

Abstract
Motivation: Underrepresentation of racial groups represents an important challenge and major gap in phenomics research. Most of the current human phenomics research is based primarily on European populations; hence it is an important challenge to expand it to consider other population groups. One approach is to utilize data from EMR databases that contain patient data from diverse demographics and ancestries. The implications of this racial underrepresentation of data can be profound regarding effects on the healthcare delivery and actionability. To the best of our knowledge, our work is the first attempt to perform comparative, population-scale analyses of disease networks across three different populations, namely Caucasian (EA), African American (AA) and Hispanic/Latino (HL).
Results: We compared susceptibility profiles and temporal connectivity patterns for 1988 diseases and 37 282 disease pairs represented in a clinical population of 1 025 573 patients. Accordingly, we revealed appreciable differences in disease susceptibility, temporal patterns, network structure and underlying disease connections between EA, AA and HL populations. We found 2158 significantly comorbid diseases for the EA cohort, 3265 for AA and 672 for HL. We further outlined key disease pair associations unique to each population as well as categorical enrichments of these pairs. Finally, we identified 51 key ‘hub’ diseases that are the focal points in the race-centric networks and of particular clinical importance. Incorporating race-specific disease comorbidity patterns will produce a more accurate and complete picture of the disease landscape overall and could support more precise understanding of disease relationships and patient management towards improved clinical outcomes.
Contacts: rong.chen@mssm.edu or joel.dudley@mssm.edu
Supplementary information: Supplementary data are available at Bioinformatics online.
1 Introduction
Design, comparison and analytics of disease networks can inform epidemiology and disease biology (Barabasi and Oltvai, 2004; Feldman et al., 2008; Zanzoni et al., 2009). Comparative network analyses and network inference have helped in understanding the relative risk of various diseases and characterize their shared disease architectures (Barabasi et al., 2011; Cassidy-Bushrow et al., 2011; Goh et al., 2007; Lee et al., 2008; Li et al., 2013, 2014, 2015b; Zhou et al., 2014). Global disease network analyses utilizing biological databases and patient data from electronic medical records (EMR) have emerged as a powerful modality for understanding the complexity of disease relationships (Jensen et al., 2012; Shameer et al., 2014). Incorporating findings from disease networks has been used to inform disease repurposing (Dudley et al., 2011c), develop therapeutics (Schadt et al., 2009) and improve patient safety (Stewart et al., 2007). Phenomics (Bilder et al., 2009) aim to map and understand the system of phenotypes and their interactions—where in clinical studies a phenotype can include a trait (e.g. height), lab test (e.g. cholesterol levels) or disease (e.g. rheumatoid arthritis). The catalog of phenome-wide associations, which evaluate phenomic correlations of genotypes, is rapidly growing and currently being leveraged for drug development and drug repositioning (Denny et al., 2010; Hall et al., 2014; Namjou et al., 2014). We recently used EMR-wide phenomic information to identify: shared genetic architectures of various diseases (Glicksberg et al., 2015; Li et al., 2014; Suthram et al., 2010), sub-types of type-2 diabetes (Li et al., 2015a), drug repurposing for various indications (Dudley et al., 2011a; Shameer et al., 2015), disease progression patterns through data stream visualization (Badgeley et al., 2016; Shameer et al., 2016), disease risk estimations (Nead et al., 2016), and genomics-informed, personalized therapy (Dudley et al., 2011b, 2015).
Similar to the current situation in genomics research, racial groups and related factors remain understudied in phenomics. Most of the current human phenomics research is based primarily on populations of European background. Thus, compiling and analyzing data from EMR databases that contains patient data from diverse demographics and racial groups remains a priority. It is clear that racial background represents an overt source of variability in disease risk and mortality (Trepka et al., 2015). Traditionally, clinicians are required to ‘bridge the inferential gap’, or make clinical decisions for one racial group based on data from another, due to lack of knowledge. Accordingly, the implications of this racial underrepresentation of data can be profound with regard to healthcare delivery and actionability. For example, a previous study found that African American women were twice as likely, and Hispanic women were 50% as likely, to be readmitted to the hospital within 30 days of vaginal or cesarean delivery, even when controlling for socioeconomic status (Aseltine et al., 2015). Systematic analysis of phenomic data represented in a racially and demographically diverse patient population could reveal precise patterns and further understanding of disease relationships, risk and comorbidity.
Previous studies put forth several approaches for the phenomic study of clinical populations. Blair et al. (2013) utilized data from the Centers for Medicare and Medicaid Services (CMS) Databases, multiple hospitals across the United States, and the population registry of Denmark (n = 110 million) to discover comorbidity patterns across complex and Mendelian diseases. This work, however, was mainly focused comparing certain types of diseases (i.e. Mendelian and complex diseases) and did not fully investigate the disease space. Hidalgo et al. (2009) created a more expansive phenotype disease network (CMS data, n = 30 million) that incorporated demographic factors, such as sex and race into the analytics. The authors revealed disparate disease patterns and network connectivity that was due to race, but only between Caucasian and African American populations. Jensen et al. (2014) extended the field of disease network research by using timescale data to define temporal disease trajectories in a Danish clinical cohort (n = 6.2 million). The researchers were successfully able to identify clusters of diseases that consistently manifested in particular order (i.e. disease trajectories). These trajectories, however, were built specifically on European population data and may not extend to other racial groups.
These studies, while powerful and pioneering, did not sufficiently address the issue of racial diversity in their disease networks partially due to limitations of their datasets. As such, a particular concern is a lack of representation of Mexican Americans and other Hispanic Americans in healthcare analytics (López-Candales et al., 2015). As indicated by Hidalgo et al., there is a significant disparity in network structure between racial populations. However, prior phenomic studies have not evaluated racially diverse populations in depth. In the current study, we propose to combine many of the powerful approaches developed in the previous studies and leverage a racially diverse hospital population to compare disease network structure and connectivity between Caucasian, African American and Hispanic/Latino populations. To the best of our knowledge, our work is the first attempt to perform comparative, population-scale analyses of disease networks across three different populations within the same hospital cohort.
2 Methods
We present a schematic of our study design and approach in Figure 1.
Fig. 1.
