

Abstract

The disulfide-based cyclic monomer, 3-methylidene-1,9-dioxa-5,12,13-trithiacyclopentadecane-2,8-dione (MTC), is statistically copolymerized with 2-hydroxypropyl methacrylate to form a range of diblock copolymer nano-objects via reversible addition–fragmentation chain transfer (RAFT) polymerization. Poly(glycerol monomethacrylate) (PGMA) is employed as the hydrophilic stabilizer block in this aqueous polymerization-induced self-assembly (PISA) formulation, which affords pure spheres, worms or vesicles depending on the target degree of polymerization for the core-forming block. When relatively low levels (<1 mol %) of MTC are incorporated, high monomer conversions (>99%) are achieved and high blocking efficiencies are observed, as judged by 1H NMR spectroscopy and gel permeation chromatography (GPC), respectively. However, the side reactions that are known to occur when cyclic allylic sulfides such as MTC are statistically copolymerized with methacrylic comonomers lead to relatively broad molecular weight distributions. Nevertheless, the worm-like nanoparticles obtained via PISA can be successfully transformed into spherical nanoparticles by addition of excess tris(2-carboxyethyl)phosphine (TCEP) at pH 8–9. Surprisingly, DLS and TEM studies indicate that the time scale needed for this order–order transition is significantly longer than that required for cleavage of the disulfide bonds located in the worm cores indicated by GPC analysis. This reductive degradation pathway may enable the use of these chemically degradable nanoparticles in biomedical applications, such as drug delivery systems and responsive biomaterials.
Introduction
Degradable polymers have been the subject of significant and sustained research, not least for their potential in the design of therapeutic devices such as temporary prostheses, scaffolds for tissue engineering and controlled drug delivery vehicles.1−4 This has resulted in the development of a diverse range of materials based on either naturally occurring or entirely synthetic feedstocks.5 The extent and rate of degradability of these materials is primarily determined by the type and number of cleavable chemical bonds that are incorporated within the polymer chains, as well as their precise location.6 Anhydride, ester, amide, and disulfide bonds have been successfully employed, enabling chemical degradation via exposure to either photo, thermal, mechanical, or chemical stimuli.1,2,5−9 To ensure that sufficiently high levels of degradability can be achieved, such labile bonds are incorporated into the polymer backbone using techniques such as step polymerization10−17 or ring-opening polymerization (ROP).15−23 Although significant progress has been made, conferring chemical degradability on vinyl polymers undoubtedly remains a significant technical challenge.24
The development of reversible deactivation radical polymerization (RDRP) techniques such as nitroxide-mediated polymerization (NMP),25 atom transfer radical polymerization (ATRP),26,27 and reversible addition–fragmentation chain transfer (RAFT) polymerization28 has led to the design of many new controlled-structure copolymers based on vinyl monomers.29,30 Moreover, formulations based on polymerization-induced self-assembly (PISA)31−33 enable the efficient synthesis of a wide range of nano-objects at high solids (up to 50% w/w)34 in either polar or non-polar solvents.34−37 A prototypical PISA formulation involves the RAFT aqueous dispersion polymerization of 2-hydroxypropyl methacrylate (HPMA) using a poly(glycerol monomethacrylate) (PGMA) macromolecular chain transfer agent (macro-CTA).38,39 Under certain conditions, PGMA–PHPMA diblock copolymer chains self-assemble in situ to produce worm-like micelles.37 These highly anisotropic nanoparticles form soft, free-standing aqueous gels at 20 °C. Moreover, a morphological transformation from worms to spheres can be induced on cooling to 5 °C.40,41 This order–order transition is fully reversible and enables convenient sterilization of such worm gels via cold ultrafiltration.38 Such worm gels are currently being evaluated for in vitro applications such as a long-term storage medium for human stem cells42 and also for the cryopreservation of red blood cells.43 Nevertheless, the nondegradability of the methacrylic backbone is a major barrier for potential in vivo biomedical applications.
Several methodologies have been explored to circumvent this important problem. For example, branched degradable vinyl copolymers have been designed using disulfide,44,45 acetal,46 or silyl ether47 comonomers. Alternatively, a central degradable unit can be introduced via ATRP by using a disulfide-based bifunctional initiator.48,49 Such approaches have been recently reviewed by Rikkou and Patrickios, who have focused on copolymers prepared via either living or pseudoliving techniques.50 Another strategy involves coupling telechelic polymers (typically via postpolymerization oxidation of thiols) to produce degradable materials.51−55 Alternatively, ring-opening polymerization (ROP) of a cyclic monomer (containing a cleavable functionality such as an ester) has been combined with vinyl polymerization. For example, Frick and co-workers56 coupled the ROP of lactide with the anionic polymerization of isoprene to produce various ABA triblock copolymers. Several groups have prepared bespoke RAFT CTAs that enable both ROP of lactide and controlled vinyl polymerization.57−59 Similar dual-functional nitroxides and ATRP initiators have also been utilized in this context.60
Mecerreyes et al. designed an acrylic monomer containing caprolactone functionality, making it suitable for both ROP and ATRP.61 This was subsequently polymerized to form cleavable branched structures. Li and Armes prepared highly branched methacrylic copolymers using a disulfide dimethacrylate comonomer that enabled the primary chains within the branched structure to be characterized via postpolymerization cleavage.62 A similar approach was used by Armes and co-workers to design chemically degradable poly(2-hydroxyethyl methacrylate)-based fibers.63
More recently, there has been growing interest in the radical ring-opening polymerization (RROP)64 of cyclic ketene acetals65−68 and cyclic allylic sulfides.69−71 Such monomers can be copolymerized with vinyl monomers to afford chemically degradable vinyl copolymers.72,73 There are a number of literature reports utilizing RDRP techniques to (co)polymerize cyclic ketene acetals, including RAFT,74,75 NMP,76,77 and ATRP.78,79 However, as far as we are aware, there is currently only a single literature example describing the RDRP of cyclic allylic sulfides.80
In the present study, a small amount of a cyclic allylic sulfide, 3-methylidene-1,9-dioxa-5,12,13-trithiacyclopentadecane-2,8-dione (MTC),80 is statistically copolymerized with 2-hydroxypropyl methacrylate using an aqueous PISA formulation.31,38,40 The MTC comonomer introduces a disulfide bond into the methacrylic backbone of the hydrophobic component of an amphiphilic diblock copolymer, which has been recently shown to exhibit excellent biocompatibility for various cell types, including human stem cells and red blood cells.40,42,43 Subsequent reductive cleavage under appropriate conditions81 leads to a significantly shorter hydrophobic block, which is sufficient to produce a change in the morphology of the diblock copolymer nano-objects produced during PISA. In principle, such an order–order transition may be sufficient to allow a renal clearance mechanism, which suggests the possibility of in vivo biomedical applications.
