

Summary
The recruitment of immune cells to sites of tissue inflammation is orchestrated by chemokine/chemokine receptor networks. Among these, the CXCL13/CXCR5 axis is thought to be involved critically in systemic lupus erythematosus (SLE) and lupus nephritis pathogenesis. Beyond B cell abnormalities, another hallmark of SLE disease is the occurrence of aberrant T cell responses. In particular, double‐negative (DN) T cells are expanded in the peripheral blood of patients with SLE and in lupus‐prone mice. DN T cells induce immunoglobulin production, secrete proinflammatory cytokines and infiltrate inflamed tissue, including kidneys. We aimed to investigate how CXCR5 deficiency changes immune cell trafficking in murine lupus. We therefore crossed CXCR5–/– mice with B6/lpr mice, a well‐established murine lupus model. B cell numbers and B cellular immune responses were diminished in CXCR5‐deficient B6/lpr mice. In addition, we observed reduced accumulation of DN T cells in spleen and lymph nodes, paralleled by reduced splenomegaly and lymphadenopathy. In‐vivo migration assays revealed reduced migration of CXCR5‐deficient DN T cells into lymph nodes, and ex‐vivo‐activated CXCR5‐deficient DN T cells failed to infiltrate kidneys of recipients. Moreover, DN T cells and B cells of CXCR5‐deficient B6/lpr mice failed to migrate towards CXCL13 in vitro. We propose that CXCR5 is involved critically in B cell trafficking and germinal cell (GC) formation in murine lupus and in guiding pathogenic DN T cells into lymphoid organs and kidneys, and we therefore describe new pathomechanisms for the CXCL13/CXCR5 axis in SLE.
Keywords: CXCR5, DN T cells, SLE
Introduction
Systemic lupus erythematosus (SLE) is a complex autoimmune disorder that affects nearly all organ systems in the human body 1. A hallmark of the disease is the production of autoantibodies by B cells, which bind to endogenous tissue and cells and result in tissue destruction via complement activation and induction of inflammation. Autoreactive B cells are eliminated in healthy people, but differentiate into plasma cells and memory B cells in germinal centres (GCs) of SLE patients 2. GC reactions and GC generated effector cells are observed generally in secondary lymphoid tissues (lymph nodes, spleen, and mucosal tissues such as the tonsils and Peyer's patches). In addition, inflammation occurring during autoimmune/inflammatory conditions results in GC/follicle formation in ectopic sites, including the kidney, in lupus nephritis 3, 4. The chemokine receptor CXCR5 orchestrates GC formation, is expressed on B cells and follicular T helper cells and is required for migration and responsiveness of cells to CXCL13 5. The peripheral blood of SLE patients shows increased expression of CXCR5‐positive T cells 6, 7. In concordance with this, CXCL13 is elevated significantly in SLE sera and correlates with disease activity in lupus nephritis 8, 9, 10. The CXCR5/CXCL13 axis is therefore thought to be involved critically in autoimmunity, primarily through its influence on B cell trafficking and the development of lymphatic organs.
In addition to B cells, T cell signalling aberrancies play an important role in disease progression and end‐organ damage in SLE 11. Several studies describe the occurrence of pathogenic double‐negative (DN) T cells in SLE and murine lupus models 11, 12, 13. In addition, an expanded DN T cell population is associated with primary Sjögren's syndrome 14 and a murine arthritis model 15. DN T cells constitute a relatively small T cell population (1–5%) in the peripheral blood and in lymphoid organs of normal rodents and humans. They are characterized by expression of CD3, but lack CD4 and CD8 or natural killer cell markers 16. Notably, DN T cells can produce inflammatory cytokines and induce immunoglobulin (Ig)G and anti‐DNA antibody production, and can contribute to tissue damage by infiltrating organs such as the kidneys of SLE patients 12, 17. DN T cells in SLE are derived most probably from CD8+ T cells by down‐regulation of CD8 surface expression – a process that is mediated by 3′,5′‐cyclic adenosine monophosphate (cAMP)‐responsive element modulator α, the transcription factor that orchestrates epigenetic remodelling of the CD8 cluster 18, 19, 20. A recent study demonstrated that loss of CD8 expression occurs after exposure to self‐antigen, indicating that DN T cells are derived from self‐reactive CD8 T cells. The resulting DN T cells express programmed death 1 (PD‐1) and Helios 21, and while expression of these inhibitor molecules restricts their function in healthy individuals, it is likely that the mechanism somehow fails under autoimmune conditions. To conclude, recent studies show that local expansion in response to inflammation drives DN T cell accumulation. However, migration patterns of this T cell population in SLE and in lupus nephritis are not well understood.
Involvement of the kidneys is one of the most severe and common manifestations of SLE and is associated with significant patient morbidity and mortality. The exact mechanisms resulting in lupus nephritis (LN) are not clear, but it is known that a deposition of immune complexes in the glomeruli as well as infiltration of activated lymphocytes into the interstitial space mediate inflammation. Chemokine/chemokine receptor interactions direct leucocyte trafficking and positioning within the tissue. CXCL13 is one of the chemokines produced in murine nephritis and expressed highly in the renal cortex of patients with lupus nephritis 8, 9, 10. CXCL13 is thought to initiate early events in LN development by recruitment of B cells to the kidneys 8, 22. In addition to B cells, T cells also infiltrate the kidneys. In particular interleukin (IL)‐17‐producing DN T cells are expanded in the inflamed kidney tissue and trigger inflammation 11, 12. However, until now it has remained unclear how DN T cells are brought to the inflamed kidneys. We therefore aimed to analyse how migration of DN T cells in autoimmune‐prone conditions is organized and, in particular, how they gain access to inflamed kidneys.
Materials and methods
Animals
Experiments were performed with B6/lpr, B6 wild‐type, Rag–/– and B6/lpr CXCR5–/– mice. The study was approved by regional governmental authorities and animal procedures were performed according to German animal protection legislation.
Assessment of lymphadenopathy
Blinded scoring of lymphadenopathy in B6/lpr and B6/lpr CXCR5–/– mice was performed by two observers on a 0–5+ scale, broadly as described previously 18, and scored as follows: 0 = no detectable lymphadenopathy; 1+ = mild submandibular adenopathy only; 2+ = moderate submandibular adenopathy only; 3+ = severe submandibular adenopathy only; 4+ = submandibular adenopathy plus one other palpable node; and 5+ = diffuse lymphadenopathy.
