

Abstract
Motivation: As ‘omics’ biotechnologies accelerate the capability to contrast a myriad of molecular measurements from a single cell, they also exacerbate current analytical limitations for detecting meaningful single-cell dysregulations. Moreover, mRNA expression alone lacks functional interpretation, limiting opportunities for translation of single-cell transcriptomic insights to precision medicine. Lastly, most single-cell RNA-sequencing analytic approaches are not designed to investigate small populations of cells such as circulating tumor cells shed from solid tumors and isolated from patient blood samples.
Results: In response to these characteristics and limitations in current single-cell RNA-sequencing methodology, we introduce an analytic framework that models transcriptome dynamics through the analysis of aggregated cell–cell statistical distances within biomolecular pathways. Cell–cell statistical distances are calculated from pathway mRNA fold changes between two cells. Within an elaborate case study of circulating tumor cells derived from prostate cancer patients, we develop analytic methods of aggregated distances to identify five differentially expressed pathways associated to therapeutic resistance. Our aggregation analyses perform comparably with Gene Set Enrichment Analysis and better than differentially expressed genes followed by gene set enrichment. However, these methods were not designed to inform on differential pathway expression for a single cell. As such, our framework culminates with the novel aggregation method, cell-centric statistics (CCS). CCS quantifies the effect size and significance of differentially expressed pathways for a single cell of interest. Improved rose plots of differentially expressed pathways in each cell highlight the utility of CCS for therapeutic decision-making.
Availability and implementation: http://www.lussierlab.org/publications/CCS/
Contact: yves@email.arizona.edu or piegorsch@math.arizona.edu
Supplementary information: Supplementary data are available at Bioinformatics online.
1 Introduction
The advent of single-cell RNA-sequencing (scRNA-seq; Liang et al., 2014; Tang et al., 2009) enables discovery of transcriptional patterns at the most fundamental unit of life. In contrast, conventional RNA-seq technologies only provide an average RNA expression across many cells, concealing much of the transcriptional heterogeneity (Schubert, 2011). Understanding individual cell uniqueness within a multicellular context offers new insights into the biological underpinning of organ ontogeny, immune response and cancer etiology, progression and drug resistance (Navin, 2015; Sandberg, 2014). Particularly, scRNA-seq has become increasingly adopted to resolve intra-tumor heterogeneity and analyze rare tumor cell populations such as circulating tumor cells (CTCs) (Aceto et al., 2014; Chen and Bai, 2015; Ramsköld et al., 2012) originating from primary solid tumors. However, analyzing whole-genome RNA expression from individual cells remains challenging (Stegle et al., 2015). These challenges include the poor sensitivity of conventional methods for studying a limited number of cells and the absence of methods for generating statistical significance at a single-cell transcriptome.
Improvements in scRNA-seq experimentation, through the additional quantitative standards of spike-in external control RNA (Jiang et al., 2011) and unique molecular identifiers (Islam et al., 2014), as well as computational methodologies (Ding et al., 2015; Grün et al., 2014; Scialdone et al., 2015; Wu et al., 2014), have reduced the impact of noisy measurements in single-cell RNA expression. However, these methods rely on expensive technologies and require many single-cell transcriptomes to adequately model noise and infer sources of variation, which preclude analysis of individual cells. To establish significance of patterns observed at a single cell, one could in theory compare cellular mRNAs with a consensus mRNA expression atlas (Kapushesky et al., 2009; Lukk et al., 2010). However, the notion that there is a consensus transcriptome is questionable as expression levels and kinetics vary over time and across tissues. In response to these shortcomings, we introduce a novel analytic framework: the analysis of aggregated cell–cell statistical distances within pathways (Fig. 1). We hypothesized that we could aggregate many analyses of pairs of single-cell transcriptomes to predict differentially expressed pathways (DEPs). In principle, this approach could even produce cell-centric statistics (CCS) that may scale down to analyze DEPs in a single cell despite the lack of true reference transcriptome and circumvent sample size requirements intrinsic to group-based statistics. By quantifying gene sets (pathways) rather than individual mRNAs, our framework is designed ab initio to reduce the noise intrinsic to scRNA-seq measurements, while providing functional interpretation of dynamic changes between cells.
Fig. 1.

Analytic framework: analysis of aggregated cell–cell statistical distances within pathways unveils cross-group, within-group and cell-centric properties of single-cell transcriptomes. Here, the four analytic strategies used in this study are presented, culminating with CCS. (A) Prior work led to the development of N-of-1-pathways MD. In this study, MD is used to find DEPs between a pair of cells. MD quantifies differential mRNA expression within a set of genes (left, illustrated as a funnel). Specifically, the average signed Mahalanobis vertical distance (MD score, ) from the equal-expression reference line (diagonal) quantifies differential pathway expression between two cells (right). One such distance is illustrated as a vertical line for gene j. A bootstrap procedure produces a P-value testing whether the expected value of the MD score, , is different from zero, indicating a DEP. This MD score can be interpreted as a covariance-adjusted log fold change of the pathway mRNA expression between the paired cells. (B) When studying single-cell RNA-seq datasets from two phenotypic groups (e.g. drug A versus drug B), pairwise statistics of DEPs can be aggregated in a cross-group, ‘many-to-many’ fashion (left). This affords discovery of phenotypic differences, anchored in mechanistic interpretation while embracing the cellular heterogeneity revealed by scRNA-seq data. On the right, a bootstrapped distribution of the median MD score (second quartile, Q2) is used to test whether a pathway is centrally differentially expressed between the cross-group pairs. The two shaded tail areas represent the cross-group P-value associated with a sample median MD score, . (C) Inspection of many-to-many pairings of cells within a group provides insight into within-group heterogeneity of pathway expression (left). Using the P-value provided by the MD pairwise procedure for a pathway of interest, every within-group pair of cells can be classified as having a DEP or the pathway is non-significantly differentially expressed (‘Non-Sig’, on right). Higher abundance of DEPs within group indicates greater pathway expression heterogeneity. (D) The pairwise, cross-group comparisons for a single cell of interest (CoI) can be aggregated and summarized to provide CCS of DEPs. This ‘one-to-many’ perspective (left) yields single-cell, pathway-anchored differences between phenotypes. CCS presents opportunities for precision medicine in developing drug targets or understanding propensity for response to therapy. On the right, the CCS distribution of MD scores for a given pathway are displayed in a histogram. A ‘central pair’ is found by retrieving the pair associated with the median MD score. The central effect size of pathway differential expression is given by the MD score for the central pair, . The corresponding P-value for the central pair provides a ‘CCS P-value’, which can be used to classify the CoI as differentially expressed for a pathway
Our aggregation framework begins by quantifying transcription dynamics for a pair of cells through the application of a gene set scoring procedure, N-of-1-pathways Mahalanobis Distance (MD), that we recently developed to predict DEPs using a single pair of transcriptomes (Schissler et al., 2015) (Fig. 1A). MD produces pathway-level significance that is readily interpretable biologically and potentially clinically actionable for pathway-targeting therapies. Originally, we applied MD to measure dynamic changes of mRNA within a single subject by exploring differential pathway expression from a baseline to a case sample (i.e. dysregulation). In this manner, two transcriptomes from a patient could be transformed into a personal pathway dysregulation profile. These patient-specific profiles are predictive of clinical outcomes, including survival and response to therapy, in cancer and viral infection (Gardeux et al., 2015; Gardeux et al., 2014a, b; Schissler et al., 2015). Moreover, N-of-1-pathways MD can also be used to measure differential pathway expression between any pair of samples. We have shown that this approach unveils DEPs between groups when traditional statistics are underpowered (Schissler et al., 2015).