Experimental Section
Materials
2-Hydroxypropyl methacrylate (HPMA, 97%) and 4,4′-azobis(4-cyanopentanoic acid) (ACVA; V-501; 99%) were purchased from Alfa Aesar (Heysham, U.K.) and used as received. Glycerol monomethacrylate (GMA, 99.8%) was kindly donated by GEO Specialty Chemicals (Hythe, U.K.) and used without further purification. 3-Methylidene-1,9-dioxa-5,12,13-trithiacyclopentadecane-2,8-dione (MTC) was synthesized as described elsewhere.80 2-Cyano-2-propyl benzodithioate (CPDB) was purchased from Strem Chemicals (Cambridge, U.K.) and tris(2-carboxyethyl)phosphine (TCEP hydrochloride, 99%) was purchased from Amresco (Solon, Ohio, U.S.A.). Deuterated methanol (CD3OD) was purchased from Goss Scientific (Nantwich, U.K.). Sodium hydroxide pellets were purchased from VWR (Lutterworth, U.K.). Deionized water was used for all dispersion polymerizations. All other solvents were of HPLC quality, purchased from Fisher Scientific (Loughborough, U.K.) and used as received.
Synthesis and Purification of PGMA56 Macro-CTA
A typical protocol for the synthesis of PGMA56 macro-CTA is as follows. To a round-bottomed flask containing CPDB RAFT agent (75% purity, 0.020 mol, 6.03 g), GMA monomer (1.268 mol, 203.0 g) and ethanol (3.38 mol, 156.0 g) was added to target a mean degree of polymerization (DP) of 63. To this, ACVA initiator (4.07 mmol, 1.14 g; CTA/ACVA molar ratio = 5.0) was added, and the resulting pink solution was sparged with N2 for 20 min before the sealed flask was immersed into an oil bath set at 70 °C. After 140 min (69% conversion as judged by 1H NMR), the GMA polymerization was quenched by immersing the flask in an ice bath and exposing the reaction solution to air. The crude polymer solution was then precipitated into a 10-fold excess of DCM (twice) and then washed three times with DCM before being dissolved in water and lyophilized overnight. 1H NMR analysis indicated a mean DP of 56 for this PGMA macro-CTA. Taking into account the target DP of 63 and the GMA conversion of 69%, this suggests a CTA efficiency of 76%. DMF GPC analysis (refractive index detector; vs a series of poly(methyl methacrylate) calibration standards) indicated Mn and Mw/Mn values of 14300 g mol–1 and 1.14, respectively.
RAFT Synthesis of PGMA56-P(HPMA180-stat-MTC0.9) Diblock Copolymer
A typical protocol for the synthesis of PGMA56-P(HPMA180-stat-MTC0.9) statistical diblock copolymer is as follows: MTC monomer (0.0036 g, 0.011 mmol) was added to a glass vial or round bottomed flask, followed by HPMA monomer (0.3388 g, 2.35 mmol), PGMA56 macro-CTA (0.12 g, 0.013 mmol), and water (4.17 g, to produce 10% w/w total solids). ACVA was then added (0.9 mg, 0.003 mmol, macro-CTA/ACVA molar ratio = 4.0), and the solution was sparged with N2 for 30 min. The flask was sealed and immersed in an oil bath set at 70 °C and stirred for 16 h to ensure complete monomer conversion. The polymerization was quenched by exposure to air and cooling the flask to 20 °C.
Addition of Reducing Agent to PGMA56-P(HPMA180-stat-MTC0.9) Diblock Copolymer
The protocol is as follows: To PGMA56-P(HPMA180-stat-MTC0.9) statistical diblock copolymer (3.00 g of 10% w/w dispersion, 0.0076 mmol of MTC) TCEP reducing agent (0.011 g, 0.038 mmol, TCEP/MTC molar ratio = 5) was added, followed by 1 M NaOH solution to adjust the final pH to between 8 and 9. The reaction solution was agitated on a roller at 20 °C and sampled as required.
RAFT Synthesis of PGMA56-PHPMA180 Diblock Copolymer
A typical protocol for the synthesis of PGMA56-PHPMA180 statistical diblock copolymer is as follows: PGMA56 macro-CTA (0.1124 g, 0.012 mmol), HPMA monomer (0.3122 g, 2.15 mmol), and water (3.79 g, to produce 10% w/w total solids) were added to a glass vial or round bottomed flask. ACVA was then added (0.8 mg, 0.003 mmol, macro-CTA/ACVA molar ratio = 4.0), and the solution was sparged with N2 for 30 min. The flask was sealed and immersed in an oil bath set at 70 °C and stirred for 16 h to ensure complete monomer conversion. The polymerization was quenched by exposure to air and cooling the flask to 20 °C.
