

Abstract
Barley (Hordeum vulgare) is an economically important species, which can be cultivated in environmentally adverse conditions due to its higher tolerance in contrast to other cereal crops. The draft of H. vulgare genome is available already for couple of years; however its functional annotation is still incomplete. All available databases were searched to expand current annotation. The improved annotation was used to describe processes and genes regulated in transgenic lines showing higher tolerance to drought in our associated article, doi:10.1016/j.nbt.2016.01.010 (Vojta et al., 2016) [1]. Here we present whole transcriptome response, using extended annotation, to severe drought stress and subsequent re-watering in wild-type barley plants in stem elongation phase of growth. Up- and down-regulated genes fall into distinct GO categories and these enriched by stress and revitalization are highlighted. Transcriptomic data were evaluated separately for root and aerial tissues.
Keywords: Barley, Drought stress, Genome annotation, Re-watering, Transcriptomics
Specifications Table
| Subject area | Biology |
| More specific subject area | RNA-seq transcriptome data of barley (Hordeum vulgare) |
| Type of data | Tables and figures |
| How data was acquired | Sequencing on Illumina HiSeq 2500 Sequencing System |
| Data format | Processed, analyzed |
| Experimental factors | Samples were exposed to severe drought stress and subsequently re-watered |
| Experimental features | RNA was extracted using RNAqueous kit and purified on magnetic beads. Sequencing libraries were prepared using the TruSeq Stranded mRNA kit from Illumina and quantified using the Kapa Library Quantification kit. Libraries were sequenced on HiSeq 2500 Illumina platform. |
| Data source location | Palacký University, Olomouc, Czech Republic |
| Data accessibility | Data are within this article |
Value of the data
-
•
Improvement of a functional annotation of Hordeum vulgare genome draft.
-
•
This dataset provides the list of all up- and down-regulated genes during one day long desiccation and subsequent re-watering separately in roots and upper part of 4-week-old barley seedlings.
-
•
Enriched gene ontology (GO) term analysis highlights processes targeted by above mentioned conditions.
-
•
The dataset can serve as a source of candidate genes for markers used for drought associated studies.
1. Data
This data consist of five high-throughput sequenced samples of barley roots (Supplementary Table 1, n=2) and upper part (Supplementary Table 2, n=3), exposed to optimal or drought conditions and subsequent re-watering, generated from an Illumina HiSeq 2500, together with GO term analysis of the most affected Biological Processes (Table 1, Table 2, Table 3). Predicted genes from the latest genome version (082214v1.25) have been annotated based on three various databases (Fig. 1) and associated to GO term categories (Fig. 2). Several GO terms have been assigned to each predicted sequence (Fig. 3).
Table 1.
The most affected GO terms from Biological Processes in the stressed roots and percentage of differentially expressed genes (adjusted p-value ≤0.05) at the GO level 6.
|
GO number |
GO term |
Total # |
% of affected genes |
|---|---|---|---|
| DOWN-REGULATED | |||
| GO:0010089 | xylem development | 13 | 69.23% |
| GO:0071103 | DNA conformation change | 104 | 59.62% |
| GO:0070726 | cell wall assembly | 11 | 54.55% |
| GO:0048544 | recognition of pollen | 91 | 50.55% |
| GO:0006915 | apoptotic process | 296 | 50.34% |
| GO:0051129 | negative regulation of cellular component organization | 10 | 50.00% |
| GO:0001666 | response to hypoxia | 16 | 50.00% |
| GO:0009664 | plant-type cell wall organization | 76 | 48.68% |
| GO:0046271 | phenylpropanoid catabolic process | 21 | 47.62% |
| GO:0007166 | cell surface receptor signaling pathway | 42 | 47.62% |
| GO:0009834 | plant-type secondary cell wall biogenesis | 19 | 47.37% |
| GO:0015851 | nucleobase transport | 13 | 46.15% |
| GO:0006002 | fructose 6-phosphate metabolic process | 11 | 45.45% |
| GO:0042886 | amide transport | 65 | 44.62% |
| GO:0000910 | cytokinesis | 106 | 43.40% |
| UP-REGULATED | |||
| GO:0071462 | cellular response to water stimulus | 11 | 63.64% |
| GO:0009407 | toxin catabolic process | 33 | 60.61% |
| GO:0072348 | sulfur compound transport | 10 | 60.00% |
| GO:1902644 | tertiary alcohol metabolic process | 22 | 59.09% |
| GO:0044242 | cellular lipid catabolic process | 97 | 57.73% |
| GO:0033015 | tetrapyrrole catabolic process | 35 | 57.14% |
| GO:0042538 | hyperosmotic salinity response | 20 | 55.00% |
| GO:0010286 | heat acclimation | 26 | 53.85% |
| GO:0046164 | alcohol catabolic process | 10 | 50.00% |
| GO:0046434 | organophosphate catabolic process | 12 | 50.00% |
| GO:0048545 | response to steroid hormone | 20 | 50.00% |
| GO:0050801 | ion homeostasis | 80 | 48.75% |
| GO:0055082 | cellular chemical homeostasis | 52 | 48.08% |
| GO:0042542 | response to hydrogen peroxide | 62 | 46.77% |
| GO:0009699 | phenylpropanoid biosynthetic process | 28 | 46.43% |
Table 2.