Flow cytometric analysis
For surface staining, single‐cell suspensions were prepared from spleens, lymph nodes and blood of B6/lpr and B6/lpr CXCR5–/– mice and stained with the following specific antibodies: anti‐CD3‐allophycocyanin (APC) (eBiosciences, San Diego, CA, USA), anti‐CD4‐fluorescein isothiocyanate (FITC) (eBiosciences), anti‐CD4‐PE‐Cy7 (eBiosciences), anti‐CD8‐Pacific Blue (eBiosciences), anti‐CXCR5‐APC (BD Biosciences, San Jose, CA, USA), anti‐CD3‐phycoerythrin (PE) (eBiosciences), anti‐CD19‐FITC (eBiosciences) and anti‐CD138‐PE (BD Biosciences, Heidelberg, Germany). For measurement of intracellular interferon (IFN)‐γ and IL‐17 production, cells were treated with phorbol myristate acetate (PMA) (20 nM), ionomycin (2 μM) (both Sigma‐Aldrich, St Louis, MO, USA) and GolgiPlug (BD Biosciences, Germany) for 6 h and intracellular staining was performed with IFN‐γ‐APC (eBiosciences) and IL‐17‐Alexa Fluor 488 (BD Biosciences, Germany) antibodies using the eBiosciences forkhead box protein 3 (FoxP3) staining buffer kit according to the manufacturer's instructions. Samples were analysed using a fluorescence activated cell sorter (FACS) Canto II (BD Biosciences, USA) and data were analysed using FCS Express software (De Novo Software, Los Angeles, CA, USA).
Immunohistochemistry
For immunofluorescence analysis, spleens were harvested and frozen immediately in embedding medium (Tissue‐Tek; Sakura, Alphen aan den Rijn, the Netherlands). Sections were cut at a thickness of 7 µm. CD3 was detected using CD3e APC antibody (eBiosciences). To detect GCs, sections were stained with biotin‐conjugated biotinylated peanut agglutinin (PNA) (Vector Laboratories, Burlingame, CA, USA) followed by DyLight488‐conjugated streptavidin (Thermo Scientific, Waltham, MA, USA), sections were mounted with Fluoromount‐G (Southern Biotech, Birmingham, AL, USA). Images were captured using a Cool SNAP HQ2 camera and an Axioplan 2 imaging microscope (Zeiss, Oberkochen, Germany) with VisiView software (Visitron Systems GmbH, Puchheim, Germany).
Life expectancy
Kaplan–Meier survival plots were used to assess the life expectancy rates of B6/lpr and B6/lpr CXCR5–/– mice with GraphPad Prism software (San Diego, CA, USA). For statistical analysis the log‐rank (Mantel–Cox) test was performed.
Enzyme‐linked immunosorbent assay (ELISA)
Total IgG was measured in sera from B6/lpr and B6/lpr CXCR5–/– mice using the Ready‐set‐go ELISA system (Affymetrix; eBioscience).
Chemotaxis assay. Transmigration of primary B cells and T cells was assessed in 6·5‐mm‐diameter 24‐Transwell chemotaxis chambers (Costar, Corning, NY, USA) with a pore size of 5 μm. Freshly isolated splenic cells were suspended in medium [RPMI‐1640, 10% fetal calf serum (FCS), 1% penicillin/streptomycin] and a total of 100 μl, containing 2 × 106 cells, was loaded into the upper chamber of the Transwell culture insert. Aliquots were examined using FACS Canto II, as described above, to determine percentages of CD4+, CD8+ and DN T cells in the CD3+ population and the percentage of CD19+ cells. After a 30‐min preincubation at 37°C in 5% CO2, filters were transferred into the wells containing 600 µl medium in the presence or absence of CXCL13 (2 µg/600 µl) (R&D Systems, Minneapolis, MN, USA). The chambers were incubated for 4 h at 37°C in 5% CO2. After incubation, cells that had migrated into the lower chamber were counted and collected for flow cytometric analysis. Results are presented as chemotactic index (CTX), which was calculated by dividing the number of migrated cells in the presence of the chemoattractant by the number of migrated cells in the absence of the chemoattractant (control). Migrated cells were then analysed using FACS Canto II to determine percentages of CD4+, CD8+ and DN T cells in the CD3+ population and the percentage of CD19+ cells to calculate the total numbers of migrated cells of each cell type.
Migration experiments
B6/lpr and B6/lpr CXCR5–/– mice were killed and the spleens harvested. Single‐cell suspensions were prepared from spleens. CD3+ T cells were separated with nylon wool (Kisker Biotech, Steinfurt, Germany) and labelled with a fluorescent proliferation dye Dye670 (eBiosciences). CD3+ T cell purity was greater than 90%, as confirmed by flow cytometric analysis. Flow cytometric analysis was performed to detect the absolute number of different T cell subtypes. The absolute number of DN T cells was defined in the CD3+ T cell population. B6 wild‐type mice were injected with 2 × 106 DN T cells from B6/lpr mice or with 2 × 106 DN T cells from B6/lpr CXCR5–/– mice in the tail vein. After 48 h mice were killed and spleens, lymph nodes and blood harvested to prepare single‐cell suspensions. Cells were stained with anti‐CD3‐PE, anti‐CD4‐FITC and anti‐CD8‐Pacific Blue antibodies, and flow cytometric analysis was performed. Migrated DNTs were identified by Dye670 staining.