Copolymer Characterization
1H NMR Spectroscopy
All NMR spectra were recorded using a 400 MHz Bruker Avance-400 spectrometer (64 scans per sample) in CD3OD or CDCl3.
Gel Permeation Chromatography (GPC)
Polymer molecular weights and polydispersities were determined using a DMF GPC instrument operating at 60 °C that comprised two Polymer Laboratories PL gel 5 μm Mixed C columns and one PL gel 5 μm guard column connected in series to an Agilent Technologies 1260 Infinity multidetector suite (refractive index detector only) and an Agilent Technologies 1260 ISO pump fitted with a 1260 ALS autosampler. The GPC eluent was HPLC-grade DMF containing 10 mM LiBr and was filtered prior to use. The flow rate used was 1.0 mL min–1 and DMSO was used as a flow-rate marker. Calibration was conducted using a series of 10 near-monodisperse poly(methyl methacrylate) standards (Mn = 625–618000 g mol–1, K = 2.094 × 10–3, α = 0.642). Chromatograms were analyzed using Agilent Technologies GPC/SEC software version 1.2.
Transmission Electron Microscopy (TEM)
Reaction mixtures were diluted at 20 °C to generate 0.60% w/w dispersions. Copper TEM grids (Agar Scientific, U.K.) were surface-coated in-house to yield a thin film of amorphous carbon. The grids were then plasma glow-discharged for 40 s to create a hydrophilic surface. Each aqueous diblock copolymer dispersion (11 μL) was placed onto a freshly glow-discharged grid for 1 min and then blotted with filter paper to remove excess solution. To stain the deposited nanoparticles, a 0.75% w/w aqueous solution of uranyl formate (11 μL) was placed via micropipette on the sample-loaded grid for 15 s and then carefully blotted to remove excess stain. Each grid was then carefully dried using a vacuum hose. Imaging was performed at 100 kV using a Phillips CM100 instrument equipped with a Gatan 1 k CCD camera.
Dynamic Light Scattering (DLS)
Intensity-average hydrodynamic diameters of the dispersions were obtained by DLS using a Malvern Zetasizer NanoZS instrument. Dilute aqueous dispersions (0.25% w/w) were analyzed using disposable cuvettes, and all data were averaged over three consecutive runs.
Results and Discussion
Paulusse et al. have reported the statistical copolymerization of MTC with methyl methacrylate, 2-hydroxyethyl methacrylate, or 2-dimethylaminoethyl methacrylate using RAFT solution polymerization in either chlorobenzene or dimethylformamide.80 Higher levels of MTC in the comonomer feed led to a gradual loss of control over the molecular weight distribution and also produced lower comonomer conversions. Bearing this prior study in mind, a small amount of MTC was statistically copolymerized with HPMA to introduce chemically degradable disulfide units into the methacrylic backbone of the predominantly PHPMA core-forming block using a RAFT aqueous dispersion polymerization formulation (see Figure 1).
Figure 1.

Synthesis of PGMA56-P(HPMAy-stat-MTCz) diblock copolymer nano-objects via RAFT statistical copolymerization of HPMA with MTC in aqueous solution at 70 °C. As the overall target DP (y + z) of the P(HPMA-stat-MTC) core-forming block is increased, polymerization-induced self-assembly (PISA) occurs to produce either spherical, worm-like, or vesicular nano-objects with cleavable disulfide bonds being located within the hydrophobic P(HPMA-stat-MTC) chains.
First, a PGMA56 macro-CTA was prepared via RAFT solution polymerization in ethanol, as previously described.82 Then the statistical copolymerization of MTC with HPMA was conducted using this macro-CTA, with 2 mol % MTC being utilized relative to the HPMA target DP of 180. The resulting diblock copolymer had a relatively broad molecular weight distribution (Mw/Mn = 1.52, as judged by DMF GPC, see Figure S1) compared to similar PGMA56-PHPMAy copolymers prepared in the absence of any MTC.39,83 Moreover, the overall comonomer conversion was only 84% after 16 h at 70 °C, as judged by 1H NMR spectroscopy. When the MTC content was reduced to 1 mol %, the final conversion exceeded 98%, but the dispersity was only slightly reduced (Mw/Mn = 1.44). Such relatively high Mw/Mn values may explain why only mixed phases (e.g., spheres plus worms or vesicles plus worms) were observed when these dispersions were analyzed using TEM (see Figure S2). Fortunately, further lowering the MTC content to 0.50 mol % led to slightly lower dispersities (Mw/Mn < 1.40), and more than 99% comonomer conversion was achieved in all cases. Moreover, relatively pure spherical, worm-like, or vesicular morphologies could be obtained (see Figure S3), although rather higher core-forming block DPs were required to produce worm and vesicle phases compared to that needed for similar PGMA–PHPMA diblock copolymers prepared in the absence of MTC (see Figure S4). Interestingly, the DP range over which the worm phase is observed appears to be significantly broader for PISA syntheses conducted in the presence of MTC.39 Both observations are most likely related to the higher copolymer dispersities that arise from side reactions (e.g., vinyl addition71) that are known to occur when cyclic allylic sulfides are statistically copolymerized with methacrylic monomers via RAFT.80 Nonetheless, in addition to the expected dependence on the degree of polymerization (DP) of the PHPMA block and the concentration at which the HPMA polymerization is conducted, it is clear that the copolymer morphology is also sensitive to the proportion of the more hydrophobic MTC comonomer.39 After conducting some scouting experiments, we targeted P(HPMA170-stat-MTC0.85), which formed a predominantly worm-like morphology. In principle, cleaving the disulfide bonds located in the methacrylic backbone should significantly reduce the core-forming block DP and hence drive a worm-to-sphere transition. Comonomer conversions typically reached more than 99% within approximately 3 h at 70 °C. However, the copolymerization was allowed to proceed for a further 13 h to ensure the highest possible conversion, since this did not appear to be detrimental to the overall level of control (see Figure S5). The relative copolymerization rates for HPMA and MTC indicated that the latter comonomer initially reacted slightly faster than HPMA but overall was incorporated more or less statistically into the core-forming block (see 1H NMR spectra in Figure 2).