In this study, we introduce and validate our aggregation framework using RNA-seq data derived from prostate cancer CTCs as a proof of concept and implicate mechanisms of resistance to androgen inhibition therapy. DEPs are identified at the individual cell level using the CCS component of the framework. Emerging biological systems properties of pathway resistance are illustrated at the level of individual cells, as well as aggregated at the level of individual patient and at the treatment group level. The accuracy of our aggregation method in prioritizing DEPs across treatment groups is contrasted to that of conventional methods such as Gene Set Enrichment Analysis (GSEA) (Subramanian et al., 2005), single-cell differential expressed genes (SCDE) (Kharchenko et al., 2014) followed by gene set enrichment (DEG + Enrichment) and weighted least squares (WLS) regression (Piegorsch, 2015). Further, novel single-cell visualization of DEP transcriptome dynamics is developed to demonstrate the utility of CCS for predicting therapeutic resistance based on a single CTC.
2 Methods
2.1 Data sets
Single-cell RNA-seq of circulating (prostate) tumor cells. RNA-seq read count data from single prostate CTCs (Miyamoto et al., 2015) were downloaded from the Gene Expression Omnibus (Edgar et al., 2002) under accession GSE67980 on September 22, 2015. A total of 108 candidate CTCs were isolated from the 13 blood samples using microfluidic CTC-iChip technology (Ozkumur et al., 2013). RNA sequences were aligned to human transcriptome (based on hg19). Further, cells that lacked epithelial gene markers or possessed gene signatures consistent with leukocytes were excluded to increase confidence that the remaining cells are truly prostate derived (Miyamoto et al., 2015). The single candidate prostate CTCs were filtered to 77 lineage-confirmed prostate CTCs with at least 100 000 uniquely aligned sequencing reads as described in Miyamoto et al. All read counts were transformed into Reads per Million (RPM) following the pipeline for normalizing CTC RNA-seq data from Aceto et al. (2014).
Androgen inhibition therapeutic response annotations for CTCs. CTCs were derived from 13 prostate cancer patients. These patients were retrospectively labeled as either ‘enzalutamide (EZT)-naïve’ (n = 8, Group N) or ‘EZT-resistant’ (n = 5, Group R). Each CTC was labeled according to the patient group label (nN = 41 cells, nR = 36 cells).
Signaling Pathways defined by Pathway Interaction Database. Gene sets were defined using the Pathway Interaction Database (PID; Schaefer et al., 2009; last update September 18, 2012). Genes were originally annotated to pathways using Universal Protein Resource (UniProt) IDs (Consortium, 2012). UniProt IDs were converted to HUGO (Povey et al., 2001) gene symbols in R (R Development Core Team, 2011) using the Bioconductor (Gentleman et al., 2004) package MyGene.Info (Wu et al., 2013). All gene symbols were retained in the case that UniProt IDs mapped non-uniquely to multiple gene symbols. Further, 349 UniProt IDs without corresponding HUGO gene symbols were removed. Finally, among the 223 PID-defined gene sets, the 187 pathways comprising >15 genes were retained for analysis, as we previously have shown this cutoff is robust against individual gene bias (Yang et al., 2012a, b; Chen et al., 2013; Perez-Rathke et al., 2013).
2.2 Pairwise statistics of DEPs via N-of-1-pathways MD
Differential pathway expression for a pair of single-cell transcriptomes is quantified by the N-of-1-pathways MD score, a covariance-adjusted log fold change of all pathway genes (Schissler et al., 2015). Here, a pathway is defined as a set of genes that function together or are related molecularly. Specifically, the pathway MD score () is computed by measuring the average signed Mahalanobis vertical distance (Mahalanobis, 1936) from an equal-expression reference line (diagonal line in Fig. 1A):
| (1) |
| (2) |
where FCj refers to the fold change of jth mRNA; C1j, C2j refers to the jth mRNA expression for the baseline cell and case cell within the cell–cell pair, respectively; j indexes the m genes (mRNAs) annotated to the pathway; s1 is the sample standard deviation of the C1j for j = 1, …, m; s2 is the sample standard deviation of the C2js and s12 = s21 is their sample covariance.
2.2.1. Pairwise significance: assessing certainty of DEPs through mRNA resampling for a cell–cell pair
The statistical significance for determining a DEP between a pair of cell transcriptomes was assessed by bootstrapping the average signed Mahalanobis vertical distance (Equation (2)) (Schissler et al., 2015) for a given gene set. A bootstrap resample (Chernick, 2008) was computed by randomly sampling with replacement the gene indices to produce a new, bootstrapped average MD score, . This was repeated B = 20 000 times to produce a bootstrap distribution for a given pathway. We view the deviation from zero as indicative of pathway differential expression. This leads us to define a proxy ‘P-value’ as the smaller of two proportions ( > 0)/B or ( < 0)/B. If the distribution of s completely separates from the origin, then the P-value is conservatively recorded as 1/(B + 1).
2.2.2. Quantifying differential pathway expression for CTC pairs
Every possible pair of the 77 CTCs was selected for a total 2926 pairs of transcriptomes. MD scores were computed for each of the 187 externally defined PID pathways for every CTC pair. RPM counts for each HUGO gene symbol were transformed to log2(RPM + 1) to stabilize variance in the expression counts, preserve zero counts, and afford log fold change interpretation of the MD scores. In the degenerative case that the Mahalanobis multiplier on the log fold change (Equation (2)) is not well defined, i.e. , the MD score for that pathway is set to zero, indicating an absence of differential pathway expression.