Adoptive transfer
B6/lpr and B6/lpr CXCR5–/– mice were killed and lymph nodes were harvested. Single‐cell suspensions were prepared and lymph node cells were stimulated using plate‐bound anti‐CD3 (10 µg/ml) and anti‐CD28 (0·8 µg/ml) antibodies (both from eBiosciences) in the presence of IL‐23 (20 ng/ml; R&D Systems) for 48 h. Expanded lymph node cells were harvested and washed with phosphate‐buffered saline (PBS), and Rag–/– mice were injected intraperitoneally (i.p.) with 5 × 106 cells from either B6/lpr or B6/lpr CXCR5–/– mice. Mice were killed after 9 weeks of transfer and kidneys were frozen immediately in embedding (Tissue‐Tek; Sakura) media for immunohistochemistry. Kidneys were cut into sections of 6 µm thickness and CD3 signal was detected using anti‐CD3e APC antibody (eBiosciences), while 4′,6‐diamidino‐2‐phenylindole (DAPI) was used for nucleus staining. Five segments in two different layers were analysed for CD3+ cells in each kidney and t‐tests were used for statistical analysis. For kidney glomerulopathy, kidneys were collected and fixed in methacarn. Fixed tissues were embedded in paraffin and stained with periodic acid Schiff reagent (PAS). Hamamatsu NDP View 1.2.36"‐Software (Hamamatsu Photonics, Herrsching am Ammersee, Germany) was used to evaluate the area of glomeruli; 115 glomeruli were evaluated in each slide.
Statistical analysis
All data are presented as mean ± standard error of the mean (s.e.m.). Differences between two groups were evaluated using unpaired two‐sided (or one‐sided, where indicated specifically) or Student's t‐test when data were distributed normally. Otherwise, a non‐parametric Mann–Whitney test was performed. Statistical analysis of the Kaplan–Meier survival plots was performed using the log‐rank (Mantel–Cox) test. All statistical analysis and subsequent graphics generation was performed using GraphPad Prism version 5·0 (GraphPad Software, San Diego, CA, USA). A P‐value < 0·05 was considered significant.
Results
Deletion of CXCR5 in a murine lupus model reduces the humoral immune response and increases life expectancy
Like CCR7, CXCR5 is a key molecule for the entry of lymphocytes and dendritic cells into secondary lymphoid organs and their homing to T cell and B cell zones therein. To analyse how CXCR5 influences disease progression in murine lupus we crossed B6/CXCR5–/– mice with B6/lpr mice, a well‐established murine lupus model. CXCR5 is expressed primarily on B cells and B6/lpr CD19+ cells also expressed high levels of CXCR5 (Fig. 1a). As expected, absolute numbers of CD19+ B cells were reduced in B6/lpr CXCR5–/– mice (Fig. 1b) and percentages were reduced significantly in the spleen and tended to be reduced in lymph nodes (Fig. 1c). As CXCR5 is involved critically in GC formation, we next analysed formation of GCs. We performed peanut agglutinin staining, which effectively stains GC B cells, in combination with CD3+ antibody for T cell staining in spleen sections. B6/lpr mice showed spontaneous secondary follicles with GCs within the white pulp of the spleen and a T cell zone situated around the central artery at one pole of the follicle. No GCs were observed in spleens from B6/lpr CXCR5–/– mice, and here the T cell zone was situated centrally but not polarized in the follicle (Fig. 2a). This is in line with the lymphoid tissue microarchitecture observed by Förster et al. in CXCR5 single‐knock‐out mice 5. Furthermore, we analysed the occurrence of plasma cells and IgGs in sera of aged B6/lpr and B6/lpr CXCR5–/– mice. Percentages (Fig. 2b) and absolute numbers (Supporting information, Fig. S1a) of plasma cells were reduced in B6/lpr CXCR5–/– mice. Consequently, aged B6/lpr mice revealed significantly more IgG in sera than their aged B6/lpr CXCR5–/– littermates (Fig. 2c). Furthermore, B6/lpr CXCR5–/– mice had a higher life expectancy, while their B6/lpr littermates tended to die after the age of 50 weeks (Fig. 2d).
Figure 1.

Deletion of CXCR5 in a murine lupus model reduces B cell accumulation in peripheral lymphoid organs. (a) CXCR5 expression on CD3–B220+ B cells (red) and isotype control (grey) in B6/lpr mice (38 weeks). (b) Absolute numbers of splenic CD19+ cells from B6/lpr and B6/lpr CXCR5–/– mice (10–11 weeks old) of four mice per group. (c) Percentages of CD19+ B cells in spleens and lymph nodes of age‐matched B6/lpr and B6/lpr CXCR5–/– mice (8–11 weeks old). Data show mean percentage ± standard error of the mean (s.e.m.) of CD19+ cells in B6/lpr and B6/lpr CXCR5–/– mice (at least six mice per group).
Figure 2.

Deletion of CXCR5 in a murine lupus model reduces the humoral immune response and increases life expectancy. (a) Microscope photos show frozen spleen sections from 15–16‐week‐old B6/lpr (left) and B6/lpr CXCR5–/– (right) mice that were stained with anti‐CD3‐allophycocyanin (APC) antibody (red) for T cells and biotinylated peanut agglutinin (PNA)/streptavidin DyLight‐488 (green) for germinal centre (GC) cells. The slides were imaged with a ×10 objective using a fluorescence microscope. Statistical analysis of frozen spleen sections stained with anti‐CD3‐allophycocyanin (APC) antibody and biotinylated PNA/streptavidin DyLight‐488. The numbers of GCs per mm2 spleen were counted in 12–16‐week‐old B6/lpr and B6/lpr CXCR5–/– mice. At least four mice per group were analysed. (b) Percentages of splenic CD138+ cells from 10–11‐week‐old B6/lpr and B6/lpr CXCR5–/– mice assessed by flow cytometry (at least four mice per group). (c) Total immunoglobulin (Ig)G enzyme‐linked immunosorbent assay (ELISA) of sera from aged (> 45 weeks) B6/lpr and B6/lpr CXCR5–/– mice. Data indicate mean values of µg/ml IgG in serum ± standard error of the mean (s.e.m.) of at least eight mice per group. (d) Kaplan–Meier survival plots were used to assess the life expectancy rates of B6/lpr and B6/lpr CXCR5–/– mice; 69 B6/lpr mice and 134 B6/lpr CXCR5–/– mice were observed.
