

Abstract
Motivation: Despite rapid progress in sequencing technology, assembling de novo the genomes of new species as well as reconstructing complex metagenomes remains major technological challenges. New synthetic long read (SLR) technologies promise significant advances towards these goals; however, their applicability is limited by high sequencing requirements and the inability of current assembly paradigms to cope with combinations of short and long reads.
Results: Here, we introduce Architect, a new de novo scaffolder aimed at SLR technologies. Unlike previous assembly strategies, Architect does not require a costly subassembly step; instead it assembles genomes directly from the SLR’s underlying short reads, which we refer to as read clouds. This enables a 4- to 20-fold reduction in sequencing requirements and a 5-fold increase in assembly contiguity on both genomic and metagenomic datasets relative to state-of-the-art assembly strategies aimed directly at fully subassembled long reads.
Availability and Implementation: Our source code is freely available at https://github.com/kuleshov/architect.
Contact: kuleshov@stanford.edu
1 Introduction
Genome sequencing technology has had an enormous impact on modern science and medicine. Information gleaned from the genome has become a crucial ingredient in numerous industrial and medical applications, such as breeding disease-resistant crops, identifying infectious microbes or diagnosing human health problems. Yet, despite rapid progress in sequencing technology, fully reconstructing de novo the genomes of new organisms or complex metagenomes still remains a major technological challenge.
The main obstacle in de novo genome assembly remains sequencing read length. Current technologies can only read short hundred-base substrings of the genome; recovering the original sequence from these substrings is impossible, as they fundamentally cannot resolve the true position of repetitive sequences that are longer than their own length. This results in highly fragmented assemblies that need to be further improved with more sophisticated and expensive techniques.
Recently, new synthetic long read (SLR) technologies have offered great promise towards making inexpensive and accurate de novo assembly a reality. These technologies exhibit read lengths in the tens of kilobases and theoretically have the power to reconstruct a large fraction of an organism’s genome.
Nonetheless, SLRs have not yet realized their full potential. Most existing approaches involve a two-stage process in which long fragments are first assembled from short reads, and then the genome is assembled from the long fragments. Such strategies typically require prohibitively large amounts of short-read sequencing for each long fragment; in some cases, attaining this high level of coverage may not even be feasible. In addition, long reads often must be complemented by short reads (e.g. to compensate for sequencing bias); yet, there are currently very few assemblers that can effectively handle both types of data.
Here, we introduce Architect, a new de novo scaffolder for SLR technologies that aims to address these shortcomings. Unlike previous assembly strategies, Architect does not require a costly subassembly step; instead it assembles genomes directly from the SLR’s underlying short reads. Moreover, by dealing only with short reads, it avoids difficulties that arise from jointly assembling reads of highly differing lengths.
In practice, Architect leads to a 4- to 20-fold reduction in sequencing requirements and up to a 5-fold increase in assembly contiguity compared with current state-of-the-art assembly strategies aimed directly at fully subassembled long reads. We demonstrate the improvements offered by Architect on the genomes of Drosophila melanogaster and Caenorhabditis elegans as well as on two metagenomic samples: the synthetic mock community from the human microbiome project, and a bona fide human gut metagenome from a healthy male individual. Our results suggest that Architect may lower the cost of accurate de novo assembly and facilitate the analysis of long-range genomic features in metagenomic samples, for example long operons or strain haplotypes.
2 Background
2.1 De novo assembly
The goal of de novo assembly is to reconstruct a target genome (viewed as a string of up to several billion letters) from sequencing reads, which can be viewed as random substrings of the genome.
Assembly paradigms. There exist two main approaches to de novo assembly and each is best suited to a particular type of data. The Ovelap-Layout-Consensus (OLC) paradigm (Myers et al., 2000) works best with long reads (> 1 kp); it involves computing overlaps between all pairs of reads and simplifying the resulting overlap graph until we obtain long, contiguous subsequences of the genome called contigs. Contigs may be further assembled into scaffolds using paired-end or mate-pair read data. The main shortcomings of OLC assemblers are high computational requirements for computing overlaps between a very large number of short-read pairs (< 200 bp). The alternative De Brujn graph (DBG) paradigm (Pevzner et al., 2001) addresses this problem by first breaking reads into k-mers (with k < 127) and then linking them in a graph. This reduces the number of vertices to consider, but loses important contiguity signal encoded in longer reads.
Finally, when using paired-end or mate-pair reads, it is common to further extend the de novo assembly via a scaffolding process. The term ‘scaffold’ refers to a genomic sequence containing subsequences of unknown nucleotides (usually denoted by N) of potentially uncertain lengths. Many assemblers include a scaffolding module that produces such sequences from paired-end reads (see e.g. Zerbino et al., 2009); in addition, there exist many standalone scaffolding tools, whose performance can often match or exceed that of more complex assemblers (Hunt et al., 2014).
The importance of read length. The main difficulty faced by either paradigm is genomic repeats (Myers et al., 2000). If a genome contains subsequences ARB and CRD (meaning that R is a repeat occurring twice; are unique sequences), and if the length of sequencing reads is smaller than R, then we cannot determine whether ARB or ARD is the correct contig ordering. In such cases, we must report as individual contigs. Thus, read length is one of the most important factors determining the quality of de novo assemblies (Chaisson et al., 2009).
2.2 SLR technologies
This work introduces tools targeted at two closely related types of sequencing technologies: SLRs and read clouds; both types of methods share a common protocol, which we illustrate in Figure 1.
Fig. 1.