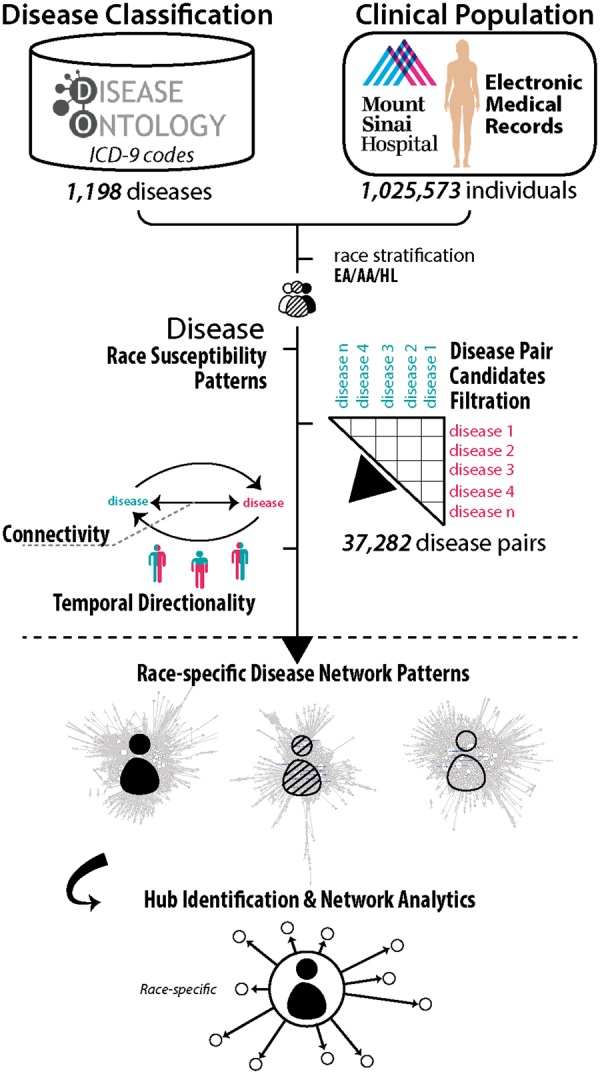
Workflow of the current study. We outline steps taken in our study from data organization and statistical methodologies to network analytics
2.1 Data sources
2.1.1 Clinical cohort
We performed disease-related analyses on patients from the Mount Sinai Hospital (MSH) located in New York City, NY. The unique location of MSH engenders a diverse racial patient population. The Mount Sinai Data Warehouse which houses all the clinical data, currently has 4 034 924 unique patients (as of February 2015), over 16 million patient visits recorded, over 1.7 billion patient encounters, and over 46 million International Classification of Diseases (ICD)-9 code cases documented. We performed the following extensive pre-processing and filtering steps of the clinical data from the EMR.
We excluded individuals that did not have a healthcare visit since 2003, when the EMR was implemented into the MSH system.
We included individuals with a reported sex and age.
We only included individuals with self-identified races of Caucasian (White) [EA], African American (Black) [AA] or Hispanic/Latino [HL].
For all individuals with recorded death, we excluded individuals without an age of death. Of these individuals, we used their date of death as their current age as to not confound subsequent analyses.
In compliance with Protected Health Information (PHI) and Health Insurance Portability and Accountability Act (HIPPA), we censored the ages of individuals <18 or >90 years old to those limits.
After these filtering steps, a total of 1 025 573 individuals remained for analysis. The mean age within the population is 47.19 ± 24.3 years. The population contained 443 816 (43.27%) Males and 581 757 (56.73%) Females. The race breakdown of the population is as follows: 621 827 (60.63%) EA 223 915 (21.83%) AA and 179 831 (17.53%) HL.
2.1.2 Disease classification sources
At the time of this analysis, the MSH EMR system used ICD-9 codes for billing and recording diagnoses. As the ICD-9-CM classification system is fraught with challenges (Hazlewood, 2003), particularly when dealing with rare and/or recently discovered diseases, we utilized a curated ontology of established and documented mappings for clinical studies. Disease Ontology (Schriml et al., 2012) (DO; July 15th, 2015 release) is an open-source repository that integrates phenotype information relating to human diseases.
The Healthcare Cost and Utilization Project (HCUP) has developed Clinical Classifications Software (CCS) (HCUP Clinical Classifications Software (CCS) for ICD-9-CM, 2006–2009), which we used to characterize the individually mapped diseases into broader categories. In the current study, we used the ‘Single-Level Diagnosis’ terms for categorization, which has 202 different categories. For enrichment analyses, we further only kept categories that contained at least 5 diseases, which left 93 categories. The full list of diseases, their respective ICD-9 codes, classification categories, as well as frequencies in our population can be found in the Supplementary materials. We present the disease frequencies (A) and category composition (B) in Figure 2.
Fig. 2.
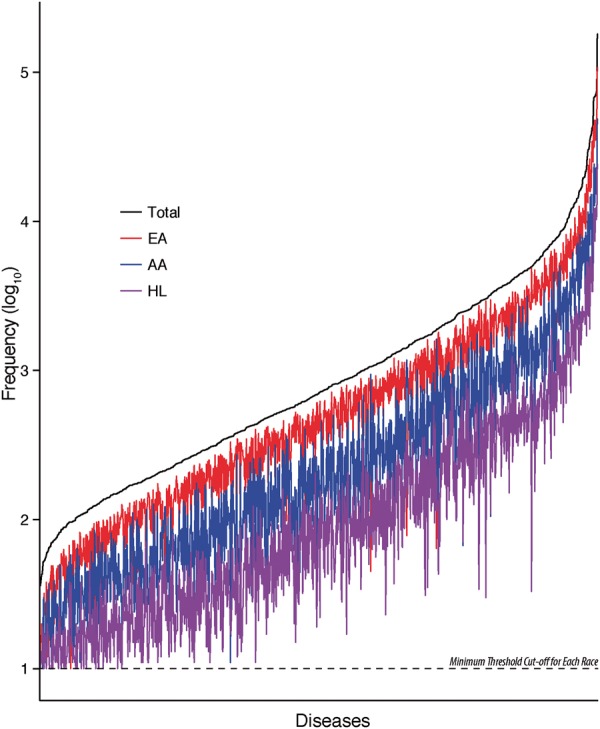
Disease and category frequency. We show for A disease counts (log10) overall and by EA, AA and HL cohorts. We show for B the distribution of the number of diseases encompassed within each of the 93 used CCS disease categories
2.1.2 Disease filtration
The primary focus of the current study is to compare temporal disease connection patterns across races. Accordingly, we performed several filtering steps on the raw list of 6545 disease terms to prioritize particular diseases of interest best suited for the analysis:
We only included diseases that mapped to at least one ICD-9 code.
