

Abstract
Alzheimer's disease (AD) is accepted nowadays as a complex neurodegenerative disorder with multifaceted cerebral pathologies, including extracellular deposition of amyloid β peptide‐containing plaques, intracellular neurofibrillary tangles, progressive loss of cholinergic neurons, metal dyshomeostasis, mitochondrial dysfunction, neuroinflammation, glutamate excitoxicity, oxidative stress and increased MAO enzyme activity. This may explain why it is currently widely accepted that a more effective therapy for AD would result from the use of multifunctional drugs, which may affect more than one brain target involved in the disease pathology. The current review will discuss the potential benefits of novel multimodal neuroprotective, brain permeable drugs, recently developed by Youdim and collaborators, as a valuable therapeutic approach for AD treatment. The pharmacological and neuroprotective properties of these multitarget‐directed ligands, which target MAO enzymes, the cholinergic system, iron accumulation and amyloid β peptide generation/aggregation are described, with a special emphasis on their potential therapeutic value for ageing and AD‐associated cognitive functions. This review is conceived as a tribute to the broad neuropharmacology work of Professor Moussa Youdim, Professor Emeritus in the Faculty of Medicine and Director of Eve Topf Center of Excellence in Technion‐Israel Institute of Technology, and Chief Scientific Officer of ABITAL Pharma Pipeline Ltd., at the occasion of his 75th birthday.
Linked Articles
This article is part of a themed section on Updating Neuropathology and Neuropharmacology of Monoaminergic Systems. To view the other articles in this section visit http://onlinelibrary.wiley.com/doi/10.1111/bph.v173.13/issuetoc
Abbreviations
- Aβ
amyloid β peptide
- AChEIs
AChE inhibitors
- AD
Alzheimer's disease
- APP
amyloid precursor proteins
- BACE 1
β‐site amyloid precursor protein cleaving enzyme 1
- EPO
erythropoietin
- GAP‐43
growth‐associated protein‐43
- GPx
glutathione peroxidase
- HFD
high‐fat diet
- NFT
neurofibrillary tangles
- PHDs
prolyl‐4‐hydroxylases
- PS1
presenilin 1
- RNS
nitrogen species
- SIN‐1
3‐morpholinosydnonimine
- STZ
streptozotocin
- TfR
transferrin receptor
- T2DM
diabetes mellitus
- VAR
- 5′UTR
5′ untranslated region
Tables of Links
| TARGETS | |
|---|---|
| Nuclear hormone receptors a | Enzymes d |
| PPAR‐γ | AChE |
| Catalytic receptors b | BChE |
| InsR | Casein kinase 1δ (ck1δ) |
| TrkB | CDK‐5 |
| Transporters c | GSK‐3β |
| GLUT‐1 | HIF‐1α |
| IRE | |
| iNOS | |
| MAO‐A | |
| MAO‐B |
These Tables list key protein targets and ligands in this article which are hyperlinked to corresponding entries in http://www.guidetopharmacology.org, the common portal for data from the IUPHAR/BPS Guide to PHARMACOLOGY (Pawson et al., 2014) and are permanently archived in the Concise Guide to PHARMACOLOGY 2013/14 (a,b,c,dAlexander et al., 2013a, 2013b, 2013c, 2013d).
Introduction
Alzheimer's disease (AD) is the most prevalent neurodegenerative disease in the elderly population (Bullock, 2004). Its predominant clinical manifestation is progressive memory deterioration and other cognitive functions, including disordered behaviour and impairment in language, comprehension and visual‐spatial skills (Tsolaki et al., 2001; Weintraub et al., 2012). The neuropathology of AD is characterized by several features, including extracellular deposition of amyloid β peptide (Aβ)‐containing plaques in the cerebral cortical regions, accompanied by the presence of intracellular neurofibrillary tangles (NFT, intracellular lesions consisting of paired helical filaments formed of hyperphosphorylated tau) and a progressive loss of basal forebrain cholinergic neurons, leading to reduction in cholinergic parameters, such as ACh and choline acetyltransferase (ChAT) levels and muscarinic and nicotinic ACh receptor binding (Selkoe and Schenk, 2003; Schliebs, 2005). Additionally, there is accumulating evidence indicating that elevated oxidative stress, increased MAO‐B enzyme activity, metal accumulation, ion dyshomeostasis, mitochondrial dysfunction, neuroinflammation, glutamate excitoxicity, gene mutations and an impaired ubiquitin–proteasome system are all involved in the dysfunctional brain network associated with AD (Figure 1) (Smith et al., 2000; Rogers and Lahiri, 2004; Joseph et al., 2005). A significant reduction in 5‐hydroxytryptaminergic and noradrenergic transmission also occurs, which might explain the relatively high incidence of depression found in AD patients (Palmer et al., 1988; Newman, 1999).
Figure 1.

Major mechanisms involved in the pathogenesis of AD. Full explanation is discussed in the text.
Due to the multifactorial aetiology of AD, it is currently widely accepted that a more effective therapy for AD would result from multifunctional drugs, able to simultaneously target the multiple processes involved in the disease pathology. Thus, based on a multimodal drug design paradigm, Youdim and collaborators have developed a series of non‐toxic brain permeable multifunctional compounds, capable of targeting the numerous mechanisms that underlie AD, including MAO enzymes, the cholinergic system, iron accumulation and Aβ generation/aggregation. In this review, we discuss the molecular mechanisms involved and the beneficial effects of these novel multitarget compounds in ageing and AD.
Pathophysiological targets in ageing and AD
The exact biochemical mechanism of the pathogenesis of AD is still unknown, but a marked loss of cholinergic cells and ACh, necessary for cognition and memory, and oxidative stress‐related pathways have been suggested to play an important role in the neurodegenerative processes associated with AD (Maccioni et al., 2001).
The cholinergic system is the primary neurotransmitter system responsible for the cognitive dysfunction in normal ageing and AD (Davies and Maloney, 1976; Perry et al., 1981; Bartus et al., 1982) and thus, restoration of cholinergic function may improve cognitive functions. There are several AChE inhibitors (AChEIs) [rivastigmine (Jann, 2000; Grossberg and Desai, 2001; Machado, 2009), donepezil (Dooley and Lamb, 2000; Doody et al., 2012) and galantamine (Prvulovic et al., 2010)] for the treatment of AD, but despite their mild effectiveness as cognitive enhancers, these drugs have not been shown to significantly affect the neurodegenerative process (Francis et al., 1999; Giacobini, 2002; Lopez et al., 2002; Racchi et al., 2004). Thus, it is widely accepted nowadays that an effective therapeutic strategy in AD is likely to be more complex than simply replacement of lost ACh.
Many lines of evidence suggest that mitochondrial dysfunction and oxidative stress play an important role in the development of neurodegenerative processes in ageing and AD (Schulz et al., 2000; Fukui and Moraes, 2008; Bar‐Am et al., 2015). The apparent imbalance between the generation and removal of reactive oxygen species (ROS) and/or nitrogen species (RNS) potentially leads to free radical‐mediated processes, that target cellular proteins, DNA, lipids and polysaccharides (Andreyev et al., 2005; Zorov et al., 2014). The most consistent defects in mitochondria in ageing and AD are functional deficits in several key mitochondrial respiratory enzymes, mainly mediated by a functional decline in complexes I and IV, and reduced mitochondrial membrane potential and ATP levels (Keil et al., 2006; Su et al., 2010). Iron‐induced lipid peroxidation may mediate alterations in mitochondrial function, as a result of damage to the mitochondrial membrane, resulting in the generation/accumulation of ROS in synaptic mitochondria and an impaired synaptic function (Stauch et al., 2014).
Dysregulation of brain iron homeostasis and enhanced iron accumulation along with reduced antioxidant defence mechanism [e.g. catalase, superoxide dismutase (SOD) and glutathione peroxidase (GPx)] and increased basal lipid peroxidation have been reported in patients with AD and in ageing (Reiter, 1995; Arlt et al., 2002; Montine et al., 2002; Zecca et al., 2004; Raven et al., 2013). It was suggested that increased concentrations of iron might be caused by several factors, including increased blood brain barrier (BBB) permeability, inflammation, redistribution of iron within the brain and modifications in iron homeostasis (Mesquita et al., 2012; Ward et al., 2014). A defective iron homeostasis has been implicated in the misfolding process associated with the two major pathologies in AD: Aβ generation and tau hyperphosphorylation, which may generate oxidative stress by inducing the production of ROS and RNS (Castellani et al., 2004; Moreira et al., 2005; Ward et al., 2014). Indeed, it was found that senile plaques and NFTs contain redox‐active transition metals and may exert pro‐oxidant/antioxidant activities, depending on the balance between neuronal antioxidants and reductants (Sayre et al., 2000).
A direct link between iron metabolism and Aβ pathologies was previously provided by Rogers and collaborators (2002) who demonstrated the presence of an iron‐responsive element (IRE) in the 5′ untranslated region (5′UTR) of the amyloid precursor proteins (APP) transcript. Consequently, APP 5′UTR is selectively responsive to intracellular iron levels in a pattern that reflects iron‐dependent regulation of intracellular APP synthesis. Indeed, iron levels were shown to regulate mRNA translation of APP in neuronal cells, similarly to the mRNAs of ferritin‐L and ferritin‐H (Rogers et al., 2002; Rogers et al., 2008). Overall, these findings led to the hypothesis that chelation therapy should be considered as a valuable therapeutic strategy for AD, to prevent iron‐induced ROS, oxidative stress and Aβ generation/aggregation in specific brain regions (Youdim, 2006).
Furthermore, it was suggested that various redox‐sensitive transcription factors, regulated by oxygen accessibility might bind specific DNA consensus sequences and activate the expression of various genes, particularly those controlling adaptive cellular homeostasis and known to compensate for oxidative stress in the brain (Bergeron et al., 1999; Semenza, 2001). Among such factors, the hypoxia‐inducible factor (HIF)‐1, whose activation state is differentially regulated by the levels of oxygen and iron within its vicinity, is particularly important (Wang and Semenza, 1995; Bergeron et al., 1999; Zaman et al., 1999; Semenza, 2001), since its activation and nuclear translocation may enhance the transcription of specific neuroprotective‐target genes that encode proteins involved in various adaptive/survival cascades (Hewitson and Schofield, 2004; Siddiq et al., 2008). During ageing, HIF‐1α accumulation and signalling activation in the brain are reduced in response to hypoxia and ischaemia (Frenkel‐Denkberg et al., 1999; Chavez and LaManna, 2003). Decreased HIF‐1 was also demonstrated in AD brain (Liu et al., 2008).
An additional contribution to oxidative stress in the brain may arise from the increased activity of the mitochondrial enzymes MAO‐A and MAO‐B, which catalyse the oxidative deamination of various biogenic and xenobiotic amines, including monoamine neurotransmitters, such as 5‐HT, noradrenaline and dopamine (Youdim and Riederer, 2004; Finberg, 2014), with concomitant generation of aldehydes and H2O2 (Halliwell and Gutteridge, 1986). Previously, it was reported that MAO‐B activity is significantly increased in specific brain regions in ageing (Fowler et al., 1980; Strolin Benedetti and Dostert, 1989; Cohen, 2000) and AD (Hirvonen et al., 2009) and may contribute to neurodegenerative processes, secondary to ROS production. Furthermore, MAO‐A activity and gene expression have been found to be up‐regulated in different brain areas of AD patients (Burke et al., 1999; Emilsson et al., 2002; Naoi et al., 2015). It was suggested that decreased levels of the CNS monoamines, such as dopamine, noradrenaline and 5‐HT (Adolfsson et al., 1979) and an altered monoaminergic neurotransmitter system (e.g. the 5‐hydroxytryptaminergic and dopaminergic), demonstrated in the ageing brain (Carlsson, 1987), and particularly in depressed and aggressive patients with AD (Vermeiren et al., 2014), may play an important role in the deterioration of memory and other cognitive functions in ageing and AD (Palmer and DeKosky, 1993; Engelborghs and De Deyn, 1997). This led to the therapeutic strategy of developing dual MAO‐A and MAO‐B inhibitors for the treatment of AD (Youdim and Buccafusco, 2005a; Youdim et al., 2005b), as MAO inhibition might increase amine neurotransmitters, reduce the generation of ROS and exert beneficial neuroprotective effects (Riederer et al., 2004; Finberg, 2014).
