

Abstract
Chronic activation of AMP‐activated protein kinase (AMPK) increases glycogen content in skeletal muscle. Previously, we demonstrated that a mutation in the ryanodine receptor (RyR1R615C) blunts AMPK phosphorylation in longissimus muscle of pigs with a gain of function mutation in the AMPK γ3 subunit (AMPK γ3R200Q); this may decrease the glycogen storage capacity of AMPK γ3R200Q + RyR1R615C muscle. Therefore, our aim in this study was to utilize our pig model to understand how AMPK γ3R200Q and AMPK activation contribute to glycogen storage and metabolism in muscle. We selected and bred pigs in order to generate offspring with naturally occurring AMPK γ3R200Q, RyR1R615C, and AMPK γ3R200Q + RyR1R615C mutations, and also retained wild‐type littermates (control). We assessed glycogen content and parameters of glycogen metabolism in longissimus muscle. Regardless of RyR1R615C, AMPK γ3R200Q increased the glycogen content by approximately 70%. Activity of glycogen synthase (GS) without the allosteric activator glucose 6‐phosphate (G6P) was decreased in AMPK γ3R200Q relative to all other genotypes, whereas both AMPK γ3R200Q and AMPK γ3R200Q + RyR1R615C muscle exhibited increased GS activity with G6P. Increased activity of GS with G6P was not associated with increased abundance of GS or hexokinase 2. However, AMPK γ3R200Q enhanced UDP‐glucose pyrophosphorylase 2 (UGP2) expression approximately threefold. Although UGP2 is not generally considered a rate‐limiting enzyme for glycogen synthesis, our model suggests that UGP2 plays an important role in increasing flux to glycogen synthase. Moreover, we have shown that the capacity for glycogen storage is more closely related to the AMPK γ3R200Q mutation than activity.
Keywords: Calcium, glucose 6 phosphate, glycogen synthase, skeletal muscle, UDP‐glucose pyrophosphorylase
Introduction
AMP‐activated protein kinase (AMPK) plays a key role in cellular energy homeostasis in skeletal muscle. AMPK is a heterotrimeric serine/threonine kinase composed of a catalytic α and regulatory β and γ subunits. Decreasing energy charge (ATP:AMP) enhances AMPK activation by allosteric binding of AMP to the γ subunit, and promotes phosphorylation of the activating site, αThr‐172, by upstream kinases (Stein et al. 2000; Suter et al. 2006; Gowans et al. 2013). In order to preserve cellular ATP, activated AMPK acutely inhibits anabolic pathways and stimulates catabolic pathways involved in carbohydrate, lipid, and fatty acid metabolism. AMPK also modulates long‐term adaptation by coordinating changes in gene and protein expression.
The γ3 subunit is highly expressed in glycolytic skeletal muscle and plays a key role in adaptation and fuel metabolism (Mahlapuu et al. 2004). Activating mutations in γ3 contribute to significant elevations in glycogen content in glycolytic muscles of mice (Barnes et al. 2004), pigs (Milan et al. 2000), and humans (Costford et al. 2007). Glycogen synthesis is primarily controlled by glucose transport and glycogen synthase (GS) activity (Azpiazu et al. 2000). AMPK facilitates contraction‐induced, insulin‐independent glucose uptake by promoting translocation of the muscle‐specific glucose transporter (GLUT4) to the cell membrane (Kurth‐Kraczek et al. 1999; Jørgensen et al. 2004b). Moreover, chronic activation of AMPK results in adaptive changes in glucose metabolism, including increased GLUT4 protein content (Holmes et al. 1999). However, AMPKγ3 knockout mice exhibit normal glucose tolerance and similar muscle glycogen content as wild type, but show reduced capacity to resynthesize glycogen after exercise (Barnes et al. 2004). Together, this suggests that glucose transport is not the primary step contributing to γ3‐mediated glycogen synthesis; instead, glycogen synthase activity or intermediary steps may limit storage of glucose as glycogen.
The regulation of GS is rather complex; it is negatively regulated by phosphorylation by AMPK (Jensen et al. 2006) as well as several other protein kinases. Although nine phosphorylation sites have been identified, only four sites (2, 2a, 3a, 3b) are considered the most influential for regulating GS activity in muscle (Roach et al. 2012). However, glucose 6‐phosphate (G6P) can completely overcome the inhibitory effects of phosphorylation, and thus restore full activity of GS (Roach et al. 2012). In this manner, limiting glucose transport restricts G6P available for glycolysis and glycogen synthesis, whereas higher rates of glucose transport enhance G6P levels and flux toward glycogen synthesis. In fact, Hunter et al. (Hunter et al. 2011) demonstrated that the primary molecular mechanism by which AMPK enhances glycogen storage despite GS phosphorylation is via G6P‐induced allosteric activation. Chronic activation of AMPK likely enhances G6P content by increasing hexokinase content (Leick et al. 2010) and activity (Holmes et al. 1999; Granlund et al. 2011).
Pigs with a single‐nucleotide polymorphism in the regulatory γ3 subunit of AMPK possess increased glycogen content in white skeletal muscle (Milan et al. 2000). This polymorphism results in an amino acid substitution (R200Q) in domains involved in binding AMP or ATP; ultimately, the AMPKγ3R200Q mutation results in lack of AMP dependence and elevated basal activity of AMPK (Barnes et al. 2004). However, we have shown that phosphorylation of AMPK, as well as GLUT4 protein content, are blunted in AMPKγ3R200Q pig muscle that also has a mutation in ryanodine receptor 1 (RyR1R615C), or the calcium release channel (Park et al. 2009). Thus, we anticipated that blunted AMPK phosphorylation and GLUT4 protein content in AMPKγ3R200Q + RyR1R615C muscle may also blunt hexokinase‐mediated increases in G6P and glycogen synthase activity, and thus limit glycogen storage compared to AMPKγ3R200Q . Yet, the mitochondrial content and oxidative capacity of AMPKγ3R200Q + RyR1R615C is increased and similar to AMPKγ3R200Q muscle, suggesting that the mutation is sufficient to alter fuel storage and utilization (Scheffler et al. 2014). Thus, our objective was to use our AMPK and RyR1 pig model to understand how AMPKγ3R200Q and AMPK activation contribute to glycogen storage and metabolism in muscle.