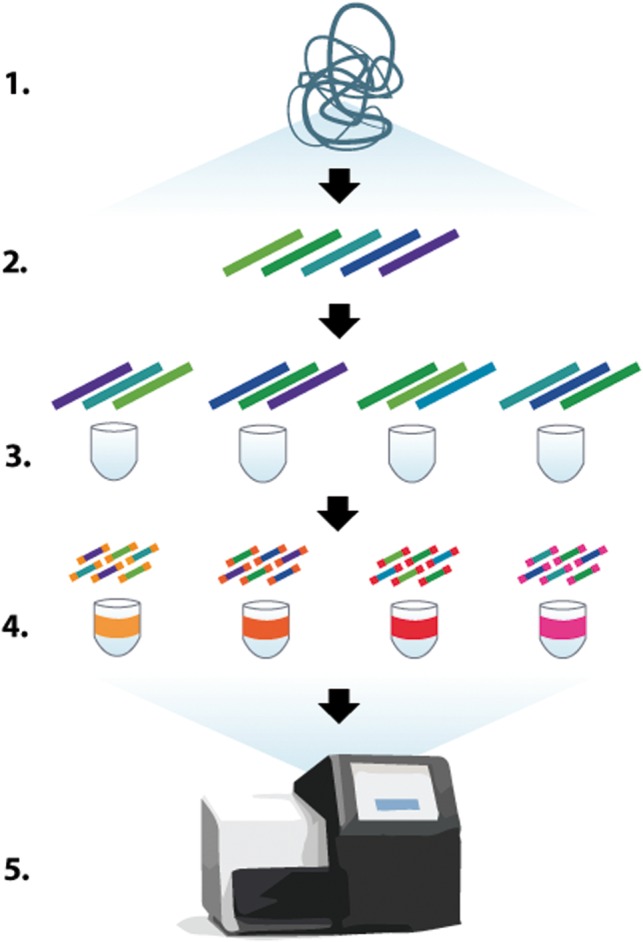
High-level overview of SLR and read cloud technologies. DNA (1) is sheared into kilobase-long fragments (2), which are then diluted and placed into multiple containers, typically with 0.1–2% of the genome per container (3). Within each container, fragments may be amplified before being cut into short fragments, and barcoded (4). The barcoded fragments are finally pooled together and sequenced (5); reads can be demultiplexed on a computer into their original compartment via the barcodes in order to form read clouds or SLRs
Synthetic long reads. The first set of technologies aims to produce ‘virtual’ long reads on standard short-read sequencers via a specialized library preparation method (Fig. 1). At a high level, input DNA is first sheared into kilobase-long fragments, which are then randomly distributed across a small number of containers. The fragments are typically diluted such that each container holds a small fraction (0.1–2%) of the target genome. The contents of each container are then sheared further into shorter fragments and are assigned a unique barcode before being pooled together for sequencing.
After sequencing, reads are demultiplexed into their containers of origin using the barcodes. Each container may be assembled separately with a short-read assembler, which produces multiple kilobase-long sequences in each well; this approach is referred to as subassembly. The resulting sequences correspond to the original long fragments. In the last step, the target genome is assembled from the long fragments using an OLC-based method.
There exist multiple instantiations of the protocol described above. Techniques that produce fully assembled SLRs include fosmid pooling (Duitama et al., 2012), long fragment reads (Peters et al., 2012) and Tru-seq SLRs (Voskoboynik et al., 2013) (TSLR), which is also one of the few technologies to be commercially available. SLRs have been applied to a wide range of problems, including genome phasing (Kuleshov, 2014; Kuleshov et al., 2014), read alignment (Bishara et al., 2015) and metagenomic analysis (Kuleshov et al., 2015; Sharon et al., 2015).
Repeat reduction. The key process that makes subassembly possible is a reduction in the repeat content of the genome within each container. Because each container holds only a small fraction (0.1–2%) of the target genome, the probability of two copies of the same repeat R finding themselves in the same container is very low. Thus, each container can be seen as containing a genome with no repeats and that is therefore relatively easy to assemble. Once each container has been assembled separately, we may merge the resulting long fragments into a final genome assembly.
Read cloud technologies. Alternatively, the contents of each container may be sequenced at a relatively low coverage, either to lower sequencing requirements, or because the laboratory protocol may not permit high-coverage sequencing for technical reasons. In such cases, we only obtain clusters of short reads that originate from long fragments. We refer to such clusters as read clouds. The term ‘cloud’ comes from the appearance of such reads when aligned to a reference genome and visualized in a genome browser: they typically form isolated clusters with an imprecise shape (Fig. 2, top).
Fig. 2.

Scaffolding using read clouds. A genome contains a repeat R flanked by unique sequences (A, B) and (C, D) (top). With short reads, the correct assembly is ambiguous (middle). If two read clouds (marked as red and orange) map, respectively, to ARB and CRD, this provides signal that may be used to correctly resolve the repeat structure (bottom).
Although they do not output long contiguous sequences, read cloud technologies contain signal which may be used for resolving genomic repeats; the focus of this work is precisely to extract this signal. Examples of read cloud methods include contiguity preserving transposase sequencing (CPT-seq; Amini et al., 2014), which produces very thin clouds, and the 10X GemCode platform, which features an adjustable cloud depth.
2.3 Related work
Most applications of SLR and read cloud technologies to de novo assembly have used a subassembly-based strategy. These methods were used to assemble the genomes of Botryllus schlosseri (Voskoboynik et al., 2013), D.melanogaster (McCoy et al., 2014), C.elegans (Li et al., 2015) as well as metagenomic samples from the human gut (Kuleshov et al., 2015) and from the environment (Sharon et al., 2015). In all cases, assemblies achieved N50 lengths below 100 kb, highlighting limitations of subassembly-based strategies.
Currently, only one method is able to use read clouds for de novo assembly, and that is FragScaff (Adey et al., 2014), a scaffolder aimed at extremely low-internal-coverage read clouds obtained via the contiguity preserving transposase sequencing (CPT-seq) technology. FragScaff produces orderings of contigs by leveraging the same signal as Architect; it differs mainly in its scaffolding algorithm, which is optimized for CPT-seq. In particular, FragScaff formulates the scaffolding problem as finding the maximum-weight spanning tree (MST) on the scaffold graph. This formulation was shown to be highly effective at scaffolding large genomes form CPT-seq data; however, it is less effective when scaffolding metagenomic data as well as read clouds obtained from alternative technologies such as TSLR, long fragment reads or fosmid clones. We further discuss differences between FragScaff and Architect in Section 5.