Multitarget‐directed compounds
Rasagiline (N‐propargyl‐1‐R‐aminoindan) (Azilect®, Teva pharmaceutical Co. Ltd, Netanya, Israel) is a new generation, highly potent irreversible MAO‐B inhibitor, anti‐Parkinsonian drug (Youdim, 2003). Rasagiline is effective as monotherapy or adjunct therapy to L‐DOPA for early and late PD patients (Parkinson Study Group, 2002; Parkinson Study Group, 2004; Parkinson Study Group, 2005; Olanow et al., 2008; Olanow et al., 2009). Previously, structure‐activity studies have demonstrated that the propargyl moiety confers not only MAO inhibitory activity but also a wide range of neuroprotective/neurorestorative activities, exerted by rasagiline (Weinreb et al., 2005; Weinreb et al., 2010a). Employing the propargyl moiety, we have designed and synthesized a series of multifunctional, non‐toxic, lipophilic brain permeable drugs, including (i) M30 (5‐[N‐methyl‐N‐propargylaminomethyl]‐8‐hydroxyquinoline) and VAR10303 (VAR) (5‐[2‐(methyl‐prop‐2‐ynyl‐amino)‐ethyl]‐quinolin‐8‐ol dihydrochloride) with iron chelating, brain‐selective MAO‐A and MAO‐B inhibitory and neuroprotective activities and HLA20 (5‐[4‐propargylpiperazin‐1‐ylmethyl]‐8‐hydroxyquinoline) with iron chelating, weak MAO inhibitory and neuroprotective activities (Figure 2A). (ii) Ladostigil [(N‐propargyl‐(3R) aminoindan‐5yl)‐ethyl methyl carbamate], which was designed to combine the AChE and butyrylcholinesterase (BChE) inhibitory activities of the anti‐AD drug rivastigmine with the neuroprotective and MAO inhibitory activities of rasagiline (Figure 2B).
Figure 2.
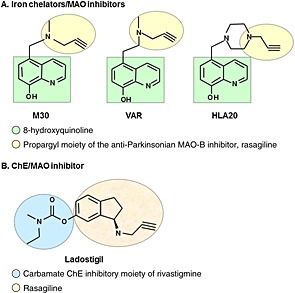
The structures of multifunctional, non‐toxic, lipophilic brain permeable drugs, including (A) M30 (5‐[N‐methyl‐N‐propargylaminomethyl]‐8‐hydroxyquinoline) and VAR10303 (VAR) (5‐[2‐(methyl‐prop‐2‐ynyl‐amino)‐ethyl]‐quinolin‐8‐ol dihydrochloride) with iron chelating, brain selective MAO‐A and MAO‐B inhibitory and neuroprotective activities and HLA20 (5‐[4‐propargylpiperazin‐1‐ylmethyl]‐8‐hydroxyquinoline) with iron chelating, weak MAO inhibitory and neuroprotective activities. The multimodal iron‐chelating compounds, M30, VAR and HLA20 were designed from the prototype brain‐permeable iron chelator, VK28 (5‐[4‐(2‐hydroxyethyl) piperazine‐1‐ylmethyl]‐quinoline‐8‐ol) and enriched with the propargyl moiety of the anti‐Parkinsonian MAO‐B inhibitor, rasagiline. (B) Ladostigil [(N‐propargyl‐(3R) aminoindan‐5yl)‐ethyl methyl carbamate] with neuroprotective, AChE and BChE and brain selective MAO‐A and MAO‐B inhibitory activities. The underlying principle of the design of ladostigil was to amalgamate the carbamate ChE inhibitory moiety of rivastigmine into the 6 position of the pharmacophore of rasagiline.
Iron‐chelating and MAO inhibitory compounds
The multimodal iron‐chelating compounds, M30, VAR and HLA20 were designed from the prototype brain‐permeable iron chelator, VK28 (5‐[4‐(2‐hydroxyethyl) piperazine‐1‐ylmethyl]‐quinoline‐8‐ol) (Varinel Inc., West Chester, PA, USA) and enriched with the propargyl moiety of rasagiline and thus, inherit some of their neuroprotective/neurorestorative properties (Ben‐Shachar et al., 2004; Gal et al., 2005; Zheng et al., 2005a; Avramovich‐Tirosh et al., 2007b; Avramovich‐Tirosh et al., 2007a; Kupershmidt et al., 2009; Avramovich‐Tirosh et al., 2010; Gal et al., 2010).
M30, HLA20 and VAR demonstrated high antioxidant activity against iron‐induced lipid peroxidation with IC50 value comparable with that of the prototype iron chelator, deferoxamine (Zheng et al., 2005a; Bar‐Am et al., 2014). It is well established that strong iron chelators could form inactive complexes with iron and interfere with the Fenton reaction, leading to a decrease in hydroxyl free radical production, and thus block lipid peroxidation. M30, HLA20 and VAR that are shown to possess high iron binding capacity (Zheng et al., 2005b; Bar‐Am et al., 2014) may also be active through this mechanism to inhibit free radical formation. In addition, electron parametric resonance spin‐trapping studies suggest that M30 and HLA20 can behave as radical scavengers to directly scavenge hydroxyl radicals (Zheng et al., 2005c).
Regarding inhibition of MAO activity, M30 and VAR were found to be highly potent brain selective (striatum, hippocampus and cerebellum) inhibitors of both MAO‐A and MAO‐B activities in vivo [IC50 values (μM) for M30: MAO‐A, 0.037 ± 0.02; MAO‐B, 0.057 ± 0.01], with little effect on peripheral MAO activities (liver and small intestine), thus limiting the adverse, potentiating effect of catecholamine releasing agents, such as tyramine, on the cardiovascular system (‘cheese reaction’) (Gal et al., 2005; Zheng et al., 2005a; Bar‐Am et al., 2014). The compound HLA20 was shown to be a weak MAO‐A and MAO‐B inhibitor [IC50 values (μM) for HLA20: MAO‐A, >200; MAO‐B, >50] (Zheng et al., 2005a).
Molecular mechanism of action of neuroprotective effects
Antioxidant/iron‐chelating activities
Our previous study demonstrated that M30 and HLA20 significantly reduced cell mortality induced by H2O2 and the peroxynitrite ion generator, 3‐morpholinosydnonimine (SIN‐1) in mouse motor neuron‐neuroblastoma fusion line, NSC‐34 cells (Kupershmidt et al., 2009). In addition, M30 and HLA20 induced a dose‐dependent increase in transferrin receptor (TfR) levels, further indicating the iron‐chelating effect of these drugs (Kupershmidt et al., 2009). Similarly, recent in vitro studies demonstrated that VAR elicited a significant neuroprotective effect against oxidative stress‐induced damage by H2O2 in human neuroblastoma SH‐SY5Y cells (Bar‐Am et al., 2014). Thus, the antioxidant activity may act synergistically with the direct iron‐chelating effects of these multifunctional iron‐chelating drugs, modulating the toxicity of H2O2‐Fe2+ reactivity. In support of this hypothesis, it was shown that three antioxidant enzymes (catalase, SOD‐1 and GPx) were up‐regulated in a brain region‐dependent manner, by chronic treatment of mice with M30 (Kupershmidt et al., 2011).
Regulation of HIF‐1α cascade
Previous studies have described several iron chelators and molecule inhibitors of prolyl‐4‐hydroxylases (PHDs) as a route to HIF‐1 activation and neuroprotection (see reviews Lee and Andersen, 2006; Harten et al., 2010; Nagel et al., 2010; Weinreb et al., 2010b). Indeed, M30 and HLA20 were found to regulate the HIF‐PHD system, up‐regulate HIF‐1α and significantly increase the levels of HIF‐1‐dependent neuroprotective genes, enolase‐1, vascular endothelial growth factor (VEGF) and brain‐derived neurotrophic factor (BDNF) in NSC‐34 cells (Kupershmidt et al., 2009). In addition, in rat primary embryonic cortical neurons, these multitarget iron chelators up‐regulated HIF‐1α and its dependent protective genes [e.g. TfR, VEGF, BDNF, the glycolytic enzyme enolase 1, the cyclin‐dependent kinase (CDK) inhibitor p21 and erythropoietin (EPO)] (Avramovich‐Tirosh et al., 2010; Maoz et al., 2012). Supporting these findings, it was demonstrated that chronic administration of M30 to adult mice resulted in up‐regulation of HIF‐1α protein levels and HIF‐1 target genes, VEGF, EPO, enolase‐1, TfR, haem oxygenase 1(HO‐1), inducible nitric oxide synthase (iNOS) and glucose transporter (GLUT)‐1 in the brain (Kupershmidt et al., 2011). Given the wide spectrum of cellular functions regulated by HIF‐1‐target genes, it is suggested that this compensatory neuroprotective pathway may be involved in many of the neuroprotective effects of M30.
APP regulation and Aβ reduction
Previous studies demonstrated that M30 reduced APP protein expression in SH‐SY5Y neuroblastoma cells (Avramovich‐Tirosh et al., 2007a) and mice hippocampus (Avramovich‐Tirosh et al., 2007b). It was suggested that this effect is mediated by chelating the intracellular iron pool, resulting in an effect on APP translation via the IRE in 5′UTR of the APP transcript (Rogers et al., 2002; Rogers and Lahiri, 2004). Indeed, M30 was found to suppress the translation of a luciferase receptor mRNA via the 5′UTR sequence that includes the APP IRE (Avramovich‐Tirosh et al., 2007b). Moreover, it was shown that M30 reduced the levels of β‐C‐terminal fragment and Aβ in the medium of Chinese hamster ovary cells, stably transfected with the APP ‘Swedish’ mutation (CHO/ΔNL) (Avramovich‐Tirosh et al., 2007a), and exerted protection against Aβ25‐35‐induced toxicity in primary cultured neurons (Avramovich‐Tirosh et al., 2010). M30 was also found to activate the non‐amyloidogenic pathway of APP processing, thus increasing the levels of secreted sAPPα and α‐C‐terminal fragment in CHO/ΔNL cells (Avramovich‐Tirosh et al., 2007a). Hence, the reduction in Aβ levels, demonstrated in response to M30, may result from both mRNA translational regulation of APP levels, as well as from activation of the non‐amyloidogenic pathway by α‐secretase cleavage. The latter will result in an increased secretion of the neuroprotective sAPPα, at the expense of the production of the neurotoxic Aβ.
Neurite outgrowth and anti‐apoptotic activity
An additional aspect of the mechanism of action of the multitarget iron chelating compounds is cell differentiation and anti‐apoptotic effects. All three compounds, M30, HLA20 and VAR, were demonstrated to stimulate neurite extension and induce neuronal differentiation, accompanied by a significant increase in the expression of the neuronal specific axonal marker of differentiation, growth‐associated protein‐43 (GAP‐43) in SH‐SY5Y and PC12 cells (Avramovich‐Tirosh et al., 2007a; Bar‐Am et al., 2014). Moreover, M30 and HLA20 were shown to increase the number of PC12 cells in G0/G1 and decrease the number in S phase and their proportional number in the G2 phase, indicating that these multitarget compounds can inhibit cell progression beyond G0/G1 phase (Avramovich‐Tirosh et al., 2007a).