Deletion of CXCR5 decreases lymphadenopathy and splenomegaly in lupus‐prone mice
With age, B6/lpr mice progressively develop severe lymphadenopathy and splenomegaly, largely attributable to an expanded pool of DN T cells. Deletion of CXCR5 in B6/lpr mice decreased lymphadenopathy, as defined by lymphadenopathy score (Fig. 3a); more specifically, the lymphadenopathy of B6/lpr CXCR5–/– remained constantly low while lymphadenopathy of B6/lpr mice increased severely with age. In addition, splenomegaly, as defined by the weight of spleens, was not altered in younger mice but was markedly lower in older B6/lpr CXCR5–/– mice (Fig. 3b,c). Lymphadenopathy and splenomegaly in lpr mice results largely from the accumulation of an unusual subset of CD4–CD8– (DN) T cells 23. This led us to investigate whether the DN T cells of our B6/lpr mice express CXCR5 on their surface, and we indeed found an even higher expression than that seen on CD4+ T cells (Supporting information, Fig. S1b). As expected, no expression of CXCR5 on any cell population was found in B6/lpr CXCR5–/– mice (Supporting information, Fig. S1c).
Figure 3.

Deletion of CXCR5 decreases lymphadenopathy and splenomegaly in lupus‐prone mice. (a) Assessment of lymphadenopathy in differently aged B6/lpr and B6/lpr CXCR5–/– mice. Data indicate mean ± standard error of the mean (s.e.m.) of at least four mice per group. (b) Assessment of splenomegaly by spleen weight of differently aged B6/lpr and B6/lpr CXCR5–/– mice. Data indicate mean ± s.e.m. of at least four mice per group. (c) Representative image of spleens from 58‐week‐old B6/lpr (left) and B6/lpr CXCR5–/– (right) mice.
Reduced lymphadenopathy and splenomegaly are associated with a lower accumulation of DN T cells
We next analysed cell populations in lymph nodes, spleens and blood from B6/lpr CXCR5–/– and B6/lpr mice. In addition to enhanced B cell numbers (as discussed above), we found an approximately threefold increase in absolute numbers of splenic DN T cells in 10‐week‐old B6/lpr compared to age‐matched B6/lpr CXCR5–/– mice (Fig. 4a). B6/lpr CXCR5–/– mice (up to 11 weeks) showed an approximately 1.4‐fold reduction in the percentage of DN T cells in spleens (Fig. 4b,c) and an approximately 1.7‐fold reduction of their frequency in lymph nodes (Fig. 4b). The frequencies of DN T cells in blood were only slightly enhanced by the deletion of CXCR5 (Fig. 4b), suggesting that the migration of DN T cells into spleen and lymph nodes is attenuated. With regard to other T cell subsets, young B6/lpr CXCR5–/– mice displayed reduced absolute numbers of CD4+ and CD8+ T cells (Supporting information, Fig. S1d), but even slightly enhanced percentages of CD3+CD4+ and no deviating percentages of CD3+CD8+ cells in spleens (Supporting information, Fig. S1e,f) and lymph nodes (Supporting information, Fig. S1e,f) compared to B6/lpr mice. Again, the distribution of CD3+CD4+ cells and CD3+CD8+ cells in the blood of young B6/lpr mice was not influenced by the deletion of CXCR5 (Supporting information, Fig. S1e,f). Our data thus demonstrate that a deletion of CXCR5 reduces percentages of DN T cells but not of CD4+ and CD8+ T cells in spleen and lymph nodes which, in addition to reduced numbers and percentages of B cells (Fig. 1a,b), is associated with decreased lymphadenopathy and splenomegaly in B6/lpr mice.
Figure 4.
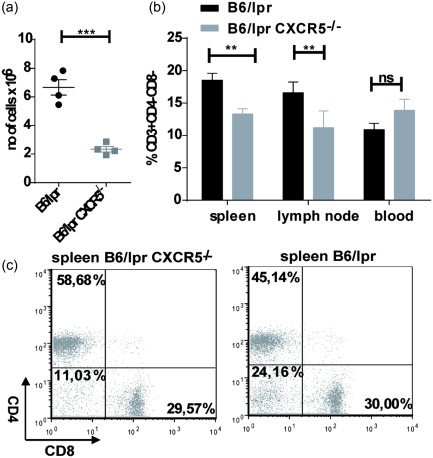
Reduced lymphadenopathy and splenomegaly are caused by lower accumulation of double‐negative (DN) T cells. (a) Absolute numbers of splenic DN T cells in 10–11‐week‐old mice. (b) Percentages of DN T cells in spleen, lymph nodes and blood of age‐matched (6–11 weeks old) B6/lpr and B6/lpr CXCR5–/– mice. Data show mean percentage ± standard error of the mean (s.e.m.) of DN T cells in B6/lpr and B6/lpr CXCR5–/– mice (at least five mice per group). (c) Representative flow cytometric dot‐plots of CD3+ T cell from spleens of 8‐week‐old B6/lpr and B6/lpr CXCR5–/– mice. All plots are shown in a CD3+ gate.
Deletion of CXCR5 in B6/lpr mice reduces occurrence of inflammatory DN T cells in spleen and lymph nodes
DN T cells have been ascribed proinflammatory and regulatory roles 12, 24. Expanded DN T cells in SLE and murine lupus are believed to be pathogenic and a major source of IL‐17 25. We aimed to analyse if CXCR5 deletion does indeed reduce numbers of pathogenic and inflammatory DN T cells in B6/lpr mice. DN T cells in both B6/lpr and CXCR5–/– B6 lpr expressed IL‐17 and IFN‐γ (Fig. 5a,c). Percentages of IL‐17+ and IFN‐γ cells within the DN T cell population were not reduced in spleens and lymph nodes. However, in terms of absolute numbers, both CD3+ IL‐17+ and DN IL‐17+ cells (Fig. 5b,d) were reduced significantly in lymph nodes of CXCR5–/– B6/lpr mice. This indicates that CXCR5 deletion does not alter the cytokine production of DN T cells themselves but clearly reduces numbers of inflammatory DN T cells in lymph nodes of B6/lpr mice.
Figure 5.

Deletion of CXCR5 in B6/lpr mice reduces occurrence of inflammatory double‐negative (DN) T cells in spleen and more so in lymph nodes. (a) Representative flow cytometric dot‐plots of interferon (IFN)‐γ‐producing CD3+CD4–CD8– cells and (c) interleukin (IL)−17‐producing CD3+CD4–CD8– cells from cervical lymph nodes of 14‐week‐old B6/lpr and B6/lpr CXCR5–/– mice. Plots show all cells alive in whole cervical lymph nodes with CD4+‐ and CD8+ cells excluded from the CD3+ gate. (b) Absolute numbers of CD3+ IFN‐γ+ and IFN‐γ+ DN T cells and (d) of CD3+ IL‐17+ and IL‐17+ DN T cells from cervical lymph nodes (left) and spleen (right) in 14‐week‐old mice. Data indicate mean standard error of the mean (s.e.m.) of at least four mice per group.