2.3 Analysis of aggregated cross-group, cell–cell statistical distances within pathways
The first application of our analytic framework quantifies effect size and determines the statistical significance for phenotypic pathway differential expression by exploring the ‘cross-group’ pairs. Cross-group pairs are defined as cell–cell pairings that involve two distinct phenotypes (Fig. 1B). As an illustrative case study, we present an analysis of aggregated cell–cell statistical distances within pathways for the CTC cross-group pairs. This analysis prioritizes EZT-resistance pathways by investigating the MD scores for every pairwise comparison between CTCs from the EZT-naïve (N) patients and CTCs from the EZT-resistant (R) patients (Section 2.1). Here, N CTCs serve as the reference baseline and R CTCs as the case sample; thereby, the log fold change of expression is positive when an mRNA is overexpressed in an R cell relative to an N cell. The pairing of all R-N CTCs results in 36 N cells times 41 R cells to yield 1476 pairs. In turn, this results in 1476 MD scores for a given pathway. Because the cross-group MD score distributions are right skewed (data not shown), pathways are prioritized by median MD scores that are statistically different from zero, indicating phenotypic pathway differential expression.
2.3.1. Cross-group significance via clustered bootstrapping: prioritizing EZT-resistance pathways using CTCs
A novel bootstrapping procedure is developed to assess the significance of cross-group DEPs in potentially correlated (i.e. ‘clustered’) single-cell RNA-seq samples. A test is constructed to assess the degree to which the median (second quartile noted as Q2) cross-group MD score of a pathway is different from zero. Specifically, the statistical hypotheses are as follows:
| (3) |
| (4) |
To construct a statistical test of these hypotheses, we begin by mimicking a null distribution with Q2truly zero, H0, by subtracting the observed sample median from every cell–cell MD score to create a ‘shifted’ distribution of . Next, bootstrapping from the null distribution is conducted to construct an approximate sampling distribution of . To account for within-patient correlation, the resampling procedure incorporates the nested structure of the data (i.e. multiple CTCs from a patient). To do so, patients are resampled: eight ‘patients’ are sampled with replacement from the naïve patients to create a bootstrapped N sample, N*, and five ‘patients’ are sampled with replacement from the resistant patients to create a bootstrapped R sample, R*. All CTCs are retained from the selected patients. The median is calculated from the shifted MD scores corresponding to all resampled pairs, .
This process was replicated 20 000 times to approximate the distribution of under H0. To assess the two-sided alternative hypothesis via a P-value, two tail probabilities were calculated from the bootstrap distribution: and . The bootstrap P-value is the sum of these two tail probabilities (Fig. 1C). This procedure was repeated for all 187 PID pathways.
2.3.2. Conventional cross-group significance: prioritizing EZT-resistance pathways by gene set testing methods
The 187 PID pathways were ranked according to differential pathway expression between EZT-resistant patients (R group) and EZT-naïve patients (N group) using an ad hoc two-sample comparison with WLS (Piegorsch, 2015), GSEA (Subramanian et al., 2005) and single-cell DEG + Enrichment (Kharchenko et al., 2014). First, an ad hoc statistical approach, WLS, was applied to determine pathways differing between treatment groups. For a given pathway, the corresponding mRNA counts were averaged (using an arithmetic mean) for each CTC to summarize the pathway-level expression. Then, a within-patient pathway score was computed by averaging the pathway means across all CTCs from a patient. The pathway scores for all 13 patients were regressed on a binary indicator for group status (1 = Resistant Group, 0 = Naïve Group) using WLS with weights corresponding to the count of CTCs for each patient (with the lm function in R), essentially mimicking a weighted, two-sample t-test. The pointwise (unadjusted) P-value testing for testing whether group N differed from group R was retained to rank the pathways for comparison with other methods.
Next, conventional gene set testing approaches, GSEA and DEG + Enrichment, were applied to rank how pathways differ between groups. The expression values of all CTCs within a patient were averaged (using an arithmetic mean) to obtain a within-patient mRNA measurement. In the GSEA analysis, the significance of pathway differential expression was assessed by completing 1000 permutations of the patients’ resistant and naïve labels to obtain a pathway P-value using publically available GSEA software in R. In the DEG + Enrichment analysis, differentially expressed genes (DEGs) were first identified by SCDE (Kharchenko et al., 2014) pointwise P < 0.05), and then each pathway was analyzed for enrichment of the identified DEGs. In total, ordering the corresponding pointwise P-values calculated by WLS, GSEA and DEG + Enrichment, respectively, produced three ranked lists for the 187 PID pathways.
2.4 Analysis of aggregated within-group pairs quantifying heterogeneity through DEP abundance
Within a priori defined gene sets, one may address whether a phenotype displays consistent mRNA expression within pathways (Fig. 4). The investigation of the prevalence of DEPs for within-group pairs (Fig. 1C) lends insight into such mechanistic variation. Continuing the illustrative example of the CTC case study, the P-values corresponding to the MD score for the N-N and R-R pairs, excluding within-patient pairs, were examined to determine the prevalence of differential expression for the five prioritized resistance-associated pathways. The proportion of instances where the given pathway was significantly differentially expressed (Benjamini and Yekutieli, 2015; false discovery rate < 5%) for either the N-N or R-R pairing type was calculated. Here, N-N pairs are arbitrarily ordered; therefore, the direction of differential pathway expression is ignored.
Fig. 4.
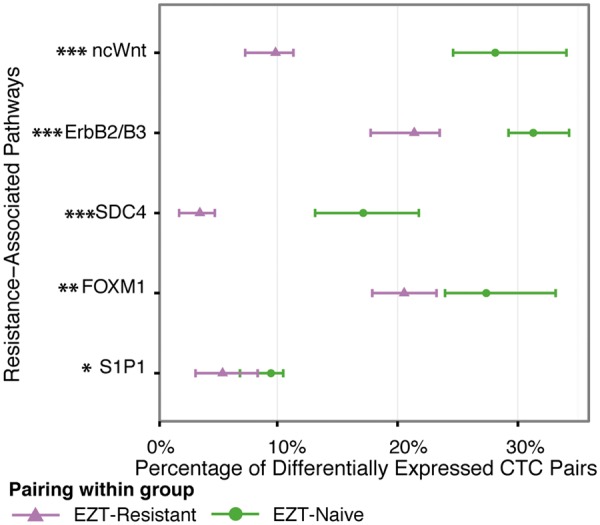
EZT exposure incurs consistent molecular expression in resistance-associated pathways for within-group pairwise CTC comparisons. For the five EZT-resistance pathways in Figure 3, effect size—as measured by MD scores (Equation (2))—was calculated for each of the 477 and 635 pairs of CTC transcriptomes using combinations within the EZT-resistance group and within the EZT-naïve group, respectively (Fig. 1C; Section 2.4), excluding CTCs paired within patient. For each pathway of a cell pair, a P-value was determined for this MD score (Section 2.2.1). Each illustrated point represents the proportion of CTC pairs that are significantly differentially expressed for a prioritized pathway (Benjamini–Yekutieli adjusted P-value < 5%). Note that the direction of DEP is arbitrary for within-group pairs by construction. The variability in the statistic is indicated by a 95% bootstrap percentile confidence interval for the proportion of differentially expressed pairs in a given pathway (Section 2.4.1). The CTCs within the EZT-naïve group (N-versus-N) exhibit greater heterogeneity than CTCs within the EZT-resistant group (R-versus-R). ***, **, * indicate P-values <0.1%, 1% and 5% respectively, for testing a non-zero difference in DEP prevalence (Section 2.4.1). Within-patient comparisons are available in Supplementary Section II. Cross-group pathway analytics, such as WLS, GSEA and DEG + Enrichment, cannot be shown here as they are not designed to generate a measure of the effect size or significance for each cell pair within group (Supplementary Section III)
2.4.1. Within-group DEP abundance variability and significance
A ‘clustered’ bootstrap (Section 2.3.1) was applied to produce 95% percentile confidence intervals on the proportion of DEPs for within-group CTC pairs. Bootstrap resampling was conducted with replacement on eight patients for the N group or on five patients for the R group. Further, we tested difference of DEP prevalence within group by similar procedure. Specifically, a bootstrap distribution was constructed for the differences of DEP prevalence between EZT-resistant and EZT-naïve groups. To assess the significance at 5%, 1% and 0.1% levels, we examined whether the 95%, 99% and 99.9% bootstrap percentile confidence intervals from the difference contained zero.