Materials and Methods
Animals
Animals were bred and reared at the Purdue University Swine Center and the Virginia Tech Swine Center, and all procedures were carried out in accordance with the guidelines of each university's Institutional Animal Care and Use Committee. Pigs heterozygous at the RyR1 and AMPKγ3 loci were bred to generate all possible genotype combinations. Female and castrated male pigs were reared under standard conditions and fed ad libitum. At approximately 120 kg, animals were transported to the university's meat science center and harvested. Immediately after exsanguination, muscle samples (~5–10 g) were collected from the lumbar region of the longissimus muscle. Samples were immediately frozen in liquid nitrogen and stored at −80°C until further analysis.
Genotype determination
Genotypes were determined using polymerase chain reaction (PCR) restriction fragment length polymorphism technique. DNA was isolated from blood or tissue and used for PCR amplification. PCR products were digested with appropriate restriction enzyme overnight and fragments were separated on an agarose gel stained with ethidium bromide for visualization. For determination of AMPKγ3 genotype, the primers were (5′–3′) AAATGTGCAGACAAGGATCTC (forward) and CCCACGAAGCTCTGCTT (reverse). AMPKγ3 products were digested with restriction enzyme BsrBI. Pigs were evaluated for RyR1 genotype (Fujii et al. 1991) following the procedures outlined by O'Brien et al. (O'Brien et al. 1993). Those that were homozygous “normal” (wild type) at both RyR1 and AMPK loci were considered control while those pigs designated RyR1R615C were homozygous mutant. RyR1 heterozygotes were excluded because they tend to exhibit an intermediate phenotype. In contrast, AMPKγ3 mutation is dominant, so both homozygous mutant and heterozygotes were utilized (designated AMPKγ3R200Q). Finally, pigs denoted as AMPKγ3 + RyR1 mutants were either heterozygous or homozygous mutant at AMPKγ3 locus, and homozygous mutant at the RyR1 locus.
Glycogen synthase and phosphorylase activity
Enzyme activities were determined in muscle homogenates. Muscles were powdered in liquid nitrogen and homogenized on ice in buffer containing protease and phosphatase inhibitors (50 mmol/L Tris pH 7.8, 10 mmol/L EDTA, 10 mmol/L EGTA, 100 mmol/L NaF, 35 mg/mL tosyllysine chloromethyl ketone hydrochloride, 2 mmol/L benzamidine, 10 mg/mL leupeptin, and 0.5 mmol/L β‐mercapthoethanol). Glycogen synthase activity was quantified by the incorporation of [U‐14C]glucose from UDP[U‐14C]glucose into glycogen. The assay was conducted in the absence or presence of 10 mmol/L G6P (−G6P or +G6P), and activity ratio (−G6P/+G6P) was also determined. Glycogen phosphorylase activity was measured in the direction of glycogen synthesis from [U‐14C]glucose 1 phosphate (G1P) in the absence or presence of 3 mmol/L 5′AMP. Glycogen phosphorylase activity ratio was determined by dividing activity without AMP by activity with AMP (−AMP/+AMP).
Glycogen
Glycogen content was determined as previously described (Scheffler et al. 2013). Briefly, frozen muscle was powdered in liquid nitrogen and subjected to acid hydrolysis at 95°C. Another portion of frozen muscle was homogenized in perchloric acid, centrifuged, and the resulting supernatant was used for glucose, glucose‐6‐phosphate, and lactate analysis. Muscle glucose (free glucose and glucose resulting from glycogen hydrolysis), glucose‐6‐phosphate, and lactate concentrations were determined using enzyme analytical methods (Bergmeyer 1974). These metabolite concentrations were used to calculate total glycogen content (in glucose equivalents) using the formula: glycogen = glucose + G6P + (lactate/2).
Western blotting
Procedures for sample processing were determined based on preliminary testing. For detection of hexokinase 2 (HK2), UGP2, and GS, muscle samples were homogenized and sonicated in HEPES buffer (25 mmol/L HEPES, 1 mmol/L EDTA, 1 mmol/L benzamidine, 1 mmol/L 4‐(2‐aminoethyl)‐benzene + sulfonyl fluoride, 1 μmol/L aprotinin, 1 μmol/L leupeptin, 1 μmol/L pepstatin, pH 7.5) and retained as a total homogenate. Protein concentration was determined using BCA protein assay kit (Pierce, Rockford, IL) and samples were diluted to yield equal protein concentration and mixed with Laemmli buffer. Protein samples were separated using SDS‐PAGE and transferred to nitrocellulose membranes. Membranes were blocked in StartingBlock Blocking Buffer (Thermo Scientific). Blots were probed with primary antibody (UGP2 and HK2, Sigma, St. Louis, MO) and GS (Bioss; Woburn, MA) prepared in blocking buffer with 0.05% tween and washed in Tris‐buffered saline with 0.05% tween. Then, blots were incubated with the appropriate IRDye® 680 or 800 conjugated anti‐IgG antibody (LI‐COR® Biosciences, Lincoln, NE). Bands were visualized using Odyssey ® Infrared Imaging System (LI‐COR® Biosciences) and quantified using the manufacturer's software.
Statistical analysis
Data were analyzed using SAS‐JMP (Statistical Analysis Software; Cary, NC). The model was a two‐way ANOVA with main effects of AMPK (wild type or mutated) and RyR1 (wild type or mutated) genotypes and their interaction (AMPK*RyR1). Data are represented as LSM ± SE. If the interaction was significant, differences between genotype combinations were determined using Tukey's adjustment for multiple comparisons. P < 0.05 was considered significant.
Results
Previously, we reported that pigs with both AMPK and RyR1 mutations have blunted AMPK phosphorylation and decreased GLUT4 protein expression compared to pigs possessing only AMPK mutation (Park et al. 2009). Yet, despite these differences, AMPK + RyR1 mutant muscle exhibits some traits associated with AMPK activation, such as increased oxidative capacity (Scheffler et al. 2014). Therefore, our goal was to understand the contribution of AMPKγ3R200Q and AMPK activation to glycogen storage and metabolism in pig longissimus muscle.
Muscle glycogen (sum of equivalents), glycogen, G6P, and lactate
AMPK genotype influenced (P < 0.0001) glycogen content in longissimus muscle (Fig. 1A); In contrast, RyR1R615C decreased glycogen (P = 0.0003), consistent with increased lactate content (Fig. 1B, P < 0.001), possibly linked to increased phosphorylase activation (see below). In addition, both AMPK and RyR mutations influenced G6P levels (P < 0.0001 and P = 0.005, respectively). To better reflect total initial glycogen levels in muscle, we calculated the sum of major glycolytic metabolites (glucose equivalents, Fig. 1D). AMPKγ3R00Q muscle had approximately 70% greater glucose equivalents, regardless of RyR genotype. RyR1R615C genotype tended to decrease glycogen (P = 0.08), but the change was relatively small (~5%).