The most affected GO terms from Biological Processes in the stressed aerial part and percentage of differentially expressed genes (adjusted p-value ≤0.05) at the GO level 6.
|
GO number |
GO term |
Total # |
% of affected genes |
|---|---|---|---|
| DOWN-REGULATED | |||
| GO:0009765 | photosynthesis, light harvesting | 32 | 87.50% |
| GO:0019750 | chloroplast localization | 67 | 71.64% |
| GO:0051667 | establishment of plastid localization | 67 | 71.64% |
| GO:0009668 | plastid membrane organization | 123 | 70.73% |
| GO:0009658 | chloroplast organization | 146 | 67.12% |
| GO:0016226 | iron-sulfur cluster assembly | 70 | 62.86% |
| GO:0019682 | glyceraldehyde-3-phosphate metabolic process | 216 | 62.04% |
| GO:0051156 | glucose 6-phosphate metabolic process | 121 | 61.16% |
| GO:0033014 | tetrapyrrole biosynthetic process | 102 | 59.80% |
| GO:0042727 | flavin-containing compound biosynthetic process | 12 | 58.33% |
| GO:0010374 | stomatal complex development | 69 | 53.62% |
| GO:0009767 | photosynthetic electron transport chain | 57 | 50.88% |
| GO:0006720 | isoprenoid metabolic process | 255 | 49.02% |
| GO:0006778 | porphyrin-containing compound metabolic process | 138 | 47.83% |
| GO:0016143 | S-glycoside metabolic process | 60 | 46.67% |
| UP-REGULATED | |||
| GO:0042538 | hyperosmotic salinity response | 20 | 50.00% |
| GO:0009962 | regulation of flavonoid biosynthetic process | 11 | 36.36% |
| GO:0010647 | positive regulation of cell communication | 15 | 33.33% |
| GO:0006026 | aminoglycan catabolic process | 18 | 33.33% |
| GO:0046348 | amino sugar catabolic process | 18 | 33.33% |
| GO:1901071 | glucosamine-containing compound metabolic process | 18 | 33.33% |
| GO:0060548 | negative regulation of cell death | 29 | 31.03% |
| GO:0010583 | response to cyclopentenone | 14 | 28.57% |
| GO:0046271 | phenylpropanoid catabolic process | 21 | 28.57% |
| GO:0033015 | tetrapyrrole catabolic process | 35 | 28.57% |
| GO:0009414 | response to water deprivation | 68 | 27.94% |
| GO:1902644 | tertiary alcohol metabolic process | 22 | 27.27% |
| GO:0043067 | regulation of programmed cell death | 63 | 25.40% |
| GO:0006662 | glycerol ether metabolic process | 32 | 25.00% |
| GO:0009407 | toxin catabolic process | 33 | 24.24% |
Table 3.