With regard to the anti‐apoptotic activity, M30 caused a marked decrease in the level of apoptotic cells, down‐regulated the expression of phosphorylated histone H2A.X (a marker of apoptosis) and cleaved caspase‐3, increased the levels of Bcl‐2 and conversely decreased Bax expression in SH‐SY5Y cells exposed to long‐term serum deprivation (Avramovich‐Tirosh et al., 2007a).
In addition, in recent studies it was demonstrated that HLA20 has neuroprotective potential as it reduced NMDA and non‐NMDA glutamatergic‐mediated excitoxicity in rat primary hippocampal and cortical cultures (Maoz et al., 2012). It was suggested that the neuroprotective effect of HLA20 may be associated, at least in part, with its down‐regulating effect on caspase‐3 and annexin V, and inactivation of the apoptotic pathway involving nuclear translocation of GAPDH and consequent activation of MAO‐B gene transcription. Furthermore, it was shown that HLA20 activated the HIF‐1‐related neuroprotective deacetylase, sirtuin and increased the nuclear content of its downstream effector, the mitochondrial biogenesis‐related PPAR‐γ coactivator‐1α (Maoz et al., 2012).
Neuroprotective/neurorestorative effects in ageing and AD
As regards the ageing process, chronic treatment of aged mice with M30 exerted significant positive effects on neuropsychiatry functions and cognitive age‐related deficits: thus the drug reduced levels of anxiety and aggression, induced locomotor activity and improved memory and nest behaviour (Kupershmidt et al., 2012a). The observed improvement in behavioural deficits was accompanied by a significant reduction in the cerebral accumulation of iron and Aβ plaques (Kupershmidt et al., 2012a) (Table 1). Furthermore, M30 caused a significant irreversible inhibition of both MAO‐A and MAO‐B activities in aged mice brain (Kupershmidt et al., 2012a). The other multifunctional compound in the M30 series, VAR, was also shown to induce neuroprotective effects on age‐related changes in neurobehavioural functions; treatment with VAR in aged rats exerted a significant beneficial effect on depressive‐like behaviour, induced locomotor activity and improved cognitive deficits (Bar‐Am et al., 2014). In addition, assessment of the neuroprotective effects of VAR revealed that the drug enhanced mRNA expression levels of the growth factors, glial cell line‐derived neurotrophic factor (GDNF), nerve growth factor (NGF), BDNF and its receptor Trk B; synaptic plasticity markers (e.g. synapsin‐1 and GAP‐43) and the anti‐apoptotic, Bcl‐2 in the brain of aged rats (Bar‐Am et al., 2014) (Table 1). These findings in aged animals indicate that the novel MAO inhibitor/iron‐chelating drugs, M30 and VAR, acting against multiple brain targets and age‐associated memory impairments, may provide a potential strategic treatment against the progression of age‐related neurodegeneration.
Table 1.
In vivo pharmacological profiles of multitarget‐directed hybrid drugs in ageing
| Multitarget compounds | Species and strain | Dose/experimental design | Behavioural alterations | Molecular alterations | Ref. |
|---|---|---|---|---|---|
| Iron chelators/MAO inhibitors | |||||
| M30 | C57BL mice; male (15 months‐old) | 1 and 5 mg·kg−1, oral gavage 4 times a week for 6 months | Reversed age‐associated memory impairments | Reduced cerebral accumulation of iron and Aβ plaques | (Kupershmidt et al., 2012a) |
| VAR | Sprague‐Dawley rats; male (18 months‐old) | 1 mg·kg−1, s.c. 4 times a week for 6 months | Improved cognitive behavioural deficits | Increased expression levels of GDNF, BDNF, NGF and Bcl‐2; decreased expression levels of Bax | (Bar‐Am et al., 2014) |
| ChE/MAO inhibitor | |||||
| Ladostigil | Wistar rats; male | 1 mg·kg−1·day−1 in drinking water for 4 months | Prevented the loss of novel object discrimination; improved reference memory | Reduced CD11b expression and microglial neuroinflammation in the fornix and parietal cortex | (Weinstock et al., 2013) |
| Rhesus aged macaques; male and female | Chronic escalating regimen, 1–7 mg·kg−1 p.o. | Improved working memory; enhanced motivational aspects | (Buccafusco et al., 2003) | ||
| Wistar rats; male (27 months‐old) | 1 mg·kg−1·day−1, gavage for 30 days | Induced significant changes in hippocampal expression of various mitochondrial and key regulator genes involved in neurodegeneration, cell survival synaptogenesis and oxidation | (Weinreb et al., 2007b) | ||
| Ladostigil | Wistar rats; male (16 months‐old) | 1 mg·kg−1·day−1 in drinking water for 6 months | Prevented the development of spatial memory deficits | Decreased gene expression of IL‐1β, IL‐6, TNF‐α and iNOS in the parietal cortex | (Panarsky et al., 2012) |
| Wistar rats; male (16 months‐old) | 1 mg·kg−1·day−1 in drinking water for 6 months | Improved spatial memory | Prevented increase in activated astrocytes and microglia in hippocampus and white matter areas; increased pro‐NGF in hippocampus; prevented reduction in cortical AChE activity and increase in hippocampal BChE activity; reduced gliosis | (Weinstock et al., 2011) | |
Further studies in AD animal model (Table 2) demonstrated that M30 markedly attenuated various cognitive deficits, including spatial learning and memory retention, working memory, learning abilities, anxiety levels and memory for novel food recognition and nesting behaviour in APP/presenilin‐1 (PS1) transgenic mice (Kupershmidt et al., 2012b). In addition, M30 was found to reduce brain iron accumulation and induce a significant reduction in a number of cerebral AD‐like phenotypes, such as APP, phospho‐APP, Aβ levels, Aβ plaques formation and aggregation, phospho‐tau and CDK‐5, as well as elevation of the levels of phospho‐PKB and phospho‐glycogen synthase kinase (GSK‐3β) (Kupershmidt et al., 2012b). Chronic treatment with M30 significantly elevated cortical insulin (Ins) and insulin receptor (InsR) mRNA and protein expressions, and increased the levels of cerebral HIF‐1α and the expression of HIF‐1‐target genes involved in glycolysis, including aldolase A, enolase‐1 and GLUT‐1 in APP/PS1 mice (Mechlovich et al., 2014a). Treatment with M30 also increased the hepatic protein expression levels of InsR and GLUT‐1 and attenuated the increase in blood glucose levels following glucose tolerance test in APP/PS1 mice (Mechlovich et al., 2014a). These findings indicate that iron chelating drugs, such as M30 may also affect impaired neuronal Ins signalling and GLUT‐1 expression, implicated in AD (Solano et al., 2000; Salkovic‐Petrisic et al., 2006), presumably through its regulation of glucose metabolism.
Table 2.
In vivo pharmacological profiles of multitarget‐directed hybrid drugs in AD
| Multitarget compounds | Species and strain | Dose/experimental design | Behavioural alterations | Molecular alterations | Ref. |
|---|---|---|---|---|---|
| Iron chelators/MAO inhibitors | |||||
| M30 | APP/PS1 mice; male | 1 and 5 mg·kg−1, oral gavage 4 times a week for 9 months | Improved cognitive behaviour deficits; improved memory loss | Reduced APP levels and Aβ levels/plaques aggregation; reduced cerebral iron; modulated cerebral HIF‐1α‐related glycolytic genes and insulin signalling | (Kupershmidt et al., 2012b); (Mechlovich et al., 2014a) |
| STZ‐i.c.v. Wistar rats; male | 10 mg·kg−1·day−1, oral gavage for 5 days | Neuroprotection: prevented development of spatial memory impairment | Decreased catalase activity | (Knezovic et al., 2012); (Sofic et al., 2014) | |
| 2 and 10 mg·kg−1, oral gavage 3 times a week for 11 weeks | Neurorescue: ameliorated already developed cognitive deficits | ||||
| HFD C57BL mice; male | 5 mg·kg−1, oral gavage 3 times a week for 5 months | Modulated cerebral HIF‐1α‐related glucolytic Modulated cerebral HIF‐1a‐related glycolytic‐neuroprotective‐neurogenesis and oxidative stress genes | (Mechlovich et al., 2014b) | ||
| Ob/ob mice; male | 1 mg·kg−1 oral gavage 3 times a week for 5 months | ||||
| HLA20 | STZ‐i.c.v. Wistar rats; male | 5 mg·kg−1·day−1 oral gavage for 5 days | Neuroprotection: prevented development of spatial memory impairment | (Šalković‐Petrišić et al., 2015) | |
| ChE/MAO inhibitor | |||||
| Ladostigil | Adult rats | 25–100 µmol·kg−1; oral gavage (90 min before scopolamine) | Antagonized the impairment of spatial memory | Increased brain ACh | (Weinstock et al., 2000a) |
| STZ‐i.c.v. Sprague‐Dawley rats; male | 1 mg·kg−1·day−1; oral gavage 1 week before until 8 weeks after STZ | Prevented memory deficits | Prevented glial changes and the increase in nitrotyrosine immunoreactivity | (Shoham et al., 2007) | |
| NaN3‐induced cytochrome oxidase inhibition in Sprague‐Dawley rats; male | 17 mg·kg−1·day−1; oral gavage for 5 weeks | Prevented spatial memory deficits | Prevented the decrease in ChAT and the compensatory increase in synaptic plasticity in the subgranular layer of the dentate gyrus and the increase in TfR expression in interneurons in the hilus | (Luques et al., 2007) | |
Considering the close correlation between AD and type 2 diabetes mellitus (T2DM) (Roriz‐Filho et al., 2009; Cholerton et al., 2011), additional studies have recently demonstrated that M30 exerted neuroprotective regulatory effects in high‐fat diet (HFD) and ob/ob transgenic mouse models of T2D (Mechlovich et al., 2014b) (Table 2). Indeed, M30 treatment increased cerebral levels of Ins/InsR and p‐GSK‐3β, produced a significant up‐regulation of cerebral HIF‐1α protein levels and induced the expression of several HIF‐1‐target genes involved in neuroprotection, glycolysis, neurogenesis, oxidative stress and anti‐inflammation in these T2DM mice models (Mechlovich et al., 2014b). In addition, M30 caused a significant inhibition of brain MAO‐A and MAO‐B activities, reduced brain levels of the metabolites of dopamine and increased the levels of 5‐HT and noradrenaline (Mechlovich et al., 2014b). Glucose tolerance was also improved after M30 treatment in both models of T2DM (Mechlovich et al., 2014b). These findings suggest that M30 exerts various beneficial neuroprotective regulatory effects that may act synergistically and delay or prevent AD‐related neurodegenerative processes associated with T2DM.
In the rat model of a sporadic form of AD, developed by i.c.v. administration of streptozotocin (STZ) (Knezovic et al., 2015), M30 was found to exert neuroprotective activity by completely preventing the development of, or ameliorating already developed, cognitive deficits in the preventive or neurorestoration paradigms respectively (Knezovic et al., 2012) (Table 2). M30 was shown to prevent the STZ (administered i.c.v.)‐induced decrease in catalase activity, in a region‐dependent manner (Sofic et al., 2014). Further experiments showed that the multifunctional iron chelator HLA20 also exerted neuroprotective effects in STZ (i.c.v.)‐induced memory impairments (Šalković‐Petrišić et al., 2015) (Table 2).