Figure 2.
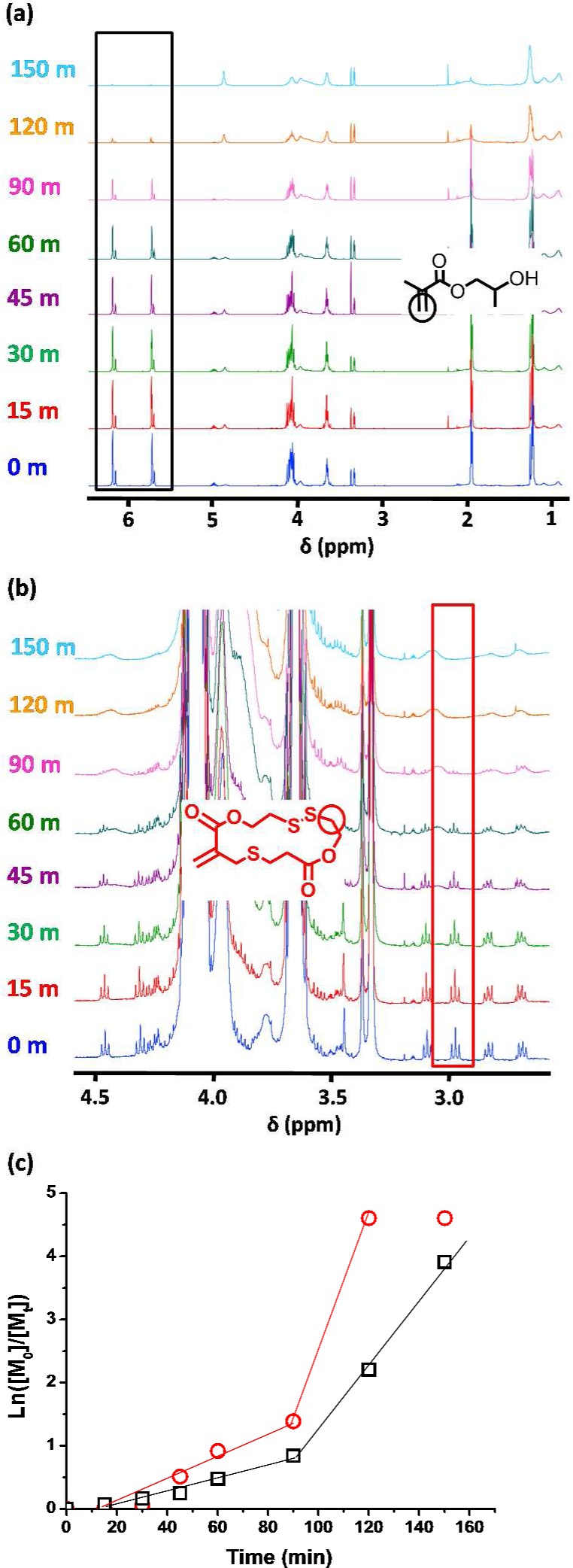
(a, b) 1H NMR spectra obtained for a PGMA56-P(HPMA170-stat-MTC0.85) copolymer synthesis sampled at various time periods during RAFT statistical copolymerization of HPMA with MTC at 70 °C and 10% w/w solids in aqueous solution, indicating the signals utilized to produce the graph shown in (c). (c) Rate of consumption (as judged by 1H NMR) of HPMA (black squares) and MTC (red circles) for a PGMA56-P(HPMA170-stat-MTC0.85) copolymer sampled at various time periods during RAFT polymerization at 70 °C and 10% w/w in water.
Addition of tris(2-carboxyethyl)phosphine (TCEP; TCEP/MTC molar ratio = 5.0) to a stirred 10% w/w aqueous dispersion of PGMA56-P(HPMA170-stat-MTC0.85) for 16 h at pH 8–9 led to a marked reduction in Mn from 40000 to 27900 g mol–1 with a concomitant increase in Mw/Mn from 1.36 to 1.51, see Figure 3a. This reduction in copolymer Mn is consistent with the relatively low level of MTC that is (approximately) statistically incorporated into the hydrophobic core-forming block. In contrast, no molecular weight reduction was observed in a control experiment whereby a PGMA56-PHPMA180 copolymer prepared in the absence of any MTC was treated with TCEP under the same conditions (see Figure S6).
Figure 3.

(a) DMF GPC curves recorded for PGMA56-P(HPMA170-stat-MTC0.85) [denoted as G56-(H170-M0.85) for brevity] copolymers prepared via RAFT copolymerization of MTC with HPMA using a PGMA56 macro-CTA at 70 °C before (black curve) and after (red curve) exposure to TCEP (TCEP/MTC molar ratio = 5.0) at pH 8–9 for 8 days at 20 °C. (b) Evolution of intensity-average particle size distributions (determined for 0.20% w/w aqueous copolymer dispersions) before and after a 10% w/w aqueous dispersion of PGMA56-P(HPMA170-stat-MTC0.85) was exposed to TCEP (TCEP/MTC molar ratio = 5.0) at pH 8–9 for 1, 5, or 8 days at 20 °C. The corresponding DLS polydispersities (PDI) are indicated in brackets. (c) DLS data plotted vs time (days after TCEP addition), demonstrating the observed reduction in the intensity-average diameter and count rate.