Figure 1.

Influence of AMPK and RyR1 mutations on glycogen (A); glucose 6‐phosphate (B); lactate (C); and glucose equivalents (D) in longissimus muscle. (all are expressed in μmol/g wet weight). Glucose equivalents was calculated as Gly + Glc + G6P + 1/2lactate; or ½ GP. Data are LSM ± SE. (n = 7–9 pigs per genotype).
Glycogen synthase activity and content
Glycogen synthase is considered to be the rate‐limiting enzyme for glycogen synthesis. Phosphorylation contributes to down‐regulation of GS activity, but inactivation can be completely overcome by increasing the allosteric activator, G6P. Thus, GS is typically assayed in the absence and presence of G6P, and the resulting ratio (−/+ G6P) is used as an indication of the phosphorylation state of GS. An AMPK × RyR1 genotype interaction (P = 0.04) influenced GS activation in the absence of G6P, as demonstrated by the lower GS activity (P < 0.05) in AMPKγ3R200Q muscle relative to all other genotypes (Fig. 2A). However, G6P increased (P < 0.0001) GS activity about twofold in muscle with AMPKγ3R200Q, regardless of the RyR1 mutation. For GS activity ratios, AMPKγ3R200Q muscle exhibited the lowest activity, whereas AMPKγ3R200Q + RyR1R615C was intermediate, and control and RyR1R615C were the highest. The low activity ratio of AMPKγ3R200Q muscle reflects both inactivation of GS by phosphorylation in the absence of G6P and increased activity in the presence of G6P, whereas the intermediate value of AMPKγ3R200Q + RyR1R615C muscle is due exclusively to increased activity with G6P.
Figure 2.
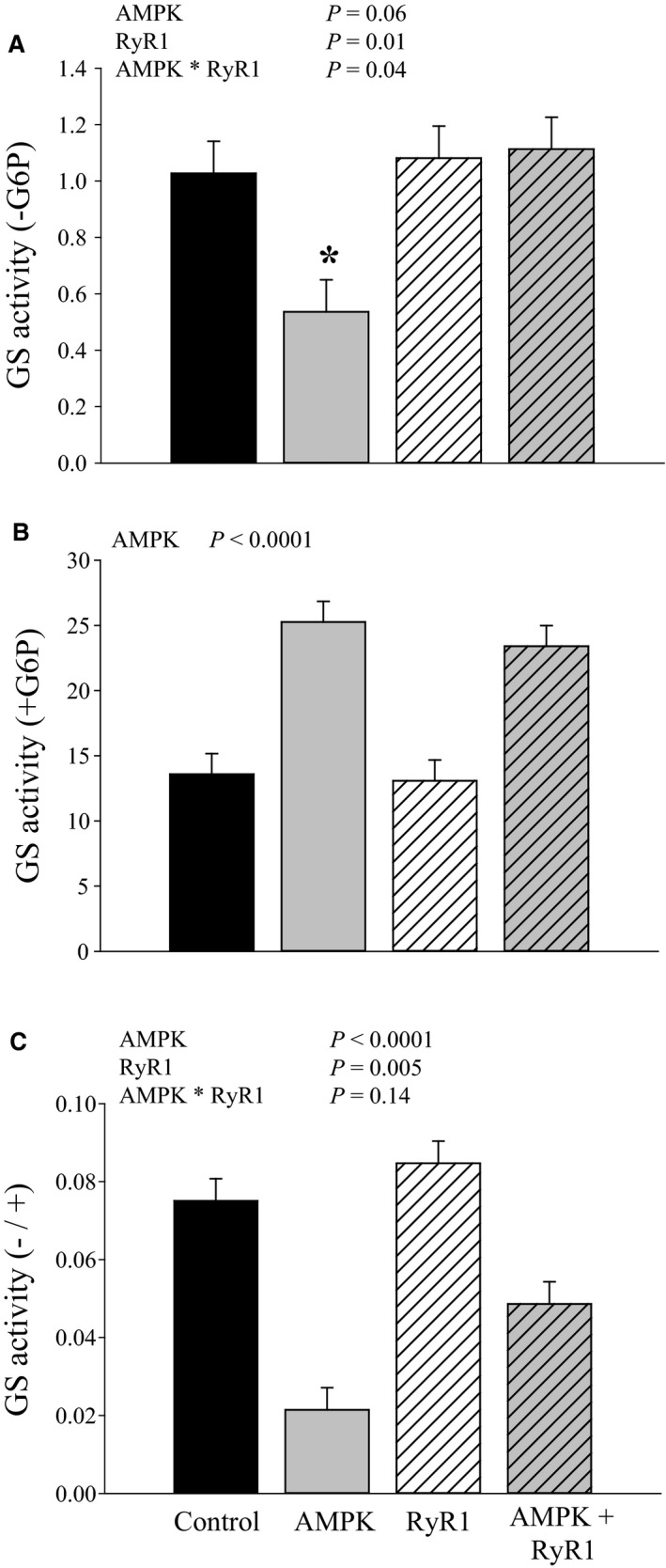
Glycogen synthase activity in AMPK and RyR genotypes. (A) Glycogen synthase activity in the absence of glucose 6‐phosphate. (B) Glycogen synthase activity in the presence of glucose 6‐phosphate. (C) Activity of glycogen synthase expressed as ratio (−/+ G6P). Data are LSM ± SE (n = 5 pigs per genotype). *indicates significantly different than other genotypes (P < 0.05).
Because total activity (+G6P) is often related to protein content (Manchester et al. 1996), we evaluated GS protein content by western blot. Unexpectedly, GS content was about 20% higher (P < 0.05) in wild‐type pigs relative to all other genotypes (Fig. 3).
Figure 3.