Deletion of CXCR5 in B6/lpr mice reduces the migration of DN T cells in vivo and in vitro
To clarify whether reduced occurrence of DN T cells in CXCR5–/– B6/lpr is simply an epiphenomenon or is caused by a migratory defect of these cells, we performed in‐vivo migration assays. B6 wild‐type mice were injected intravenously (i.v.) with equal numbers of labelled lpr or lpr CXCR5–/– DN T cells. After 48 h mice were killed and spleen, lymph nodes and blood were analysed for DN T cells. Significantly more DN T cells from B6/lpr mice migrated into lymph nodes of B6/lpr wild‐type mice than DN T cells from B6/lpr CXCR5–/– mice (Fig. 6a), while DN T levels in blood or spleen remained unaltered. This suggests that CXCR5 expression is required for migration into the lymph nodes.
Figure 6.

Deletion of CXCR5 in B6/lpr mice reduces the migration of double‐negative (DN) T cells into lymph nodes of B6 wild‐type mice and into kidneys of Rag–/– mice. (a) Percentages of Dye‐670‐positive cells derived from either B6/lpr or B6/lpr CXCR5–/– 9‐week‐old mice in the CD3+CD4–CD8– gate in lymph nodes (left) in spleen (middle) and blood (right) of B6 wild‐type mice (at least four mice per group). Data show mean percentage standard error of the mean (s.e.m.) of Dye‐670‐labelled DN T cells in B6 wild‐type mice from two independent experiments. (b) Frozen kidney sections from Rag–/– mice with expanded CD3 +CD4–CD8– cells transferred from 13‐week‐old B6/lpr (upper image) or B6/lpr CXCR5–/– (lower image) mice were stained with anti‐CD3‐allophycocyanin (APC) antibody (red) and 4',6‐diamidino‐2‐phenylindole (DAPI) (blue). The slides were analysed with a ×40 objective and imaged using a fluorescence microscope. White arrows point to individual cells representing the DN T cells. Statistical analysis of frozen kidney sections stained with anti‐CD3‐APC antibody (right). Five segments in two different layers were analysed for CD3+ cells. (c) Periodic acid Schiff (PAS) staining of kidney sections from Rag−/− mice injected with either B6/lpr (upper image) or B6/lpr CXCR5–/– (lower image) derived lymphocytes, original magnification ×400. The glomerular area from kidney sections was measured as described in Materials and methods. The mean ± s.e.m. of the area of 115 glomeruli (derived from three mice in both groups) is plotted. (d) In‐vitro migration assay was performed using Transwell chambers containing CXCL13 (2 µg/600 µl) or no chemoattractant (control). Single‐cell suspensions from spleens of either B6/lpr or B6/lpr CXCR5–/– mice (47–48 weeks old) were added to the upper chamber and subjected to a 4‐h migration period. Data of at least three mice per group are expressed as chemotactic index CTX (mean ± s.e.m.) compared to their respective control. (e) Chemotactic index of CD19+ cells from spleen of B6/lpr and B6/lpr CXCR5–/– mice (47–48 weeks old) in relation to their respective control (without chemoattractant). Unpaired two‐sided Student's t‐tests were performed and a P‐value < 0·05 was considered significant. (f) Chemotactic index of CD3+CD4+, CD3+CD8+ and DN T cells from spleen of B6/lpr and B6/lpr CXCR5–/– 47–48‐week‐old mice in relation to their respective control (at least three mice per group). Unpaired one‐sided Student's t‐tests were performed and a P‐value < 0·05 was considered significant.
We next asked if a deletion of CXCR5 also prevents DN T cells from infiltrating the kidneys. Previous work by Zhang et al. indicated that expanded DN T cells derived from MRL/lpr mice produce IL‐17, infiltrate the kidneys and contribute to nephritis 13. We transferred expanded DN T cells from lymph nodes, either from B6/lpr or B6/lpr CXCR5–/– mice, into lymphopenic Rag–/– mice. After 9 weeks mice were killed and kidneys were harvested for immunofluorescence analysis. Kidney sections from Rag–/– mice transferred with DN T cells from B6/lpr mice clearly showed CD3+ cells engrafted in their kidney structure, while almost no CD3+ cells were found in kidney sections of Rag–/– mice transferred with DN T cells from B6/lpr CXCR5–/– mice (Fig. 6b). In addition, we measured the average glomerular area in paraffin‐embedded and PAS‐stained kidney sections. We found that the transfer of activated lymphocytes derived from B6/lpr CXCR5–/– mice led to a decrease in the average glomerular area compared to the transfer of B6/lpr lymphocytes with CXCR5 expression (Fig. 6c). However, only some of the animals developed proteinuria and there was no significant difference between the two groups. Nevertheless, we conclude that a deletion of CXCR5 in B6/lpr mice inhibits infiltration of DN T cells into the kidneys and possibly protects B6/lpr CXCR5–/– mice from developing nephritis.
Our findings, that DN T cells from B6/lpr mice express CXCR5 and that CXCR5–/– B6/lpr mice have reduced accumulation of DN T cells in peripheral lymph organs, suggest that the CXCR5/CXCL13 axis promotes the chemotactic migration of DN T cells. However, when we analysed expression of CD62L, an adhesion molecule also responsible for lymphocyte homing into peripheral lymph organs, we found reduced CD62L expression on CXCR5–/– DN T cells in the spleen (Supporting information, Fig. S1g). To clarify whether CXCR5 deletion indeed prevents DN T cell migration, we performed in‐vitro cell migration assays with CXCL13. The migratory capacity of splenic B6/lpr and CXCR5–/– B6/lpr cells in response to CXCL13 or controls was assayed using a Transwell migration device. CXCL13 showed a chemotactic effect on all splenic B6/lpr cells, while the chemotactic response toward CXCL13 was reduced strongly and nearly absent in CXCR5–/– B6/lpr splenic cells (Fig. 6d). We then further analysed the chemotactic response of particular lymphocyte populations using flow cytometry. As expected, the migratory capacity of B cells was diminished strongly in the absence of CXCR5 (Fig. 6e). Moreover, CXCR5–/– B6/lpr DN T cells also revealed a reduced chemotactic response towards CXCL13 (Fig. 6f).