The ChE and MAO inhibitory compound, ladostigil
The underlying principle of the design of ladostigil was to amalgamate the carbamate ChE inhibitory moiety of the anti‐AD drug, rivastigmine ((S)‐3‐[1‐(dimethylamino)ethyl]phenyl N‐ethyl‐N‐methylcarbamate) into the 6 position of the pharmacophore of rasagiline (Weinstock et al., 2000b; Weinstock et al., 2000a; Weinstock et al., 2001; Sterling et al., 2002). The resulting molecule, ladostigil is a dual AChE‐BChE inhibitor (the inhibitory effect is ~100 times more potent against AChE than BChE) and brain‐selective MAO‐A and MAO‐B inhibitor, with little or no MAO inhibitory effects in the liver and small intestine; an important property that enables this drug to exert only limited potentiation of blood pressure in response to oral tyramine (Weinstock et al., 2001; Youdim and Weinstock, 2001; Weinstock et al., 2002; Weinstock et al., 2006) (presently in stages of completion of Phase IIb clinical study). Previous in vivo studies have shown that administration of ladostigil (26 mg·kg−1) to rats for 2 weeks, inhibited brain MAO‐A and MAO‐B activity by ~70%, with very little or no effect in the intestine (Weinstock et al., 2000a; Weinstock et al., 2002). Chronic treatment of rats with ladostigil for 3 weeks (52 mg·kg−1) was shown to inhibit cerebral ChE activity by ~50% (Sagi et al., 2005).
Molecular mechanism of action of neuroprotective effects
Regulation of cell survival signalling pathways and neurotrophic factors
Previous studies have demonstrated that in apoptotic cells, AChE aggregates in the nucleus, suggesting that AChE might play an important role in cell apoptosis and favour the association between AChE and neuronal apoptosis in AD (Yang et al., 2002). Thus, it is suggested that cholinergic neurons expressing AChE are considerably more vulnerable to apoptosis (Toiber and Soreq, 2005). In agreement with this, it was reported that various AChEIs exhibited neuroprotective effects by increasing the expression levels of anti‐apoptotic/pro‐apoptotic genes and proteins (Villarroya et al., 2004; Wang and Tang, 2005). Indeed, in the neurotoxic high‐density cultured SK‐N‐SH cells, ladostigil was shown to have significant neuroprotective activity, including inhibition of caspase‐3 activation, induction of Bcl‐2 and reduction of Bad and Bax genes and protein expression (Yogev‐Falach et al., 2006). In addition, ladostigil was reported to prevent the fall in mitochondrial membrane potential and thus inhibit the initiation of the apoptotic cascade in SH‐SY5Y cells exposed to SIN‐1 (Maruyama et al., 2003). These protective properties of ladostigil may be associated with its AChE inhibitory activity. Nonetheless, previous studies demonstrated that the propargyl moiety of rasagiline also promoted neuronal survival, mediated by PKC‐dependent and MAPK‐dependent activation, associated with Bcl‐2 family members (Weinreb et al., 2004) and mitochondrial membrane stabilization (Maruyama et al., 2003; Yogev‐Falach et al., 2006), and thus this effect might have also a crucial role in the neuroprotective activity of ladostigil.
In addition, it was demonstrated that the neuroprotective effects of ladostigil involve or result from activation of BDNF/GDNF‐MAPK pathways (Weinreb et al., 2007a). Thus, the elevation of neurotrophic factors, induced by ladostigil, may possibly initiate respective cell signalling cascades, suggesting the involvement of neurotrophic factors may be involved in the molecular mechanism of action of ladostigil.
Antioxidant activity
Ladostigil was found to exert a significant neuroprotective effect against H2O2‐induced damage in SH‐SY5Y cells (Bar‐Am et al., 2009), demonstrating an antioxidant activity via direct scavenging effect on free radicals overproduced in H2O2‐treated neuronal cells, and an indirect effect by stimulating the expression and activity of cellular antioxidant enzymes, such as catalase and glutathione reductase (Weinreb et al., 2008). Similarly, results obtained from high density cytotoxic model of SK‐N‐SH cells, also revealed that ladostigil induced an increase in the mRNA expression levels of these antioxidant enzymes (Bar‐Am et al., 2009). In support of these findings, previous studies showed that ladostigil has neuroprotective effects against other in vitro insults associated with oxidative damage: SIN‐1, serum withdrawal (Yogev‐Falach et al., 2003) and glucose‐oxygen deprivation (Weinstock et al., 2001).
Regulation of APP processing
An essential molecular mechanism of action of ladostigil may involve its ability to regulate the processing of APP by the non‐amyloidogenic α‐secretase pathway (Vetrivel and Thinakaran, 2006; Bar‐Am et al., 2010). In support of this, previous findings demonstrated that ladostigil markedly suppressed holo‐APP protein levels and elevated sAPPα release (Yogev‐Falach et al., 2002; Bar‐Am et al., 2004; Yogev‐Falach et al., 2006), indicating that this drug can be of clinical value for accelerating the non‐amyloidogenic APP processing, thereby reducing Aβ generation. The observation that ladostigil did not alter APP mRNA levels suggests that the decrease in APP protein and Aβ levels can be attributed to suppression of APP translation (Yogev‐Falach et al., 2006). Furthermore, the stimulation of sAPPα release induced by ladostigil was blocked by a hydroxamic acid‐based metalloprotease inhibitor, indicating that α‐secretase and metalloprotease are involved in this effect (Yogev‐Falach et al., 2002). Using several signalling inhibitors it was demonstrated that PKC and MAPK signalling pathways might be involved in the enhancement of sAPPα release by ladostigil (Yogev‐Falach et al., 2006). In accordance with inhibitory studies, it was shown that ladostigil stimulated ERK1‐MAPK and ERK2‐MAPK phosphorylation (Yogev‐Falach et al., 2002). In vivo studies revealed that ladostigil reduced the levels of holo‐APP and up‐regulated the levels of p‐PKC, PKCα and PKCε in the mice hippocampus (Bar‐Am et al., 2004).
Neuroprotective/neurorestoration effects in ageing and AD
Regarding the effect of ladostigil on age‐related alterations, previous in vivo studies demonstrated that the drug improved cognitive deficits in aged monkeys (Buccafusco et al., 2003) and prevented the development of age‐related memory deficits in aged rats (Weinstock et al., 2011; Panarsky et al., 2012; Weinstock et al., 2013) (Table 1). These beneficial effects may be associated with immunomodulatory (microglial activation) and antioxidant effects of ladostigil (Weinreb et al., 2008; Weinstock et al., 2013). In agreement with this hypothesis, aged rats treated with ladostigil showed an increase in pro‐NGF in various brain regions, presumably resulting from enhanced neuronal activity (Weinstock et al., 2011). It was also shown that ladostigil can restore gene expression levels of iNOS, IL‐1β, IL‐6 and TNF‐α in the brain of ageing rats to the respective levels of cognitively intact adult rats (Panarsky et al., 2012).
Additional studies on the neuroprotective activity of ladostigil in aged rats identified significant changes in various hippocampal genes and proteins, related to the iron‐mediated oxidative stress pathway, such as areduction in antioxidant enzymes and induction of ferritin (Weinreb et al., 2007b). It was also found that ladostigil reversed the effect of ageing on mRNA expression levels of various genes associated with metabolism and oxidation processes in the hippocampus of old rats, including the GPx precursor, glutathione S‐transferase, glutathione synthetase, thioredoxin peroxidase and glucose‐6‐phosphate dehydrogenase (Weinreb et al., 2008) (Table 1).
With regard to the potential beneficial effects of ladostigil in AD (Table 2), it was shown that the drug antagonized the spatial memory deficits induced by scopolamine in rats, indicating that the drug caused an increase in brain ACh, sufficient to compete with scopolamine for muscarinic receptors subserving memory (Weinstock et al., 2000a; Weinstock et al., 2001). In addition, ladostigil was shown to prevent gliosis, oxidative‐nitrative stress and memory impairments in the STZ (i.c.v.) rat model of AD (Shoham et al., 2007). Ladostigil also prevented the decrease in ChAT in the diagonal band, and the compensatory increase in synaptic plasticity and TfR and restored spatial memory deficits, induced in rats by the cytochrome oxidase inhibitor, sodium azide (a chronic AD animal model) (Luques et al., 2007). Genomic studies demonstrated that ladostigil significantly down‐regulated rat hippocampal levels of the familial AD‐linked PS1 gene (Weinreb et al., 2007b). This effect can be of value in reducing Aβ formation, because PS1 is a major component of the γ‐secretase complex, which facilitates the generation of Aβ peptide via intramembranous proteolysis of APP (Kern et al., 2006; Vetrivel and Thinakaran, 2006). Furthermore, ladostigil down‐regulated mRNA expression of casein kinase 1δ, associated with pathological hallmarks in several neurodegenerative diseases (Weinreb et al., 2007b).
Concluding remarks and future perspectives
The complex pathology of AD led to the development of single drug molecules with the ability to address multiple pathophysiological targets for the treatments of dementia and AD. This approach may also reduce the risk of drug–drug interactions in AD patients and enhance patient compliance. Accordingly, many research groups have recently designed and synthesized a variety of multifunctional molecules that can interact with multiple symptomatic or AD modifying targets by combining different pharmacophores, mainly derived from existing drugs (e.g. donepezil, tacrine, rivastigmine, rasagiline and selegiline).
An example of these is the series of multitarget MAO‐A and MAO‐B/AChE/BChE inhibitors, which have been designed as hybrids from the AChE inhibitor, donepezil and the selective potent MAO‐B inhibitor, PF9601N (Samadi et al., 2011; Samadi et al., 2012). Among these multitarget hybrid compounds, ASS234 has been shown to retain the anti‐apoptotic and antioxidant properties of PF9601N and to cross the BBB (Bolea et al., 2011; Bolea et al., 2013; Esteban et al., 2014). In vitro results showed that further to its MAO/ChE inhibitory properties, ASS234 can inhibit Aβ1‐42 self‐aggregation and block AChE‐mediated Aβ1‐40/Aβ1‐42 aggregation (Bolea et al., 2013). Additionally, ASS234 reduced Aβ1‐42‐induced toxicity and apoptosis and prevented Aβ1‐42‐mediated depletion of the antioxidant enzymes catalase and SOD‐1 (Bolea et al., 2013). In an experimental model of vascular dementia, it was demonstrated that rats treated with ASS234 tended to perform various working and reference memories better than the untreated control rats (Stasiak et al., 2014). Another multitarget‐directed ligand of AChEI/β‐secretase‐1 (the amyloidogenic β‐site amyloid precursor protein cleaving enzyme 1 (BACE 1) enzyme) inhibitor and anti‐Aβ aggregation (Cavalli et al., 2007; Bolognesi et al., 2009) is the quinone‐bearing polyamine, memoquin (2,5‐bis‐(6‐,[ethyl‐(2‐methoxy‐benzyl)‐amino]‐hexylamino)‐[1,4] benzo‐quinone). Memoquin was shown to restore cholinergic impairments and behavioural deficits linked to attention and memory and to reduce Aβ expression and accumulation and tau hyperphosphorylation and deposition in several AD animal models (Bolognesi et al., 2009). It was also reported that memoquin was able to ameliorate various aspects of cognitive impairments (e.g. spatial, episodic and short‐ term and long‐term memory) in scopolamine‐induced and Aβ‐induced amnesia mouse models (Capurro et al., 2013). In addition, several hybrid multifunctional compounds have been recently developed, synthesized and evaluated as potential multitarget drug candidates for AD, including: a chimeric molecule of tacrine and the selective MAO‐B inhibitor, selegiline (Lu et al., 2013b); o‐hydroxyl and o‐amino benzylamine‐tacrine hybrids (Mao et al., 2012), a series of resveratrol derivatives (Lu et al., 2013a) and tetrahydrobenzo[h][1,6]naphthyridine‐6‐chlorotacrine hybrids, possessing ChE inhibitory and Aβ and tau anti‐aggregation activities (Di Pietro et al., 2014).