The most affected GO terms from Biological Processes in the aerial parts 12 h after re-watering and percentage of differentially expressed genes (adjusted p-value ≤0.05) at the GO level 6.
|
GO number |
GO term |
Total # |
% of affected genes |
|---|---|---|---|
| DOWN-REGULATED | |||
| GO:0009765 | photosynthesis, light harvesting | 32 | 81.25% |
| GO:0071462 | cellular response to water stimulus | 11 | 63.64% |
| GO:0051156 | glucose 6-phosphate metabolic process | 121 | 53.72% |
| GO:0009767 | photosynthetic electron transport chain | 57 | 52.63% |
| GO:0019750 | chloroplast localization | 67 | 50.75% |
| GO:0051667 | establishment of plastid localization | 67 | 50.75% |
| GO:0072525 | pyridine-containing compound biosynthetic process | 20 | 50.00% |
| GO:0009637 | response to blue light | 49 | 48.98% |
| GO:0019682 | glyceraldehyde-3-phosphate metabolic process | 216 | 46.30% |
| GO:0010109 | regulation of photosynthesis | 13 | 46.15% |
| GO:0043085 | positive regulation of catalytic activity | 79 | 45.57% |
| GO:0016143 | S-glycoside metabolic process | 60 | 45.00% |
| GO:0006778 | porphyrin-containing compound metabolic process | 138 | 44.93% |
| GO:0009668 | plastid membrane organization | 123 | 44.72% |
| GO:0033014 | tetrapyrrole biosynthetic process | 102 | 43.14% |
| UP-REGULATED | |||
| GO:0042273 | ribosomal large subunit biogenesis | 14 | 71.43% |
| GO:0000741 | karyogamy | 25 | 64.00% |
| GO:0072528 | pyrimidine-containing compound biosynthetic process | 97 | 59.79% |
| GO:0000085 | mitotic G2 phase | 23 | 56.52% |
| GO:0043572 | plastid fission | 11 | 54.55% |
| GO:0006518 | peptide metabolic process | 498 | 49.20% |
| GO:0043604 | amide biosynthetic process | 512 | 48.44% |
| GO:0007292 | female gamete generation | 69 | 46.38% |
| GO:0007006 | mitochondrial membrane organization | 11 | 45.45% |
| GO:0051604 | protein maturation | 47 | 44.68% |
| GO:0006026 | aminoglycan catabolic process | 18 | 44.44% |
| GO:0046348 | amino sugar catabolic process | 18 | 44.44% |
| GO:1901071 | glucosamine-containing compound metabolic process | 18 | 44.44% |
| GO:0051169 | nuclear transport | 125 | 44.00% |
| GO:0009553 | embryo sac development | 110 | 43.64% |
Fig. 1.
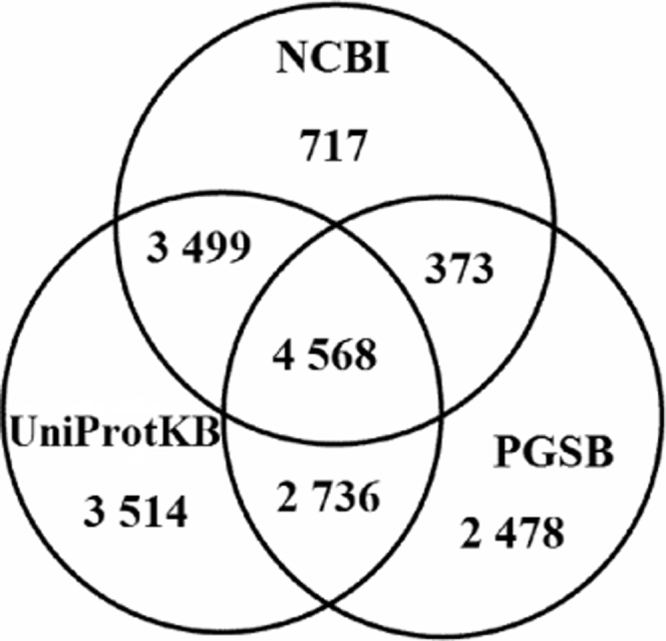
Venn diagram showing numbers of genes due to the source database used for their annotation.