There also exist multiple de novo assembly methodologies that provide an alternative to read clouds. Burton et al. (2013) showed that contigs can be effectively scaffolded using chromatin-level contact probability maps generated by the high-throughput chromosome conformation capture (Hi-C) technology; however, Hi-C has high input-DNA requirements and its ability to scaffold high-complexity metagenomes remains relatively limited (Burton et al., 2014). An alternative technology, single-molecule real-time (SMRT) sequencing, has been shown to be highly effective at assembling bacterial genomes (Koren et al., 2012; Chin et al., 2013) and was recently scaled to handle entire human genomes (Chaisson et al., 2015). Its shortcomings include requiring specialized sequencing instruments as well as significant reagent costs relative to the more standard Illumina platform; also, SMRT technologies may exhibit lower accuracy when assembling highly heterozygous genomic regions, especially in the context of metagenomics (Kuleshov et al., 2015). We further compare SMRT and read cloud technologies in Section 5.
3 Results
3.1 High-level overview of Architect
Current SLR-based approaches to de novo assembly have several important shortcomings. First, subassembly requires very deep sequencing, since each long fragment must be covered to a sufficiently high depth in order to be assembled; in some cases, attaining this high level of coverage may not even be feasible due to inherent technical limitations of the library preparation protocol. Secondly, long reads often work best in combination with standard shotgun reads; however, neither the OCL nor the DBG assembly paradigm is effective at assembling the two types of data jointly. Below, we introduce the Architect scaffolder, which implements a solution to both of these limitations.
Read clouds. Rather than adopting a two-stage subassembly approach, Architect attempts to scaffold the genome using low internal coverage read clouds. Recall that we use the term read cloud to denote the set of short reads derived from shearing a long fragment within a given container (Fig. 2, top).
Local and global coverage. To better explain how our approach differs from subassembly, we introduce the concepts of local and global coverage. Local coverage refers to the average coverage of a long fragment with short reads; it is formally defined as the total number of base pairs in short reads obtained from sequencing a given container, divided by the number of total number of base pairs of genomic content originally placed in the container. Global coverage refers to the coverage of the original genome with long fragments. It is obtained by dividing the number of base pairs placed in all containers by the size of the target genome.
Read cloud-based scaffolding. The high-level intuition for how low-local coverage read clouds may be used for scaffolding genomes is illustrated in Figure 2. Consider a genome with a repeat R flanked by unique sequences (A, B) and (C, D) (Fig. 2, top). If the length of R is longer than the read length, the assembly graph will contain a characteristic X-shaped structure that cannot be resolved (Fig. 2, middle). However, if there are two read clouds that map to ARB and CRD in different containers, we can align the clouds to the contigs and observe that read clouds from the same container align to A and B. This indicates how the contigs should be scaffolded.
Contig orderings. A crucial distinction between Architect and regular scaffolders is that read clouds provide relatively little signal about the distance between adjacent contigs. This is partly due to the greatly varying lengths of read clouds previously reported for certain technologies. Another cause is the relatively shallow internal coverage of read clouds, which makes it difficult to estimate where the cloud starts and ends.
Because of these complications, Architect reports orderings of contigs instead of scaffolds. The main difference between the two is that orderings offer no guarantees about the relative distance of two consecutive contigs. Although the contiguity of assemblies provided by Architect significantly exceeds that of alternative approaches, the reader should still keep in mind this important distinction when evaluating our results.
Algorithm overview. The Architect scaffolder takes as input pre-assembled scaffolds or contigs from a standard short-read assembler as well as an alignment of read clouds to these scaffolds. In addition to read clouds, Architect is also able to leverage paired-end are mate-pair reads to guide scaffolding in cases where the original assembly is ambiguous. Architect uses alignment of read clouds and paired-end reads to form a scaffold graph; this graph is then simplified to produce orderings of scaffolds. The simplification process is guided by an algorithm which is explained in detail in the next section.
3.2 Datasets
We evaluated Architect on four publicly available genomic datasets produced using the commercially available Tru-Seq SLR sequencing technology from Illumina. We obtained datasets for two genomes and two metagenomes; in each case, we had access to the sub-assembled long reads and their underlying raw short reads. We subsampled these to various percentages (from 5% to 25%), and used them as our ‘read cloud’ dataset. All read cloud datasets were complemented with standard shotgun libraries.
Drosophilamelanogaster. We used a dataset previously published by McCoy et al. (2014) (SRX447481). We library mol-32-281c for our analysis (the library contained 212M read pairs, each read being 100 bp in length), in addition to two short-read datasets published in an independent study (SRX543254).
Caenorhabditiselegans. We used a dataset made available by Illumina as part of its TSLR technology demonstration (data are available on BaseSpace). We used TSLR library no. 1 (out of 2) for our experiments; we complemented this with a standard shotgun read dataset used in benchmarking genomic assemblers (Simpson and Durbin, 2011).
Mock gut metagenome. We tested our ability to assess the accuracy of our metagenomic assemblies on the human microbiome project staggered mock community (Human Microbiome Project Consortium, 2012). This synthetic community contains 20 organisms with known reference genomes and is widely used for validation. We used a recent TSLR dataset (library 1) in addition to the accompanying short reads. In addition to helping validate the robustness of Architect to different coverages, this dataset also provides an indication of the ability of long read clouds to scaffold bacterial genomes.
Bona fide gut metagenome. Finally, we assemble a bona fide sample from the gut of a healthy male adult individual (Kuleshov et al., 2015). This dataset was previously assembled from TSLRs and was found to be extremely diverse and complex, making it a realistic and challenging benchmark dataset for Architect. We again used TSLR libraries 1–3 from the previous study as well as the entirety of the accompanying short reads.