The development of a multitarget‐directed ligand that combines more than two pharmacophores would represent an advanced strategy for the design of a more effective drug candidate that can simultaneously modulate various pathological pathways of AD. Youdim, Zheng and other collaborators have recently described a structure‐based strategy for the rational design of more advanced site‐activated multitarget ligands derived from the compound M30, which possess ChE and MAO‐A and MAO‐B inhibitory, as well as site‐activated chelating and neuroprotective activities (Zheng and Monnot, 2012). Preliminary studies have shown that these novel compounds act as pro‐chelators, being metabolized to the active chelator M30 following pseudo‐inhibition of AChE (Zheng et al., 2014). In addition, the synthesis and biological evaluation of a new class of metal chelator‐ChE and MAO inhibitor compounds, derived from donepezil, propargylamine and 8‐hydroxyquinoline for the potential prevention and treatment of AD has been reported (Wang et al., 2014).
For future therapeutic applications of multifunctional compounds that address the multifaceted nature of AD, such as MAO elevation and oxidative stress, Aβ and tau protein accumulation/aggregation, cholinergic hypofunction and iron dyshomeostasis, a detailed pharmacological evaluation should be conducted (e.g. cellular and BBB penetration, metabolite stability and undesired toxic side effects) and presumably, if needed, optimized by structural modifications.
Author contributions
M. B. H. Y., T. A., O. B. and O. W. wrote the manuscript. All authors have revised and approved the manuscript.
Conflict of interest
M. B. H. Y. is CSO of ABITAL Pharma Pipeline Ltd. He is a part owner of the company and has shares.
Acknowledgements
We are gratefully acknowledged the support of the Technion‐Research and Development and Rappaport Family Research Institute, Technion‐Israel Institute of Technology (Haifa, Israel).
Weinreb, O. , Amit, T. , Bar‐Am, O. , and Youdim, M. B. H. (2016) Neuroprotective effects of multifaceted hybrid agents targeting MAO, cholinesterase, iron and β‐amyloid in ageing and Alzheimer's disease. British Journal of Pharmacology, 173: 2080–2094. doi: 10.1111/bph.13318.
References
- Adolfsson R, Gottfries CG, Roos BE, Winblad B (1979). Changes in the brain catecholamines in patients with dementia of Alzheimer type. Br J Psychiatry 135: 216–223. [DOI] [PubMed] [Google Scholar]
- Alexander SPH, Benson HE, Faccenda E, Pawson AJ, Sharman JL, Spedding M et al. (2013a). The concise guide to PHARMACOLOGY 2013/14: nuclear hormone receptors. Br J Pharmacol 170: 1652–1675. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Alexander SPH, Benson HE, Faccenda E, Pawson AJ, Sharman JL, Spedding M et al. (2013b). The concise guide to PHARMACOLOGY 2013/14: catalytic receptors. Br J Pharmacol 170: 1676–1705. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Alexander SPH, Benson HE, Faccenda E, Pawson AJ, Sharman JL, Spedding M et al. (2013c). The concise guide to PHARMACOLOGY 2013/14: transporters. Br J Pharmacol 170: 1706–1796. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Alexander SPH, Benson HE, Faccenda E, Pawson AJ, Sharman JL, Spedding M et al. (2013d). The concise guide to PHARMACOLOGY 2013/14: enzymes. Br J Pharmacol 170: 1797–1867. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Andreyev AY, Kushnareva YE, Starkov AA (2005). Mitochondrial metabolism of reactive oxygen species. Biochemistry (Mosc) 70: 200–214. [DOI] [PubMed] [Google Scholar]
- Arlt S, Beisiegel U, Kontush A (2002). Lipid peroxidation in neurodegeneration: new insights into Alzheimer's disease. Curr Opin Lipidol 13: 289–294. [DOI] [PubMed] [Google Scholar]
- Avramovich‐Tirosh Y, Amit T, Bar‐Am O, Zheng H, Fridkin M, Youdim MB (2007a). Therapeutic targets and potential of the novel brain‐permeable multifunctional iron chelator‐monoamine oxidase inhibitor drug, M‐30, for the treatment of Alzheimer's disease. J Neurochem 100: 490–502. [DOI] [PubMed] [Google Scholar]
- Avramovich‐Tirosh Y, Bar‐Am O, Amit T, Youdim MB, Weinreb O (2010). Up‐regulation of hypoxia‐inducible factor (HIF)‐1α and HIF‐target genes in cortical neurons by the novel multifunctional iron chelator anti‐Alzheimer drug, M30. Curr Alzheimer Res 7: 300–306. [DOI] [PubMed] [Google Scholar]
- Avramovich‐Tirosh Y, Reznichenko L, Mit T, Zheng H, Fridkin M, Weinreb O et al. (2007b). Neurorescue activity, APP regulation and amyloid‐beta peptide reduction by novel multi‐functional brain permeable iron‐chelating‐antioxidants, M‐30 and green tea polyphenol, EGCG. Curr Alzheimer Res 4: 403–411. [DOI] [PubMed] [Google Scholar]
- Bar‐Am O, Amit T, Kupershmidt L, Aluf Y, Mechlovich D, Kabha H et al. (2014). Neuroprotective and neurorestorative activities of a novel iron chelator‐brain selective monoamine oxidase‐A/monoamine oxidase‐B inhibitor in animal models of Parkinson's disease and aging. Neurobiol Aging 36: 1529–1542. [DOI] [PubMed] [Google Scholar]
- Bar‐Am O, Amit T, Weinreb O, Youdim MB, Mandel S (2010). Propargylamine containing compounds as modulators of proteolytic cleavage of amyloid‐beta protein precursor: involvement of mitogen activated protein kinase and protein kinase C activation. J Alzheimers Dis 21: 361–371. [DOI] [PubMed] [Google Scholar]
- Bar‐Am O, Amit T, Youdim MB, Weinreb O (2015). Neuroprotective and neurorestorative potential of propargylamine derivatives in ageing: focus on mitochondrial targets. J Neural Transm . doi:10.1007/s00702-015-1395-3. [DOI] [PubMed] [Google Scholar]
- Bar‐Am O, Weinreb O, Amit T, Youdim MB (2009). The novel cholinesterase‐monoamine oxidase inhibitor and antioxidant, ladostigil, confers neuroprotection in neuroblastoma cells and aged rats. J Mol Neurosci 37: 135–145. [DOI] [PubMed] [Google Scholar]
- Bar‐Am O, Yogev‐Falach M, Amit T, Sagi Y, Youdim MBH (2004). Regulation of protein kinase C by the anti‐Parkinson drug, MAO‐B inhibitor, rasagiline and its derivatives, in vivo . J Neurochem 89: 1119–1125. [DOI] [PubMed] [Google Scholar]
- Bartus RT, Dean RL 3rd, Beer B, Lippa AS (1982). The cholinergic hypothesis of geriatric memory dysfunction. Science 217: 408–414. [DOI] [PubMed] [Google Scholar]
- Ben‐Shachar D, Kahana N, Kampel V, Warshawsky A, Youdim MBH (2004). Neuroprotection by a novel brain permeable iron chelator, VK‐28, against 6‐hydroxydopamine lession in rats. Neuropharmacology 46: 254–263. [DOI] [PubMed] [Google Scholar]
- Bergeron M, Yu AY, Solway KE, Semenza GL, Sharp FR (1999). Induction of hypoxia‐inducible factor‐1 (HIF‐1) and its target genes following focal ischaemia in rat brain. Eur J Neurosci 11: 4159–4170. [DOI] [PubMed] [Google Scholar]
- Bolea I, Gella A, Monjas L, Perez C, Rodriguez‐Franco MI, Marco‐Contelles J et al. (2013). Multipotent, permeable drug ASS234 inhibits Abeta aggregation, possesses antioxidant properties and protects from Abeta‐induced apoptosis in vitro. Curr Alzheimer Res 10: 797–808. [DOI] [PubMed] [Google Scholar]
- Bolea I, Juarez‐Jimenez J, de Los RC, Chioua M, Pouplana R, Luque FJ et al. (2011). Synthesis, biological evaluation, and molecular modeling of donepezil and N‐[(5‐(benzyloxy)‐1‐methyl‐1H‐indol‐2‐yl)methyl]‐N‐methylprop‐2‐yn‐1‐amine hybrids as new multipotent cholinesterase/monoamine oxidase inhibitors for the treatment of Alzheimer's disease. J Med Chem 54: 8251–8270. [DOI] [PubMed] [Google Scholar]
- Bolognesi ML, Cavalli A, Melchiorre C (2009). Memoquin: a multi‐target‐directed ligand as an innovative therapeutic opportunity for Alzheimer's disease. Neurotherapeutics 6: 152–162. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Buccafusco JJ, Terry AV Jr, Goren T, Blaugrun E (2003). Potential cognitive actions of (n‐propargly‐(3r)‐aminoindan‐5‐yl)‐ethyl, methyl carbamate (tv3326), a novel neuroprotective agent, as assessed in old rhesus monkeys in their performance of versions of a delayed matching task. Neuroscience 119: 669–678. [DOI] [PubMed] [Google Scholar]
- Bullock R (2004). Future directions in the treatment of Alzheimer's disease. Expert Opin Investig Drugs 13: 303–314. [DOI] [PubMed] [Google Scholar]
- Burke WJ, Li SW, Schmitt CA, Xia P, Chung HD, Gillespie KN (1999). Accumulation of 3,4‐dihydroxyphenylglycolaldehyde, the neurotoxic monoamine oxidase A metabolite of norepinephrine, in locus ceruleus cell bodies in Alzheimer's disease: mechanism of neuron death. Brain Res 816: 633–637. [DOI] [PubMed] [Google Scholar]
- Capurro V, Busquet P, Lopes JP, Bertorelli R, Tarozzo G, Bolognesi ML et al. (2013). Pharmacological characterization of memoquin, a multi‐target compound for the treatment of Alzheimer's disease. PLoS One 8: e56870. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Carlsson A (1987). Brain neurotransmitters in aging and dementia: similar changes across diagnostic dementia groups. Gerontology 33: 159–167. [DOI] [PubMed] [Google Scholar]
- Castellani RJ, Honda K, Zhu X, Cash AD, Nunomura A, Perry G et al. (2004). Contribution of redox‐active iron and copper to oxidative damage in Alzheimer disease. Ageing Res Rev 3: 319–326. [DOI] [PubMed] [Google Scholar]
- Cavalli A, Bolognesi ML, Capsoni S, Andrisano V, Bartolini M, Margotti E et al. (2007). A small molecule targeting the multifactorial nature of Alzheimer's disease. Angew Chem Int Ed Engl 46: 3689–3692. [DOI] [PubMed] [Google Scholar]
- Chavez JC, LaManna JC (2003). Hypoxia‐inducible factor‐1alpha accumulation in the rat brain in response to hypoxia and ischemia is attenuated during aging. Adv Exp Med Biol 510: 337–341. [DOI] [PubMed] [Google Scholar]
- Cholerton B, Baker LD, Craft S (2011). Insulin resistance and pathological brain ageing. Diabet Med 28: 1463–1475. [DOI] [PubMed] [Google Scholar]
- Cohen G (2000). Oxidative stress, mitochondrial respiration, and Parkinson's disease. Ann New York Acad Sci 899: 112–120. [DOI] [PubMed] [Google Scholar]
- Davies P, Maloney AJ (1976). Selective loss of central cholinergic neurons in Alzheimer's disease. Lancet 2: 1403. [DOI] [PubMed] [Google Scholar]