2.5 CCS: Aggregating cell-specific, cross-group pairs to produce single-cell DEPs
The final aggregation analytic method in our framework is CCS of individual cell differential pathway expression. CCS allows for exploration of individual cellular mechanistic differences from a reference population. In the CTC case study, each cell was paired with all cells of the opposing treatment group to unveil a single cell’s propensity for EZT resistance (Fig. 1D).
2.5.1. CCS significance: assessing cross-group DEPs for a single cell
We seek to determine a central DEP status for an individual CTC with respect to resistance–naïve cell comparisons. Every CTC has a distribution of DEP statuses when compared with the opposing treatment group. In particular, a naïve-labeled CTC has a pairing with each of the 41 EZT-resistance CTCs, and each EZT-resistant CTC has 36 pairings with the naïve CTCs. Thus, for a given pathway, a naïve cell has 41 DEP statuses of up (+), down (−) or a non-significantly (NS) expressed pathway. For naïve cells, the pathway median MD maps to exactly one CTC pair (‘central pair’, Fig. 1D). This pair’s DEP status is annotated as the ‘central DEP status’ of this CTC. For the resistant CTCs, there are two CTC pairs that are involved in the calculation of the median CTC. In this case, we enact the following criteria for determining the central DEP status: (1) + when both CTC pairs are up, (2) − when both CTC pairs are down, (3) + when one CTC is up and the other is NS, (4) − when one CTC is down and the other is NS and (5) NS when both CTCs are NS differentially expressed.
2.6 Modified rose plots of CCS P-values
Modified rose plots (Beniger and Robyn, 1978) were created in R using the ggplot2 package (Wickham, 2009). Rose plots illustrating the central DEP significance for two treatment-characteristic cells were constructed using cell-central statistics (CCS) (Section 2.5.1). Each rose plot is modified in that diagonally opposite ‘petals’ (pie-shaped sectors) across the horizontal line represents distinct pathways, with positive MD scores above the axis and negative ones below. To enable both visualization of effect size and statistical significance for resistance-associated pathways, the petal radii represent a transformed CCS P-value. Specifically, a cross-group, central P-value for a cell of interest is transformed by applying the negative natural logarithm. This transformation maps P-values from the unit interval to the positive real line and provides more weight to P-values near zero. Further, following principles of aspect ratio visualization, the petal radii were specified as the square root of this transformed P-value (Court, 1963).
3 Results and Discussion
3.1 Case study: aggregation of cell–cell distances within pathways of prostate CTCs unveils resistance mechanisms to androgen inhibition treatment
In metastatic prostate cancer, standard treatments target the Androgen Receptor (AR) pathway either through reduction of testosterone or by blocking the AR with an inhibitor (Trewartha and Carter, 2013). In the past few years, potent new AR inhibitors have emerged, such as EZT that provide additional clinical benefit to prostate cancer patients (Trewartha and Carter, 2013). To this end, RNA-seq data from CTCs sampled from 13 advanced prostate cancers treated with or naïve to EZT (GSE67980; Miyamoto et al., 2015) was available for analysis. After filtering of potential leukocytes and poorly sequenced cells, 77 ‘lineage-confirmed’ prostate CTCs remained (Miyamoto et al., 2015). Distributions of distances within signaling pathways for the cross-group CTC pairs were examined to discover mechanisms of EZT resistance.
3.2 Overview of aggregation framework validation and benchmarking
The aggregation framework was designed to analyze transcriptional dynamics of pathways at the single-cell level and, as such, determining a single-cell gold standard remains elusive. Despite this, the accuracy of our framework in prioritizing DEPs is assessed by comparison with traditional cross-group pathway analytics: GSEA (Subramanian et al., 2005), DEG + Enrichment (Kharchenko et al., 2014) and WLS regression (Piegorsch, 2015). Notably, none of the explored methods were able to discover dysregulated pathways while controlling for multiplicity of testing. In our approach, EZT-resistance pathways measured by MD scores at the level of individual cells are aggregated at the level of treatment groups (Section 2.3). Further, novel cell-centric visualizations of DEP transcriptome dynamics are developed to demonstrate the utility of CCS for predicting therapeutic resistance from a single CTC.
3.2.1. Aggregation of cross-group distances concurs with conventional cohort-based analytics while enabling cell-specific interpretation
We accumulated evidence of EZT-mediated dysregulation for each of the 187 PID pathways using the four analytic methods. A ranked list of the pathways was created for every method by sorting the pathways by increasing P-values (Sections 2.3.1 and 2.3.2). We assessed the concordance of these ranked lists visually (Fig. 2) and with two distinct correlation tests. The overall concordance between the ranked lists was assessed via Spearman’s rho (Spearman, 1904). Because highly ranked pathways (low P-values) are of greater interest in determining EZT-resistance mechanisms, we also computed a weighted correlation, the top-weighted overlap score (Top-WOS; Yang et al., 2006). Top-WOS is correlation metric that more heavily weights top-ranked pathways. Overall, our ‘cross-group’ and WLS ranked lists display a high level of agreement (Spearman’s ρ = 0.832). Moreover, they exhibit a strong concordance in the highly prioritized pathways (Top-WOS P < 0.001). The cross-group rankings moderately concur with GSEA globally (Spearman’s ρ = 0.481), but they do not agree in the highly ranked pathways (Top-WOS P = 0.558). GSEA and DEG + Enrichment ranked list are mildly negatively correlated (Spearman’s ρ = −0.16, P = 0.03), and they do not agree in highly ranked pathways (Top-WOS P = 0.98).