We removed all diseases that were top-level, parent disease categories (e.g. endocrine system disease).
We only kept diseases if there were ≥10 affected individuals from each racial group in our cohort.
These filtering steps resulted in a list of 1198 diseases. To assess connectivity between diseases, we then compiled pairs of diseases from this list that underwent further curation steps. We filtered the raw list of all possible 759528 disease-pair combinations as following:
4.We kept a disease pair only if there were ≥10 individuals from each racial group with both diseases in our cohort.
5.We removed disease pairs in which one disease was a complete subset of another.
There were 37 282 disease pairs remaining after these filtering steps.
2.2 Statistical analyses
2.2.1 Deriving race-specific disease dynamics
For each disease of interest, we assessed if and to what extent demographic factors, namely race, play a role in defining morbidity, when controlling for other potentially associated factors. Specifically, we ran a logistic regression adjusting for sex, age and race and assessed if any demographic covariate was significantly associated with disease risk (Eq. 1).
| (1) |
where βr is categorical piecewise (Caucasian, African American, Hispanic/Latino), βa is a continuous constant per year and βs is binary piecewise Female/Male
As such, for each disease we compared the effect of race using the EA population as a baseline for disease susceptibility.
2.2.2 Disease pair temporal patterns
With the filtered disease pairs compiled, we then sought to determine whether each disease pair had temporal directionality, or specifically whether one disease consistently preceded the other. For the overlapping individuals that are afflicted with both diseases in each pair, we tabulated per patient the ordering of their pathogeneses. Specifically, we compared the number of patient instances where one disease preceded the other and vice versa or if they were recorded during the same encounter. We took earliest instance recording date for each disease. If one disease more frequently predated the other, we calculated the cumulative binomial probability that the precedence occurs significantly more often than by chance (Eq. 2). For each disease pair, we made the assumption that there was a 50% chance that one disease can occur before the other.
| (2) |
where n is the number of individuals with both diseases, r is number of instances where one disease predates the other, p is the probability of success (0.5) and q is the probability of failure (0.5).
2.2.3 Comorbidity calculation
While one disease may statistically precede another, it does not necessarily mean they have a direct relationship. Accordingly, for each disease pair with significant directionality identified by the previous step, we next determined whether there was significant comorbidity in the clinical population. Specifically, for each of these 37 282 disease pairs, we performed a logistic regression estimating the contribution the prior disease (i.e. the ‘predictor’ disease) has for risk of developing subsequent sequelae (i.e. the ‘response’ disease) (Eq. 3) for each population separately.
| (3) |
where βd is binary Yes/No, βa is a continuous constant per year, βs is binary piecewise Female/Male
2.3 Disease network construction and comparative analytics
Using results from the previous analyses, we generated population-specific disease networks using the Cytoscape (Shannon et al., 2003) platform (v3.3.0). These networks are comprised of directed connections between source and target disease pairs found to be significant in terms of both temporal directionality and connectivity for individual race populations, using β as edge weight. We then performed network metric analyses for each population network using the NetworkAnalyzer (Doncheva et al., 2012) plugin for Cytoscape. Using these generated metric statistics, we compared each population networks to determine network structure concordance via metrics (e.g. closeness centrality). Specifically, we performed a one-way analysis of variance (ANOVA) between the metrics for race-cohort networks. We then performed Tukey HSD test on significant results to determine which race networks differed.
2.3.1 Disease hub identification and categorical enrichments
For each population, we identified ‘hubs’ of connectivity, which are focal points in the network that have many outgoing connections. We defined hubs as diseases that have at least 10 outgoing disease connections: specifically, any predictor disease in a pair (i.e. those that predate the latter) that is significantly connected to at least 10 diseases within a population.
We then evaluated the different composition of the results between populations using the 93 different categories of diseases. We first determined whether the identified hub diseases for each population were enriched for any of these categories. We then performed the same analysis on predictor (i.e. earlier) and response (i.e. later) diseases in the significant disease pairs in each population. Specifically, for hub, source and target diseases significant for each population, we performed a one-way Fisher’s exact test comparing the amount of overlap with diseases of each category.
3 Results
For the current study, we calculated disease connectivity patterns for a 1198 diseases in a large, ethnically diverse EMR cohort with 3 well-represented populations and compared across race-specific networks.
3.1 Effect of race on disease susceptibility prediction
Using the EA cohort as a baseline, we first determined how race affects susceptibility of each of the 1198 diseases while controlling for age and sex factors compared to AA and HL. In total, we found that a large portion, 968 (81%), of these diseases had some race contribution (Bonferroni corrected P < 4.2 × 10−05) to pathogenesis (Eq. 1). The corresponding trends of race association with disease risk along with selected examples are displayed in Figure 3.
Fig. 3.
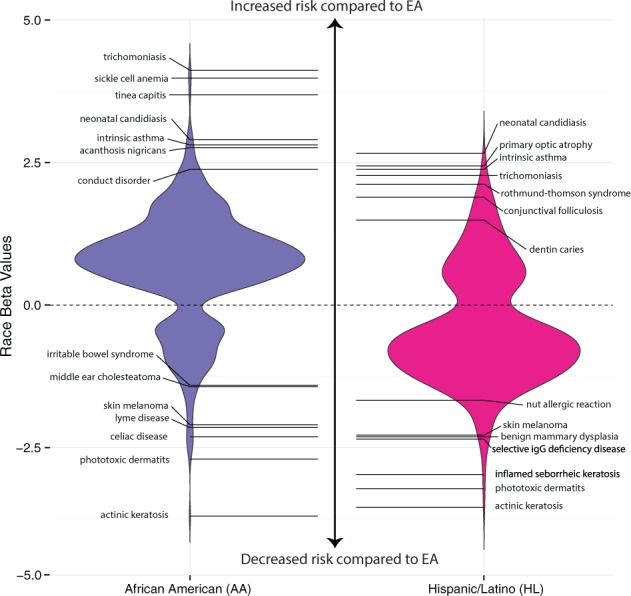
Disease susceptibility profiles based on racial group. We present here the distribution of diseases (with highlighted examples) that have statistically significant (Bonferroni corrected P < 4.2 × 10−05) differences in risk profiles for AA and HL cohorts compared to EA. The race beta values refer to effect size of race when controlling for age and sex with positive values indicating increased risk compared to EA and vice versa
We found 731 diseases (61%) for which EA and AA individuals had significantly different risks of affliction, 369 of which were not associated with the HL population. Effect sizes, in terms of β, ranged from −3.70 to 4.12 with positive values indicating increased risk for AA individuals and vice versa. Our data suggests that the AA population is more susceptible to disease acquisition overall: out of the significant associations a large proportion, 580 (79%), were positively associated with AA.