- Di Pietro O, Perez‐Areales FJ, Juarez‐Jimenez J, Espargaro A, Clos MV, Perez B et al. (2014). Tetrahydrobenzo[h][1,6]naphthyridine‐6‐chlorotacrine hybrids as a new family of anti‐Alzheimer agents targeting beta‐amyloid, tau, and cholinesterase pathologies. Eur J Med Chem 84: 107–117. [DOI] [PubMed] [Google Scholar]
- Doody RS, Cummings JL, Farlow MR (2012). Reviewing the role of donepezil in the treatment of Alzheimer's disease. Curr Alzheimer Res 9: 773–781. [DOI] [PubMed] [Google Scholar]
- Dooley M, Lamb HM (2000). Donepezil: a review of its use in Alzheimer's disease. Drugs Aging 16: 199–226. [DOI] [PubMed] [Google Scholar]
- Emilsson L, Saetre P, Balciuniene J, Castensson A, Cairns N, Jazin EE (2002). Increased monoamine oxidase messenger RNA expression levels in frontal cortex of Alzheimer's disease patients. Neurosci Lett 326: 56–60. [DOI] [PubMed] [Google Scholar]
- Engelborghs S, De Deyn PP (1997). The neurochemistry of Alzheimer's disease. Acta Neurol Belg 97: 67–84. [PubMed] [Google Scholar]
- Esteban G, Allan J, Samadi A, Mattevi A, Unzeta M, Marco‐Contelles J et al. (2014). Kinetic and structural analysis of the irreversible inhibition of human monoamine oxidases by ASS234, a multi‐target compound designed for use in Alzheimer's disease. Biochim Biophys Acta 1844: 1104–1110. [DOI] [PubMed] [Google Scholar]
- Finberg JP (2014). Update on the pharmacology of selective inhibitors of MAO‐A and MAO‐B: focus on modulation of CNS monoamine neurotransmitter release. Pharmacol Ther 143: 133–152. [DOI] [PubMed] [Google Scholar]
- Fowler CJ, Wiberg A, Oreland L, Marcusson J, Winblad B (1980). The effect of age on the activity and molecular properties of human brain monoamine oxidase. J Neural Transm 49: 1–20. [DOI] [PubMed] [Google Scholar]
- Francis PT, Palmer AM, Snape M, Wilcock GK (1999). The cholinergic hypothesis of Alzheimer's disease: a review of progress. J Neurol Neurosurg Psychiatry 66: 137–147. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Frenkel‐Denkberg G, Gershon D, Levy AP (1999). The function of hypoxia‐inducible factor 1 (HIF‐1) is impaired in senescent mice. FEBS Lett 462: 341–344. [DOI] [PubMed] [Google Scholar]
- Fukui H, Moraes CT (2008). The mitochondrial impairment, oxidative stress and neurodegeneration connection: reality or just an attractive hypothesis? Trends Neurosci 31: 251–256. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gal S, Zheng H, Fridkin M, Youdim MB (2005). Novel multifunctional neuroprotective iron chelator‐monoamine oxidase inhibitor drugs for neurodegenerative diseases. In vivo selective brain monoamine oxidase inhibition and prevention of MPTP‐induced striatal dopamine depletion. J Neurochem 95: 79–88. [DOI] [PubMed] [Google Scholar]
- Gal S, Zheng H, Fridkin M, Youdim MB (2010). Restoration of nigrostriatal dopamine neurons in post‐MPTP treatment by the novel multifunctional brain‐permeable iron chelator‐monoamine oxidase inhibitor drug, M30. Neurotox Res 17: 15–27. [DOI] [PubMed] [Google Scholar]
- Giacobini E (2002). Long‐term stabilizing effect of cholinesterase inhibitors in the therapy of Alzheimer' disease. J Neural Transm Suppl 62: 181–187. [DOI] [PubMed] [Google Scholar]
- Grossberg G, Desai A (2001). Review of rivastigmine and its clinical applications in Alzheimer's disease and related disorders. Expert Opin Pharmacother 2: 653–666. [DOI] [PubMed] [Google Scholar]
- Halliwell B, Gutteridge JM (1986). Oxygen free radicals and iron in relation to biology and medicine: some problems and concepts. Arch Biochem Biophys 246: 501–514. [DOI] [PubMed] [Google Scholar]
- Harten SK, Ashcroft M, Maxwell PH (2010). Prolyl hydroxylase domain (PHD) inhibitors: a route to HIF activation & neuroprotection. Antioxid Redox Signal 12: 459–480. [DOI] [PubMed] [Google Scholar]
- Hewitson KS, Schofield CJ (2004). The HIF pathway as a therapeutic target. Drug Discov Today 9: 704–711. [DOI] [PubMed] [Google Scholar]
- Hirvonen J, Kailajarvi M, Haltia T, Koskimies S, Nagren K, Virsu P et al. (2009). Assessment of MAO‐B occupancy in the brain with PET and [11C]‐L‐deprenyl‐D2: a dose‐finding study with a novel MAO‐B inhibitor, EVT 301. Clin Pharmacol Ther 85: 506–512. [DOI] [PubMed] [Google Scholar]
- Jann MW (2000). Rivastigmine, a new‐generation cholinesterase inhibitor for the treatment of Alzheimer's disease. Pharmacotherapy 20: 1–12. [DOI] [PubMed] [Google Scholar]
- Joseph JA, Shukitt‐Hale B, Casadesus G, Fisher D (2005). Oxidative stress and inflammation in brain aging: nutritional considerations. Neurochem Res 30: 927–935. [DOI] [PubMed] [Google Scholar]
- Keil U, Hauptmann S, Bonert A, Scherping I, Eckert A, Muller WE (2006). Mitochondrial dysfunction induced by disease relevant AbetaPP and tau protein mutations. J Alzheimers Dis 9: 139–146. [DOI] [PubMed] [Google Scholar]
- Kern A, Roempp B, Prager K, Walter J, Behl C (2006). Down‐regulation of endogenous amyloid precursor protein processing due to cellular aging. J Biol Chem 281: 2405–2413. [DOI] [PubMed] [Google Scholar]
- Knezovic A, Knapic M, Osmanovic‐Barilar J, Mandel S, Youdim M, Riederer P et al. (2012). Therapeutic potential of a novel multifunctional iron chelator on cognitive deficits and insulin degrading enzyme expression in a rat model of sporadic Alzheimer's disease. BMC Pharmacol Toxicol 13 (Suppl 1): A65. [Google Scholar]
- Knezovic A, Osmanovic‐Barilar J, Curlin M, Hof PR, Simic G, Riederer P et al. (2015). Staging of cognitive deficits and neuropathological and ultrastructural changes in streptozotocin‐induced rat model of Alzheimer's disease. J Neural Transm 122: 577–592. [DOI] [PubMed] [Google Scholar]
- Kupershmidt L, Amit T, Bar‐Am O, Youdim MB, Weinreb O (2012a). Neuroprotection by the multitarget iron chelator M30 on age‐related alterations in mice. Mech Ageing Dev 133: 267–274. [DOI] [PubMed] [Google Scholar]
- Kupershmidt L, Amit T, Bar‐Am O, Youdim MB, Weinreb O (2012b). The novel multi‐target iron chelating‐radical scavenging compound m30 possesses beneficial effects on major hallmarks of Alzheimer's disease. Antioxid Redox Signal 17: 860–877. [DOI] [PubMed] [Google Scholar]
- Kupershmidt L, Weinreb O, Amit T, Mandel S, Bar‐Am O, Youdim MB (2011). Novel molecular targets of the multi‐functional brain‐permeable iron chelating drug M30 in mouse brain. Neuroscience 189: 345–358. [DOI] [PubMed] [Google Scholar]
- Kupershmidt L, Weinreb O, Amit T, Mandel S, Carri MT, Youdim MB (2009). Neuroprotective and neuritogenic activities of novel multimodal iron‐chelating drugs in motor‐neuron‐like NSC‐34 cells and transgenic mouse model of amyotrophic lateral sclerosis. FASEB J 23: 3766–3779. [DOI] [PubMed] [Google Scholar]
- Lee DW, Andersen JK (2006). Role of HIF‐1 in iron regulation: potential therapeutic strategy for neurodegenerative disorders. Curr Mol Med 6: 883–893. [DOI] [PubMed] [Google Scholar]
- Liu Y, Liu F, Iqbal K, Grundke‐Iqbal I, Gong CX (2008). Decreased glucose transporters correlate to abnormal hyperphosphorylation of tau in Alzheimer disease. FEBS Lett 582: 359–364. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lopez OL, Becker JT, Wisniewski S, Saxton J, Kaufer DI, DeKosky ST (2002). Cholinesterase inhibitor treatment alters the natural history of Alzheimer's disease. J Neurol Neurosurg Psychiatry 72: 310–314. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lu C, Guo Y, Yan J, Luo Z, Luo HB, Yan M et al. (2013a). Design, synthesis, and evaluation of multitarget‐directed resveratrol derivatives for the treatment of Alzheimer's disease. J Med Chem 56: 5843–5859. [DOI] [PubMed] [Google Scholar]
- Lu C, Zhou Q, Yan J, Du Z, Huang L, Li X (2013b). A novel series of tacrine‐selegiline hybrids with cholinesterase and monoamine oxidase inhibition activities for the treatment of Alzheimer's disease. Eur J Med Chem 62: 745–753. [DOI] [PubMed] [Google Scholar]
- Luques L, Shoham S, Weinstock M (2007). Chronic brain cytochrome oxidase inhibition selectively alters hippocampal cholinergic innervation and impairs memory: prevention by ladostigil. Exp Neurol 206: 209–219. [DOI] [PubMed] [Google Scholar]
- Maccioni RB, Munoz JP, Barbeito L (2001). The molecular bases of Alzheimer's disease and other neurodegenerative disorders. Arch Med Res 32: 367–381. [DOI] [PubMed] [Google Scholar]
- Machado JC (2009). Review: rivastigmine reduces rate of cognitive decline and improves performance in mild to moderate Alzheimer's. Evid Based Ment Health 12: 113. [DOI] [PubMed] [Google Scholar]
- Mao F, Huang L, Luo Z, Liu A, Lu C, Xie Z et al. (2012). O‐Hydroxyl‐ or o‐amino benzylamine‐tacrine hybrids: multifunctional biometals chelators, antioxidants, and inhibitors of cholinesterase activity and amyloid‐beta aggregation. Bioorg Med Chem 20: 5884–5892. [DOI] [PubMed] [Google Scholar]
- Maoz E, Bar‐am O, Amit T, Mandel S, Youdim MBH (2012). Activation of the mitochondrial biogenesis‐related factor PGC‐1alpha by the multitarget neurotrophic drug, HLA20, in excitotoxicity‐induced damage of primary hippocampal and cortical neurons. J Mol Neurosci 48 (Suppl 1): S76. [Google Scholar]
- Maruyama W, Weinstock M, Youdim MB, Nagai M, Naoi M (2003). Anti‐apoptotic action of anti‐Alzheimer drug, TV3326 [(N‐propargyl)‐(3R)‐aminoindan‐5‐yl]‐ethyl methyl carbamate, a novel cholinesterase‐ monoamine oxidase inhibitor. Neurosci Lett 341: 233–236. [DOI] [PubMed] [Google Scholar]
- Mechlovich D, Amit T, Bar‐Am O, Mandel S, Youdim MB, Weinreb O (2014a). The novel multi‐target iron chelator, M30 modulates HIF‐1alpha‐related glycolytic genes and insulin signaling pathway in the frontal cortex of APP/PS1 Alzheimer's disease mice. Curr Alzheimer Res 11: 119–127. [DOI] [PubMed] [Google Scholar]
- Mechlovich D, Amit T, Bar‐Am O, Weinreb O, Youdim MB (2014b). Molecular targets of the multifunctional iron‐chelating drug, M30, in the brains of mouse models of type 2 diabetes mellitus. Br J Pharmacol 171: 5636–5649. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mesquita SD, Ferreira AC, Sousa JC, Santos NC, Correia‐Neves M, Sousa N et al. (2012). Modulation of iron metabolism in aging and in Alzheimer's disease: relevance of the choroid plexus. Front Cell Neurosci 6: 25. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Montine TJ, Neely MD, Quinn JF, Beal MF, Markesbery WR, Roberts LJ et al. (2002). Lipid peroxidation in aging brain and Alzheimer's disease. Free Radic Biol Med 33: 620–626. [DOI] [PubMed] [Google Scholar]
