyl cellulose and sodium lauryl sulfate | Singare et al. (2010) |
| Apigenin (flavonoid) | Media milling and subsequent high pressure homogenization (smartCrystal combination technology – CT) | Water | Plantacare 2000® (alkyl polyglycoside) | Shaal et al. (2010) |
| Fenofibrate | Antisolvent precipitation coupled with immediate spray drying | Active in ethanol and excipients in water | Hydroxylpropyl methyl cellulose and sodium dodecyl sulfate | Hu et al. (2011) |
| Phenytoin, pranlukast hydrate and nifedipine | Media milling (and subsequent freeze drying for characterization) | Water | Polyvinyl pyrrolidine and sodium lauryl sulfate | Niwa et al. (2011) |
| Nevirapine | Comparison of high pressure homogenization and media milling (and subsequent air drying for characterization) | Water | Polyvinyl pyrrolidine, poloxamer 188, tween 80, volpoL4, and plasdone | Shegokar et al. (2011) |
| Nitrendipine | Precipitation-homogenization combination and further spray drying | Active in acetone and excipients in water | Polyvinyl alcohol | Quan et al. (2011) |
| NVS-102 (model drug) | Media milling (and subsequent freeze drying for characterization) | Water | d-α-tocopheryl polyethylene glycol 1000 succinate and hydroxylpropyl methyl cellulose | Ghosh et al. (2012) |
Acknowledgment
The first author is grateful to the Director, IIT (BHU) for providing institutional PhD fellowship in the form of teaching assistantship.
Footnotes
Peer review under responsibility of King Saud University.

References
- Abdelwahed W., Degobert G., Stainmesse S., Fessi H. Freeze-drying of nanoparticles formulation, process and storage considerations. Adv. Drug Deliv. Rev. 2006;58:1688–1713. doi: 10.1016/j.addr.2006.09.017. [DOI] [PubMed] [Google Scholar]
- Ali H.S.M., York P., Ali A.M.A., Blagden N. Hydrocortisone nanosuspensions for ophthalmic delivery: a comparative study between microfluidic nanoprecipitation and wet milling. J. Control Release. 2011;149:175–181. doi: 10.1016/j.jconrel.2010.10.007. [DOI] [PubMed] [Google Scholar]
- Cerdeira A.M., Mazzotti M., Gander B. Miconazole nanosuspensions: influence of formulation variables on particle size reduction and physical stability. Int. J. Pharm. 2010;396:210–218. doi: 10.1016/j.ijpharm.2010.06.020. [DOI] [PubMed] [Google Scholar]
- Chan H., Kwok P.C.L. Production methods for nanodrug particles using the bottom-up approach. Adv. Drug Deliv. Rev. 2011;63:406–416. doi: 10.1016/j.addr.2011.03.011. [DOI] [PubMed] [Google Scholar]
- de Waard H., Grasmeijer N., Hinrichs W.L.J., Eissens A.C., Pfaffenbach P.P.F., Frijlink H.W. Preparation of drug nanocrystals by controlled crystallization: application of a 3-way nozzle to prevent premature crystallization for large scale production. Eur. J. Pharm. Sci. 2009;38:224–229. doi: 10.1016/j.ejps.2009.07.005. [DOI] [PubMed] [Google Scholar]
- Deschamps B., Musaji N., Gillespie J.A. Food effect on the bioavailability of two distinct formulations of megesterol acetate oral suspension. Int. J. Nanomed. 2009;4:185–192. doi: 10.2147/ijn.s6308. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dong Y., Ng W.K., Hu J., Shen S., Tan R.B.H. A continuous and highly effective static mixing process for antisolvent precipitation of nanoparticles of poorly water-soluble drugs. Int. J. Pharm. 2010;386:256–261. doi: 10.1016/j.ijpharm.2009.11.007. [DOI] [PubMed] [Google Scholar]