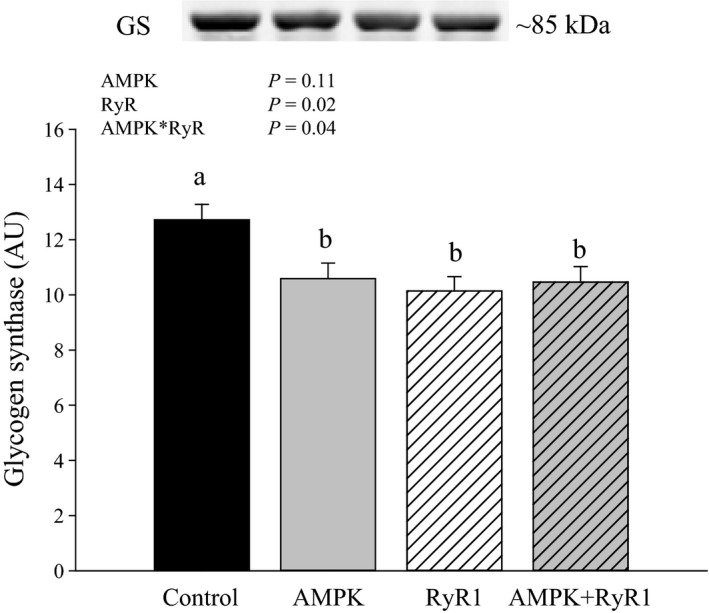
Glycogen synthase protein content of AMPK and RyR genotypes. Data are LSM ± SE (n = 6–7 pigs per genotype). a,bGenotypes not sharing a common superscript are significantly different (P < 0.05).
Enzymes in glycogen synthesis pathway
Although GS is considered the rate‐limiting enzyme, glycogen synthesis rate is dependent on glucose supply. The first step is the uptake of glucose into the cell via glucose transporters. Previously, we determined that GLUT4 protein content in muscle with AMPKγ3R200Q was enhanced relative to control, whereas GLUT4 in AMPKγ3R200Q + RyR1R615C muscle was similar to control (Park et al. 2009). Enzymes downstream of glucose entry are HK2, phosphoglucomutase, and UGP2. Hexokinase 2 protein content was not affected by genotype (Fig. 4). However, AMPKγ3R200Q increased (P < 0.0001) the content of UGP2 approximately threefold (Fig. 5).
Figure 4.
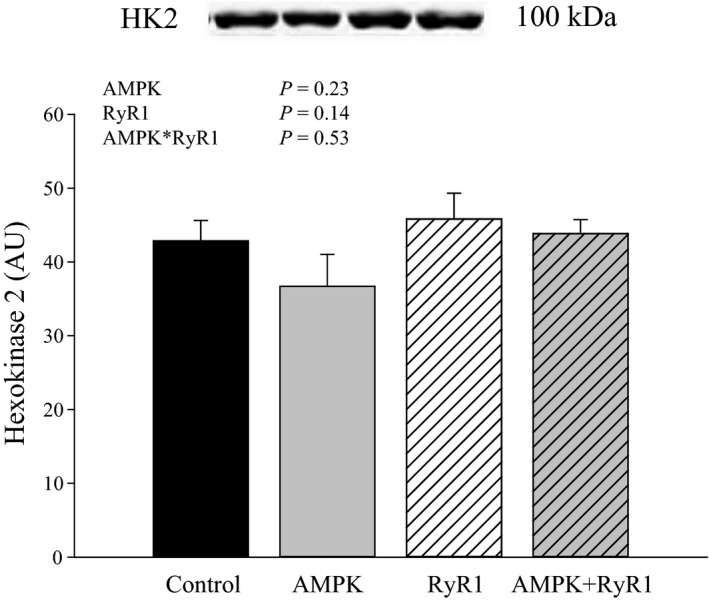
Hexokinase 2 protein content of AMPK and RyR genotypes. Data are LSM ± SE (n = 6–7 pigs per genotype).
Figure 5.
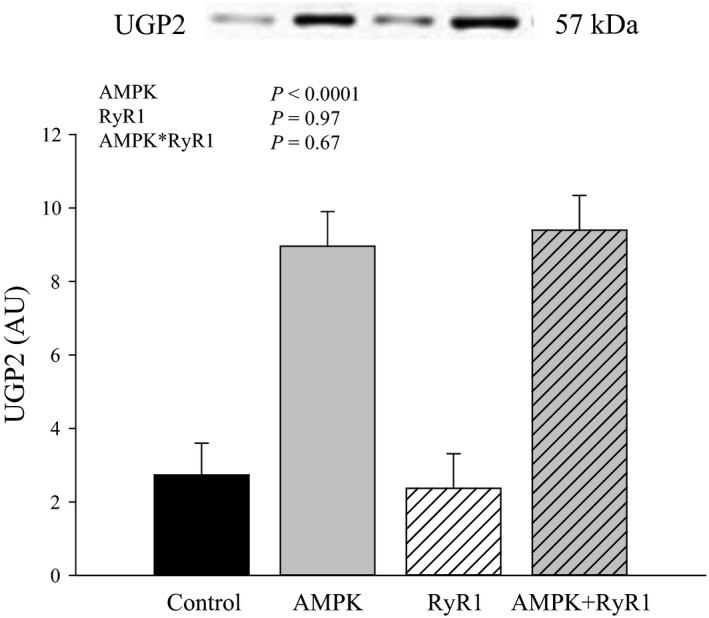
UDP‐glucose pyrophosphorylase 2 protein content of AMPK and RyR genotypes. Data are LSM ± SE (n = 6–7 pigs per genotype).
Glycogen phosphorylase
Since glycogen content is the net result of glycogen synthesis and degradation, we evaluated the activity of glycogen phosphorylase (GP), the rate‐limiting enzyme for glycogen breakdown. In the absence of AMP, the AMPK mutation contributed to increased GP activity (P = 0.002; Fig. 6), suggesting greater phosphorylation (activation) of GP in muscle with AMPKγ3R200Q. AMPK and RyR1 genotype also exerted significant (P < 0.05 and P = 0.03, respectively), but rather small effects on GP activity (+AMP). Although AMPKγ3R200Q was associated with increased activity, RyR1R615C decreased GP (+AMP) activity. Therefore, the GP activity ratio (−/+AMP) was lowest in control and highest in AMPKγ3R200Q + RyR1R615C muscle.
Figure 6.