Compared to the AA population, there were fewer diseases significantly associated with altered risk profiles for HL individuals. Only 599 (30%) of the diseases were associated with HL cohort and 237 of which were not associated with AA risk. The effect sizes ranged from −3.55 to 2.66, with a fewer number of diseases, 182 (30%), at increased prevalence in HL which is the opposite of the trend for the AA population.
3.2 Directionality of race-specific temporal disease pairs
We first determined (Eq. 2) which of the 37 282 disease pairs had significant temporal directionality (i.e. a pair in which one disease significantly precedes the other) for EA, AA and HL populations separately (P < 1.42 × 10−06). For EA, we found 2333 (6.61%) significant temporally related disease pairs, 3311 (9.38%) for AA and 691 (1.96%) for HL. In total, across all population, we found 6336 (5.99%) disease pairs that were significantly related temporally.
3.3 Race-specific disease pair connectivity patterns
Within each population, for each disease pair that we determined to have significant directionality, we then evaluated (Eq. 3.) whether and to what extent they were connected (P < 1.42 × 10−06). We present the relative distribution of significant disease pairs between each population in Figure 4. We also highlight select pairs unique to each race in Table 1.
Fig. 4.
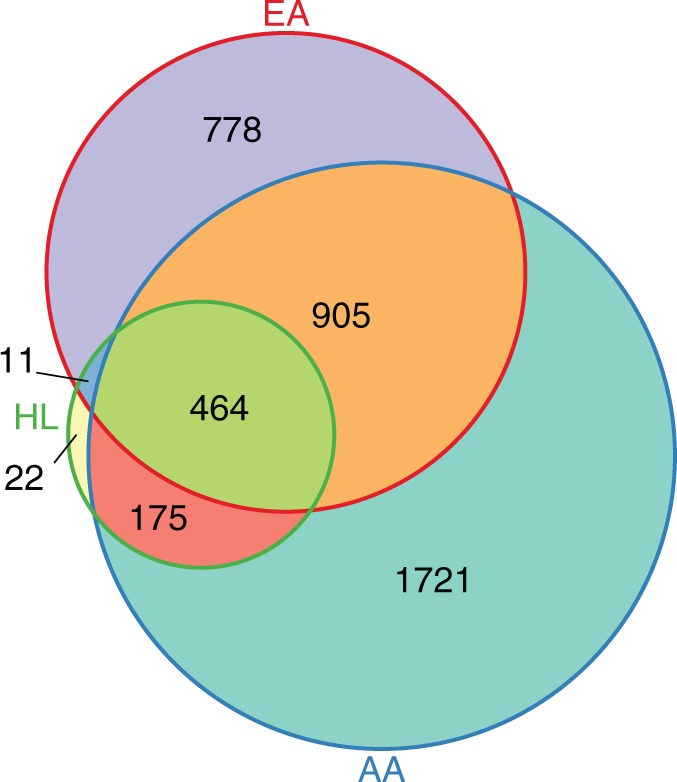
Distribution of significantly connected disease pairs by racial cohort. We show the amount of disease pairs that were significantly temporally related and comorbid for all racial groups (P < 1.42 × 10−06 criteria for both)
Table 1.
Temporal directionality and connectivity significance of selected disease pairs unique to each race cohort
| Pop. | Disease 1 | Disease 2 | P-val | β |
|---|---|---|---|---|
| EA | Thyroid cancer | Postsurgical hypothyroidism | <6.4E−324 | 5.25 |
| EA | Lymphosarcoma | Aplastic anemia | <6.4E−324 | 3.42 |
| EA | Ulcerative colitis | Intestinal obstruction | <6.4E−324 | 3.27 |
| EA | Toxic diffuse goiter | Postsurgical hypothyroidism | 1.3E−153 | 3.11 |
| EA | Familial hypercholesterolemia | Acute cystitis | <6.4E−324 | 3.09 |
| AA | Diabetes mellitus, type 2 | Diabetic cataract | 2.1E−16 | 5.73 |
| AA | Hyperthyroidism | Toxic diffuse goiter | <6.4E−324 | 5.10 |
| AA | Chronic ulcer of skin | Osteomyelitis | 1.4E−235 | 4.96 |
| AA | Hypertension | IgA glomerulonephritis | 6.4E−75 | 4.09 |
| AA | HIV disease | Esophageal candidiasis | <6.4E−324 | 3.87 |
| HL | Diabetes mellitus, type 1 | Clostridium difficile colitis | 3.3E−73 | 2.51 |
| HL | Benign essential hypertension | Phobic disorder | 5.1E−28 | 2.25 |
| HL | Coronary artery disease | ARDS | 1.7E−61 | 1.89 |
| HL | Generalized anxiety disorder | Anemia | 3.1E−64 | 1.72 |
| HL | Major depressive disorder | Decubitus ulcer | 2.1E−42 | 1.67 |
For each population, we determined which temporally related disease pairs had Bonferroni-corrected significant connectivity (P < 1.42 × 10−06). We present particular disease pairs of interest from among the top-25 associations for each population, ranked by effect size. Effect size, or β, can be interpreted as the odds ratio of disease 2 occurring given disease 1, holding age and sex constant.
We further determined the relative timescale of the latencies between disease pairs across all populations. Specifically, for all significantly comorbid disease pairs common among all racial groups (n = 464), we determined the average latency from the pathogenesis of the first disease to developing the latter within each racial group. The average latency between diseases was 1.67 ± 0.62 years for EA, 2.35 ± 0.94 years for AA and 1.75 ± 0.76 years for HL.
3.4 Race-specific network dynamics
Using the results from the previous sections, we generated unique disease networks for each race cohort as displayed in Figure 5A/B/C. In addition to the varying disease patterns across the cohorts, there were also significant differences in the composition of the networks, which we show in Table 2. Full descriptions of these metrics can be found in the documentation for the Cytoscape NetworkAnalyzer package.
Fig. 5.

Network structure patterns for each racial cohort and hub connectivity. We provide race-specific networks for EA (A), AA (B) and HL (C) populations for disease pairs that were significantly temporally related and comorbid for each group (P < 1.42 × 10−06 criteria for both). Effect size, shown as edge weight, is the increased risk of developing the target disease when having the source, controlling for sex and age. Node size reflects number of directed, outgoing connections. The larger text refers to diseases identified as hubs for the population
Table 2.