Fig. 2.

Distribution of GO terms in whole transcriptome on the level 2 for Biological Processes (A), Molecular Function (B) and Cellular Component (C).
Fig. 3.

GO terms distribution per sequence annotated in improved Hordeum vulgare reference genome.
2. Experimental design, materials and methods
2.1. Plant material
Spring barley plants, cultivar Golden Promise, were grown in a phytotron with a photoperiod of 15 °C/16 h light and 12 °C/8 h dark in soil or in hydroponic tanks containing aerated Hoagland nutrient solution. Samples of root tissue 4 weeks after germination were collected from hydroponically grown plants due to the inability to collect root tissues from soil without initiation of mechanical stress. The stress was applied by removing the nutrient solution off the tank. Control samples were collected just before stress induction; stressed root samples were collected 24 h later. Aerial part samples were collected from 4 week old plants cultivated in the shallow soil. Watering on daily basis was interrupted for four days and stressed samples were collected in the end of the drought period. Revitalization samples were collected 12 h after re-watering. Each sequencing library was prepared from pool of 3 individual plants.
2.2. Annotation
Additional annotation of predicted genes was mined using Blast2GO version 3.0 program to improve raw reference genome available at Ensembl (http://plants.ensembl.org/index.html, version 082214v1.25). Gene description from the National Center for Biotechnology Information database (NCBI; version b2g_Jan15) were mined using the BLAST module from program Blast2GO with parameters blastn and e-value ≤10−5. The other step in annotation process was mapping predicted genes to other databases using Blast2GO with default parameters. Additional annotation of other predicted genes was extracted from The UniProt Knowledgebase database (http://www.uniprot.org/, version 2015_02) and the Plant Genome and Systems Biology database (PGSB; http://pgsb.helmholtz-muenchen.de/plant/, version 2014_07_31) for hits with blastn stringency of e-value ≤10−5. Finally, annotation information was obtained for 17,885 genes from a total number of 26,072 predicted genes in Hordeum vulgare genome (Fig. 1).
Gene ontology analysis was performed using the Blast2GO v.3.0 [2], firstly for all predicted genes and then specifically for significantly up-regulated and down-regulated genes with adjusted p-value (padj) ≤0.05. Total number of 70,719 GO terms was assigned to 20,991 predicted genes. Out of these 40.87%, 42.12% and 17.01% were assigned to Biological Processes, Molecular Function and Cellular Component GO categories, respectively (Fig. 2). Number of GO terms assigned to one predicted sequence was in range from 1 to 35 (Fig. 3). Differentially expressed genes were categorized to Biological Processes (BP), Cellular Components (CC) and Molecular Functions (MF) on the level 6. Number of differentially expressed genes for particular GO terms was compared with total number of genes assigned to the term and enriched GO terms were highlighted (Table 1, Table 2, Table 3). Supplementary Appendix A, Appendix A contains GO terms at the level 6 with associated 10 or more genes in roots and aerial part, respectively. GO terms with associated 9 or less genes were filtered out and are not listed. GO terms are sorted due to increased percentage in category of differentially expressed genes with adjusted p-value ≤0.05 from total number of genes with the same assigned GO term. The 30 most affected Biological Processes are shown for stressed root (Table 1), stressed aerial part (Table 2) and the aerial part after re-watering (Table 3).
2.3. RNA-extraction and sequencing
Total RNA was extracted and cDNA sequencing library was prepared and sequenced as described elsewhere [1], [3].
2.4. RNA-seq analysis
Single end reads generated by the sequencing were mapped to the reference genome and quantified the same way as described in Ref. [1]. The comparison for differentially expressed genes among 3 time-points (before stress, during stress, 12 h after re-watering) was conducted using the DESeq2 package [4] implemented in R (R Development Core Team, 2008). Normalized RPKM (reads per kilobase of transcript per million reads mapped) were subjected to principle components analysis (PCA) in order to control quality of replicates. The PCA analysis shows good accordance between replicates, which cluster together (Fig. 4). The log2fold value is calculated for each gene and genes are sorted according to adjusted p-value. Positive log2fold values are for up-regulated and negative for down-regulated genes. The base mean value represents mean of normalized RPKM for all comparisons and thus expresses transcript abundance of each gene in particular organ.