- Eerdenbrugh B.V., Mooter G.V., Augustijns P. Top-down production of drug nanocrystals nanosuspension stabilization, miniaturization and transformation into solid products. Int. J. Pharm. 2008;364:64–75. doi: 10.1016/j.ijpharm.2008.07.023. [DOI] [PubMed] [Google Scholar]
- Eerdenbrugh B.V., Stuyven B., Froyen L., Humbeeck J.V., Martens J.A., Augustijns P., Mooter G.V.D. Downscaling drug nanosuspension production: processing aspects and physicochemical characterization. AAPS PharmSciTech. 2009;10:44–53. doi: 10.1208/s12249-008-9170-5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- El-Gendy N., Pornputtapitak W., Berkland C. Nanoparticle agglomerates of fluticasone propionate in combination with albuterol sulfate as dry powder aerosols. Eur. J. Pharm. Sci. 2011;44:522–533. doi: 10.1016/j.ejps.2011.09.014. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Faure A., York P., Rowe R.C. Process control and scale-up of pharmaceutical wet granulation processes: a review. Eur. J. Pharm. Biopharm. 2001;52:269–277. doi: 10.1016/s0939-6411(01)00184-9. [DOI] [PubMed] [Google Scholar]
- Ganta S., Paxton J.W., Baguley B.C., Garg S. Formulation and pharmacokinetic evaluation of an asulacrine nanocrystalline suspension for intravenous delivery. Int. J. Pharm. 2009;367:179–186. doi: 10.1016/j.ijpharm.2008.09.022. [DOI] [PubMed] [Google Scholar]
- Gao L., Zhang D., Chen M., Duan C., Daia W., Jia L., Zhao W. Studies on pharmacokinetics and tissue distribution of oridonin nanosuspensions. Int. J. Pharm. 2008;355:321–327. doi: 10.1016/j.ijpharm.2007.12.016. [DOI] [PubMed] [Google Scholar]
- Gao L., Zhang D., Chen M. Drug nanocrystals for the formulation of poorly soluble drugs and its application as a potential drug delivery system. J. Nanopart. Res. 2008;10:845–862. [Google Scholar]
- Gasper, R.S., 2010. Nanomedicines on the market and in development: Nanoparticles Ist international workshop on nanomedicine, European medicines agency, 2nd, 3rd, September 2010. London, UK. Available at: <http://www.ema.europa.eu/docs/en_GB/document_library/Presentation/2010/09/WC500096193.pdf>. (accessed 28.01.14).
- Ghosh I., Schenck D., Bose S., Ruegger C. Optimization of formulation and process parameters for the production of nanosuspension by wet media milling technique: effect of vitamin E TPGS and nanocrystal particle size on oral absorption. Eur. J. Pharm. Sci. 2012;47:718–728. doi: 10.1016/j.ejps.2012.08.011. [DOI] [PubMed] [Google Scholar]
- Hanafy A., Spahn-Langguth H., Vergnault G., Grenier P., Tubic Grozdanis M., Lenhardt T., Langguth P. Pharmacokinetic evaluation of oral fenofibrate nanosuspensions and SLN in comparison to conventional suspensions of micronized drug. Adv. Drug Deliv. Rev. 2007;59:419–426. doi: 10.1016/j.addr.2007.04.005. [DOI] [PubMed] [Google Scholar]
- Hu J., Ng W.K., Dong Y., Shen S., Tan R.B.H. Continuous and scalable process for water-redispersible nanoformulation of poorly aqueous soluble APIs by antisolvent precipitation and spray-drying. Int. J. Pharm. 2011;404:198–204. doi: 10.1016/j.ijpharm.2010.10.055. [DOI] [PubMed] [Google Scholar]