3.3 Assembly strategies
We compared Architect to four alternative assembly strategies.
Shotgun reads were assembled using a standard short-read assembler into contigs or scaffolds. In our experiments, we used SPAdes 3.5.0 (Bankevich et al., 2012) on the D.melanogaster dataset and Soapdenovo2 rc240 (Luo et al., 2012) on the other three datasets (we found this choice to produce the highest quality contigs). Overall, these two assemblers have been shown in previous studies to achieve state-of-the-art performance on a variety of genomes (Salzberg et al., 2012).
Long reads. Next, we used the Celera assembler (Myers et al., 2000) directly on subassembled SLRs. The Celera assembler has been previously used to obtain high-quality assemblies from TSLRs on both genomes and metagenomes (Voskoboynik et al., 2013).
Shotgun and long reads were jointly assembled using SPAdes 3.5.0 for the D.melanogaster and C.elegans datasets and Minimus2 (Sommer et al., 2007) for the metagenomic datasets. Minimus2 is a tool that merges independent shotgun and long read assemblies in a post-processing stage; we found that it assembled two times more sequence that SPAdes on both metagenomic datasets.
Shotgun reads and read clouds were assembled with Architect. We aligned shotgun and raw TSLR short-read libraries to contigs assembled from shotgun reads (using the first strategy above); Architect used this data to produce long scaffold orderings.
FragScaff. Finally, we compared Architect with an alternative scaffolding program that uses a different algorithm to perform scaffolding based on the same type of data (Adey et al., 2014). We ran FragScaff multiple times varying the two parameters specified in the documentation to have the largest effect on assembly quality; we report the best results obtained across these runs.
The exact scripts used for running our experiments are available in the GitHub repository of Architect.
3.4 Evaluation metrics
We evaluated performance using standard metrics reported by Quast 3.1, a popular tool for assessing the quality of genome and metagenome assemblies (Gurevich et al., 2013). The N50 length of a set of contigs is a measure of assembly contiguity: we say that contigs have an N50 of x if at least 50% of the total assembled sequence is in contigs of length x or longer. The genome NA50 is defined as the N50 of scaffolds that have been broken at every major misassembly. Major misassemblies are said to occur when a contig substring aligns 1 kb away or further from its neighbouring sequence. We refer the reader to the documentation of Quast for more details.
3.5 Results
3.5.1. Assembly quality
A summary of our results can be found in Table 1. At a high level, Architect outperforms alternative assembly strategies and produces genome assemblies that are up to five times longer than approaches based on shotgun and subassembled SLRs. Moreover, Architect achieves this performance with only 25% of the sequencing requirements of standard long read-based methods.
Table 1.
Assembly evaluation of Architect on four de novo assembly datasets.
| Genome + sequencing method | Scaffolds | Largest scaffold (kb) | Mb assembled | % assembled | N50 (kb) | NA50 (kb) | Misassemblies |
|---|---|---|---|---|---|---|---|
| Drosophila melanogaster | |||||||
| Shotgun reads | 65 510 | 314.5 | 143.7 | 100.0 | 44.8 | 43.1 | 2265 |
| Long reads | 5064 | 341.5 | 127.5 | 88.7 | 45.3 | 43.2 | 1742 |
| Shotgun and long reads | 29 809 | 649.4 | 117.4 | 81.7 | 123.9 | 115.1 | 2024 |
| FragScaff† | 63 018 | 567.8 | 55.3 | 38.6 | 56.8 | 55.2 | 2289 |
| Shotgun and read clouds† | 57 567 | 1767.4 | 143.7 | 100.0 | 262.8 | 252.2 | 2341 |
| Caenorhabditis elegans | |||||||
| Shotgun reads | 32 092 | 383.1 | 100.1 | 99.9 | 35.6 | 31.9 | 307 |
| Long reads | 2345 | 555.0 | 96.3 | 96.4 | 81.2 | 76.0 | 363 |
| Shotgun and long reads | 2423 | 569.0 | 83.3 | 83.5 | 95.6 | 68.7 | 771 |
| FragScaff† | 29 320 | 510.2 | 40.3 | 40.4 | 51.1 | 50.2 | 321 |
| Shotgun and read clouds† | 4235 | 630.9 | 99.6 | 99.7 | 120.2 | 113.4 | 331 |
| Mock metagenome | |||||||
| Shotgun reads | 36 081 | 414.0 | 34.0 | 41.1 | 19.1 | 18.8 | 34 |
| Long reads | 914 | 405.1 | 17.6 | 21.2 | 24.6 | 24.2 | 29 |
| Shotgun and long reads | 22 562 | 553.3 | 42.5 | 51.2 | 35.1 | 34.3 | 113 |
| FragScaff† | 33 180 | 510.1 | 10.2 | 12.3 | 33.2 | 31.1 | 37 |
| Shotgun and read clouds† | 17 688 | 743.4 | 34.0 | 41.1 | 173.7 | 173.7 | 39 |
| Bona fide metagenome | |||||||
| Shotgun reads | 128 131 | 34.1 | 230.1 | — | 5.3 | — | — |
| Long reads | 12 432 | 89.2 | 170.2 | — | 8.2 | — | — |
| Shotgun and long reads | 121 319 | 101.9 | 289.5 | — | 15.3 | — | — |
| FragScaff† | 127 943 | 40.2 | 100.3 | — | 6.2 | — | — |
| Shotgun and read clouds† | 123 975 | 91.4 | 288.1 | — | 13.3 | — |
Note: Note that metrics reported for FragScaff and Architect correspond to orderings of contigs rather than scaffolds (this is indicated by a†)
As an example, on the Drosophila dataset, Architect produces scaffolds of 253 kb in length, compared with a 124 kb SPAdes assembly of shotgun and long reads. Relative to subassembled long read sequencing, the output of Architect contains about 23% more errors; this indicates that assembling short reads separately in each container is less error-prone than assembling them jointly. However, note that the number of misassemblies produced by Architect is essentially the same as that of the purely short-read assembly (¡4% difference), indicating that the errors are primarily introduced during the initial short-read assembly stage, rather than during Architect scaffolding. Similar observations can be made for other genomes as well.