This reduction in molecular weight was also sufficient to produce an irreversible worm-to-sphere transition (see DLS and TEM data shown in Figures 3b,c and 4, respectively). The final morphology is in good agreement with the relatively small spherical particles obtained for a PGMA56-PHPMA85 copolymer prepared in the absence of MTC (see Figure S7, DLS diameter = 26 nm and PDI = 0.10). This reference copolymer was selected because its core-forming block DP is approximately half that of the original PGMA56-P(HPMA170-stat-MTC0.85). Interestingly, the worm-to-sphere transformation is relatively slow at 20 °C, requiring 5–8 days at pH 8–9 for the initial “sphere-equivalent” particle diameter of 136 nm for the worms to be reduced to a final pseudo-spherical particle diameter of 35 nm. Significant reductions in count rate (from 74300 to 27300 kcps) and DLS polydispersity (from 0.30 to 0.14) were also observed for this morphological transition, as expected (see Figure S3c). It is not yet clear why the experimental time scales for disulfide bond cleavage and the corresponding change in morphology are so different, but it is perhaps worth emphasizing that this phenomenon proved to be reproducible. Relatively fast reductive cleavage of the disulfide bonds was anticipated: PHPMA chains are known to be highly plasticized with water in similar PGMA–PHPMA worms, which should enable rapid ingress of the TCEP reagent.40 However, the change in copolymer morphology from worms to (mainly) spheres as a result of the reduction in the packing parameter is remarkably slow. This may indicate some degree of recombination of free thiols to form disulfides within the worms. Alternatively, the relatively high dispersity of the copolymer chains may play a role: the statistical distribution of the MTC residues along the core-forming block (see copolymerization kinetic data in Figure 2) means that there is minimal change in the packing parameter for a significant fraction of the copolymer chains.84 In fact, assuming a Poisson distribution it is estimated that up to 43% of the copolymer chains may not contain any MTC comonomer. Nevertheless, TEM studies in Figure 4 confirm the DLS data shown in Figure 3: the original worms are indeed eventually converted into spheres (plus some dimers and trimers) on addition of excess TCEP at pH 8–9.
Figure 4.

TEM images obtained for a 0.20% w/w aqueous dispersion of PGMA56-P(HPMA170- stat-MTC0.85) before and after exposure to TCEP (TCEP/MTC molar ratio = 5.0) at pH 8–9 for 8 days at 20 °C. Cartoon representation of the worm-to-sphere transition observed for a 10% w/w aqueous dispersion of PGMA56-P(HPMA170-stat-MTC0.85) worms on exposure to excess TCEP (TCEP/MTC molar ratio = 5.0) at pH 8–9 for 8 days at 20 °C and the corresponding reduction in the packing parameter.
Conclusions
MTC has been statistically copolymerized with HPMA using an aqueous PISA formulation to afford a series of chemically degradable diblock copolymer nano-objects. RAFT control was gradually lost and the overall comonomer conversion was reduced when using higher levels of MTC comonomer and only a relatively low level of MTC (<1 mol %) could be tolerated if relatively well-defined spherical, worm-like or vesicular phases were required. Despite these synthetic limitations, using MTC as a comonomer enabled disulfide bonds to be incorporated into the methacrylic backbone of the hydrophobic core-forming block. In the case of the worm morphology, subsequent cleavage of these disulfide bonds using excess TCEP resulted in a sufficient reduction in Mn to induce an irreversible worm-to-sphere transition, which was confirmed using TEM and DLS. In principle, this chemical degradation pathway could produce spherical nanoparticles that are sufficiently small to allow renal clearance from the body, boding well for the use of these diblock copolymer nano-objects in biomedical applications.
Acknowledgments
EPSRC is thanked for postdoctoral support of LPDR (EP/K030949/1). S.P.A. thanks the European Research Council for a five-year Advanced Investigator Grant (PISA 320372). We thank the four reviewers of this manuscript for their helpful comments.
Supporting Information Available
The Supporting Information is available free of charge on the ACS Publications website at DOI: 10.1021/acs.biomac.6b00540.
GPC traces for PGMA56-P(HPMA180-stat-MTCz) containing 0.5, 1.0, or 2.0 mol % MTC, TEM images of PGMAx-P(HPMAy-stat-MTCz) with 0.5 or 1.0 mol % MTC, TEM images, and GPC data for PGMA56-P(HPMA180-stat-MTC0.9) and PGMA56-PHPMA180. GPC traces for PGMA56-P(HPMA180-stat-MTC0.9) and PGMA56-PHPMA180 before and after addition of excess TCEP, TEM, and GPC data for PGMA56-PHPMA85 and 1H and 13C NMR spectra for MTC monomer (PDF).
The authors declare no competing financial interest.