- Jacobs C., Müller R.H. Production and characterization of a budesonide nanosuspension for pulmonary administration. Pharm. Res. 2002;19:189–194. doi: 10.1023/a:1014276917363. [DOI] [PubMed] [Google Scholar]
- Kakrana M., Shegokar R., Sahoo N.G., Shaal L.A., Li L., Müller R.H. Fabrication of quercetin nanocrystals: comparison of different methods. Eur. J. Pharm. Biopharm. 2012;80:113–121. doi: 10.1016/j.ejpb.2011.08.006. [DOI] [PubMed] [Google Scholar]
- Kassem M.A., Rahman A.A.A., Ghorab M.M., Ahmed M.B., Khalil R.M. Nanosuspension as an ophthalmic delivery system for certain glucocorticoid drugs. Int. J. Pharm. 2007;340:126–133. doi: 10.1016/j.ijpharm.2007.03.011. [DOI] [PubMed] [Google Scholar]
- Kayser O. A new approach for targeting to Cryptosporidium parvum using mucoadhesive nanosuspensions: research and applications. Int. J. Pharm. 2001;214:83–85. doi: 10.1016/s0378-5173(00)00640-2. [DOI] [PubMed] [Google Scholar]
- Kayser O., Olbrich C., Yardley V., Kiderlen A.F., Croft S.L. Formulation of amphotericin B as nanosuspension for oral administration. Int. J. Pharm. 2003;254:73–75. doi: 10.1016/s0378-5173(02)00686-5. [DOI] [PubMed] [Google Scholar]
- Keck C.M., Müller R.H. Drug nanocrystals of poorly soluble drugs produced by high pressure homogenization. Eur. J. Pharm. Biopharm. 2006;62:3–16. doi: 10.1016/j.ejpb.2005.05.009. [DOI] [PubMed] [Google Scholar]
- Keck C., Kobierski S., Mauludin R., Müller R.H. Second generation of drug nanocrystals for delivery of poorly soluble drugs: smartCrystals technology. Dosis. 2008;24:124–128. [Google Scholar]
- Kipp J.E. The role of solid nanoparticle technology in the parenteral delivery of poorly water-soluble drugs. Int. J. Pharm. 2004;284:109–122. doi: 10.1016/j.ijpharm.2004.07.019. [DOI] [PubMed] [Google Scholar]
- Kocbek P., Baumgartner S., Kristl J. Preparation and evaluation of nanosuspensions for enhancing the dissolution of poorly soluble drugs. Int. J. Pharm. 2006;312:179–186. doi: 10.1016/j.ijpharm.2006.01.008. [DOI] [PubMed] [Google Scholar]
- Levin M. Marcel Dekker; New York: 2002. Pharmaceutical Process Scale-up. [Google Scholar]
- Lieberman H.A., Rioger M.M., Banker G.S. Marcel Dekker; New York: 1998. Pharmaceutical dosage forms: disperse systems, vol. 3. [Google Scholar]
- Mauludin R., Müller R.H., Keck C.M. Development of an oral rutin nanocrystal formulation. Int. J. Pharm. 2009;370:202–209. doi: 10.1016/j.ijpharm.2008.11.029. [DOI] [PubMed] [Google Scholar]
- Merisko-Liversidge E., Liversidge G. Drug nanoparticles: formulating poorly water-soluble compounds. Toxicol. Pathol. 2008;36:43–48. doi: 10.1177/0192623307310946. [DOI] [PubMed] [Google Scholar]
- Merisko-Liversidge E., Liversidge G.G. Nanosizing for oral and parenteral drug delivery: a perspective on formulating poorly-water soluble compounds using wet media milling technology. Adv. Drug Deliv. Rev. 2011;63:427–440. doi: 10.1016/j.addr.2010.12.007. [DOI] [PubMed] [Google Scholar]
- Merisko-Liversidge E., Liversidge G.G., Cooper E.R. Nanosizing: a formulation approach for poorly-water-soluble compounds. Eur. J. Pharm. Sci. 2003;18:113–120. doi: 10.1016/s0928-0987(02)00251-8. [DOI] [PubMed] [Google Scholar]
- Mishra P.R., Shaal L.A., Müller R.H., Keck C.M. Production and characterization of hesperetin nanosuspensions for dermal delivery. Int. J. Pharm. 2009;371:182–189. doi: 10.1016/j.ijpharm.2008.12.030. [DOI] [PubMed] [Google Scholar]