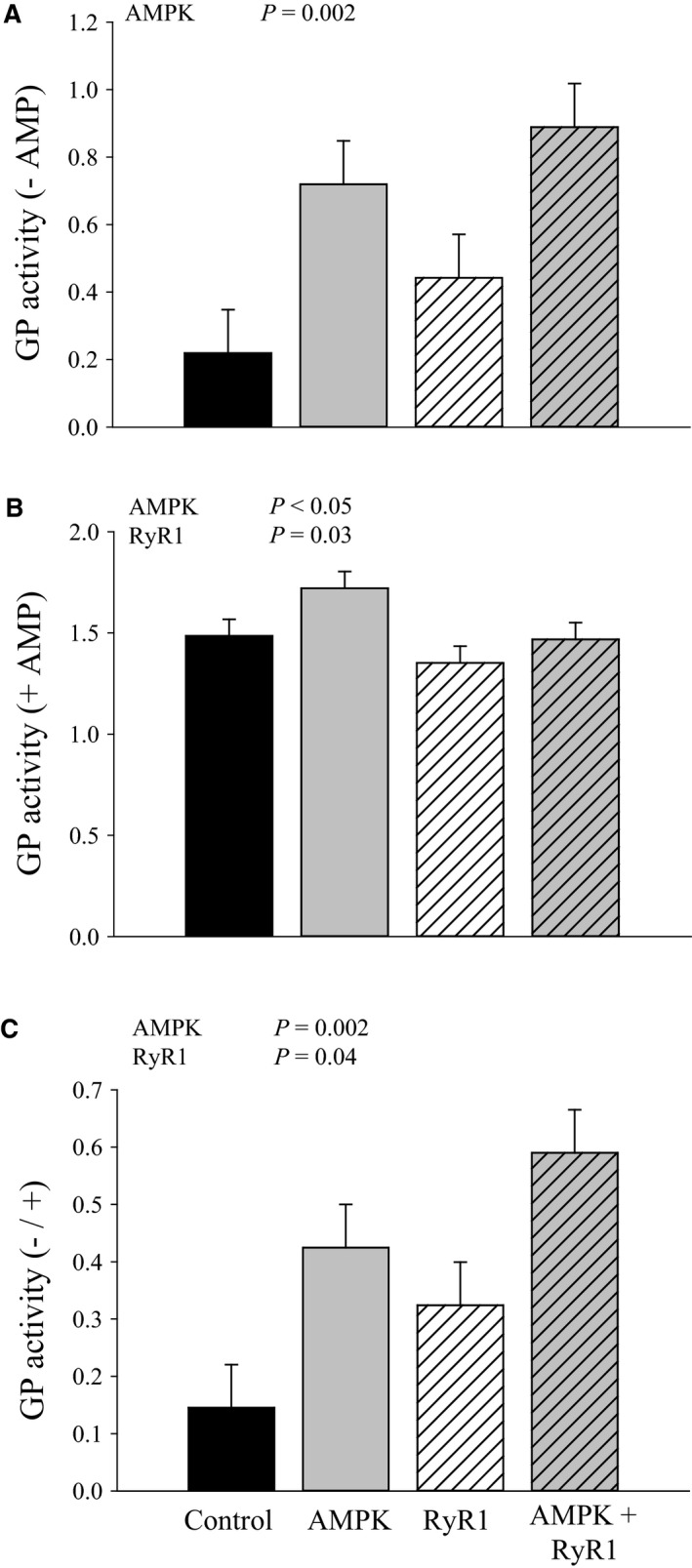
Glycogen phosphorylase activity in AMPK and RyR genotypes. (A) Glycogen phosphorylase activity in the absence of AMP. (B) Glycogen synthase activity in the presence of AMP. (C) Activity of glycogen phosphorylase expressed as ratio (−/+ AMP). Data are LSM ± SE (n = 5 pigs per genotype).
Discussion
AMPK signaling plays important roles in acute and adaptive regulation of muscle metabolism. Under acute energy stress, AMPK is well‐documented to protect cellular energy levels by down‐regulating anabolic pathways, such as glycogen synthesis. Yet, AMPK's role in limiting glycogen synthase activity is not consistent with the dramatic increase in muscle glycogen in pigs and mice with an AMPK‐activating mutation. Increases in muscle glycogen despite inactivation of GS are largely mediated by allosteric activation of GS by G6P (Bouskila et al. 2010; Hunter et al. 2011). Enhanced glucose transport contributes to elevated G6P, which overcomes the inhibitory effect of phosphorylation on glycogen synthase, and thus promotes glycogen storage (Hunter et al. 2011). However, we previously demonstrated that pigs with AMPKγ3R200Q exhibit enhanced AMPK activity and a concordant increase in GLUT4 content, whereas these effects are blunted in muscle with AMPKγ3R200Q + RyR1R615C (Park et al. 2009). In turn, we expected these differences in AMPK activity and GLUT4 content would be associated with altered glycogen storage capacity and metabolism.
Curiously, AMPKγ3R200Q + RyR1R615C muscle contains similar glycogen as AMPKγ3R200Q muscle, which is approximately 70% greater than wild‐type and RyR1R615C littermates. In muscle, control of glycogen synthesis is distributed primarily between glucose transport and glycogen synthase (Azpiazu et al. 2000). GLUT4 content and translocation contribute significantly to glucose uptake into the cell, and initially, these steps were considered rate‐limiting for glycogen synthesis (Ren et al. 1993). Consistent with this concept, transgenic overexpression of GLUT4 in muscle increases glucose transport activity and enhances glycogen storage, without affecting activation of glycogen synthase (Hansen et al. 1995). However, GLUT4 knockout mice also possess greater muscle glycogen in the fasted state despite markedly reduced glucose transport (Kim et al. 2005), suggesting that glucose transport is not always rate‐limiting for glycogen synthesis. Along these lines, RyR1R615C blunted AMPKγ3R200Q ‐induced increases in GLUT4 content (Park et al. 2009), but did not diminish glycogen storage. Although GLUT4 content significantly contributes to glucose transport, other mechanisms may permit glycogen storage in AMPKγ3R200Q + RyR1R615C muscle.