Metric statistic results across race-specific networks
| Metric | EA | AA | HL | P-value | EA/AA (p) | EA/HL (p) | AA/HL (p) | Trend |
|---|---|---|---|---|---|---|---|---|
| Closeness centrality | 0.27±0.38 | 0.21±0.35 | 0.16±0.37 | 2.0E−03 | 0.04 | 2.00E−03 | 0.28 |  |
| Clustering coefficient | 0.05±0.09 | 0.08±0.1 | 0.01±0.05 | 1.07E−14 | 1.00E-03 | 2.10E−06 | 4.94E−324 |  |
| Eccentricity | 0.78±1.18 | 0.69±1.21 | 0.21±0.50 | 1.80E−08 | 0.42 | 1.37E−08 | 9.95E−07 |  |
| Edge count | 11.34±23.94 | 13.3±33.54 | 6.86±15.06 | 2.30E−02 | 0.55 | 0.16 | 0.02 |  |
| In-degree | 5.67±7.89 | 6.65±8.16 | 3.43±3.1 | 1.98E−06 | 0.13 | 1.73E−03 | 9.03E−07 |  |
| Neighborhood connectivity | 109.22±66.37 | 289.76±111.47 | 69.08±34.72 | 2.97E−77 | 4.94E-324 | 5.16E−07 | 4.94E−324 |  |
| Out-degree | 5.67±23.46 | 6.65±33.31 | 3.43±15.48 | 0.38 | – | – | – | – |
| Stress | 8.55±43.08 | 13.13±64.38 | 0.29±1.7 | 1.1E−02 | 0.38 | 0.15 | 8.00E−03 |  |
We determined significant differences (italicized) in network structure across EA, AA and HL networks using a one-ANOVA to compare average metric statistics for race-cohort networks (P < 0.05). We then performed Tukey HSD test on significant results to determine specifically which races differed from one another (P < 0.05).
3.5 Population-specific disease hubs
In total, across all populations, we identified 51 unique diseases that were hubs. Many of these hubs were so in multiple populations with 9 being hubs in all 3 populations. We found 7 diseases that were hubs only in the EA population, 24 only in AA population and none that were unique to the HL population. We present the sub-network of hub diseases significant to each population, along with their first neighbor connections in Figure 5D.
3.6 Disease categorical enrichment of connectivity results between populations
From our network connectivity results, we determined whether hub, source (i.e. predictor) and target (i.e. response) diseases significant to each population were enriched for any of the 93 disease categories.
3.6.1 Hub disease categorical enrichment
In total, we found 14 nominally significant (p < 0.05) disease category-hub enrichments. The hubs of all 3 cohorts were most highly enriched for ‘Diabetes mellitus with complications’ (EA: P = 7.0 × 10−04, odds ratio = 23.42; AA: P = 8.0 × 10−04, OR = 28.2; HL: P = 7.0 × 10−03, OR = 84.9).
Furthermore, the EA hubs were enriched for ‘Mood disorders’ (P = 0.01, OR = 18.72), ‘Esophageal disorders’ (P = 0.01, OR = 18.7) and ‘Thyroid disorders’ (P = 0.04, OR = 7.7). While the AA hubs were similarly enriched for ‘Mood disorders’ (P = 0.03, OR = 11.0) and ‘Esophageal disorders’ (P = 0.03, OR = 11.0), they were also enriched for ‘Asthma disorders’ (P = 0.01, OR = 11.0), ‘Allergic reactions’ (P = 0.03, OR = 9.1), ‘Anxiety disorders’ (P = 0.03, OR = 9.1) and ‘Other gastrointestinal disorders’ (P = 0.04, OR = 7.8). The HL hubs only were enriched for ‘Asthma disorders’ (P = 0.04, OR = 37.1) and ‘Complications of surgical procedures or medical care’ (P = 0.04, OR = 37.1).
3.6.2 Source disease categorical enrichment
Next, we determined categorical enrichment for source diseases in significant pairs in each race population. Within the EA disease network, there were 136 significant source diseases, 144 for AA and 33 for HL. The source diseases of each race were significantly enriched for ‘Diabetes mellitus with complications’ (EA: P = 0.02, odds ratio = 8.0; AA: P = 0.02, OR = 15.1; HL: P = 4.0 × 10−04, OR = 38.9) and ‘Mood disorders’ (EA: P = 0.04, odds ratio = 6.0; AA: P = 5.0 × 10−03, OR = 10.1; HL: P = 6.0 × 10−04, OR = 29.2).
For the EA cohort, source diseases were also enriched for ‘Diseases of white blood cells’ (P = 0.02, OR = 8.0), ‘Esophageal disorders’ (P = 0.04, OR = 6.0) and ‘Thyroid disorders’ (P = 0.02, OR = 4.5). The source diseases of the AA population were likewise enriched for ‘Thyroid disorders’ (P = 3.4 × 10−03, OR = 5.8) but also for ‘Epilepsy/Convulsions’ (P = 5.0 × 10−03, OR = 10.1), ‘Mycoses’ (P = 0.04, OR = 3.15) and ‘Pulmonary heart diseases’ (P = 0.01, OR = 11.3). Like the EA cohort, HL source diseases were enriched for ‘Esophageal disorders’ (P = 0.01, OR = 15.0). Additionally, we found enrichment for ‘Asthma diseases’ (P = 7.0 × 10−03, OR = 25.1) and ‘Other inflammatory skin conditions’ (P = 0.04, OR = 7.5).
3.6.3 Target disease categorical enrichment
Finally, we analyzed categorical enrichment of target diseases, which are the direct connections from source diseases. Overall there were more target diseases than source: 319 target diseases for EA, 454 for AA and 178 for HL. The only disease category significantly enriched in target diseases of all races was ‘Mycoses’ (EA: P = 7.0 × 10−04, odds ratio = 23.42; AA: P = 8.0 × 10−04, OR = 28.2; HL: P = 7.0 × 10−03, OR = 84.9).