- Moreira PI, Honda K, Liu Q, Santos MS, Oliveira CR, Aliev G et al. (2005). Oxidative stress: the old enemy in Alzheimer's disease pathophysiology. Curr Alzheimer Res 2: 403–408. [DOI] [PubMed] [Google Scholar]
- Nagel S, Talbot NP, Mecinovic J, Smith TG, Buchan AM, Schofield CJ (2010). Therapeutic manipulation of the HIF hydroxylases. Antioxid Redox Signal 12: 481–501. [DOI] [PubMed] [Google Scholar]
- Naoi M, Riederer P, Maruyama W (2015). Modulation of monoamine oxidase (MAO) expression in neuropsychiatric disorders: genetic and environmental factors involved in type A MAO expression. J Neural Transm. [DOI] [PubMed] [Google Scholar]
- Newman SC (1999). The prevalence of depression in Alzheimer's disease and vascular dementia in a population sample. J Affect Disord 52: 169–176. [DOI] [PubMed] [Google Scholar]
- Olanow CW, Hauser RA, Jankovic J, Langston W, Lang A, Poewe W et al. (2008). A randomized, double‐blind, placebo‐controlled, delayed start study to assess rasagiline as a disease modifying therapy in Parkinson's disease (the ADAGIO study): rationale, design, and baseline characteristics. Mov Disord 23: 2194–2201. [DOI] [PubMed] [Google Scholar]
- Olanow CW, Rascol O, Hauser R, Feigin PD, Jankovic J, Lang A et al. (2009). A double‐blind, delayed‐start trial of rasagiline in Parkinson's disease. N Engl J Med 361: 1268–1278. [DOI] [PubMed] [Google Scholar]
- Palmer AM, DeKosky ST (1993). Monoamine neurons in aging and Alzheimer's disease. J Neural Transm Gen Sect 91: 135–159. [DOI] [PubMed] [Google Scholar]
- Palmer AM, Stratmann GC, Procter AW, Bowen DM (1988). Possible neurotransmitter basis of behavioral changes in Alzheimer's disease. Ann Neurol 23: 616–620. [DOI] [PubMed] [Google Scholar]
- Panarsky R, Luques L, Weinstock M (2012). Anti‐inflammatory effects of ladostigil and its metabolites in aged rat brain and in microglial cells. J Neuroimmune Pharmacol 7: 488–498. [DOI] [PubMed] [Google Scholar]
- Parkinson Study Group (2002). A controlled trial of rasagiline in early Parkinson disease: the TEMPO study. Arch Neurol 59: 1937–1943. [DOI] [PubMed] [Google Scholar]
- Parkinson Study Group (2004). A controlled, randomized, delayed‐start study of rasagiline in early Parkinson disease. Arch Neurol 61: 561–566. [DOI] [PubMed] [Google Scholar]
- Parkinson Study Group (2005). A randomized placebo‐controlled trial of rasagiline in levodopa‐treated patients with Parkinson disease and motor fluctuations: the PRESTO study. Arch Neurol 62: 241–248. [DOI] [PubMed] [Google Scholar]
- Pawson AJ, Sharman JL, Benson HE, Faccenda E, Alexander SP, Buneman OP et al. (2014). The IUPHAR/BPS Guide to PHARMACOLOGY: an expert‐driven knowledgebase of drug targets and their ligands. Nucl. Acids Res. 42 (Database Issue): D1098–106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Perry EK, Tomlinson BE, Blessed G, Perry RH, Cross AJ, Crow TT (1981). Noradrenergic and cholinergic systems in senile dementia of Alzheimer type. Lancet 2: 149. [DOI] [PubMed] [Google Scholar]
- Prvulovic D, Hampel H, Pantel J (2010). Galantamine for Alzheimer's disease. Expert Opin Drug Metab Toxicol 6: 345–354. [DOI] [PubMed] [Google Scholar]
- Racchi M, Mazzucchelli M, Porrello E, Lanni C, Govoni S (2004). Acetylcholinesterase inhibitors: novel activities of old molecules. Pharmacol Res 50: 441–451. [DOI] [PubMed] [Google Scholar]
- Raven EP, Lu PH, Tishler TA, Heydari P, Bartzokis G (2013). Increased iron levels and decreased tissue integrity in hippocampus of Alzheimer's disease detected in vivo with magnetic resonance imaging. J Alzheimers Dis 37: 127–136. [DOI] [PubMed] [Google Scholar]
- Reiter RJ (1995). Oxidative processes and antioxidative defense mechanisms in the aging brain. FASEB J 9: 526–533. [PubMed] [Google Scholar]
- Riederer P, Danielczyk W, Grunblatt E (2004). Monoamine oxidase‐B inhibition in Alzheimer's disease. Neurotoxicology 25: 271–277. [DOI] [PubMed] [Google Scholar]
- Rogers JT, Bush AI, Cho HH, Smith DH, Thomson AM, Friedlich AL et al. (2008). Iron and the translation of the amyloid precursor protein (APP) and ferritin mRNAs: riboregulation against neural oxidative damage in Alzheimer's disease. Biochem Soc Trans 36 (Pt 6): 1282–1287. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rogers JT, Lahiri DK (2004). Metal and inflammatory targets for Alzheimer's disease. Curr Drug Targets 5: 535–551. [DOI] [PubMed] [Google Scholar]
- Rogers JT, Randall JD, Cahill CM, Eder PS, Huang X, Gunshin H et al. (2002). An iron‐responsive element type II in the 5'‐untranslated region of the Alzheimer's amyloid precursor protein transcript. J Biol Chem 277: 45518–45528. [DOI] [PubMed] [Google Scholar]
- Roriz‐Filho JS, Sa‐Roriz TM, Rosset I, Camozzato AL, Santos AC, Chaves ML et al. (2009). (Pre)diabetes, brain aging, and cognition. Biochim Biophys Acta 1792: 432–443. [DOI] [PubMed] [Google Scholar]
- Sagi Y, Drigues N, Youdim MB (2005). The neurochemical and behavioral effects of the novel cholinesterase‐monoamine oxidase inhibitor, ladostigil, in response to L‐dopa and L‐tryptophan, in rats. Br J Pharmacol 146: 553–560. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Šalković‐Petrišić M, Knezovic A, Osmanović‐Barilar J, Smailovic U, Trkulja V, Riederer P et al. (2015). Improve memory loss in a rat model of sporadic Alzheimer's disease. Life Sci 136: 108–119. [DOI] [PubMed] [Google Scholar]
- Salkovic‐Petrisic M, Tribl F, Schmidt M, Hoyer S, Riederer P (2006). Alzheimer‐like changes in protein kinase B and glycogen synthase kinase‐3 in rat frontal cortex and hippocampus after damage to the insulin signalling pathway. J Neurochem 96: 1005–1015. [DOI] [PubMed] [Google Scholar]
- Samadi A, Chioua M, Bolea I, de Los RC, Iriepa I, Moraleda I et al. (2011). Synthesis, biological assessment and molecular modeling of new multipotent MAO and cholinesterase inhibitors as potential drugs for the treatment of Alzheimer's disease. Eur J Med Chem 46: 4665–4668. [DOI] [PubMed] [Google Scholar]
- Samadi A, de los Rios C, Bolea I, Chioua M, Iriepa I, Moraleda I et al. (2012). Multipotent MAO and cholinesterase inhibitors for the treatment of Alzheimer's disease: synthesis, pharmacological analysis and molecular modeling of heterocyclic substituted alkyl and cycloalkyl propargyl amine. Eur J Med Chem 52: 251–262. [DOI] [PubMed] [Google Scholar]
- Sayre LM, Perry G, Harris PL, Liu Y, Schubert KA, Smith MA (2000). In situ oxidative catalysis by neurofibrillary tangles and senile plaques in Alzheimer's disease: a central role for bound transition metals. J Neurochem 74: 270–279. [DOI] [PubMed] [Google Scholar]
- Schliebs R (2005). Basal forebrain cholinergic dysfunction in Alzheimer's disease–interrelationship with beta‐amyloid, inflammation and neurotrophin signaling. Neurochem Res 30: 895–908. [DOI] [PubMed] [Google Scholar]
- Schulz JB, Lindenau J, Seyfried J, Dichgans J (2000). Glutathione, oxidative stress and neurodegeneration. Eur J Biochem 267: 4904–4911. [DOI] [PubMed] [Google Scholar]
- Selkoe DJ, Schenk D (2003). Alzheimer's disease: molecular understanding predicts amyloid‐based therapeutics. Annu Rev Pharmacol Toxicol 43: 545–584. [DOI] [PubMed] [Google Scholar]
- Semenza GL (2001). Hypoxia‐inducible factor 1: oxygen homeostasis and disease pathophysiology. Trends Mol Med 7: 345–350. [DOI] [PubMed] [Google Scholar]
- Shoham S, Bejar C, Kovalev E, Schorer‐Apelbaum D, Weinstock M (2007). Ladostigil prevents gliosis, oxidative‐nitrative stress and memory deficits induced by intracerebroventricular injection of streptozotocin in rats. Neuropharmacology 52: 836–843. [DOI] [PubMed] [Google Scholar]
- Siddiq A, Aminova LR, Ratan RR (2008). Prolyl 4‐hydroxylase activity‐responsive transcription factors: from hydroxylation to gene expression and neuroprotection. Front Biosci 13: 2875–2887. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Smith MA, Rottkamp CA, Nunomura A, Raina AK, Perry G (2000). Oxidative stress in Alzheimer's disease. Biochim Biophys Acta 1502: 139–144. [DOI] [PubMed] [Google Scholar]
- Sofic E, Salkovic‐Petrisic M, Tahirovic I, Sapcanin A, Mandel S, Youdim M et al. (2014). Brain catalase in the streptozotocin‐rat model of sporadic Alzheimer's disease treated with the iron chelator‐monoamine oxidase inhibitor, M30. J Neural Transm 122: 559–564. [DOI] [PubMed] [Google Scholar]
- Solano DC, Sironi M, Bonfini C, Solerte SB, Govoni S, Racchi M (2000). Insulin regulates soluble amyloid precursor protein release via phosphatidyl inositol 3 kinase‐dependent pathway. FASEB J 14: 1015–1022. [DOI] [PubMed] [Google Scholar]
- Stasiak A, Mussur M, Unzeta M, Samadi A, Marco‐Contelles JL, Fogel WA (2014). Effects of novel monoamine oxidases and cholinesterases targeting compounds on brain neurotransmitters and behavior in rat model of vascular dementia. Curr Pharm Des 20: 161–171. [DOI] [PubMed] [Google Scholar]
- Stauch KL, Purnell PR, Fox HS (2014). Aging synaptic mitochondria exhibit dynamic proteomic changes while maintaining bioenergetic function. Aging (Albany NY) 6: 320–334. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sterling J, Herzig Y, Goren T, Finkelstein N, Lerner D, Goldenberg W et al. (2002). Novel dual inhibitors of AChE and MAO derived from hydroxy aminoindan and phenethylamine as potential treatment for Alzheimer's disease. J Med Chem 45: 5260–5279. [DOI] [PubMed] [Google Scholar]
- Strolin Benedetti M, Dostert P (1989). Monoamine oxidase, brain ageing and degenerative diseases. Biochem Pharmacol 38: 555–561. [DOI] [PubMed] [Google Scholar]
- Su B, Wang X, Zheng L, Perry G, Smith MA, Zhu X (2010). Abnormal mitochondrial dynamics and neurodegenerative diseases. Biochim Biophys Acta 1802: 135–142. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Toiber D, Soreq H (2005). Cellular stress reactions as putative cholinergic links in Alzheimer's disease. Neurochem Res 30: 909–919. [DOI] [PubMed] [Google Scholar]