These data indicate that CXCR5 indeed plays an important role in the correct homing of activated B cells and T cell subtypes into secondary follicles and is required for an effective antibody production in lpr mice.
Discussion
SLE is a multi‐system autoimmune disease characterized by the production of numerous autoantibodies and involvement of several organs and tissues, such as skin, joints, kidneys, brain, serosal surfaces, blood vessels, blood cells, lungs and heart. Mechanisms of organ and tissue damage in patients with SLE involve autoantibody and immune complex deposition, as well as infiltration of lymphocytes. T lymphocytes from patients with SLE display a complex array of cellular, molecular and signalling anomalies. An obvious anomaly is the expanded population of DN T cells 12. The origin of this rare cell population in healthy humans and rodents is not known completely, but recent studies suggest that DN T cells in SLE are not generated in the thymus and derive from CD8+ T cells through down‐regulation of the CD8 receptor after TCR stimulation 19, 20. There is strong evidence that DN T cells infiltrate the kidneys, produce proinflammatory cytokines (including IL‐1β and IL‐17) and promote B cell antibody production 12, 13, 17. LN is one of the most frequent and severe manifestations of SLE and increases patient morbidity and mortality. Conventional immunosuppressive therapy has increased the life expectancy of patients diagnosed with LN, but only 70–80% of the patients respond sufficiently to treatment and the adverse effects of therapy are considerable. Therefore, analyses of the mechanisms that drive autoimmune inflammation are a current matter of intensive research. CXCL13 is expressed highly in the renal cortex from patients with LN and in kidneys of lupus‐prone mice developing lupus nephritis 8, 9, 10, 26. CXCL13 shows potential as a marker for diagnosis of lupus nephritis, but is also seen as a promising therapeutic approach for SLE. Nevertheless, an in‐depth analysis of the CXCL13/CXCR5 signalling mechanism in SLE, target cells and potential aberrancies in autoimmune conditions are still lacking. We deleted CXCR5 in B6/lpr mice to investigate the cellular effects of CXCR5 in vivo. Our data suggest that in addition to attracting B cells, CXCL13 induces infiltration of DN T cells into inflamed areas. In detail, our data indicate that CXCR5 is expressed on DN T cells from lpr mice and facilitates migration of these cells to lymphoid organs and kidneys. CXCR5‐deficient lpr mice show reduced lymphadenopathy and develop less splenomegaly with age, and this is associated with reduced percentages and numbers of DN T cells as well as of B cells. Although absolute numbers of B cells and DN T cells are already reduced in spleens of younger mice, an obvious phenotype with reduced splenic weight occurs later in life in aged mice. DN T cells are considered to be a T cell population with an inflammatory phenotype in SLE. However, a regulatory function is also described for DN T cells, in that they suppress excessive immune responses, for instance, on allograft rejection or in graft‐versus‐host‐ and autoimmune diseases 16. DN T cells in the B6/lpr mice described here have an inflammatory phenotype characterized by the expression of cytokines such as IFN‐γ and IL‐17. CXCR5 deletion clearly down‐regulates accumulation of cytokine‐expressing DN T cells in lymph nodes.
In‐vivo migration experiments underscore reduced migration of DN T cells lacking CXCR5 into lymph nodes and into kidneys and reduced development of nephritis. Former studies showed that a significant proportion of DN T cells that infiltrate kidneys in LN and produce IL‐17 are DN T cells 12. Lymph node cells from lupus‐prone mice treated in vitro with IL‐23 induce nephritis when transferred to non‐autoimmune, lymphocyte‐deficient Rag−/− mice 13. Our data indicate that CXCR5 is involved in these processes: first, the size of the average glomerular area was decreased after the transfer of B6/lpr CXCR5–/– lymphocytes compared to the transfer of B6/lpr lymphocytes expressing CXCR5. Secondly, almost no CD3+ cells were found in kidney sections of Rag–/– mice with DN T cells transferred from B6/lpr CXCR5–/– mice. Thirdly, in‐vitro migration assays provide further proof that CXCR5 directs DN T cell migration, as CXCR5‐deficient DN T cells failed to migrate towards CXCL13 in vitro. Lower expression of CD62L on B6/lpr CXCR5–/– DN T cells in spleen might contribute further to the reduced entry of DN T cells into spleen and lymph nodes. However, we consider it more likely that reduced expression of CD62L is secondary to the decreased disease severity in the B6/lpr CXCR5–/– mice. Reduced CD62L expression might be caused by lower B and plasma cell activation in B6/lpr CXCR5–/– mice. CD62L expression correlates with disease activity in human SLE 27, and our model appears analogous to human disease.
This leads to the second major finding of this study: CXCR5 deletion also resulted in reduced IgG levels, reduced GC formation and reduced B cell numbers in spleens of B6/lpr mice. These findings were expected, as the role of CXCR5 for B cell migration and follicle formation has been studied intensively. CXCR5 is required for B cell migration to splenic follicles and also enables follicular T helper cells to gain access to the follicles 5 and GCs 28. Consequently, mice lacking CXCL13 or the CXCL13 receptor CXCR5 fail to form lymphoid follicles 5, 29. With regard to SLE, current studies suggest that CXCR5 expression on CD4+ T cells and B cells plays a pathogenic role in SLE. Peripheral blood B cells bearing CXCR5 are more abundant in SLE patients 10. Furthermore, studies also show increases in circulating follicular T helper cells in SLE patients, which correlate with autoantibody titres, frequencies of circulating GC B cells, plasma cells and disease severity in SLE 7, 30. Our data thereby underpin the relevance of CXCR5 expression for humoral responses in lupus in vivo. Finally, CXCR5 deletion also prolonged the life expectancy of lpr mice. We hypothesize that this results from reduced B cell migration, GC formation and IgG production and reduced DN T cell migration and infiltration of the kidneys by DN T cells, B cells and CD4+ T cells.