We found that ‘Diseases of white blood cells’ (P = 4.5 × 10−02, OR = 5.6) and ‘Retinal detachments/defects/vascular occlusions/retinopathies’ (P = 0.01, OR = 2.7) were the only other categories enriched for EA target diseases. For AA, we discovered that ‘Retinal detachments/defects/vascular occlusions/retinopathies’ (P = 3.0 × 10−04, OR = 4.6) was also significantly enriched along with ‘Cataract diseases’ (P = 0.03, OR = 8.4) ‘Glaucoma diseases’ (P = 7.0 × 10−03, OR = 6.8) and ‘Other diseases of kidney and ureters’ (P = 0.02, OR = 4.5). The HL population had a similar target disease enrichment profile to AA with categorical enrichments of ‘Cataract diseases’ (P = 4.5 × 10−02, OR = 5.8), ‘Glaucoma diseases’ (P = 8.8 × 10−03, OR = 5.9). The HL cohort also had enrichments in ‘Diabetes mellitus with complications’ (P = 4.5 × 10−02, OR = 5.8), ‘Gastritis and duodenitis’ (P = 0.03, OR = 8.8) and ‘Hypertension with complications and secondary hypertension’ (P = 0.01, OR = 7.9).
4 Discussion
The results from the current study provide illustrative examples of the extent disease susceptibility and connectivity patterns differ between race cohorts, formalizing the need for race-specific risk assessment. Overall, the cross-race individual disease profiles are consistent with known data and expectations (Fig. 3), which is important for implications that can inform follow-up studies.
More importantly, our results are in line with findings from related studies. In particular, our findings for the disease temporal patters in the EA cohort are consistent with the disease trajectories identified by Jensen et al. (2014). Direct comparison of results between our study and theirs is difficult, however, namely due to use of non-identical statistical methodologies and ontological disease mappings (ICD-9 versus ICD-10). Regardless, several similarities are apparent: firstly, the raw number of disease pairs with significant temporal directionality is consistent between the two studies: there were 4014 disease pairs with significant temporal directionality in their study and 2333 in ours; the small difference of which can partially be explained by sample size discrepancies. Furthermore, the authors identified related clusters of trajectories that are akin to hubs of the current study. While there are discrepancies, many focal disease points overlap: Type 2 Diabetes (T2D), for instance, was involved in many trajectories in their study and was a central hub in our EA cohort with 101 Bonferroni-corrected sequellae. Outcome diseases in the Jensen et al. T2D network included ‘retinal disorder’ which corresponds to many target diseases found in ours, including dry-eye syndrome, retinal drusen, peripheral retinal degeneration, and retinal edema. ‘Chronic renal failure’ and ‘Unspecified renal failure’ were also outcome diseases, which overlap with target diseases such as impaired renal function disease, benign hypertensive renal disease and secondary hyperparathyroidism of renal origin found in our network. Chronic obstructive pulmonary disease, another cluster disease, was found to have similar temporal patterns as well, including ‘Angina’ as a predictor disease (angina pectoris in our results) and ‘Unspecified chronic bronchitis’ as an outcome disease (bronchitis in ours).
Comparing our results to those of the Hidalgo et al. study using network approaches to study human phenotype serves as a source to validate our AA and EA networks. In their study, they found certain disease combinations that were differentially comorbid in black (AA) and white (EA) populations, many of which are validated in our findings. They demonstrate that heart diseases, including ‘mitral valve disorders’ and ‘mitral and aortic valve stenosis’ were more comorbid in white males than black males. Similarly, we found both aortic valve disease and mitral valve disease to be hubs in only the EA network. Interestingly, they show that ‘other peripheral vascular disease’ was connected to diseases across networks for both races and we also identified that the same disease is a hub for both these populations. Hidalgo et al. further demonstrate that ‘diabetes’ and ‘hypertension’ were more comorbid in black males than white males. While, in our study, both diseases were found to be hubs for both EA and AA networks, they were more highly connected (diabetes mellitus: 102 connections for EA versus 187 for AA and hypertension: 233 connections for EA versus 377 for AA) in our AA cohort than the EA network. ‘Respiratory abnormality’ was also more comorbid for black males in their study, which can be seen as corresponding to asthma-related disorder categorical enrichments in our identified AA hubs. Taken together, the concordance of results between these two studies and ours is extremely encouraging and provides support for the methodologies employed in the current paper and the ensuing HL cohort network discovery.
4.1 Race-centric disease connectivity and network composition disparities
As shown in Table 2, there are noticeable differences between the disease networks, not only between EA and AA or HL but also between AA and HL. Metric differences in average clustering coefficient and eccentricity (which reflects maximum length between one node and its connections) between all races reaffirms that disease patterns vary considerably in different racial backgrounds, despite population size. Significant differences between AA and HL network composition emphasizes that it is not enough to merely compare EA versus ‘other’ races: disease networks of each racial group requires substantial, individualized investigation.
Another interesting component of comparative analyses of these three networks is the identification of what diseases are common sequelae for each race. We reveal categories of diseases that are enriched only for the HL population. Of particular interest is ‘gastritis and duodenitis’ which is known to have higher incidence in Hispanic populations, which could possibly be due to increased rates of H. pylori infection (Dehesa et al., 1991). As gastritis can lead to gastric carcinoma, the diseases that predate it can serve as early warning signs.
4.2 Impact on healthcare delivery
Identifying diseases that are hubs within a network, especially those that are specific to certain racial groups, can highlight focal areas that warrant particular clinical attention. We show that while diseases may be abundant in multiple populations (Fig. 5D), some diseases are hubs only for certain populations. It is clear that the AA population has the most hub diseases both overall and unique to the population, which reflects an increased disease burden.
We found, for example, that Type 1 Diabetes (T1D) is a hub disease for only the AA cohort, although there were some interesting associations in the other populations (e.g. T1D to clostridium difficile colitis in HL). There has been thorough and extensive research on the impact of T1D on the AA population and which has shown that the AA population indeed has higher incidence rates (Mayer-David et al., 2009). Results from our hub analysis can extend beyond the simple observation of increased T1D risk in AA to actually illuminate the subsequent disease pathogeneses specific to AA including many eye-related diseases, such as background diabetic retinopathy, blindness, borderline glaucoma, dry eye syndrome, retinal edema and senile cataract. Knowledge of these associations can be passed along to patients in this group so they can be aware of increased risk for such complications. Furthermore, there are several AA-specific disease hubs that are not as well established in the literature: diaper rash, constipation and diarrhea are all seemingly mild conditions but, as we show, can lead to a number of disorders, particularly in the AA population.