Glycogen synthase is considered the key regulatory enzyme in glycogen synthesis. Due to the complex regulation of GS, including multiple phosphorylation sites, GS activity is typically assayed in the absence and presence of G6P. GS activity (‐G6P) was reduced only in AMPKγ3R200Q muscle, which is consistent with increased AMPK phosphorylation and activity in AMPKγ3R200Q but not AMPKγ3R200Q + RyR1R615C muscle (Park et al. 2009), and with activated AMPK functioning as a glycogen synthase kinase (Jørgensen et al. 2004a). However, GS activity (+G6P) was nearly double in AMPK mutated muscle (AMPKγ3R200Q and AMPKγ3R200Q + RyR1R615C) compared to wild type and RyR1R615C. Activity with G6P, also referred to as total GS activity, is thought to parallel GS content. Increasing GS content is sufficient to augment muscle glycogen synthesis and glycogen level without altering glucose transport (Manchester et al. 1996). However, AMPKγ3R200Q did not increase GS protein content, as evidenced by western blot quantification. Similarly, Yu et al. (Yu et al. 2006) observed that muscles from AMPKγ3R225Q mice exhibit increased GS activity compared to wild type, but do not have greater GS protein content. The disconnect between GS activity in the presence of G6P and GS protein may reflect an as yet unappreciated, stable mechanism to modify the enzyme activity. It is also possible that other mechanisms may make GS more sensitive to G6P in AMPKγ3R200Q and AMPKγ3R200Q + RyR1R615C muscle.
Allosteric regulation of GS, and hence increased total GS activity, is the primary mechanism by which AMPK (Hunter et al. 2011) and insulin (Bouskila et al. 2010) promote glycogen accumulation. The increased flux through glucose transporters and HK2 contributes to elevated G6P and allosteric activation of GS. The AMPK and RyR mutations increased G6P levels in muscle. Elevated G6P in RyR1R615C muscle is likely due to increased glycolytic flux, which is also consistent with the elevated lactate levels observed in muscle; pigs with this mutation are susceptible to stress and rapid metabolism (MacLennan and Phillips 1993). On the other hand, increased G6P in AMPK pigs may be associated with greater glucose transport. Others have shown that chronic activation of AMPK increases GLUT4 and increased HK2 activity (Holmes et al. 1999; Granlund et al. 2011). In AMPKγ3R200Q + RyR1R615C muscle, neither GLUT4 (Park et al. 2009) nor HK2 content (this study) was different from control. We did not determine HK2 activity, although it is possible that AMPKγ3R200Q enhances G6P levels by increasing HK2 activity but not content.
High GS activity ratio (−/+ G6P) indicates that dephosphorylated (active) GS predominates. In mice genetically engineered to have altered expression of GS or its phosphatase, there is a positive relationship between GS activity ratio and glycogen content (Roach 2002). However, in our model, high activity ratio is a poor predictor of glycogen content because both control and RyR1R615C muscle exhibit the highest activity ratios, but contain less glycogen than AMPKγ3R200Q and AMPKγ3R200Q + RyR1R615C muscle. The inverse relationship between GS activity ratio and glycogen content is consistent with other reports incorporating genetic (Yu et al. 2006), exercise (Manabe et al. 2013), and fasting/refeeding (Jensen et al. 2006) approaches. Depending on the model, changes in the GS activity ratios may result from more adaptive changes, such as GS protein content, or by acute mechanisms involving regulation of glycogen synthase. For example, exercise training in rats promotes glycogen storage and coincides with a decrease in GS activity ratio, which is due almost exclusively to elevated total GS activity and increases in GS content (Manabe et al. 2013). On the other hand, in rats exposed to a fasting/refeeding approach, GS activity ratio was inversely related to glycogen content, but there were no significant differences in GS content (Jensen et al. 2006).
Elevated glycogen content in AMPKγ3R200Q and AMPKγ3R200Q + RyR1R615C muscle parallels total GS activity, suggesting that capacity to stimulate glycogen synthase plays a major role in glycogen storage in our model. In turn, glycogen synthase activity is also a function of the metabolites and enzymatic activity in the steps preceding glycogen synthesis. Traditionally, other enzymes in the glycogen synthesis pathway, including phosphoglucomutase and UGP2, are not considered rate‐limiting. Overexpression of UGP2 in muscle does not affect glycogen content, glucose tolerance, or insulin‐stimulated rates of glucose incorporation into glycogen (Reynolds et al. 2005). Yet, various genetic models used to manipulate glycogen content indicate a link between UGP2 expression and glycogen level. In models manipulating GS, glycogen content is inversely related to UGP2 expression; UGP2 mRNA is increased in GS knockout mice lacking muscle glycogen, but decreased in mice overexpressing glycogen synthase (Parker et al. 2006). On the other hand, in models that manipulate AMPK, glycogen is positively related to UGP2 expression. AMPKγ3R225Q mice exhibit greater UGP2 expression and deposit more muscle glycogen, whereas AMPKγ3 knockout mice have decreased UGP2 expression and possess less muscle glycogen. Increased UGP2 protein content in AMPKγ3R200Q pig muscle reported herein and elsewhere (Hedegaard et al. 2004) is consistent with gene expression patterns in AMPK mice. Interestingly, UGP2 levels are strongly upregulated in hearts of mice with a mutation in AMPKγ2, the predominant cardiac isoform (Luptak et al. 2007); and upregulation of UGP2 precedes glycogen accumulation. These authors suggested that UGP2 helps “pull” glucose toward glycogen in cardiac muscle with mutated γ2. The equilibrium of the phosphoglucomutase reaction (G6P ↔ glucose 1‐phosphate) favors G6P; however, increasing utilization of glucose 1‐phosphate by UGP2 would help promote flux toward glucose 1‐phosphate, and in turn, UDP‐glucose. Moreover, Manchester et al. (Manchester et al. 1996) showed that GS overexpression decreases UDP‐glucose concentrations and this lack of substrate likely restricts glycogen accumulation. Therefore, increasing UGP2 content in skeletal muscle may be an important means of enhancing flux to glycogen synthase and promoting glycogen accumulation in AMPKγ3R200Q and AMPKγ3R200Q + RyR1R615C muscle under some conditions.
Glycogen content in muscle is the net result of glycogen synthesis and degradation. Although certain glycogen storage diseases are caused by defects in glycogen degradation and/or glycolysis, mutations in the γ3 and γ2 subunits of AMPK have not been linked to compromised glycogenolysis (Estrade et al. 1994; Luptak et al. 2007). In fact, elevated glycogen in AMPKγ3R225Q mouse muscle enhances capacity for anaerobic exercise compared to wild type (Barnes et al. 2005). In our hands, AMPKγ3R200Q increases glycogen phosphorylase activity; Granlund (Granlund et al. 2011) also reported elevated GP activity in longissimus muscle of AMPKγ3R200Q pigs compared to wild type. Clearly, glycogen phosphorylase activity does not help explain elevated glycogen content in our pig model; however, it does indicate that glycogen turnover is increased. Manchester et al. (Manchester et al. 1996) speculated that glycogen content influences GS and GP levels, but other models are not consistent with this concept.