Our study thereby uncovers one new mechanism by which CXCR5 may guide DN T cells in SLE and confirms in‐vivo findings of recent studies indicating involvement of CXCR5/CXCL13 in the humoral immune response in SLE. CXCL13 displays a strong association with SLE, which is highlighted in several studies with patients of different ages and ethnicities and by in‐vivo research with different murine lupus models 8. However, pathomechanisms, signalling pathways and target cells of CXCL13 in SLE have not been elucidated fully. B cells are central to the pathogenesis of SLE and are therefore an attractive therapeutic target. The B cell‐depleting agent rituximab could be a potent SLE drug. However, two randomized controlled trials failed to meet efficacy end‐points 31. As the potential consequences of prolonged B cell depletion are not known, great interest is being focused upon alternative strategies to modulate B cell function in autoimmune diseases.
In summary, our experiments provide in‐vivo evidence that CXCR5 plays a role in disease progression of lupus by regulating B cell as well as DN T cell trafficking. Our results provide further evidence that targeting of the CXCL13/CXCR5 signalling pathway may present a promising approach to limitation of pathogenic B and T cell responses in SLE.
Disclosure
The authors declare no commercial or financial disclosures.
Author contributions
A. W., N. H., T. O. and K. O. performed the experiments; A. S., N. W. and K. T. designed the study and wrote the paper; F. T. provided B6/CXCR5–/– mice and designed the study; K. O. designed the study and wrote the paper.
Supporting information
Additional Supporting information may be found in the online version of this article at the publisher's web‐site:
Fig. S1. (a) Absolute numbers of splenic CD138+ cells from B6/lpr and B6/lpr CXCR5–/– mice (10–11 weeks old). (b) CXCR5 expression on double‐negative (DN) T cells (red) compared to CXCR5 expression on CD4+ T cells (black) and isotype control (grey) in B6/lpr mice (38 weeks). (c) CXCR5 expression on DN T cells (red) compared to CXCR5 expression on CD4+ T cells (black) and isotype control (grey) in B6/lpr CXCR5–/– mice (11 weeks). (d) Absolute numbers of splenic CD3+CD4+ and CD3+CD8+ cells from B6/lpr and B6/lpr CXCR5–/– mice (10–11 weeks old). (e) Cells were isolated from cervical lymph nodes, spleen and blood and flow cytometric analysis was performed. Percentages of CD3+CD4+ cells in spleens, in lymph nodes and in blood of age‐matched (6–11 weeks old) B6/lpr and B6/lpr CXCR5–/– mice. Data show mean percentage ± standard error of the mean (s.e.m.) of CD3+CD4+ cells in B6/lpr and B6/lpr CXCR5–/– mice (at least five mice per group). (f) Percentages of CD3+CD8+ cells in spleens and lymph nodes and in blood of age‐matched (6–11 weeks old) B6/lpr and B6/lpr CXCR5–/– mice. Data show mean percentage ± s.e.m. of CD3+CD8+ cells in B6/lpr and B6/lpr CXCR5–/– mice (at least five mice per group). (g) Percentages of CD62L‐expressing DN T cells from spleen of 10–14‐week‐old B6/lpr and B6/lpr CXCR5–/– mice (at least six mice per group).
Acknowledgements
We thank Lilia Görtz for technical assistance.
References
- 1. Tsokos GC. Systemic lupus erythematosus. N Engl J Med 2011; 365:2110–21. [DOI] [PubMed] [Google Scholar]
- 2. Pugh‐Bernard AE, Silverman GJ, Cappione AJ et al Regulation of inherently autoreactive VH4‐34 B cells in the maintenance of human B cell tolerance. J Clin Invest 2001; 108:1061–70. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3. Dorner T, Giesecke C, Lipsky PE. Mechanisms of B cell autoimmunity in SLE. Arthritis Res Ther 2011; 13:243. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4. Weyand CM, Kurtin PJ, Goronzy JJ. Ectopic lymphoid organogenesis: a fast track for autoimmunity. Am J Pathol 2001; 159:787–93. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5. Forster R, Mattis AE, Kremmer E, Wolf E, Brem G, Lipp M. A putative chemokine receptor, BLR1, directs B cell migration to defined lymphoid organs and specific anatomic compartments of the spleen. Cell 1996; 87:1037–47. [DOI] [PubMed] [Google Scholar]
- 6. Simpson N, Gatenby PA, Wilson A et al Expansion of circulating T cells resembling follicular helper T cells is a fixed phenotype that identifies a subset of severe systemic lupus erythematosus. Arthritis Rheum 2010; 62:234–44. [DOI] [PubMed] [Google Scholar]
- 7. Zhang X, Lindwall E, Gauthier C et al Circulating CXCR5+CD4+helper T cells in systemic lupus erythematosus patients share phenotypic properties with germinal center follicular helper T cells and promote antibody production. Lupus 2015; 24:909–17. [DOI] [PubMed] [Google Scholar]
- 8. Schiffer L, Worthmann K, Haller H, Schiffer M. CXCL13 as a new biomarker of systemic lupus erythematosus and lupus nephritis – from bench to bedside? Clin Exp Immunol 2015; 179:85–9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9. Worthmann K, Gueler F, von Vietinghoff S et al Pathogenetic role of glomerular CXCL13 expression in lupus nephritis. Clin Exp Immunol 2014; 178:20–7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10. Lee HT, Shiao YM, Wu TH et al Serum BLC/CXCL13 concentrations and renal expression of CXCL13/CXCR5 in patients with systemic lupus erythematosus and lupus nephritis. J Rheumatol 2010; 37:45–52. [DOI] [PubMed] [Google Scholar]
- 11. Crispin JC, Kyttaris VC, Terhorst C, Tsokos GC. T cells as therapeutic targets in SLE. Nat Rev Rheumatol 2010; 6:317–25. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12. Crispin JC, Oukka M, Bayliss G et al Expanded double negative T cells in patients with systemic lupus erythematosus produce IL‐17 and infiltrate the kidneys. J Immunol 2008; 181:8761–6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13. Zhang Z, Kyttaris VC, Tsokos GC. The role of IL‐23/IL‐17 axis in lupus nephritis. J Immunol 2009; 183:3160–9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14. Alunno A, Carubbi F, Bistoni O et al CD4(–)CD8(–) T‐cells in primary Sjogren's syndrome: association with the extent of glandular involvement. J Autoimmun 2014; 51:38–43. [DOI] [PubMed] [Google Scholar]