On the mock metagenomic data, we observed a 5-fold increase in N50 from 35 kb to more than 170. This suggests that Architect is robust to variation in coverage across scaffolds. Our approach also improved performance on the bona fide gut metagenome, with Architect matching the performance of the strategies involving full subassembly. Although the resulting scaffolds are still much shorter than ones obtained on the mock metagenome data, they are of a sufficient length to capture many interesting long-range genomic features such as operons or strain haplotypes.
All of the above findings suggest that the potential of SLRs is not fully realized using existing joint assembly strategies. Architect is able to use the signal from read clouds more efficiently, as it side-steps the difficulties of working with different classes of read data. Another advantage of our approach relative is that it can leverage fragments that could not be subassembled (e.g. due to sequencing biases introducing gaps in internal coverage). More generally, it is applicable to read cloud technologies that subsequence long fragments to very shallow depths, and where subassembly cannot be performed in principle.
Another observation to be made is that subassembled long reads by themselves do not outperform shotgun reads on multiple datasets. Past work has attributed this to sequencing biases in the Tru-seq technology (Kuleshov et al., 2015). This again motivates the need for an assembly approach like Architect.
3.5.2. Sensitivity to coverage
Next, we measured the effects of internal read cloud coverage on the quality of assemblies produced by Architect. More specifically, we subsampled the read cloud library for the D.melanogaster genome to 5%, 10% and 15% of the original coverage, in addition to the 25% subsampled dataset examined above.
Table 2 shows the results of our subsampling procedure. Even at very low coverages, accuracy and N50 length do not degrade significantly. This indicates that users may trade off internal coverage for increased external coverage of the genome in applications where this is necessary, for example when dealing with larger genomes. Moreover, these results suggest that Architect should scale to alternative read cloud technologies whose internal coverage is relatively sparse.
Table 2.
Effect cloud sparsity on assembly quality
| Subsample | Number of reads (M) | N50 (kb) | NA50 (kb) | Size (Mb) | Max (kb) |
|---|---|---|---|---|---|
| 25% | 53.1 | 262.8 | 252.2 | 143.7 | 1767.4 |
| 15% | 31.9 | 261.4 | 250.8 | 143.7 | 1340.2 |
| 10% | 21.2 | 242.2 | 224.5 | 143.7 | 961.3 |
| 5% | 10.6 | 178.8 | 160.4 | 143.7 | 611.3 |
Note: Results are reported for orderings of Drosophila input scaffolds produced by Architect.
3.5.3. Running times
Overall, the main computational bottlenecks in our scaffolding process are the preprocessing stages: the de novo assembly of the input contigs and the alignment of reads back to these contigs. For larger genomes, these may take on the order of days to run. The Architect algorithm itself runs on the order of tens of minutes; on our machine, its running times on D.melanogaster, C.elegans, the mock and the bona fide metagenomes were 7, 6, 13 and 24 min, respectively.
4 Methods
We now proceed to explain the details of the scaffolding algorithm implemented in Architect. The algorithm takes as input pre-assembled contigs or scaffolds from a standard shotgun assembler, as well as alignments between the scaffolds and two sets of reads: paired-end shotgun sequences and read clouds. Then, it follows a three-stage protocol whose final output is accurate orderings of the input scaffolds.
At the first stage, Architect uses the input alignments to build a scaffold graph. Nodes in the graph correspond to scaffolds; links are placed between scaffolds whenever there appears to be evidence that they might be in close proximity in the target genome.
Then, the graph is iteratively pruned in order to remove spurious edges. Pruning occurs in three steps: first, we use paired-end link information to identify the highest-confidence connections; next, we use read cloud alignments to resolve cases where paired-links could not be pruned with sufficient confidence; finally, we use information contained solely in read clouds to make decisions about edges which have no evidence from paired-end reads. These decisions are made using a model that determines the probability of a spurious assignment given observed read cloud evidence.
Finally, in the third and last step, we use the remaining unpruned edges to order the scaffolds. We now give more details about each procedure.
4.1 Scaffolding algorithm
Algorithm 1.
Architect scaffolding algorithm
Input: Set of input contigs or scaffolds. Paired-end alignment . Read cloud alignment . Threshold parameters
- Construct graph :
- Let
- Add edges for which
- Add edges for which
- Prune spurious edges:
- Paired-end pruning. Go through vertices in decreasing order of length. For each incident to v, let . If and , then select e as being the correct edge and prune from the graph.
- Joint read cloud and paired-end pruning. Go again through vertices in decreasing order of length. For each incident to v such that , let . If , then prune from the graph.
- Read-cloud pruning. Prune for which
Determine scaffold orderings via edge contraction in G.
Output: Set of orderings .
We now give a high-level overview of our scaffolding strategy. The input to our procedure is a set of scaffolds and two sets of alignments (in BAM format): a paired-end read alignment and a read cloud alignment. We also let K denote the total number of read cloud containers; for the TSLR data used in our experiments, K = 384 per library.
4.1.1. Graph construction
We start by forming the scaffold graph . The vertices of G correspond to scaffolds augmented with indicators c that represent either the head (c = h) or tail (c = t) of the node. The edge set E is restricted to ‘consistent’ pairs where or (t, h). Edges are constructed from the paired-end and read cloud alignments as follows.
Paired-end link detection. We introduce an edge between if there are at least three paired-end links connecting them. Each paired-end read must have a mapping score of ; also, the average inter-scaffold distance over all read pairs in a link must fall within three standard deviations of the average library insert size. We will use to denote the number of paired-end links between the corresponding scaffolds.