Findings from these network analytics can suggest clinical practice considerations. The AA network, for example, has significantly higher local interconnectivity scores (e.g. clustering coefficient, neighborhood connectivity) compared to both EA and HL. Furthermore, we have shown that AA individuals have, on average, relatively longer latencies between disease comorbidity onsets. While one could interpret the longer latencies between diseases to slower progression or attribute them to less frequent patient visits, these findings nonetheless could inform clinical treatment strategies: if an AA patient is diagnosed with a highly interconnected disease, such as Hepatitis C, the clinician might strongly urge proximate follow-up visits for active screening of comorbid diseases and consideration of preventative or prophylactic treatment. Similar practices are already implemented in the clinic for certain diseases: the 2016 American Diabetes Association Guidelines (American Diabetes Association, 2016) recommend recurrent T2D screening visits for individuals who have hypertension and/or are of a ‘high-risk race/ethnicity’, even if they are completely asymptomatic.
The HL network, on the other hand, consistently has lower scores relating to connectivity and clustering (e.g. clustering coefficient, in-degree, neighborhood connectivity and stress). While this pattern may reflect a unique, sparser phenotypic landscape and lower overall disease burden, it may serve as a reminder of clinical underrepresentation and the need for community outreach (see: ‘Limitations’).
4.3 Limitations
An obvious limitation of the current study is the respective size of the HL population in our analysis. Although the AA cohort was not much larger (21.8% versus 17.5%), it is clear that the AA population was more represented in the disease space (Fig. 2). Another limitation is the type of information available in the EMR data. Our results highlight differences in population disease risk patterns, that in some cases are likely indicative of other potentially confounding factors not captured in the EMR data, such as language barriers, access to healthcare or important environmental or socioeconomic factors.
Another possible reason for the sparser HL disease network could be due to a higher heterogeneity of the underlying HL population structure. Studies have indeed shown that while Hispanic/Latino populations are traditionally combined into a single ethnic group (as in the current study), there is extensive diversity in terms of cultural backgrounds and genetic ancestry (Gonzalez et al., 2005), which might be masking associations in our networks.
4.4 Future directions
The discovery of unique disease sequelae between race populations is a promising start, but it reveals how much more has to be done to generate a broader understanding of disease susceptibility patterns across diverse populations. An obvious, urgent need is to introduce and incorporate data from population groups that are almost completely absent in phenomics space, such as Native Americans and Pacific Islanders. As illustrated by the aforementioned example of HL population diversity, there is a clear need to better stratify potentially overgeneralized cohorts. This can be facilitated by increased sample sizes, more accurate demographic reporting in the EMR and incorporating genetic ancestry.
Many other population-scale factors would be important to compare across race-centric disease networks. One particular direction warranting further investigation is an examination of the latencies between disease pairs across populations beyond overall average. Accordingly, by including encounter information and visit frequency, we might be able to identify factors underlying racial group latency discrepancies for each particular pair of comorbid diseases, which may help inform clinical practices.
Furthermore, researchers (Patel et al., 2015) recently demonstrated intricate links between socioeconomic factors, health outcomes and disease risk. Environment-Wide Association Studies (EWAS) (Patel et al., 2010) have shown the dynamic relationship between disease risk, environmental exposures and genetic profiles. Combining phenomics, subtleties of phenocopies, disease genetics and environmental exposures by zip code within the current dataset can bring us further towards a framework for establishing stratified precise comorbidity networks for personalized medicine.
Supplementary Material
Acknowledgements
This work was supported in part through the computational resources and staff expertise provided by the Department of Scientific Computing at the Icahn School of Medicine at Mount Sinai.
Funding
This study was supported by funding from the following grants from the National Institute of Health (NIH): National Institute of Diabetes and Digestive and Kidney Diseases (NIDDK) (R01DK098242); National Cancer Institute (NCI) (U54CA189201); Illuminating the Druggable Genome (IDG), Knowledge Management Center sponsored by NIH Common Fund, National Cancer Institute (NCI, U54-CA189201-02) and National Center for Advancing Translational Sciences (NCATS, UL1TR000067); Clinical and Translational Science Award (CTSA). The funders had no role in the study design, data collection and analysis, decision to publish or preparation of the manuscript. The content is solely the responsibility of the authors and does not necessarily represent the official views of the NIDDK, NCI or NIH.
Conflict of Interest: J.T.D. has received consulting fees or honoraria from Janssen Pharmaceuticals, GSK, AstraZeneca and Hoffman-La Roche. JTD holds equity in NuMedii Inc., Ayasdi, Inc. and Ontomics, Inc. No writing assistance was utilized in the production of this manuscript.
Reference
- American Diabetes Association. (2016) Standards of medical care in diabetes-2016. Diabetes Care, 39, S1–106.26696671 [Google Scholar]
- Aseltine R.H., Jr. et al. (2015) Racial and ethnic disparities in hospital readmissions after delivery. Obstet. Gynecol., 126, 1040–1047. [DOI] [PubMed] [Google Scholar]
- Badgeley M.A. et al. (2016) EHDViz: clinical dashboard development using open-source technologies. BMJ Open, 6, e010579. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Barabasi A.L., Oltvai Z.N. (2004) Network biology: understanding the cell's functional organization. Nat. Rev. Genet., 5, 101–113. [DOI] [PubMed] [Google Scholar]
- Barabasi A.L. et al. (2011) Network medicine: a network-based approach to human disease. Nat. Rev. Genet., 12, 56–68. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bilder R.M. et al. (2009) Phenomics: the systematic study of phenotypes on a genome-wide scale. Neuroscience, 164, 30–42. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Blair D.R. et al. (2013) A nondegenerate code of deleterious variants in Mendelian loci contributes to complex disease risk. Cell, 155, 70–80. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cassidy-Bushrow A.E. et al. (2011) Shared genetic architecture in the relationship between adult stature and subclinical coronary. Atherosclerosis, 219, 679–683. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dehesa M. et al. (1991) High prevalence of Helicobacter pylori infection and histologic gastritis in asymptomatic Hispanics. J. Clin. Microbiol., 29, 1128–1131. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Denny J.C. et al. (2010) PheWAS: demonstrating the feasibility of a phenome-wide scan to discover gene-disease associations. Bioinformatics, 26, 1205–1210. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Doncheva N.T. et al. (2012) Topological analysis and interactive visualization of biological networks and protein structures. Nat. Protoc., 7, 670–685. [DOI] [PubMed] [Google Scholar]
- Dudley J.T. et al. (2011a) Exploiting drug-disease relationships for computational drug repositioning. Brief. Bioinf., 12, 303–311. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dudley J.T. et al. (2011b) Matching cancer genomes to established cell lines for personalized oncology. Pac. Symp. Biocomput., 16, 243–252. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dudley J.T. et al. (2011c) Computational repositioning of the anticonvulsant topiramate for inflammatory bowel disease. Sci. Transl. Med., 3, 96ra76. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dudley J.T. et al. (2015) Personalized medicine: from genotypes, molecular phenotypes and the quantified self, towards improved medicine. Pac. Symp. Biocomput., 20, 342–346. [PMC free article] [PubMed] [Google Scholar]
- Feldman I. et al. (2008) Network properties of genes harboring inherited disease mutations. Proc. Natl. Acad. Sci. USA, 105, 4323–4328. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Glicksberg B.S. et al. (2015) An integrative pipeline for multi-modal discovery of disease relationships. Pac. Symp. Biocomput., 20, 407–418. [PMC free article] [PubMed] [Google Scholar]
- Goh K.I. et al. (2007) The human disease network. Proc. Natl. Acad. Sci. USA, 104, 8685–8690. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gonzalez B.E. et al. (2005) Latino populations: a unique opportunity for the study of race, genetics, and social environment in epidemiological research. Am. J. Public Health, 95, 2161–2168. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hall M.A. et al. (2014) Detection of pleiotropy through a Phenome-wide association study (PheWAS) of epidemiologic data as part of the Environmental Architecture for Genes Linked to Environment (EAGLE) study. PLoS Genet., 10, e1004678. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hazlewood, A. (2003) “ICD-9-CM to ICD-10-CM: Implementation Issues and Challenges.” AHIMA's 75th Anniversary National Convention and Exhibit Proceedings, October 2003. Available at http://library.ahima.org/xpedio/groups/public/documents/ahima/bok3_005426.hcsp?dDocName=bok3_005426.