Altogether, we have shown that AMPKγ3R200Q and AMPKγ3R200Q + RyR1R615C muscle exhibit differences in GLUT4 as well as AMPK and GS activation; however, both clearly possess elevated total GS activity and high muscle glycogen. Our model demonstrates that the capacity for glycogen storage is more closely related to the AMPK mutation than activity. Additionally, we show that UGP2, previously considered a minor player in glycogen storage pathway, may be an important contributor to glycogen storage by promoting flux toward glycogen synthase.
Conflict of Interest
None declared.
Scheffler T. L., Park S., Roach P. J., Gerrard D. E.. Gain of function AMP‐activated protein kinase γ3 mutation (AMPK γ3R200Q) in pig muscle increases glycogen storage regardless of AMPK activation. Physiol Rep, 4 (11), 2016, e12802, doi: 10.14814/phy2.12802
Funding Information
No funding information provided.
References
- Azpiazu, I. , Manchester J., Skurat A. V., Roach P. J., and Lawrence J. C.. 2000. Control of glycogen synthesis is shared between glucose transport and glycogen synthase in skeletal muscle fibers. Am. J. Physiol. Endocrinol. Metab. 278:E234–E243. [DOI] [PubMed] [Google Scholar]
- Barnes, B. R. , Marklund S., Steiler T. L., Walter M., Hjälm G., Amarger V., et al. 2004. The 5′‐AMP‐activated protein kinase γ3 isoform has a key role in carbohydrate and lipid metabolism in glycolytic skeletal muscle. J. Biol. Chem. 279:38441–38447. [DOI] [PubMed] [Google Scholar]
- Barnes, B. R. , Glund S., Long Y. C., Hjälm G., Andersson L., and Zierath J. R.. 2005. 5′‐AMP‐activated protein kinase regulates skeletal muscle glycogen content and ergogenics. FASEB J. 19:773–779. [DOI] [PubMed] [Google Scholar]
- Bergmeyer, H. U. 1974. Methods of enzymatic analysis. Academic Press, New York. [Google Scholar]
- Bouskila, M. , Hunter R. W., Ibrahim A. F. M., Delattre L., Peggie M., Van Diepen J. A., et al. 2010. Allosteric regulation of glycogen synthase controls glycogen synthesis in muscle. Cell Metab. 12:456–466. [DOI] [PubMed] [Google Scholar]
- Costford, S. R. , Kavaslar N., Ahituv N., Chaudhry S. N., Schackwitz W. S., Dent R., et al. 2007. Gain‐of‐function R225W mutation in human AMPKγ3 causing increased glycogen and decreased triglyceride in skeletal muscle. PLoS ONE 2:1–9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Estrade, M. , Ayoub S., Talmant A., and Monin G.. 1994. Enzyme activities of glycogen metabolism and mitochondrial characteristics in muscles of RN‐ carrier pigs (Sus scrofa domesticus). Comp. Biochem. Physiol. 104B:295–301. [DOI] [PubMed] [Google Scholar]
- Fujii, J. , Otsu K., Zorzato F., De Leon S., Khanna V. K., Weiler J. E., et al. 1991. Identification of a mutation in porcine ryanodine receptor associated with malignant hyperthermia. Science 253:448–451. [DOI] [PubMed] [Google Scholar]
- Gowans, G. J. , Hawley S. A., Ross F. A., and Hardie D. G.. 2013. AMP is a true physiological regulator of amp‐activated protein kinase by both allosteric activation and enhancing net phosphorylation. Cell Metab. 18:556–566. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Granlund, A. , Jensen‐Waern M., and Essén‐Gustavsson B.. 2011. The influence of the PRKAG3 mutation on glycogen, enzyme activities and fibre types in different skeletal muscles of exercise trained pigs. Acta Vet. Scand. 53:20. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hansen, P. A. , Gulve E. A., Marshall B. A., Gao J., Pessin J. E., Holloszy J. O., et al. 1995. Skeletal muscle glucose transport and metabolism are enhanced in transgenic mice overexpressing the Glut4 glucose transporter. J. Biol. Chem. 270:1679–1684. [DOI] [PubMed] [Google Scholar]
- Hedegaard, J. , Horn P., Lametsch R., Møller H. S., Roepstorff P., Bendixen C., et al. 2004. UDP‐glucose pyrophosphorylase is upregulated in carriers of the porcine RN‐ mutation in the AMP‐activated protein kinase. Proteomics 4:2448–2454. [DOI] [PubMed] [Google Scholar]
- Holmes, B. F. , Kurth‐Kracek E. J., and Winder W. W.. 1999. Chronic activation of 5′‐AMP‐activated protein kinase increases GLUT‐4, hexokinase, and glycogen in muscle. J. Appl. Physiol. 87:1990–1995. [DOI] [PubMed] [Google Scholar]
- Hunter, R. W. , Treebak J. T., Wojtaszewski J. F. P., and Sakamoto K.. 2011. Molecular mechanism by which AMP‐activated protein kinase activation promotes glycogen accumulation in muscle. Diabetes 60:766–774. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jensen, J. , Jebens E., Brennesvik E. O., Ruzzin J., Soos M. A., Engebretsen E. M. L., et al. 2006. Muscle glycogen inharmoniously regulates glycogen synthase activity, glucose uptake, and proximal insulin signaling. Am. J. Physiol. Endocrinol. Metab. 290:E154–E162. [DOI] [PubMed] [Google Scholar]
- Jørgensen, S. B. , Nielsen J. N., Birk J. B., Olsen G. S., Viollet B., Andreelli F., et al. 2004a. The α2‐5′AMP‐activated protein kinase is a site 2 glycogen synthase in skeletal muscle and is responsive to glucose loading. Diabetes 53:3074–3081. [DOI] [PubMed] [Google Scholar]
- Jørgensen, S. B. , Viollet B., Andreelli F., Frøsig C., Birk J. B., Schjerling P., et al. 2004b. Knockout of the α2 but not α1, 5′‐AMP‐activated protein kinase isoform abolishes 5‐aminoimidazole‐4‐carboxamide‐1‐β‐4‐ribofuranoside‐ but not contraction‐induced glucose uptake in skeletal muscle. J. Biol. Chem. 279:1070–1079. [DOI] [PubMed] [Google Scholar]