Fig. 2.
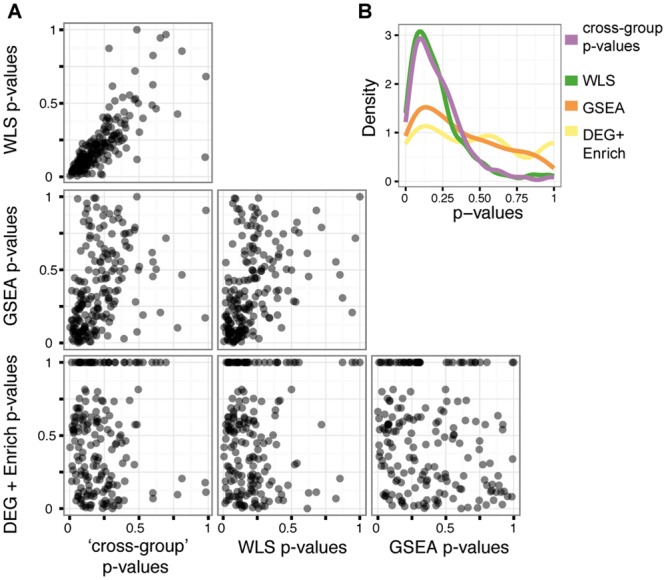
EZT-resistant prioritized pathways concur with established statistical and gene set testing procedures. MD scores were calculated for each of the 1476 cross-group pairs of cells (Fig. 1B) to obtain a median MD score for each of the 187 pathways of the PID (Section 2.1). For each pathway, a ‘cross-group’ P-value was determined for this median MD score (Section 2.3.1). For comparison, pathway P-values were also calculated for the three cross-group pathway analysis methods (WLS, GSEA, DEG + Enrichment, Section 2.3.2). Panel A contains scatterplots displaying the bivariate relationship between P-values generated from every pairwise combination of methods. Each dot represents a single pathway. GSEA correlates with cross-group, WLS and DEG + Enrichment. DEG + Enrichment shows poor concordance with cross-group and WLS. Panel B displays estimated densities of the P-values corresponding to each method. CCS and WLS show a similar distribution of P-values; GSEA and DEG + Enrichment are similar in P-value distribution. WLS = Weighted Least Squares; GSEA = Gene Set Enrichment Analysis; DEG + Enrichment = Differentially Expressed Genes followed by Enrichment
Further, a computational literature evaluation approach (Yang et al., 2010) using Pubmatrix (Becker et al., 2003) was conducted to determine the degree to which the top-hit pathways prioritized by each method were relevant to prostate cancer and treatment (resampling details in Supplementary Section I). The aggregated cross-group distances-identified pathways display strong literature support (OR = 56, P = 0.002). However, GSEA, WLS and DEG + Enrichment did not have strong literature support for their top pathways with OR = 20 (P = 0.16), OR = 2.3 (P = 0.77) and OR = 2.2 (P = 0.81), respectively.
Notably, none of the other three methods are able to specify cell-specific transcriptional dynamics. This unique advantage of our approach allows for interpretation and discovery at the single cell, within patient, within treatment group while still enabling powerful cross-group comparisons. The following sections highlight the types of discoveries possible through the analysis of aggregated cell–cell distances.
3.2.2. Pathway dysregulation of resistant–naïve CTC pairings identify potential EZT-resistance mechanisms
Our investigation of the CTC pairs begins with a focus on the R-N pairing subtype. By exploring the MD score distribution for these pairs, we aimed to identify EZT-resistance pathways. There are 41 and 36 cells from the N Group and R Group, respectively. This yields 1476 R-N CTC pairs. MD is applied to each of these pairs to compute scores for each of the 187 signaling pathways defined by the PID with at least 15 genes annotated to the gene set (Schaefer et al., 2009). PID pathways are manually curated based on well-established biomolecular interactions and cellular processes. The curating effort was enabled through a collaboration of the National Cancer Institute and the Nature Publishing Group. PID signaling pathways are not viewed as a comprehensive ontology of biological pathways, but key pathways implicated in cancer and other diseases. To compensate for the fact that multiple CTCs were derived from a single patient, we developed a ‘clustered’ bootstrapping procedure (Section 2.3.1) to assess whether the median MD score for a given pathway is larger than zero, an indication of pathway differential expression. Figure 3 displays the five pathways whose pointwise P-values were calculated to be <2%. All five pathways are higher expressed in the EZT-resistant CTCs. The five prioritized pathways are Non-canonical Wnt signaling pathway (ncWnt), ErbB2/ErbB3 signaling events (ErbB2/B3), Syndecan-4-mediated signaling events (SDC4), FOXM1 transcription factor network (FoxM1) and S1P1 signaling pathway (S1P1).
Fig. 3.