Fig. 4.

The PCA analysis for replicates from root samples before stress (Root_Ctrl) and during drought stress period (Root_Stress), and from the upper part before stress (Upper_Ctrl), during stress (Upper_Stress) and 12 h after re-watering (Upper_R12H).
Acknowledgments
This work was supported by the Czech Science Foundation (Grant no. 14-12355 S) and the National Program for Sustainability (Project no. LO1204).
Footnotes
Transparency data associated with this article can be found in the online version at 10.1016/j.dib.2016.05.051.
Supplementary data associated with this article can be found in the online version at 10.1016/j.dib.2016.05.051.
Transparency document. Supplementary material
Supplementary material
Appendix A. Supplementary material
Table S1. Comparative transcriptomics of Hordeum vulgare roots during the drought stress. Average expression level (baseMean) and change due to optimal conditions (log2FoldChange) with statistical significance (padj) are presented. Table S2. Comparative transcriptomics of Hordeum vulgare aerial part during the drought and 12 h after re-watering. Average expression level (baseMean) and change due to optimal conditions (log2FoldChange) with statistical significance (padj) are presented. Table S3. GO analysis at the level 6 of differentially expressed genes (adjusted p-value ≤0.05) in Hordeum vulgare roots during the drought stress. Data are categorized according to Biological Processes (BP). Molecular Function (MF) and Cellular Component (CC) and sorted by percentage of affected genes. Table S4. GO analysis at the level 6 of differentially expressed genes (adjusted p-value ≤0.05) in Hordeum vulgare aerial part during the drought and 12 h after re-watering. Data are categorized according to Biological Processes (BP). Molecular Function (MF) and Cellular Component (CC) and sorted by percentage of affected genes.
References
- 1.Vojta P., Kokáš F., Husičková A., Grúz J. Whole transcriptome analysis of transgenic barley with altered cytokinin homeostasis and increased tolerance to drought stress. New Biotechnol. 2016 doi: 10.1016/j.nbt.2016.01.010. [DOI] [PubMed] [Google Scholar]
- 2.Conesa A., Götz S., García-Gómez J.M., Terol J. Blast2GO: a universal tool for annotation. visualization and analysis in functional genomics research. Bioinformatics. 2005;21:3674–3676. doi: 10.1093/bioinformatics/bti610. [DOI] [PubMed] [Google Scholar]
- 3.Pospíšilová H., Jiskrová E., Vojta P., Mrízová K. Transgenic barley overexpressing a cytokinin dehydrogenase gene shows greater tolerance to drought stress. New Biotechnol. 2016 doi: 10.1016/j.nbt.2015.12.005. [DOI] [PubMed] [Google Scholar]
- 4.Love M.I., Huber W., Anders S. Moderated estimation of fold change and dispersion for RNA-Seq data with DESeq2. Genome Biol. 2014;15:550. doi: 10.1186/s13059-014-0550-8. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Supplementary material
Table S1. Comparative transcriptomics of Hordeum vulgare roots during the drought stress. Average expression level (baseMean) and change due to optimal conditions (log2FoldChange) with statistical significance (padj) are presented. Table S2. Comparative transcriptomics of Hordeum vulgare aerial part during the drought and 12 h after re-watering. Average expression level (baseMean) and change due to optimal conditions (log2FoldChange) with statistical significance (padj) are presented. Table S3. GO analysis at the level 6 of differentially expressed genes (adjusted p-value ≤0.05) in Hordeum vulgare roots during the drought stress. Data are categorized according to Biological Processes (BP). Molecular Function (MF) and Cellular Component (CC) and sorted by percentage of affected genes. Table S4. GO analysis at the level 6 of differentially expressed genes (adjusted p-value ≤0.05) in Hordeum vulgare aerial part during the drought and 12 h after re-watering. Data are categorized according to Biological Processes (BP). Molecular Function (MF) and Cellular Component (CC) and sorted by percentage of affected genes.