- Mitri K., Shegokar R., Gohla S., Anselmi C., Müller R.H. Lutein nanocrystals as antioxidant formulation for oral and dermal delivery. Int. J. Pharm. 2011;420:141–146. doi: 10.1016/j.ijpharm.2011.08.026. [DOI] [PubMed] [Google Scholar]
- Moeschwitzer J. Nanotechnology: particle size reduction technologies in the pharmaceutical development process. Am. Pharm. Rev. 2010;13:54–59. [Google Scholar]
- Moeschwitzer J. The role of particle size analysis in the development process of nanosized drug products. Am. Pharm. Rev. 2010;13:95–101. [Google Scholar]
- Möschwitzer J.P. Drug nanocrystals in the commercial pharmaceutical development process. Int. J. Pharm. 2013;453:142–156. doi: 10.1016/j.ijpharm.2012.09.034. [DOI] [PubMed] [Google Scholar]
- Muller R.H., Jacobs C. Buparvaquone mucoadhesive nanosuspension: preparation, optimisation and long-term stability. Int. J. Pharm. 2002;237:151–161. doi: 10.1016/s0378-5173(02)00040-6. [DOI] [PubMed] [Google Scholar]
- Müller R.H., Jacobs C., Kayser O. Nanosuspensions as particulate drug formulations in therapy: rationale for development and what we can expect for the future. Adv. Drug Deliv. Rev. 2001;47:3–19. doi: 10.1016/s0169-409x(00)00118-6. [DOI] [PubMed] [Google Scholar]
- Niwa T., Miura S., Danjo K. Universal wet-milling technique to prepare oral nanosuspension focused on discovery and preclinical animal studies – development of particle design method. Int. J. Pharm. 2011;405:218–227. doi: 10.1016/j.ijpharm.2010.12.013. [DOI] [PubMed] [Google Scholar]
- Patravale V.B., Date A.A., Kulkarni R.M. Nanosuspensions a promising drug delivery strategy. J. Pharm. Pharmacol. 2004;56:827–840. doi: 10.1211/0022357023691. [DOI] [PubMed] [Google Scholar]
- Quan P., Xia D., Piao H., Piao H., Shi K., Jia Y., Cui F. Nitrendipine nanocrystals: its preparation, characterization, and in vitro–in vivo evaluation. AAPS PharmSciTech. 2011;12:1136–1143. doi: 10.1208/s12249-011-9682-2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rabinow B., Kipp J., Papadopoulos P., Wong J., Glosson J., Gass J., Sun C.S., Wielgos T., White R., Cook C., Barker K., Wood K. Itraconazole IV nanosuspension enhances efficacy through altered pharmacokinetics in the rat. Int. J. Pharm. 2007;339:251–260. doi: 10.1016/j.ijpharm.2007.02.030. [DOI] [PubMed] [Google Scholar]
- Salazar J., Ghanem A., Müller R.H., Möschwitzer J.P. Nanocrystals: comparison of the size reduction effectiveness of a novel combinative method with conventional top-down approaches. Eur. J. Pharm. Biopharm. 2012;81:82–90. doi: 10.1016/j.ejpb.2011.12.015. [DOI] [PubMed] [Google Scholar]
- Salazar J., Müller R.H., Möschwitzer J.P. Application of the combinative particle size reduction technology H42 to produce fast dissolving glibenclamide tablets. Eur. J. Pharm. Sci. 2013;49:565–577. doi: 10.1016/j.ejps.2013.04.003. [DOI] [PubMed] [Google Scholar]
- Shaal L.A., Müller R.H., Shegokar R. SmartCrystal combination technology – scale up from lab to pilot scale and long term stability. Pharmazie. 2010;65:877–884. [PubMed] [Google Scholar]