- HCUP Clinical Classifications Software (CCS) for ICD-9-CM. (2006. –2009) Healthcare Cost and Utilization Project (HCUP). Agency for Healthcare Research and Quality [www.hcup-us.ahrq.gov/toolssoftware/ccs/ccs.jsp]. [PubMed]
- Hidalgo C.A. et al. (2009) A dynamic network approach for the study of human phenotypes. PLoS Comput. Biol., 5, e1000353. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jensen P.B. et al. (2012) Mining electronic health records: towards better research applications and clinical care. Nat. Rev. Genet., 13, 395–405. [DOI] [PubMed] [Google Scholar]
- Jensen A.B. et al. (2014) Temporal disease trajectories condensed from population-wide registry data covering 6.2 million patients. Nat. Commun., 5, 4022. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lee D.S. et al. (2008) The implications of human metabolic network topology for disease comorbidity. Proc. Natl. Acad. Sci. USA, 105, 9880–9885. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Li L. et al. (2013) Systematic identification of risk factors for Alzheimer's disease through shared genetic architecture and electronic medical records. Pac. Symp. Biocomput., 18, 224–235. [PubMed] [Google Scholar]
- Li L. et al. (2014) Disease risk factors identified through shared genetic architecture and electronic medical records. Sci. Trans. Med., 6, 234ra257. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Li L. et al. (2015a) Identification of type 2 diabetes subgroups through topological analysis of patient similarity. Sci. Trans. Med., 7, 311ra174. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Li Y.R. et al. (2015b) Meta-analysis of shared genetic architecture across ten pediatric autoimmune diseases. Nat. Med., 21, 1018–1027. [DOI] [PMC free article] [PubMed] [Google Scholar]
- López-Candales A. et al. (2015) The racial, cultural and social makeup of hispanics as potential profile risk for intensifying the need for including this ethnic group in clinical trial. Bol. Asoc. Med. Proc. R., 107, 17–23. [PubMed] [Google Scholar]
- Mayer-David E.J. et al. (2009) Diabetes in African American youth: prevalence, incidence, and clinical characteristics: the SEARCH for Diabetes in Youth Study. Diabetes Care, 32, S112–S122. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mount Sinai Data Warehouse. [https://msdw.mountsinai.org/]
- Namjou B. et al. (2014) Phenome-wide association study (PheWAS) in EMR-linked pediatric cohorts, genetically links PLCL1 to speech language development and IL5-IL13 to Eosinophilic Esophagitis. Front. Genet., 5, 401. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nead K.T. et al. (2016) Androgen deprivation therapy and future Alzheimer's Disease Risk. J. Clin. Oncol., 34, 566–571. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Patel C.J. et al. (2010) An Environment-Wide Association Study (EWAS) on type 2 diabetes mellitus. PLoS ONE, 5, e10746.. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Patel C.J. et al. (2015) Systematic assessment of the correlations of household income with infectious, biochemical, physiological, and environmental factors in the United States, 1999–2006. Am. J. Epidemiol., 181, 171. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schadt E.E. et al. (2009) A network view of disease and compound screening. Nat. Rev. Drug Disc., 8, 286–295. [DOI] [PubMed] [Google Scholar]
- Schriml L.M. et al. (2012) Disease Ontology: a backbone for disease semantic integration. Nucleic Acids Res., 40, 940. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shameer K. et al. (2014) A genome- and phenome-wide association study to identify genetic variants influencing platelet count and volume and their pleiotropic effects. Hum. Genet., 133, 95–109. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shameer K. et al. (2015) Computational and experimental advances in drug repositioning for accelerated therapeutic stratification. Curr. Top. Med. Chem., 15, 5–20. 2015; [DOI] [PubMed] [Google Scholar]
- Shameer K. et al. (2016) Translational bioinformatics in the era of real-time biomedical, health care and wellness data streams. Brief. Bioinf. https://www.ncbi.nlm.nih.gov/pubmed/26876889. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shannon P. et al. (2003) Cytoscape: a software environment for integrated models of biomolecular interaction networks. Genome Res., 13, 2498–2504. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Stewart W.F. et al. (2007) Bridging the inferential gap: the electronic health record and clinical evidence. Health Aff., 26, 181–191. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Suthram S. et al. (2010) Network-based elucidation of human disease similarities reveals common functional modules enriched for pluripotent drug targets. PLoS Comput. Biol., 6, e1000662. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Trepka M.J. et al. (2015) Sex and racial/ethnic differences in premature mortality due to HIV: Florida, 2000–2009. Public Health Rep., 130, 505–513. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zanzoni A. et al. (2009) A network medicine approach to human disease. FEBS Lett., 583, 1759–1765. [DOI] [PubMed] [Google Scholar]
- Zhou X. et al. (2014) Human symptoms-disease network. Nat. Commun., 5, 4212. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.