Supplementary Material
References
- Nair L. S.; Laurencin C. T. Prog. Polym. Sci. 2007, 32, 762–798. 10.1016/j.progpolymsci.2007.05.017. [DOI] [Google Scholar]
- Uhrich K. E.; Cannizzaro S. M.; Langer R. S.; Shakesheff K. M. Chem. Rev. 1999, 99, 3181–3198. 10.1021/cr940351u. [DOI] [PubMed] [Google Scholar]
- Goldberg M.; Langer R.; Jia X. J. Biomater. Sci., Polym. Ed. 2007, 18, 241–268. 10.1163/156856207779996931. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Guo B.; Glavas L.; Albertsson A.-C. Prog. Polym. Sci. 2013, 38, 1263–1286. 10.1016/j.progpolymsci.2013.06.003. [DOI] [Google Scholar]
- Chandra R.; Rustgi R. Prog. Polym. Sci. 1998, 23, 1273–1335. 10.1016/S0079-6700(97)00039-7. [DOI] [Google Scholar]
- Göpferich A. Biomaterials 1996, 17, 103–114. 10.1016/0142-9612(96)85755-3. [DOI] [PubMed] [Google Scholar]
- Edlund U.; Albertsson A. C. Adv. Drug Delivery Rev. 2003, 55, 585–609. 10.1016/S0169-409X(03)00036-X. [DOI] [PubMed] [Google Scholar]
- Pasparakis G.; Manouras T.; Vamvakaki M.; Argitis P. Nat. Commun. 2014, 5, n/a. 10.1038/ncomms4623. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhang Q.; Re Ko N.; Kwon Oh J. Chem. Commun. 2012, 48, 7542–7552. 10.1039/c2cc32408c. [DOI] [PubMed] [Google Scholar]
- Paramonov S. E.; Bachelder E. M.; Beaudette T. T.; Standley S. M.; Lee C. C.; Dashe J.; Fréchet J. M. J. Bioconjugate Chem. 2008, 19, 911–919. 10.1021/bc7004472. [DOI] [PubMed] [Google Scholar]
- Bettinger C. J.; Bruggeman J. P.; Borenstein J. T.; Langer R. S. Biomaterials 2008, 29, 2315–2325. 10.1016/j.biomaterials.2008.01.029. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tamada J.; Langer R. J. Biomater. Sci., Polym. Ed. 1992, 3, 315–353. 10.1163/156856292X00402. [DOI] [PubMed] [Google Scholar]
- Freed L. E.; Vunjak-Novakovic G.; Biron R. J.; Eagles D. B.; Lesnoy D. C.; Barlow S. K.; Langer R. Bio/Technology 1994, 12, 689–693. 10.1038/nbt0794-689. [DOI] [PubMed] [Google Scholar]
- Wang Y.; Ameer G. A.; Sheppard B. J.; Langer R. Nat. Biotechnol. 2002, 20, 602–606. 10.1038/nbt0602-602. [DOI] [PubMed] [Google Scholar]
- Lendlein A.; Langer R. Science 2002, 296, 1673–1676. 10.1126/science.1066102. [DOI] [PubMed] [Google Scholar]
- Williams C. K. Chem. Soc. Rev. 2007, 36, 1573–1580. 10.1039/b614342n. [DOI] [PubMed] [Google Scholar]
- Nederberg F.; Zhang Y.; Tan J. P. K.; Xu K.; Wang H.; Yang C.; Gao S.; Guo X. D.; Fukushima K.; Li L.; Hedrick J. L.; Yang Y.-Y. Nat. Chem. 2011, 3, 409–414. 10.1038/nchem.1012. [DOI] [PubMed] [Google Scholar]
- Gref R.; Minamitake Y.; Peracchia M.; Trubetskoy V.; Torchilin V.; Langer R. Science 1994, 263, 1600–1603. 10.1126/science.8128245. [DOI] [PubMed] [Google Scholar]
- Barrera D. A.; Zylstra E.; Lansbury P. T.; Langer R. Macromolecules 1995, 28, 425–432. 10.1021/ma00106a004. [DOI] [Google Scholar]
- Govender T.; Stolnik S.; Garnett M. C.; Illum L.; Davis S. S. J. Controlled Release 1999, 57, 171–185. 10.1016/S0168-3659(98)00116-3. [DOI] [PubMed] [Google Scholar]
- Albertsson A.-C.; Varma I. K. Biomacromolecules 2003, 4, 1466–1486. 10.1021/bm034247a. [DOI] [PubMed] [Google Scholar]
- Odelius K.; Plikk P.; Albertsson A.-C. Biomacromolecules 2005, 6, 2718–2725. 10.1021/bm050190b. [DOI] [PubMed] [Google Scholar]
- Coulembier O.; Degée P.; Hedrick J. L.; Dubois P. Prog. Polym. Sci. 2006, 31, 723–747. 10.1016/j.progpolymsci.2006.08.004. [DOI] [Google Scholar]
- Delplace V.; Nicolas J. Nat. Chem. 2015, 7, 771–784. 10.1038/nchem.2343. [DOI] [PubMed] [Google Scholar]
- Hawker C. J.; Barclay G. G.; Orellana A.; Dao J.; Devonport W. Macromolecules 1996, 29, 5245–5254. 10.1021/ma951905d. [DOI] [Google Scholar]
- Kato M.; Kamigaito M.; Sawamoto M.; Higashimura T. Macromolecules 1995, 28, 1721–1723. 10.1021/ma00109a056. [DOI] [Google Scholar]
- Wang J.-S.; Matyjaszewski K. J. Am. Chem. Soc. 1995, 117, 5614–5615. 10.1021/ja00125a035. [DOI] [Google Scholar]
- Chiefari J.; Chong Y. K.; Ercole F.; Krstina J.; Jeffery J.; Le T. P. T.; Mayadunne R. T. A.; Meijs G. F.; Moad C. L.; Moad G.; Rizzardo E.; Thang S. H. Macromolecules 1998, 31, 5559–5562. 10.1021/ma9804951. [DOI] [Google Scholar]