- Shaal L.A., Shegokar R., Müller R.H. Production and characterization of antioxidant apigenin nanocrystals as a novel UV skin protective formulation. Int. J. Pharm. 2011;420:133–140. doi: 10.1016/j.ijpharm.2011.08.018. [DOI] [PubMed] [Google Scholar]
- Sharma P., Zujovic Z.D., Bowmaker G.A., Denny W.A., Garg S. Evaluation of a crystalline nanosuspension: polymorphism, process induced transformation and in vivo studies. Int. J. Pharm. 2011;408:138–151. doi: 10.1016/j.ijpharm.2011.01.032. [DOI] [PubMed] [Google Scholar]
- Shegokar R., Müller R.H. Nanocrystals: industrially feasible multifunctional formulation technology for poorly soluble actives. Int. J. Pharm. 2010;399:129–139. doi: 10.1016/j.ijpharm.2010.07.044. [DOI] [PubMed] [Google Scholar]
- Shegokar R., Singh K.K., Müller R.H. Nevirapine nanosuspension: comparative investigation of production methods. Nanotechnol. Dev. 2011;1:16–22. [Google Scholar]
- Shen L., Wu F.L. Nanomedicines in renal transplant rejection—focus on sirolimus. Int. J. Nanomed. 2007;2:25–32. doi: 10.2147/nano.2007.2.1.25. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Singare D.S., Marella S., Gowthamrajan K., Kulkarni G.T., Vooturi R., Rao P.S. Optimization of formulation and process variable of nanosuspension: an industrial perspective. Int. J. Pharm. 2010;402:213–220. doi: 10.1016/j.ijpharm.2010.09.041. [DOI] [PubMed] [Google Scholar]
- Singh S.K., Srinivasan K.K., Gowthamarajan K., Singare D.S., Prakash D., Gaikwad N.B. Investigation of preparation parameters of nanosuspension by top-down media milling to improve the dissolution of poorly water-soluble glyburide. Eur. J. Pharm. Biopharm. 2011;78:441–446. doi: 10.1016/j.ejpb.2011.03.014. [DOI] [PubMed] [Google Scholar]
- Sinha B., Müller R.H., Möschwitzer J.P. Bottom-up approaches for preparing drug nanocrystals: formulations and factors affecting particle size. Int. J. Pharm. 2013;453:126–141. doi: 10.1016/j.ijpharm.2013.01.019. [DOI] [PubMed] [Google Scholar]
- Srivalli K.M.R. and Mishra B., 2014 Drug nanocrystals: Basic quadruple prerequisites for formulation development and upscalability, submitted for publication, Manuscript number SPJ-D-14-00281.
- Venkataraman S., Hedrick J.L., Ong Z.Y., Yang C., Rachel Ee P.L., Hammond P.T., Yang Y.Y. The effects of polymeric nanostructure shape on drug delivery. Adv. Drug Deliv. Rev. 2011;63:1228–1246. doi: 10.1016/j.addr.2011.06.016. [DOI] [PubMed] [Google Scholar]
- Wu Y., Loper A., Landis E., Hettrick L., Novak L., Lynn K., Chen C., Thompson K., Higgins R., Batra U., Shelukar S., Kwei G., Storey D. The role of biopharmaceutics in the development of a clinical nanoparticle formulation of MK-0869: a Beagle dog model predicts improved bioavailability and diminished food effect on absorption in human. Int. J. Pharm. 2004;285:135–146. doi: 10.1016/j.ijpharm.2004.08.001. [DOI] [PubMed] [Google Scholar]
- Yang W., Johnston K.P., Williams R.O. Comparison of bioavailability of amorphous versus crystalline itraconazole nanoparticles via pulmonary administration in rats. Eur. J. Pharm. Biopharm. 2010;75:33–41. doi: 10.1016/j.ejpb.2010.01.011. [DOI] [PubMed] [Google Scholar]
- Zhang J., Wu L., Chan H., Watanabe W. Formation, characterization, and fate of inhaled drug nanoparticles. Adv. Drug Deliv. Rev. 2011;63:441–455. doi: 10.1016/j.addr.2010.11.002. [DOI] [PubMed] [Google Scholar]