- 15. Adipue IA, Wilcox JT, King C et al Characterization of a novel and spontaneous mouse model of inflammatory arthritis. Arthritis Res Ther 2011; 13:R114. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16. Juvet SC, Zhang L. Double negative regulatory T cells in transplantation and autoimmunity: recent progress and future directions. J Mol Cell Biol 2012; 4:48–58. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17. Shivakumar S, Tsokos GC, Datta SK. T cell receptor alpha/beta expressing double‐negative (CD4–/CD8–) and CD4+ T helper cells in humans augment the production of pathogenic anti‐DNA autoantibodies associated with lupus nephritis. J Immunol 1989; 143:103–12. [PubMed] [Google Scholar]
- 18. Ohl K, Wiener A, Schippers A, Wagner N, Tenbrock K. Interleukin‐2 treatment reverses effects of cAMP‐responsive element modulator alpha‐over‐expressing T cells in autoimmune‐prone mice. Clin Exp Immunol 2015; 181:76–86. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19. Hedrich CM, Rauen T, Crispin JC et al cAMP‐responsive element modulator alpha (CREMalpha) trans‐represses the transmembrane glycoprotein CD8 and contributes to the generation of CD3+CD4–CD8– T cells in health and disease. J Biol Chem 2013; 288:31880–7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20. Hedrich CM, Crispin JC, Rauen T et al cAMP responsive element modulator (CREM) alpha mediates chromatin remodeling of CD8 during the generation of CD3+ CD4– CD8– T cells. J Biol Chem 2014; 289:2361–70. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21. Rodriguez‐Rodriguez N, Apostolidis SA, Penaloza‐MacMaster P et al Programmed cell death 1 and Helios distinguish TCR‐alphabeta+ double‐negative (CD4–CD8–) T cells that derive from self‐reactive CD8 T cells. J Immunol 2015; 194:4207–14. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22. Steinmetz OM, Velden J, Kneissler U et al Analysis and classification of B‐cell infiltrates in lupus and ANCA‐associated nephritis. Kidney Int 2008; 74:448–57. [DOI] [PubMed] [Google Scholar]
- 23. Cohen PL, Eisenberg RA. Lpr and gld: single gene models of systemic autoimmunity and lymphoproliferative disease. Annu Rev Immunol 1991; 9:243–69. [DOI] [PubMed] [Google Scholar]
- 24. Zhang ZX, Yang L, Young KJ, DuTemple B, Zhang L. Identification of a previously unknown antigen‐specific regulatory T cell and its mechanism of suppression. Nat Med 2000; 6:782–9. [DOI] [PubMed] [Google Scholar]
- 25. Mizui M, Koga T, Lieberman LA et al IL‐2 protects lupus‐prone mice from multiple end‐organ damage by limiting CD4‐CD8‐ IL‐17‐producing T cells. J Immunol 2014; 193:2168–77. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26. Ishikawa S, Sato T, Abe M et al Aberrant high expression of B lymphocyte chemokine (BLC/CXCL13) by C11b+CD11c+ dendritic cells in murine lupus and preferential chemotaxis of B1 cells towards BLC. J Exp Med 2001; 193:1393–402. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27. Font J, Pizcueta P, Ramos‐Casals M et al Increased serum levels of soluble L‐selectin (CD62L) in patients with active systemic lupus erythematosus (SLE). Clin Exp Immunol 2000; 119:169–74. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28. Crotty S. Follicular helper CD4 T cells (TFH). Annu Rev Immunol 2011; 29:621–63. [DOI] [PubMed] [Google Scholar]
- 29. Ansel KM, Ngo VN, Hyman PL et al A chemokine‐driven positive feedback loop organizes lymphoid follicles. Nature 2000; 406:309–14. [DOI] [PubMed] [Google Scholar]
- 30. Feng X, Wang D, Chen J et al Inhibition of aberrant circulating Tfh cell proportions by corticosteroids in patients with systemic lupus erythematosus. PLOS ONE 2012; 7:e51982. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31. Gregersen JW, Jayne DR. B‐cell depletion in the treatment of lupus nephritis. Nat Rev Nephrol 2012; 8:505–14. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Additional Supporting information may be found in the online version of this article at the publisher's web‐site:
Fig. S1. (a) Absolute numbers of splenic CD138+ cells from B6/lpr and B6/lpr CXCR5–/– mice (10–11 weeks old). (b) CXCR5 expression on double‐negative (DN) T cells (red) compared to CXCR5 expression on CD4+ T cells (black) and isotype control (grey) in B6/lpr mice (38 weeks). (c) CXCR5 expression on DN T cells (red) compared to CXCR5 expression on CD4+ T cells (black) and isotype control (grey) in B6/lpr CXCR5–/– mice (11 weeks). (d) Absolute numbers of splenic CD3+CD4+ and CD3+CD8+ cells from B6/lpr and B6/lpr CXCR5–/– mice (10–11 weeks old). (e) Cells were isolated from cervical lymph nodes, spleen and blood and flow cytometric analysis was performed. Percentages of CD3+CD4+ cells in spleens, in lymph nodes and in blood of age‐matched (6–11 weeks old) B6/lpr and B6/lpr CXCR5–/– mice. Data show mean percentage ± standard error of the mean (s.e.m.) of CD3+CD4+ cells in B6/lpr and B6/lpr CXCR5–/– mice (at least five mice per group). (f) Percentages of CD3+CD8+ cells in spleens and lymph nodes and in blood of age‐matched (6–11 weeks old) B6/lpr and B6/lpr CXCR5–/– mice. Data show mean percentage ± s.e.m. of CD3+CD8+ cells in B6/lpr and B6/lpr CXCR5–/– mice (at least five mice per group). (g) Percentages of CD62L‐expressing DN T cells from spleen of 10–14‐week‐old B6/lpr and B6/lpr CXCR5–/– mice (at least six mice per group).