Container hit detection. We say that a ‘hit’ for container k occurs in scaffold si when a read cloud from that container maps to si. If two si, sj are close to each other in the target genome, we expect to observe multiple hits from the same containers in both of them.
To avoid false positives due to incorrect read alignments, we call a hit when at least reads from a container map to si ( by default for TSLR data). Also, when there is a hit from container k to scaffold si, we associate that hit with an interval that indicates the coordinates to which the read cloud mapped on si; we define (respectively, ) as the start (respectively, the end) position of the 10-th short-read aligning to si from container k, starting from the left (respectively, from the right). This again encourages robustness to read misalignments.
We will use to denote the set of hits in si at endpoint ci; we also use
to denote the fraction of hits shared between . New edges in G are created whenever .
4.1.2. Pruning
Paired-end pruning. First, we determine edges that have strong support from paired-end reads and prune ones that don’t. Specifically, we identify edges that have stronger support than all alternatives in the sense that .
where and by default. In such cases, we identify e as a correct and prune away from the graph. Note that this stage is meant to emulate of SSPACE (Boetzer et al., 2011), a popular and widely used standalone scaffolder. Although the algorithm we use is very simple—especially compared with more complex, multi-stage scaffolding procedures implemented in the Celera (Myers et al., 2000) or Velvet (Zerbino et al., 2009) assemblers—it has been shown to be one of the best overall scaffolding methods in a recent empirical study (Hunt et al., 2014).
Joint paired-end and read-cloud pruning. Next, we find edges with support from both paired-end reads and read clouds, and eliminate alternatives. In particular, we find all edges such that and and for all alternatives
where is a user-specified parameter. In such cases, we identify e as correct and prune away from the graph. This step attempts to resolve paired-end link ambiguities via read clouds; it considers links with insufficient cloud support to be spurious.
Read-cloud pruning. Finally, we discard all link data, and prune away edges that have insufficient support from read clouds. In particular we prune all edges E for which
Again, is a user-specified parameter.
The parameters are set by default to (0.2, 0.33); we found that these values produced the best NA50 in our experiments. Decreasing these values (i.e. increasing the recall), did not result in any improvements in NA50, while increasing them by more than 25% (thus increasing precision), produced a considerable decrease in assembly contiguity at the cost of a relatively modest improvement in accuracy. The default parameters for were chosen to match those of SSPACE (Boetzer et al., 2011); in our experiments, performance was relatively robust relative to these parameters, mainly because most edges from paired-end links were unambiguous. Finally, we specified some parameters directly as constants (e.g. 3 minimum paired-end links to form an edge); we found that tweaking these parameters led to little improvements, and in some cases resulted in a large degradation of performance.
4.1.3. Ordering
Once we have pruned the graph G, we proceed to order and orient scaffolds. Two scaffolds can be oriented relative to each other if they are connected by an edge e, and there is no other edge that touches or . In such cases, we can contract e and merge into a new scaffold. We repeat this procedure for all edges that can be contracted and output the sequences of the scaffold in the final, simplified graph.
4.2 Evaluation methodology
We use the standard metrics of Quast (Gurevich et al., 2013) to measure misassemblies. Quast determines true contig positions by mapping them to the reference via MUMmer; it defines a major miassembly as an alignment in which a contig subsequence maps 1 kb or further from its neighbouring subsequences. We define the genome NA50 as the N50 of scaffolds that have been broken at every major misassembly.
In order to evaluate the quality of the output of Architect, we developed our own evaluation script, which is available on Github. In brief, we first map to the reference the scaffolds provided as input to Architect. Then, given a set of output orderings, we determine the number of misassemblies in the orderings as a sum (1) the number of misassemblies in the input contigs and (2) the number of misorderings introduced by Architect. The latter is defined as follows. Given two scaffolds a, b mapping to intervals (a1, a2), (b1, b2) in the reference, we say that b follows a if a, b map to the same strand of the same chromosome and the following two criteria hold: and . Given an ordering of contigs (each ci corresponding to an interval), we determine the number of misorderings as the number of consecutive pairs that are not adjacent. We consider both left-to-right and right-to-left orderings, and take the correct one to be the one with the fewest errors. Note that this procedure generalizes the methodology used in Quast.
5 Discussion
Comparison to FragScaff. FragScaff and Architect leverage the same read cloud signal to perform scaffolding; they mainly differ in their scaffolding algorithm. FragScaff generally adopts a top-to-bottom approach: it determines edge scores by fitting a normal distribution to across the entire graph. Users specify a z-score as a cutoff for pruning edges; true connections are then determined using a greedy MST algorithm. Architect on the other hand combines scaffolds from the bottom up: it computes a local score for each edge, which depends only on local signal between si and sj. Architect then prunes edges locally: it discards edges within a neighbourhood if that neighbourhood contains a single best connection. Unlike FragScaff, it does not attempt to resolve the remaining edges via a MST.
We believe the latter approach has several advantages in the context of metagenomes and high-coverage low-dilution technologies like TSLR. First, the cutoff for pruning reads ought to depend on the neighbourhood: a correct edge may have low support, but if alternative edges are even less supported, we may still choose to select and discard the alternatives. This is especially true for read cloud technologies that exhibit coverage biases across different regions of the genome. Conversely, the greedy MST approach must select a connected tree in the scaffold graph. Thus, if two edges are equally good candidates, it must choose one over the other.
Finally, Architect is able to leverage standard paired-end links to improve scaffold contiguity. These links are particularly helpful when initial short-read assemblies are fragmented; generally, we want the initial shotgun-based scaffolds to be sufficiently long, so that a sufficient number of read clouds may align to them. Paired-end links may help bootstrap scaffolding with read clouds when the starting contigs are too short.