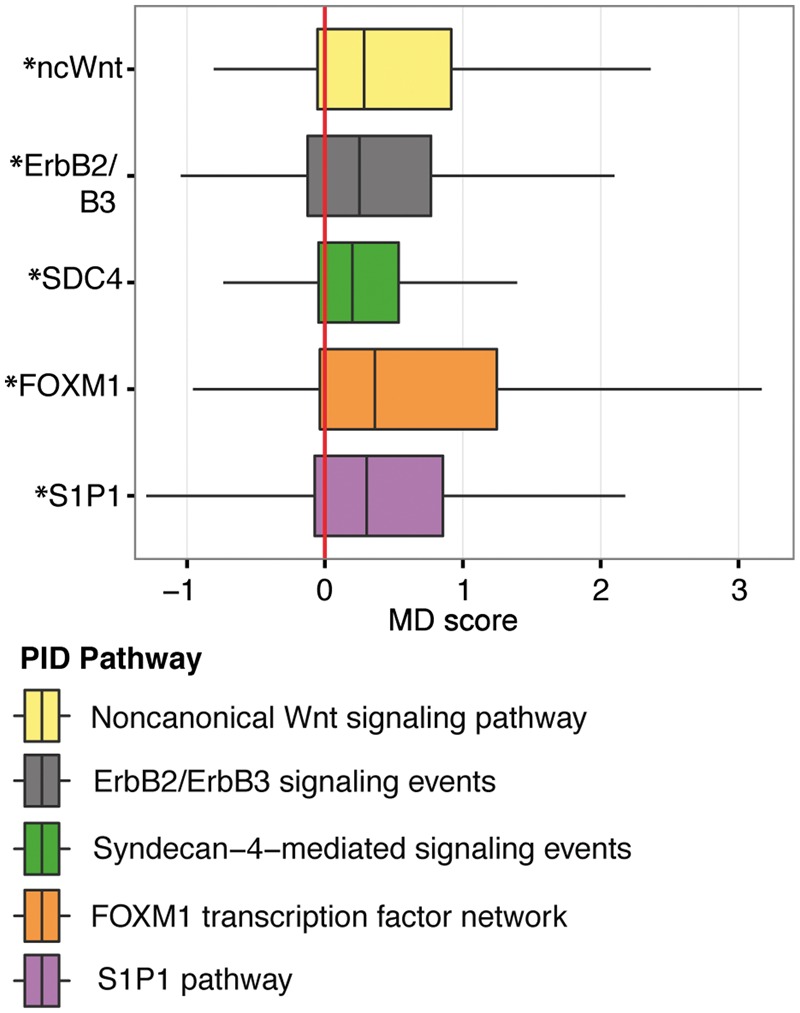
Cross-group pairwise CTC comparisons implicate five molecular pathways of EZT resistance. MD scores (Equation (2)) were calculated for each of the 1476 cross-group pairs of cells (Fig. 1B) to obtain a median MD score (Section 2.3) for each of the 187 pathways of the PID (Section 2.1). For each pathway, a cross-group P-value was determined for this median MD score (Section 2.3.1). For the five pathways with pointwise P-values < 2%, boxplots illustrate the distribution of effect size as measured by MD scores for all pairwise comparisons of transcriptomes from EZT-resistant (R) versus EZT-naïve (N) CTCs. MD scores greater than zero indicate overexpression of a pathway within EZT-resistant CTCs. ‘*’ indicates a P-value < 5% when testing median MD score different from zero. WLS and GSEA could also prioritize pathways at the 2% significance level, whereas DEG + Enrichment could not
3.2.3. Top prioritized pathways recapitulate mechanisms of EZT-resistance and highlights SDC4 as a potential targetable pathway
A number of studies have reported the implications of these oncogenetic pathways/receptors in tumorigenesis via cell cycle development, proliferation or progression (Dc et al., 2014; Akao et al., 2006; Pyne et al., 2012; Koo et al., 2012). Notably, the non-canonical Wnt signaling pathway has been implicated in antiandrogen resistance in these data (Miyamoto et al., 2015) and has been prioritized by our method. Indeed, FoxM1 has been implicated in androgen resistance in vitro (Ketola et al., 2014) and in other endocrine responsive tumors (Sanders et al., 2013). Similarly, the ERBB2/ERBB3 axis has been well-established as a marker of poorly prognostic prostate cancer (Craft et al., 1999) and ongoing clinical trials are underway targeting this pathway (Vaishampayan et al., 2015). Under similar early development is the targeting of the sphingolipid metabolism pathway (S1P1) that appears to have reasonable responsiveness in in vitro models of hormone-refractory prostate cancer (Venant et al., 2015). Perhaps this is the most novel discovery is the prioritization of the SDC4 pathway. SDC4 is under the regulation of non-canonical WNT signaling pathway (Carvallo et al., 2010), and thus may serve as another downstream mechanism of abrogating aberrant WNT signaling alone or in combination with WNT inhibition.
3.2.4. Within-group aggregation reveals homogeneity across EZT-resistant cells and heterogeneity for naïve cells within prioritized pathways
Investigating the prevalence of significant DEPs for the within-group pairs shows consistent expression of implicated pathways for the EZT-resistant group and more variable expression within the EZT-naïve group (Fig. 4). Remarkable mRNA expression heterogeneity is observed for these CTCs (average mRNA Pearson correlation, rN-N = 0.313, average rR-R = 0.373). As expected within the five prioritized pathways, biological resistance is tightly regulated as quantified by little differential pathway expression for the R-R pairs (average DEP = 12%, Section 2.4). In contrast, the naïve group demonstrates more heterogeneity within these pathways (Fig. 4), which suggests some EZT-naïve CTC may harbor intrinsic propensity for resistance. In addition, the proportion of DEP is different between EZT-resistant group and EZT-naïve group for all of the five pathways (P-value < 5%, Section 2.4.1). Collectively, these results indicate that EZT-resistance is likely to be mediated through regulated expression of these prioritized pathways in exposed CTCs.
3.2.5. Transcriptome dynamics between EZT-resistant and -naïve groups for individual CTCs elucidates patient-specific relationships
A cell-centric perspective provides interpretation of transcriptional dynamics at the single cell level. In the prostate cancer CTC data set, each cell can be classified as up-expressed, down-expressed or NS differentially expressed with respect to the opposing treatment group (Section 2.5.1). For example, a naïve-classified CTC is compared with all EZT-resistant CTC. Figure 5A illustrates the distribution of DEP status for cell by patient. It can be seen that EZT-resistant patients are relatively higher expressed in the prioritized pathways. However, inspection of a few naïve patients reveals a propensity for EZT-resistance based on even higher pathway expression than EZT-resistant cells. Figure 5, Panel B contains novel modified rose plots that visualize the magnitude and statistical significance of individual CTC cross-group dysregulation. These plots provide readily interpretable depictions of single-cell drug resistance (e.g. naïve CTCs with higher expression in resistance-associated pathways exhibit innate resistance). This color-coded, area-preserving plot affords rapid recognition of patterns when scanning many cells. Traditional cohort-based statistics cannot provide insight at this level of granularity. As such, our analysis of single-cell differential expression provides a framework for interpreting MD scores with a (clinical) translational intent.
Fig. 5.