- Braunecker W. A.; Matyjaszewski K. Prog. Polym. Sci. 2007, 32, 93–146. 10.1016/j.progpolymsci.2006.11.002. [DOI] [Google Scholar]
- Zetterlund P. B.; Thickett S. C.; Perrier S.; Bourgeat-Lami E.; Lansalot M. Chem. Rev. 2015, 115, 9745–9800. 10.1021/cr500625k. [DOI] [PubMed] [Google Scholar]
- Warren N. J.; Armes S. P. J. Am. Chem. Soc. 2014, 136, 10174–10185. 10.1021/ja502843f. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Charleux B.; Delaittre G.; Rieger J.; D’Agosto F. Macromolecules 2012, 45, 6753–6765. 10.1021/ma300713f. [DOI] [Google Scholar]
- Derry M. J.; Fielding L. A.; Armes S. P. Prog. Polym. Sci. 2016, 52, 1–18. 10.1016/j.progpolymsci.2015.10.002. [DOI] [Google Scholar]
- Ratcliffe L. P. D.; McKenzie B. E.; Le Bouëdec G. M. D.; Williams C. N.; Brown S. L.; Armes S. P. Macromolecules 2015, 48, 8594–8607. 10.1021/acs.macromol.5b02119. [DOI] [Google Scholar]
- Zehm D.; Ratcliffe L. P. D.; Armes S. P. Macromolecules 2013, 46, 128–139. 10.1021/ma301459y. [DOI] [Google Scholar]
- Ratcliffe L. P. D.; Blanazs A.; Williams C. N.; Brown S. L.; Armes S. P. Polym. Chem. 2014, 5, 3643–3655. 10.1039/c4py00203b. [DOI] [Google Scholar]
- Kang Y.; Pitto-Barry A.; Maitland A.; O’Reilly R. K. Polym. Chem. 2015, 6, 4984–4992. 10.1039/C5PY00617A. [DOI] [Google Scholar]
- Li Y.; Armes S. P. Angew. Chem., Int. Ed. 2010, 49, 4042–4046. 10.1002/anie.201001461. [DOI] [PubMed] [Google Scholar]
- Blanazs A.; Ryan A. J.; Armes S. P. Macromolecules 2012, 45, 5099–5107. 10.1021/ma301059r. [DOI] [Google Scholar]
- Blanazs A.; Verber R.; Mykhaylyk O. O.; Ryan A. J.; Heath J. Z.; Douglas C. W. I.; Armes S. P. J. Am. Chem. Soc. 2012, 134, 9741–9748. 10.1021/ja3024059. [DOI] [PubMed] [Google Scholar]
- Verber R.; Blanazs A.; Armes S. P. Soft Matter 2012, 8, 9915–9922. 10.1039/c2sm26156a. [DOI] [Google Scholar]
- Canton I.; Warren N. J.; Chahal A.; Amps K.; Wood A.; Weightman R.; Wang E.; Moore H.; Armes S. P. ACS Cent. Sci. 2016, 2, 65–74. 10.1021/acscentsci.5b00370. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mitchell D. E.; Lovett J. R.; Armes S. P.; Gibson M. I. Angew. Chem., Int. Ed. 2016, 55, 2801–2804. 10.1002/anie.201511454. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Oh J. K.; Siegwart D. J.; Lee H.-i.; Sherwood G.; Peteanu L.; Hollinger J. O.; Kataoka K.; Matyjaszewski K. J. Am. Chem. Soc. 2007, 129, 5939–5945. 10.1021/ja069150l. [DOI] [PubMed] [Google Scholar]
- Tsarevsky N. V.; Matyjaszewski K. Macromolecules 2005, 38, 3087–3092. 10.1021/ma050020r. [DOI] [Google Scholar]
- Cohen J. L.; Almutairi A.; Cohen J. A.; Bernstein M.; Brody S. L.; Schuster D. P.; Fréchet J. M. J. Bioconjugate Chem. 2008, 19, 876–881. 10.1021/bc700414j. [DOI] [PubMed] [Google Scholar]
- Themistou E.; Patrickios C. S. Macromolecules 2004, 37, 6734–6743. 10.1021/ma049618+. [DOI] [Google Scholar]
- Tsarevsky N. V.; Matyjaszewski K. Macromolecules 2002, 35, 9009–9014. 10.1021/ma021061f. [DOI] [Google Scholar]
- Li C.; Madsen J.; Armes S. P.; Lewis A. L. Angew. Chem., Int. Ed. 2006, 45, 3510–3513. 10.1002/anie.200600324. [DOI] [PubMed] [Google Scholar]
- Rikkou M. D.; Patrickios C. S. Prog. Polym. Sci. 2011, 36, 1079–1097. 10.1016/j.progpolymsci.2011.01.004. [DOI] [Google Scholar]
- Phillips D. J.; Gibson M. I. Biomacromolecules 2012, 13, 3200–3208. 10.1021/bm300989s. [DOI] [PubMed] [Google Scholar]
- Nicolaÿ R.; Marx L.; Hémery P.; Matyjaszewski K. Macromolecules 2007, 40, 9217–9223. 10.1021/ma701923q. [DOI] [Google Scholar]
- You Y.-Z.; Manickam D. S.; Zhou Q.-H.; Oupický D. J. Controlled Release 2007, 122, 217–225. 10.1016/j.jconrel.2007.04.020. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Whittaker M. R.; Goh Y.-K.; Gemici H.; Legge T. M.; Perrier S.; Monteiro M. J. Macromolecules 2006, 39, 9028–9034. 10.1021/ma061070e. [DOI] [Google Scholar]
- Gemici H.; Legge T. M.; Whittaker M.; Monteiro M. J.; Perrier S. J. Polym. Sci., Part A: Polym. Chem. 2007, 45, 2334–2340. 10.1002/pola.21985. [DOI] [Google Scholar]