Comparison to SLR subassembly. Architect produced in most cases assemblies that were longer than ones obtained from subassembled long reads, even though it required substantially less sequencing. This in part due to the fact that the TSLRs used in our experiments did not adequately cover the entirety of the target genome, owing primarily to sequencing biases. In fact, high-coverage shotgun reads typically produced assemblies with comparable N50 lengths to the SLR assemblies, indicating that repeats were not the bottleneck factor limiting the effectiveness of the long reads. This hypothesis is further supported by the fact that jointly assembling the long read and shotgun datasets produced substantially better results than with either technology by itself.
However, jointly assembling read clouds and shotgun reads produced significantly longer assemblies that even this latter approach. We believe there are two explanations behind this. First, current assembly paradigms are targeted at either short or long reads, and there are currently no effective tools for combining both types of assemblies. Architect side-steps this issue by using the read clouds only as an indirect signals during the scaffolding process. Furthermore, read cloud technologies such as TSLR often exhibit biases in their internal coverage, and as a consequence some clouds may not subassemble into long reads. By side-stepping subassembly, our approach is able to leverage these low-quality clouds.
Comparison to SMRT sequencing. Multiple authors have shown that SMRT sequencing is highly effective at assembling bacterial (Koren et al., 2012; Chin et al., 2013), eukaryotic (Berlin et al., 2015) and even human (Chaisson et al., 2015) genomes. The SMRT technology produces reads of up to a dozen of kilobases in length which exhibit very low sequence bias; these may probe genomic regions that are difficult to access even with standard shotgun sequencing reads (Chaisson et al., 2015). The same cannot be said for SLRs, which often involve an amplification step (based on e.g. PCR), which may result in highly non-uniform genomic coverage (Peters et al., 2012; Kuleshov et al., 2015). The SMRT technology is also more effective at assembling tandem repeat regions, which may confuse subassembly-based approaches. The main shortcomings of SMRT include specialized sequencing instruments and increased reagent costs relative to the Illumina platform. Moreover, SMRT technologies typically exhibit lower accuracy; although this can often be mitigated by error-correction algorithms, such algorithms may inadvertently correct real genomic variation, especially in the context of heterozygous genomes or multiple closely related metagenomic strains (Kuleshov et al., 2015).
Drosophilamelanogaster assembly analysis. To further analyze the differences between the two technologies, we compared the SMRT assembly of D.melanogaster by Berlin et al. (2015) with our assembly based on Tru-seq SLR clouds whose local coverage was subsampled to 25% of the original data. The SMRT assembly was substantially more contiguous than ours (21 Mbp versus 252 kb N50); furthermore, SMRT produced contiguous genomic sequences, whereas the output of our method consists of contig orderings.
The difference in performance between the two methods can be attributed to a significantly higher coverage of the target genome (90x versus 17x global coverage) and substantially longer read lengths (average length of 9317 kb versus 4800 kb; the latter number refers to the average length of subassembled long reads, which we use as a proxy for the average length of useful long fragments). Most importantly, Tru-seq SLRs exhibit important sequencing bias (Kuleshov et al., 2015), which leaves many genomic regions uncovered; this is partly evidenced by the fact that standard shotgun assemblies match the contiguity of assemblies based on fully subassembled long reads, even though the latter are ∼100× longer. In fact, we observed that regions in the Drosophila reference genome to which orderings produced by Architect could be aligned with MUMmer (v. 3.0 with default parameters) had an average GC content of 47%, compared with 38% for other regions. The average GC content in D.melanogaster was 42.23%. In addition, we reproduced the analysis of Berlin et al. (2015) and found that our assembly placed 4690 (86%) of 5425 annotated transposable element repeats in a single contig ordering, compared with 5274 (97%) for the SMRT assembly. This difference can be attributed to the difficulty of SLRs in handling tandem and nested repeats, as well as to the shorter fragment length used in our assembly.
Although, our assembly of D.melanogaster was less complete than that based on SMRT reads, we want to emphasize that alternative technologies (e.g. the 10X platform) may yield substantially longer read clouds with less sequencing bias than ones we used for our assembly. Since our technique is applicable to such technologies, we expect it to produce competitive assemblies when the underlying read cloud technologies become more mature.
Alternative technologies and larger genomes. Although we used Architect to assemble small- and medium-sized genomes, our high-level approach extends naturally to larger genomes. Larger genomes typically have longer repeats, and resolving them requires longer read clouds. While commercial technologies enabling such read clouds are starting to become commercially available, there are yet few publicly available read cloud datasets, which motivates us to focus our evaluation on TSLR data.
Our approach may also be complementary to alternative technologies such as chromatin-level contact probability maps or SMRT sequencing. The latter technology could be used to generate substantially longer initial contigs, which could then be further scaffolded with long read clouds. Chromatin-level contact probability maps could be used to further scaffold the ordered contigs produced by Architect into chromosome-long maps. This idea was shown to be highly effective in combination with the output of FragScaff and should be expected to work with the output of Architect as well.
Finally, an important advantage of our subassembly approach is its modularity: the base contigs can be produced using any shotgun assembler, and as a consequence our methodology can be improved by better assemblers and by additional sequencing data such as mate-pair reads.
6 Conclusion
In conclusion, we have shown that shallow read clouds can be used to effectively produce long-range scaffolds on both genomic and metagenomic data without actually forming subassembled long reads. This produces 5- to 20-fold savings in sequencing requirements while at the same time increasing the N50 length of scaffolds by up to five times compared with current state-of-the-art methods.
Furthermore, our tool Architect improves over an existing scaffolder, FragScaff, by being able to handle read clouds produced from other technologies besides CPT-seq as well as by handling metagenomic datasets. These were noted as important limitations of the read cloud scaffolding approach in the original FragScaff paper.
Our results suggest that Architect may lower the cost of accurate de novo assembly and facilitate the analysis of metagenomic samples. Scaffolds on the order of tens of kilobases are sufficient to capture many interesting long-range genomic features in metagenomes, for example long operons or strain haplotypes. Lowering the sequencing requirements needed to access these features is particularly important, since high coverage is needed to capture low abundance strains. By reducing by 10-fold the internal coverage of read clouds, we may correspondingly increase external coverage by 10-fold, which in turns let us sample species whose level of abundance is 10 times smaller than what was previously accessible.