Patient-specific transcriptome dynamics of therapeutic-resistance pathways unveiled by CCS in individual CTCs. (A) Stacked bar plot of central differential pathway expression. Using MD scores (Equation (2)), the median pathway differential expression effect size of a single CTC was estimated by comparing the pathway mRNAs of this cell of interest with that of all other cross-group CTCs (Fig. 1D). For each of the five pathways of a single cell, a ‘central DEP status’ was determined for the corresponding MD score (Section 2.5). The majority of significantly DEPs within EZT-naïve CTCs are relatively lower than the resistant CTCs in these five pathways (and, conversely for the resistant CTCs). However, greater heterogeneity in DEPs exists within the EZT-naïve patients. In particular, Pr17 and Pr9 exhibit both up- and down-regulated CTCs. Pr6 is up-regulated in three of the pathways compared with the resistant group, indicating innate resistant to therapy. Non-sig = non-significant (pointwise P-value > 5%). Pathway names are to the right of each bar graph. Group-based pathway analytics such as WLS, GSEA and DEG + Enrichment cannot be shown here, as they are not designed to generate a measure of the effect size or significance for each cell pair (Supplementary Section III). Legend: Pr## = patient identifier (##). (B) Modified rose plots of CCS of two characteristic CTCs from the EZT-naïve (top) and EZT-resistant (middle) groups. Each rose plot displays five ‘petals’, one for each of the five resistance-associated pathways. The petal area corresponds to the negative log of the P-value for the central pair (Section 2.6). The petals above the horizontal axis are higher expressed (in blue) in that CTC relative to the opposing treatment group (conversely for the lower expressed, in red). The legend (bottom) indicates the pathway and direction color-coding. The rose plots highlight the opportunity to infer treatment sensitivity or resistance for a single cell
3.4 Limitations and future studies
This approach provides a framework for exploring alternate aggregation methods in future studies between single cells grouped in distinct phenotypes and can be generalized to more than two phenotypic groups. As our aggregation framework proceeds through pairwise comparisons of a single-cell transcriptome to many distinct cells, an inherent minimum number of compared cells are required. Additional insight in the required sample size of these compared population should be investigated via simulations under distinct experimental conditions such as background measurement noise, batch effect noise, fold changes of pathway genes and percentage of differentially expressed genes between two cells. Biologically validated positive and negative control datasets of truly dysregulated pathways between subsets of CTCs would enable clearer evaluations. However, conducting the biology in this leading edge field is rate limiting, expensive and, likely, technologically challenging at the single cell. To our knowledge, no such dataset exists for single-cell RNA-seq of CTCs, and such studies should be completed as the data become available in the future. Many single-cell analytic methods pool cells data together and use population statistics such as correlation-based approaches to describe transcriptional diversity across cells. Such methods (e.g. Treutlein et al., 2014) and others seek to cluster cells into subpopulations for cell type classification. A subset of our proposed method (Section 2.2) is designed to account for pairwise correlation within the framework of finding DEPs between known phenotypes, but does not currently identify novel subpopulations of cells, a key ambition of single-cell ‘omics’.
4 Conclusion
scRNA-seq offers insight into transcriptional diversity of individual cells and presents unprecedented actionable opportunities in biology and medicine. The described methodologies of aggregation of cell–cell statistical distances within pathways including CCS and cell-centric visualizations bridge an analytical gap between cohort-based statistics and single-cell expression signals. We provide evidence that our framework accurately identifies DEPs in an individual cell. Specifically, treatment-resistance pathways of individual CTCs were differentially expressed in distinctive patterns when comparing prostate cancer subjects treated with EZT to those treated without it. Furthermore, single CTCs of some patients never exposed to EZT presented a higher heterogeneity of treatment-resistance pathway expression, with some CTCs strikingly similar to those of resistant subjects. As many therapies target pathways, these single-cell analyses may provide biologically meaningful interpretations and clinically actionable metrics. These observations suggest the utility of CCS to identify subjects likely to present resistance to future therapies as well as transcriptome dynamics of resistance over time.
Supplementary Material
Acknowledgements
The authors thank Dr Nima Pouladi for his helpful discussions and support and the ISMB 2016 reviewers for their critique and subsequent improvements.
Funding
The study was supported in part by the University of Arizona Center for Biomedical Informatics and Biostatistics, The University of Arizona Health Sciences, and the grants NIH K22LM008308 and NIH NCI P30CA023074.
Conflict of Interest: none declared.
References
- Aceto N. et al. (2014) Circulating tumor cell clusters are oligoclonal precursors of breast cancer metastasis. Cell, 158, 1110–1122. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Akao Y. et al. (2006) High expression of sphingosine kinase 1 and S1P receptors in chemotherapy-resistant prostate cancer PC3 cells and their camptothecin-induced up-regulation. Biochem. Biophys. Res. Commun, 342, 1284–1290. [DOI] [PubMed] [Google Scholar]
- Becker K.G. et al. (2003) PubMatrix: a tool for multiplex literature mining. BMC Bioinformatics, 4, 61. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Beniger J.R., Robyn D.L. (1978) Quantitative graphics in statistics: a brief history. Am. Stat., 32, 1–11. [Google Scholar]
- Benjamini Y, Yekutieli D. (2015) The control of the false discovery rate in multiple testing under dependency. Ann. Stat., 29, 1165–1188. [Google Scholar]
- Carvallo L. et al. (2010) Non-canonical Wnt signaling induces ubiquitination and degradation of Syndecan4. J. Biol. Chem., 285, 29546–29555. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chen J.L. et al. (2013) Curation-free biomodules mechanisms in prostate cancer predict recurrent disease. BMC Med. Genomics, 6(Suppl. 2), S4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chen X., Bai F. (2015) Single-cell analyses of circulating tumor cells. Cancer Biol. Med, 15, 184–192. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chernick M.R. (2008) Bootstrap Methods: A Guide for Practitioners and Researchers. John Wiley & Sons, Inc., Hoboken, New Jersey. [Google Scholar]
- Consortium,T.U. (2012) Reorganizing the protein space at the Universal Protein Resource (UniProt). Nucleic Acids Res., 40, D71–D75. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Court B.A. (1963) Wind roses. Weather, 18, 106–114. [Google Scholar]
- Craft N. et al. (1999) A mechanism for hormone-independent prostate cancer through modulation of androgen receptor signaling by the HER-2/neu tyrosine kinase. Nat. Med., 5, 280–285. [DOI] [PubMed] [Google Scholar]