- Frick E. M.; Hillmyer M. A. Macromol. Rapid Commun. 2000, 21, 1317–1322. . [DOI] [Google Scholar]
- Themistou E.; Battaglia G.; Armes S. P. Polym. Chem. 2014, 5, 1405–1417. 10.1039/C3PY01446K. [DOI] [Google Scholar]
- You Y.; Hong C.; Wang W.; Lu W.; Pan C. Macromolecules 2004, 37, 9761–9767. 10.1021/ma048444t. [DOI] [Google Scholar]
- Pratt R. C.; Lohmeijer B. G. G.; Long D. A.; Lundberg P. N. P.; Dove A. P.; Li H.; Wade C. G.; Waymouth R. M.; Hedrick J. L. Macromolecules 2006, 39, 7863–7871. 10.1021/ma061607o. [DOI] [Google Scholar]
- Bernaerts K. V.; Du Prez F. E. Prog. Polym. Sci. 2006, 31, 671–722. 10.1016/j.progpolymsci.2006.08.007. [DOI] [Google Scholar]
- Mecerreyes D.; Humes J.; Miller R. D.; Hedrick J. L.; Detrembleur C.; Lecomte P.; Jérôme R.; San Roman J. Macromol. Rapid Commun. 2000, 21, 779–784. . [DOI] [Google Scholar]
- Li Y.; Armes S. P. Macromolecules 2005, 38, 8155–8162. 10.1021/ma051121s. [DOI] [Google Scholar]
- Wang L.; Li C.; Ryan A. J.; Armes S. P. Adv. Mater. 2006, 18, 1566–1570. 10.1002/adma.200600314. [DOI] [Google Scholar]
- Sanda F.; Endo T. J. Polym. Sci., Part A: Polym. Chem. 2001, 39, 265–276. . [DOI] [Google Scholar]
- Bailey W. J.; Ni Z.; Wu S. R. Macromolecules 1982, 15, 711–714. 10.1021/ma00231a006. [DOI] [Google Scholar]
- Schulze T.; Klemm E. Polym. Bull. 1993, 31, 409–414. 10.1007/BF00329876. [DOI] [Google Scholar]
- Jin S.; Gonsalves K. E. Macromolecules 1997, 30, 3104–3106. 10.1021/ma961590h. [DOI] [Google Scholar]
- d’Ayala G. G.; Malinconico M.; Laurienzo P.; Tardy A.; Guillaneuf Y.; Lansalot M.; D’Agosto F.; Charleux B. J. Polym. Sci., Part A: Polym. Chem. 2014, 52, 104–111. 10.1002/pola.26976. [DOI] [Google Scholar]
- Evans R. A.; Moad G.; Rizzardo E.; Thang S. H. Macromolecules 1994, 27, 7935–7937. 10.1021/ma00104a062. [DOI] [Google Scholar]
- Evans R. A.; Rizzardo E. Macromolecules 1996, 29, 6983–6989. 10.1021/ma960573p. [DOI] [Google Scholar]
- Phelan M.; Aldabbagh F.; Zetterlund P. B.; Yamada B. Polymer 2005, 46, 12046–12056. 10.1016/j.polymer.2005.11.006. [DOI] [Google Scholar]
- Agarwal S. Polym. Chem. 2010, 1, 953–964. 10.1039/c0py00040j. [DOI] [Google Scholar]
- Chaumont P.; Asgarzadeh F.; Colombani D.; Arotcarena M.; Baudouin A. Macromol. Chem. Phys. 1998, 199, 2577–2582. . [DOI] [Google Scholar]
- He T.; Zou Y.-F.; Pan C.-Y. Polym. J. 2002, 34, 138–143. 10.1295/polymj.34.138. [DOI] [Google Scholar]
- Hedir G. G.; Bell C. A.; Ieong N. S.; Chapman E.; Collins I. R.; O’Reilly R. K.; Dove A. P. Macromolecules 2014, 47, 2847–2852. 10.1021/ma500428e. [DOI] [Google Scholar]
- Delplace V.; Tardy A.; Harrisson S.; Mura S.; Gigmes D.; Guillaneuf Y.; Nicolas J. Biomacromolecules 2013, 14, 3769–3779. 10.1021/bm401157g. [DOI] [PubMed] [Google Scholar]
- Delplace V.; Guegain E.; Harrisson S.; Gigmes D.; Guillaneuf Y.; Nicolas J. Chem. Commun. 2015, 51, 12847–12850. 10.1039/C5CC04610F. [DOI] [PubMed] [Google Scholar]
- Lutz J.-F.; Andrieu J.; Üzgün S.; Rudolph C.; Agarwal S. Macromolecules 2007, 40, 8540–8543. 10.1021/ma7021474. [DOI] [Google Scholar]
- Huang J.; Gil R.; Matyjaszewski K. Polymer 2005, 46, 11698–11706. 10.1016/j.polymer.2005.09.048. [DOI] [Google Scholar]
- Paulusse J. M. J.; Amir R. J.; Evans R. A.; Hawker C. J. J. Am. Chem. Soc. 2009, 131, 9805–9812. 10.1021/ja903245p. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Warren N. J.; Rosselgong J.; Madsen J.; Armes S. P. Biomacromolecules 2015, 16, 2514–2521. 10.1021/acs.biomac.5b00767. [DOI] [PubMed] [Google Scholar]
- Cunningham V. J.; Ratcliffe L. P. D.; Blanazs A.; Warren N. J.; Smith A. J.; Mykhaylyk O. O.; Armes S. P. Polym. Chem. 2014, 5, 6307–6317. 10.1039/C4PY00856A. [DOI] [Google Scholar]
- Blanazs A.; Madsen J.; Battaglia G.; Ryan A. J.; Armes S. P. J. Am. Chem. Soc. 2011, 133, 16581–16587. 10.1021/ja206301a. [DOI] [PubMed] [Google Scholar]
- Blanazs A.; Armes S. P.; Ryan A. J. Macromol. Rapid Commun. 2009, 30, 267–277. 10.1002/marc.200800713. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.