- Tsolaki M, Kokarida K, Iakovidou V, Stilopoulos E, Meimaris J, Kazis A (2001). Extrapyramidal symptoms and signs in Alzheimer's disease: prevalence and correlation with the first symptom. Am J Alzheimers Dis Other Demen 16: 268–278. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Vermeiren Y, Van Dam D, Aerts T, Engelborghs S, De Deyn PP (2014). Monoaminergic neurotransmitter alterations in postmortem brain regions of depressed and aggressive patients with Alzheimer's disease. Neurobiol Aging 35: 2691–2700. [DOI] [PubMed] [Google Scholar]
- Vetrivel KS, Thinakaran G (2006). Amyloidogenic processing of beta‐amyloid precursor protein in intracellular compartments. Neurology 66 (2 Suppl 1): S69–S73. [DOI] [PubMed] [Google Scholar]
- Villarroya M, Garcia AG, Marco JL (2004). New classes of AChE inhibitors with additional pharmacological effects of interest for the treatment of Alzheimer's disease. Curr Pharm Des 10: 3177–3184. [DOI] [PubMed] [Google Scholar]
- Wang GL, Semenza GL (1995). Purification and characterization of hypoxia‐inducible factor 1. J Biol Chem 270: 1230–1237. [DOI] [PubMed] [Google Scholar]
- Wang L, Esteban G, Ojima M, Bautista‐Aguilera OM, Inokuchi T, Moraleda I et al. (2014). Donepezil + propargylamine + 8‐hydroxyquinoline hybrids as new multifunctional metal‐chelators, ChE and MAO inhibitors for the potential treatment of Alzheimer's disease. Eur J Med Chem 80: 543–561. [DOI] [PubMed] [Google Scholar]
- Wang R, Tang XC (2005). Neuroprotective effects of huperzine A. A natural cholinesterase inhibitor for the treatment of Alzheimer's disease. Neurosignals 14: 71–82. [DOI] [PubMed] [Google Scholar]
- Ward RJ, Zucca FA, Duyn JH, Crichton RR, Zecca L (2014). The role of iron in brain ageing and neurodegenerative disorders. Lancet Neurol 13: 1045–1060. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Weinreb O, Amit T, Bar‐Am O, Chillag‐Talmor O, Youdim MB (2005). Novel neuroprotective mechanism of action of rasagiline is associated with its propargyl moiety: interaction of Bcl‐2 family members with PKC pathway. Ann N Y Acad Sci 1053: 348–355. [DOI] [PubMed] [Google Scholar]
- Weinreb O, Amit T, Bar‐Am O, Youdim MB (2007a). Induction of neurotrophic factors GDNF and BDNF associated with the mechanism of neurorescue action of rasagiline and ladostigil: new insights and implications for therapy. Ann N Y Acad Sci 1122: 155–168. [DOI] [PubMed] [Google Scholar]
- Weinreb O, Amit T, Bar‐Am O, Youdim MB (2010a). Rasagiline: a novel anti‐Parkinsonian monoamine oxidase‐B inhibitor with neuroprotective activity. Prog Neurobiol 92: 330–344. [DOI] [PubMed] [Google Scholar]
- Weinreb O, Amit T, Mandel S, Kupershmidt L, Youdim MB (2010b). Neuroprotective multifunctional iron chelators: from redox‐sensitive process to novel therapeutic opportunities. Antioxid Redox Signal 13: 919–949. [DOI] [PubMed] [Google Scholar]
- Weinreb O, Bar‐Am O, Amit T, Chillag‐Talmor O, Youdim MBH (2004). Neuroprotection via pro‐survival protein kinase C isoforms associated with Bcl‐2 family members. FASEB J 18: 1471–1473. [DOI] [PubMed] [Google Scholar]
- Weinreb O, Bar‐Am O, Amit T, Drigues N, Sagi Y, Youdim MB (2008). The neuroprotective effect of ladostigil against hydrogen peroxide‐mediated cytotoxicity. Chem Biol Interact 175: 318–326. [DOI] [PubMed] [Google Scholar]
- Weinreb O, Drigues N, Sagi Y, Reznick AZ, Amit T, Youdim MB (2007b). The application of proteomics and genomics to the study of age‐related neurodegeneration and neuroprotection. Antioxid Redox Signal 9: 169–179. [DOI] [PubMed] [Google Scholar]
- Weinstock M, Bejar C, Schorer‐Apelbaum D, Panarsky R, Luques L, Shoham S (2013). Dose‐dependent effects of ladostigil on microglial activation and cognition in aged rats. J Neuroimmune Pharmacol 8: 345–355. [DOI] [PubMed] [Google Scholar]
- Weinstock M, Bejar C, Wang RH, Poltyrev T, Gross A, Finberg J et al. (2000a). TV3326, a novel neuroprotective drug with cholinesterase and monoamine oxidase inhibitory activities for the treatment of Alzheimer's disease. J Neural Transm 60 (Suppl): S157–S170. [DOI] [PubMed] [Google Scholar]
- Weinstock M, Goren T, Youdim MBH (2000b). Development of a novel neuroprotective drug (TV3326) for the treatment of Alzheimer's disease, with cholinesterase and monoamine oxidase inhibitory activities. Drug Dev Res 50: 216–222. [DOI] [PubMed] [Google Scholar]
- Weinstock M, Gorodetsky E, Wang RH, Gross A, Weinreb O, Youdim MBH (2002). Limited potentiation of blood pressure response to oral tyramine by brain‐selective monoamine oxidase A‐B inhibitor, TV‐3326 in conscious rabbits(1). Neuropharmacology 43: 999–1005. [DOI] [PubMed] [Google Scholar]
- Weinstock M, Kirschbaum‐Slager N, Lazarovici P, Bejar C, Youdim MBH, Shoham S (2001). Neuroprotective effects of novel cholinesterase inhibitors derived from rasagiline as potential anti‐Alzheimer drugs. Ann N Y Acad Sci 939: 148–161. [DOI] [PubMed] [Google Scholar]
- Weinstock M, Luques L, Bejar C, Shoham S (2006). Ladostigil, a novel multifunctional drug for the treatment of dementia co‐morbid with depression. J Neural Transm Suppl 70: 443–446. [DOI] [PubMed] [Google Scholar]
- Weinstock M, Luques L, Poltyrev T, Bejar C, Shoham S (2011). Ladostigil prevents age‐related glial activation and spatial memory deficits in rats. Neurobiol Aging 32: 1069–1078. [DOI] [PubMed] [Google Scholar]
- Weintraub S, Wicklund AH, Salmon DP (2012). The neuropsychological profile of Alzheimer disease. Cold Spring Harb Perspect Med 2: a006171. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yang L, He HY, Zhang XJ (2002). Increased expression of intranuclear AChE involved in apoptosis of SK‐N‐SH cells. Neurosci Res 42: 261–268. [DOI] [PubMed] [Google Scholar]
- Yogev‐Falach M, Amit T, Bar‐Am O, Sagi Y, Weinstock M, Youdim MBH (2002). The involvement of mitogen‐activated protein (MAP) kinase in the regulation of amyloid precursor protein processing by novel cholinesterase inhibitors derived from rasagiline. FASEB J 16: 1674–1676. [DOI] [PubMed] [Google Scholar]
- Yogev‐Falach M, Amit T, Bar‐AM O, Youdim MBH (2003). The importance of propargylamine moiety in the anti‐Parkinson drug rasagiline and its derivatives for MAPK‐dependent amyloid precursor protein processing. FASEB J 17: 2325–2327. [DOI] [PubMed] [Google Scholar]
- Yogev‐Falach M, Bar‐Am O, Amit T, Weinreb O, Youdim MB (2006). A multifunctional, neuroprotective drug, ladostigil (TV3326), regulates holo‐APP translation and processing. FASEB J 20: 2177–2179. [DOI] [PubMed] [Google Scholar]
- Youdim MB (2006). The path from anti Parkinson drug selegiline and rasagiline to multifunctional neuroprotective anti Alzheimer drugs ladostigil and M30. Curr Alzheimer Res 3: 541–550. [DOI] [PubMed] [Google Scholar]
- Youdim MB, Buccafusco JJ (2005a). Multi‐functional drugs for various CNS targets in the treatment of neurodegenerative disorders. Trends Pharmacol Sci 26: 27–35. [DOI] [PubMed] [Google Scholar]
- Youdim MB, Fridkin M, Zheng H (2005b). Bifunctional drug derivatives of MAO‐B inhibitor rasagiline and iron chelator VK‐28 as a more effective approach to treatment of brain ageing and ageing neurodegenerative diseases. Mech Ageing Dev 126: 317–326. [DOI] [PubMed] [Google Scholar]
- Youdim MB, Weinstock M (2001). Molecular basis of neuroprotective activities of rasagiline and the anti‐Alzheimer drug TV3326 [(N‐propargyl‐(3R)aminoindan‐5‐yl)‐ethyl methyl carbamate]. Cell Mol Neurobiol 21: 555–573. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Youdim MBH (2003). Rasagiline: an anti‐Parkinson drug with neuroprotective activity. Future Drugs 3: 737–749. [DOI] [PubMed] [Google Scholar]
- Youdim MBH, Riederer PF (2004). A review of the mechanisms and role of monoamine oxidase inhibitors in Parkinson's disease. Neurology 63 (7 Suppl 1): S32–S35. [DOI] [PubMed] [Google Scholar]
- Zaman K, Ryu H, Hall D, O'Donovan K, Lin KI, Miller MP et al. (1999). Protection from oxidative stress‐induced apoptosis in cortical neuronal cultures by iron chelators is associated with enhanced DNA binding of hypoxia‐inducible factor‐1 and ATF‐1/CREB and increased expression of glycolytic enzymes, p21(waf1/cip1), and erythropoietin. J Neurosci 19: 9821–9830. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zecca L, Stroppolo A, Gatti A, Tampellini D, Toscani M, Gallorini M et al. (2004). The role of iron and copper molecules in the neuronal vulnerability of locus coeruleus and substantia nigra during aging. Proc Natl Acad Sci U S A 101: 9843–9848. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zheng H, Fridkin M, Youdim M (2014). From single target to multitarget/network therapeutics in Alzheimer's therapy. Pharmaceuticals (Basel) 7: 113–135. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zheng H, Gal S, Weiner LM, Bar‐Am O, Warshawsky A, Fridkin M et al. (2005a). Novel multifunctional neuroprotective iron chelator‐monoamine oxidase inhibitor drugs for neurodegenerative diseases: in vitro studies on antioxidant activity, prevention of lipid peroxide formation and monoamine oxidase inhibition. J Neurochem 95: 68–78. [DOI] [PubMed] [Google Scholar]
- Zheng H, Weiner LM, Bar‐Am O, Epsztejn S, Cabantchik ZI, Warshawsky A et al. (2005b). Design, synthesis, and evaluation of novel bifunctional iron‐chelators as potential agents for neuroprotection in Alzheimer's, Parkinson's, and other neurodegenerative diseases. Bioorg Med Chem 13: 773–783. [DOI] [PubMed] [Google Scholar]
- Zheng H, Youdim MB, Weiner LM, Fridkin M (2005c). Novel potential neuroprotective agents with both iron chelating and amino acid‐based derivatives targeting central nervous system neurons. Biochem Pharmacol 70: 1642–1652. [DOI] [PubMed] [Google Scholar]
- Zheng W, Monnot AD (2012). Regulation of brain iron and copper homeostasis by brain barrier systems: implication in neurodegenerative diseases. Pharmacol Ther 133: 177–188. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zorov DB, Juhaszova M, Sollott SJ (2014). Mitochondrial reactive oxygen species (ROS) and ROS‐induced ROS release. Physiol Rev 94: 909–950. [DOI] [PMC free article] [PubMed] [Google Scholar]