- Kim, Y.‐B. , Peroni O. D., Aschenbach W. G., Minokoshi Y., Kotani K., Zisman A., et al. 2005. Muscle‐specific deletion of the Glut4 glucose transporter alters multiple regulatory steps in glycogen metabolism. Mol. Cell. Biol. 25:9713–9723. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kurth‐Kraczek, E. J. , Hirshman M. F., Goodyear L. J., and Winder W. W.. 1999. 5′ AMP‐activated protein kinase activation causes GLUT4 translocation in skeletal muscle. Diabetes 48:1667–1671. [DOI] [PubMed] [Google Scholar]
- Leick, L. , Fentz J., Bienso R. S., Knudsen J. G., Jeppesen J., Kiens B., et al. 2010. PGC‐1α is required for AICAR‐induced expression of GLUT4 and mitochondrial proteins in mouse skeletal muscle. Am. J. Physiol. Endocrinol. Metab. 299:E456–E465. [DOI] [PubMed] [Google Scholar]
- Luptak, I. , Shen M., He H., Hirshman M. F., Musi N., Goodyear L. J., et al. 2007. Aberrant activation of AMP‐activated protein kinase remodels metabolic network in favor of cardiac glycogen storage. J. Clin. Invest. 117:1432–1439. [DOI] [PMC free article] [PubMed] [Google Scholar]
- MacLennan, D. H. , and Phillips M. S.. 1993. Malignant hyperthermia. Science 256:789–794. [DOI] [PubMed] [Google Scholar]
- Mahlapuu, M. , Johansson C., Lindgren K., Hjalm G., Barnes B. R., Krook A., et al. 2004. Expression profiling of the γ‐subunit isoforms of AMP‐activated protein kinase suggests a major role for γ3 in white skeletal muscle. Am. J. Physiol. Endocrinol. Metab. 286:E194–E200. [DOI] [PubMed] [Google Scholar]
- Manabe, Y. , Gollisch K. S. C., Holton L., Kim Y. B., Brandauer J., Fujii N. L., et al. 2013. Exercise training‐induced adaptations associated with increases in skeletal muscle glycogen content. FEBS J. 280:916–926. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Manchester, J. , Skurat A. V., Roach P., Hauschka S. D., and Lawrence J. C.. 1996. Increased glycogen accumulation in transgenic mice overexpressing glycogen synthase in skeletal muscle. Proc. Natl Acad. Sci. 93:10707–10711. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Milan, D. , Jeon J.‐T., Looft C., Amarger V., Robic A., Thelander M., et al. 2000. A mutation in PRKAG3 associated with excess glycogen content in pig skeletal muscle. Science 288:1248–1251. [DOI] [PubMed] [Google Scholar]
- O'Brien, P. J. , Shen H., Cory C. R., and Zhang X.. 1993. Use of a DNA‐based test for the mutation associated with porcine stress syndrome (malignant hyperthermia) in 10,000 breeding swine. J. Am. Vet. Med. Assoc. 203:842–851. [PubMed] [Google Scholar]
- Park, S. , Scheffler T. L., Gunawan A. M., Shi H., Zeng C., Hannon K. M., et al. 2009. Chronic elevated calcium blocks AMPK‐induced GLUT‐4 expression in skeletal muscle. Am. J. Physiol. Cell Physiol. 296:C106–C115. [DOI] [PubMed] [Google Scholar]
- Parker, G. E. , Pederson B. A., Obayashi M., Schroeder J. M., Harris R. A., and Roach P. J.. 2006. Gene expression profiling of mice with genetically modified muscle glycogen content. Biochem. J. 395:137–145. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ren, J. M. , Marshall B. A., Gulve E. A., Gao J., Johnson D. W., Holloszy J. O., et al. 1993. Evidence from transgenic mice that glucose transport is rate‐limiting for glycogen deposition and glycolysis in skeletal muscle. J. Biol. Chem. 268:16113–16115. [PubMed] [Google Scholar]
- Reynolds, T. H. IV , Pak Y., Harris T. E., Manchester J., Barrett E. J., and Lawrence J. C.. 2005. Effects of insulin and transgenic overexpression of UDP‐glucose pyrophosphorylase on UDP‐glucose and glycogen accumulation in skeletal muscle fibers. J. Biol. Chem. 280:5510–5515. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Roach, P. 2002. Glycogen and its metabolism. Curr. Mol. Med. 2:101–120. [DOI] [PubMed] [Google Scholar]
- Roach, P. J. , Depaoli‐Roach A. A., Hurley T. D., and Tagliabracci V. S.. 2012. Glycogen and its metabolism: some new developments and old themes. Biochem. J. 441:763–787. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Scheffler, T. L. , Scheffler J. M., Kasten S. C., Sosnicki A. A., and Gerrard D. E.. 2013. High glycolytic potential does not predict low ultimate pH in pork. Meat Sci. 95:85–91. [DOI] [PubMed] [Google Scholar]
- Scheffler, T. L. , Scheffler J. M., Park S., Kasten S. C., Wu Y., McMillan R. P., et al. 2014. Fiber hypertrophy and increased oxidative capacity can occur simultaneously in pig glycolytic skeletal muscle. Am. J. Physiol. Cell Physiol. 306:C354–C363. [DOI] [PubMed] [Google Scholar]
- Stein, S. C. , Woods A., Jones N. A., Davison M. D., and Carling D.. 2000. The regulation of AMP‐activated protein kinase by phosphorylation. Biochem. J. 345(Pt 3):437–443. [PMC free article] [PubMed] [Google Scholar]
- Suter, M. , Riek U., Tuerk R., Schlattner U., Wallimann T., and Neumann D.. 2006. Dissecting the role of 5′‐AMP for allosteric stimulation, activation, and deactivation of AMP‐activated protein kinase. J. Biol. Chem. 281:32207–32216. [DOI] [PubMed] [Google Scholar]
- Yu, H. , Hirshman M. F., Fujii N., Pomerleau J. M., Peter L. E., and Goodyear L. J.. 2006. Muscle‐specific overexpression of wild type and R225Q mutant AMP‐activated protein kinase γ3‐subunit differentially regulates glycogen accumulation. Am. J. Physiol. Endocrinol. Metab. 291:E557–E565. [DOI] [PubMed] [Google Scholar]