Finally, we would like to note the fact that our approach is highly modular and can be expected to lead to improvements when combined with better shotgun read assemblers as well as alternative sequencing methods such as chromatin-level contact probability maps, SMRT sequencing or mate pairs.
Funding
This work was supported by US National Institutes of Health/National Human Genome Research Institute (NIH/NHGRI) grant T32 HG000044. V.K. was supported by a Natural Sciences and Engineering Research Council of Canada (NSERC) post-graduate fellowship. We thank Illumina, Inc. for their assistance in providing some of the datasets used in this work.
Conflict of Interest: V.K. is a consultant for Illumina Inc. S.B. is a co-founder of DNAnexus and a member of the scientific advisory boards of 23 and Me and Eve Biomedical. M.S. is a co-founder of Personalis and a member of the scientific advisory boards of Personalis, AxioMx and Genapsys.
References
- Adey A. et al. (2014) In vitro, long-range sequence information for de novo genome assembly via transposase contiguity. Genome Res., 24, 2041–2049. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Amini S. et al. (2014) Haplotype-resolved whole-genome sequencing by contiguity-preserving transposition and combinatorial indexing. Nat. Genet., 46, 1343–1349. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bankevich A. et al. (2012) Spades: A new genome assembly algorithm and its applications to single-cell sequencing. J. Comput. Biol., 19, 455–477. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Berlin K. et al. (2015) Assembling large genomes with single-molecule sequencing and locality-sensitive hashing. Nat. Biotechnol., 33, 623–630. [DOI] [PubMed] [Google Scholar]
- Bishara A. et al. (2015) Read clouds uncover variation in complex regions of the human genome. Genome Res., 25, 1570–1580. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Boetzer M. et al. (2011) Scaffolding pre-assembled contigs using S SPACE. Bioinformatics, 27, 578–579. [DOI] [PubMed] [Google Scholar]
- Burton J.N. et al. (2013) Chromosome-scale scaffolding of de novo genome assemblies based on chromatin interactions. Nat. Biotechnol., 31, 1119–1125. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Burton J.N. et al. (2014) Species-level deconvolution of metagenome assemblies with hi-c-based contact probability maps. G3, 4, 1339–1346. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chaisson M. et al. (2009) De novo fragment assembly with short mate-paired reads: Does the read length matter? Genome Res., 19, 336.. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chaisson M.J.P. et al. (2015) Resolving the complexity of the human genome using single-molecule sequencing. Nature, 517, 608–611. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chin C.S. et al. (2013) Nonhybrid, finished microbial genome assemblies from long-read SMRT sequencing data. Nat. Methods, 10, 563–569. [DOI] [PubMed] [Google Scholar]
- Duitama J. et al. (2012) Fosmid-based whole genome haplotyping of a HapMap trio child: Evaluation of single individual haplotyping techniques. Nucleic Acids Res., 40, 2041–2053. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gurevich A. et al. (2013) QUAST: Quality assessment tool for genome assemblies. Bioinformatics, 29, 1072–1075. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Human Microbiome Project Consortium. (2012) A framework for human microbiome research. Nature, 486, 215–221. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hunt M. et al. (2014) A comprehensive evaluation of assembly scaffolding tools. Genome Biol., 15, 1–15. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Koren S. et al. (2012) Hybrid error correction and de novo assembly of single-molecule sequencing reads. Nat. Biotechnol., 30, 693–700. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kuleshov V. (2014) Probabilistic single-individual haplotyping. Bioinformatics, 30, 379–385. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kuleshov V. et al. (2014) Whole-genome haplotyping using long reads and statistical methods. Nat. Biotechnol., 32, 261–266. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kuleshov V. et al. (2015) Synthetic long-read sequencing reveals intraspecies diversity in the human microbiome. Nat. Biotech., 34, 64–69. [DOI] [PMC free article] [PubMed]
- Li R. et al. (2015) Illumina synthetic long read sequencing allows recovery of missing sequences even in the finished C. elegans genome. Sci. Rep., 5, 10814.. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Luo R. et al. (2012) Soapdenovo2: An empirically improved memory-efficient short-read de novo assembler. GigaScience, 1, 1.. [DOI] [PMC free article] [PubMed] [Google Scholar]
- McCoy R.C. et al. (2014) Illumina truseq synthetic long-reads empower de novo assembly and resolve complex, highly repetitive transposable elements. PLoS One, 9, e106689. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Myers E.W. et al. (2000) A whole-genome assembly of drosophila. Science, 287, 2196–2204. [DOI] [PubMed] [Google Scholar]
- Peters B.A. et al. (2012) Accurate whole-genome sequencing and haplotyping from 10 to 20 human cells. Nature, 487, 190–195. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pevzner P.A. et al. (2001) An Eulerian path approach to DNA fragment assembly. Proc. Natl. Acad. Sci. USA, 98, 9748–9753. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Salzberg S.L. et al. (2012) GAGE: A critical evaluation of genome assemblies and assembly algorithms. Genome Res., 22, 557–567. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sharon I. et al. (2015) Accurate, multi-kb reads resolve complex populations and detect rare microorganisms. Genome Res., 25, 534–543. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Simpson J.T., Durbin R. (2011) Efficient de novo assembly of large genomes using compressed data structures. Genome Res., 22, 126953.111–126556.gr. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sommer D.D. et al. (2007) Minimus: A fast, lightweight genome assembler. BMC Bioinformatics, 8, 64.. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Voskoboynik A. et al. (2013) The genome sequence of the colonial chordate, Botryllus schlosseri. eLife, 2, e00569. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zerbino D.R. et al. (2009) Pebble and rock band: Heuristic resolution of repeats and scaffolding in the velvet short-read de novo assembler. PLoS One, 4, e8407.. [DOI] [PMC free article] [PubMed] [Google Scholar]