- Dc W. et al. (2014) Sphingosine-1-phosphate promotes lymphangiogenesis by stimulating S1P1/G i/PLC/Ca 2+ signaling pathways. Blood, 112, 1129–1138. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ding B. et al. (2015) Gene expression normalization and noise reduction for single cell RNA-seq experiments. Bioinformatics, 31, 2225–2227. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Edgar R. et al. (2002) Gene expression omnibus: NCBI gene expression and hybridization array data repository. Nucleic Acids Res., 30, 207–210. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gardeux V. et al. (2014a) Concordance of deregulated mechanisms unveiled in underpowered experiments: PTBP1 knockdown case study. BMC Med. Genomics, 7(Suppl. 1), S1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gardeux V. et al. (2014b) ‘N-of-1-pathways’ unveils personal deregulated mechanisms from a single pair of RNA-Seq samples: towards precision medicine. J. Am. Med. Inform. Assoc, 21, 1015–1025. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gardeux V. et al. (2015) Towards a PBMC ‘virogram assay’ for precision medicine: concordance between ex vivo and in vivo viral infection transcriptomes. J. Biomed. Inform., 55, 94–103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gentleman R.C. et al. (2004) Bioconductor: open software development for computational biology and bioinformatics. Genome Biol., 5: R80. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Grün D. et al. (2014) Validation of noise models for single-cell transcriptomics. Nat. Methods, 11, 637–640. [DOI] [PubMed] [Google Scholar]
- Islam S. et al. (2014) Quantitative single-cell RNA-seq with unique molecular identifiers. Nat. Methods, 11, 163–166. [DOI] [PubMed] [Google Scholar]
- Jiang L. et al. (2011) Synthetic spike-in standards for RNA-seq experiments. Genome Res., 1543–1551. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kapushesky M. et al. (2009) Gene expression atlas at the European bioinformatics institute. Nucleic Acids Res., 38, D690–D698. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ketola K. et al. (2014) Inhibition of FOXM1 targets both high and low PSA expressing prostate cancer cells resistant to Enzalutamide. Cancer Res., 74, 676. [Google Scholar]
- Kharchenko P.V. et al. (2014) Bayesian approach to single-cell differential expression analysis. Nat. Methods, 11, 740–742. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Koo C.Y. et al. (2012) FOXM1: from cancer initiation to progression and treatment. Biochim. Biophys. Acta, 1819, 28–37. [DOI] [PubMed] [Google Scholar]
- Liang J. et al. (2014) Single-cell sequencing technologies: current and future. J. Genet. Genomics, 41, 513–528. [DOI] [PubMed] [Google Scholar]
- Lukk M. et al. (2010) A global map of human gene expression. Nat. Biotechnol., 28, 322–324. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mahalanobis P.C. (1936) On the generalized distance in statistics. Proc. Natl. Inst. Sci., 2, 49–55. [Google Scholar]
- Miyamoto D.T. et al. (2015) RNA-Seq of single prostate CTCs implicates noncanonical Wnt signaling in antiandrogen resistance. Science, 349, 1351–1357. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Navin N.E. (2015) The first five years of single-cell cancer genomics and beyond. Genome Res., 25, 1499–1507. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ozkumur E. et al. (2013) Inertial focusing for tumor antigen-dependent and -independent sorting of rare circulating tumor cells. Sci. Transl. Med., 5, 179ra47. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Perez-Rathke A. et al. (2013) Interpreting personal transcriptomes: personalized mechanism-scale profiling of RNA-seq data. Pac. Symp. Biocomputing, 159–170. [PMC free article] [PubMed] [Google Scholar]
- Piegorsch W.W. (2015) Statistical Data Analytics: Foundations for Data Mining, Informatics, and Knowledge Discovery John Wiley & Sons, Chichester. [Google Scholar]
- Povey S. et al. (2001) The HUGO Gene Nomenclature Committee (HGNC). Hum. Genet., 109, 678–680. [DOI] [PubMed] [Google Scholar]
- Pyne N.J. et al. (2012) Sphingosine 1-phosphate signalling in cancer. Biochem. Soc. Trans., 40, 94–100. [DOI] [PubMed] [Google Scholar]
- R Development Core Team (2011) R: a language and environment for statistical computing. R Found. Stat. Comput, 1, 409. [Google Scholar]
- Ramsköld D. et al. (2012) Full-length mRNA-Seq from single-cell levels of RNA and individual circulating tumor cells. Nat. Biotechnol., 30, 777–782. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sandberg R. (2014) Entering the era of single-cell transcriptomics in biology and medicine. Nat. Methods, 11, 22–24. [DOI] [PubMed] [Google Scholar]
- Sanders D.A. et al. (2013) Genome-wide mapping of FOXM1 binding reveals co-binding with estrogen receptor alpha in breast cancer cells. Genome Biol., 14, R6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schaefer C.F. et al. (2009) PID: the pathway interaction database. Nucleic Acids Res., 37, D674–D679. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schissler A.G. et al. (2015) Dynamic changes of RNA-sequencing expression for precision medicine: N-of-1-pathways Mahalanobis distance within pathways of single subjects predicts breast cancer survival. Bioinformatics, 31, i293–i302. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schubert C. (2011) Single-cell analysis: the deepest differences. Nature, 480, 133–137. [DOI] [PubMed] [Google Scholar]
- Scialdone A. et al. (2015) Computational assignment of cell-cycle stage from single-cell transcriptome data. Methods, 85, 54–61. [DOI] [PubMed] [Google Scholar]
- Spearman C. (1904) The proof and measurement of association between two things. Am. J. Psychol., 100, 441–471. [PubMed] [Google Scholar]
- Stegle O. et al. (2015) Computational and analytical challenges in single-cell transcriptomics. Nat. Rev. Genet, 16, 133–145. [DOI] [PubMed] [Google Scholar]
- Subramanian A. et al. (2005) Gene set enrichment analysis: a knowledge-based approach for interpreting genome-wide expression profiles. Proc. Natl. Acad. Sci. U S. A., 102, 15545–15550. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tang F. et al. (2009) mRNA-Seq whole-transcriptome analysis of a single cell. Nat. Methods, 6, 377–382. [DOI] [PubMed] [Google Scholar]
- Treutlein B. et al. (2014) Reconstructing lineage hierarchies of the distal lung epithelium using single-cell RNA-seq. Nature, 509, 371–375. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Trewartha D., Carter K. (2013) Advances in prostate cancer treatment. Nat. Rev. Drug Discov., 23, 8–12. [DOI] [PubMed] [Google Scholar]
- Vaishampayan U. et al. (2015) Phase I Study of Anti-CD3 x Anti-Her2 bispecific antibody in metastatic castrate resistant prostate cancer patients. Prostate Cancer, d2015/2015/285193. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Venant H. et al. (2015) The Sphingosine kinase 2 inhibitor ABC294640 reduces the growth of prostate cancer cells and results in accumulation of dihydroceramides in vitro and in vivo. Mol. Cancer Ther., 14, 2744–2752., [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wickham H. (2009) ggplot2: Elegant Graphics for Data Analysis. Springer, New York. [Google Scholar]
- Wu A.R. et al. (2014) Quantitative assessment of single-cell RNA-sequencing methods. Nat. Methods, 11, 41–46. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wu C. et al. (2013) BioGPS and MyGene.info: organizing online, gene-centric information. Nucleic Acids Res., 41, D561–D565. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yang X. et al. (2010) Kinase inhibition-related adverse events predicted from in vitro kinome and clinical trial data. J. Biomed. Inform., 43, 376–384. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yang X. et al. (2006) Similarities of ordered gene lists. J. Bioinform. Comput. Biol., 4, 693–708. [DOI] [PubMed] [Google Scholar]
- Yang X. et al. (2012a) Single sample expression-anchored mechanisms predict survival in head and neck cancer. PLoS Comput. Biol, 8, e1002350. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yang X. et al. (2012b) Towards mechanism classifiers: expression-anchored Gene Ontology signature predicts clinical outcome in lung adenocarcinoma patients. AMIA Annu. Symp. Proc., 2012, 1040–1049. [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.