

Abstract
Identifying “druggable” targets and their corresponding therapeutic agents are two fundamental challenges in drug discovery research. The one-bead-one-compound (OBOC) combinatorial library method has been developed to discover peptides or small molecules that bind to a specific target protein or elicit a specific cellular response. The phage display cDNA expression proteome library method has been employed to identify target proteins that interact with specific compounds. Here, we combined these two high-throughput approaches, efficiently interrogated approximately 1013 possible molecular interactions, and identified 91 small molecule compound beads that interacted strongly with the phage library. Of 19 compounds resynthesized, 4 were cytotoxic against cancer cells; one of these compounds was found to interact with EIF5B and inhibit protein translation. As more binding pairs are confirmed and evaluated, the “library-against-library” screening approach and the resulting small molecule–protein domain interaction database may serve as a valuable tool for basic research and drug development.
Keywords: library-against-library screening, one-bead-one-compound combinatorial library, phage display cDNA expression proteome library, molecular interactions, small molecule compound beads
Introduction
The one-bead-one-compound (OBOC) combinatorial library technology enables rapid and economical generation of huge libraries of peptides or small molecule compounds and efficient screening of such libraries against protein or cellular targets for compounds of biological significance.1−11 When screening against protein targets, often one target protein is used as the screening probe. Similarly, phage display random peptide libraries, fragment antigen binding (Fab) region protein libraries, and cDNA expression proteome libraries are usually screened with one single target protein or small molecule.12−21 Even in cell-based screening, one biological end point is often employed, such as cell binding, cell uptake, and specific signal transduction.
We previously reported the use of total cell lysate to screen OBOC small molecule libraries and succeeded in identifying small molecule ligands against β-actin22 as well as other unknown proteins.23 However, retrieval and identification of each unknown binding protein is an arduous and challenging task. Furthermore, the range of expression levels of various cellular proteins is huge, and the use of total cell lysates to screen compound libraries will be biased toward ligands against proteins with a high expression level. To address these issues, we performed a novel high throughput screening of an OBOC library against a phage display cDNA expression proteome library in which the cDNA library has been size-fractionated and normalized to exclude highly abundant cDNA. The protein domain displayed on the phages that bind to a small molecule can be easily identified by sequencing the cDNA insert in the phage.
In consideration of some general OBOC library screening issues, such as multivalent effects and steric hindrance from the linker that connects the small molecule to the bead,24 when making the OBOC library, we first down-substituted the bead surface25 to reduce the surface concentration of the small molecules to increase the screening stringency and therefore reduce the nonspecific binding during this library-on-library screening. Please see Scheme S1 in the Supporting Information for details on the OBOC library synthesis. The structures of the compounds that bind to the phage display protein domains were decoded by the mass spectrometric (MS) method. We then resynthesized each decoded small molecule hit on beads for multiple biopannings with their corresponding phage display proteome binding partners to enrich the population of the phages that had the strongest binding to the small molecule hits. Because we are looking for anticancer applications in this study, we resynthesized the decoded small molecule hits in soluble form to test their antiproliferation effect without the influence from the linker. We also conjugated a linker-lysine-biotin (LKB) moiety to the small molecule hits for performing lysate protein pull-down assays to confirm the protein–small molecule binding results from the library–library screening and while excluding binding that was affected by this LKB moiety.
From the results of the above assays tailored to each protein–small molecule binding pair, we were able to rule out nonspecific bindings and protein–small molecule interactions that did not elicit biological effects. This systematic approach of library-against-library screening, chemical decoding, phage retrieval, DNA sequencing, and specific binding/biological assay confirmation has allowed us to create a small molecule–protein domain interaction database, which is a valuable resource for the discovery of probes for basic research and drug discovery.
Results and Discussion
Identification of 91 Small Molecule Compounds from the OBOC Library that Bound to the Phage Display Proteome
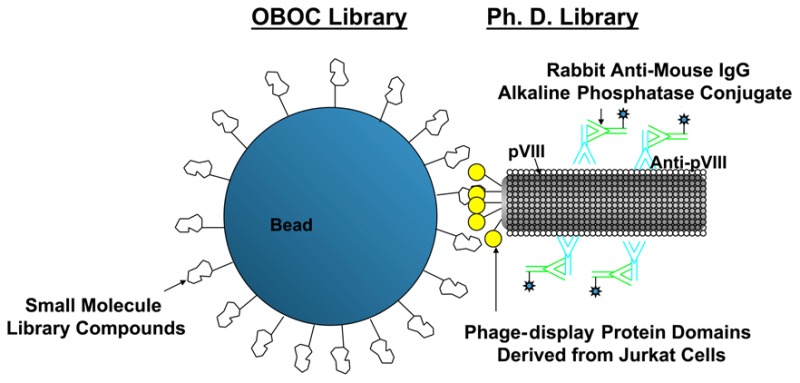
We screened 109 cfu of the M13 phage display Jurkat cell cDNA library against ∼100,000 beads of a 3-diversity point OBOC small molecule library (permutations = 16,896). The OBOC library was prepared by sequentially coupling one of the 24 amine building blocks (R1, Table S1), one of the 22 aldehyde building blocks (R2, Table S2), and one of the 32 acid/isocyanate building blocks (R3, Table S3) to the topographically segregated bilayer beads via the “split-and-mix”26,27 procedure between each coupling step, such that multiple copies of the same unique small molecule compound were displayed on the outer layer of each bead, and the coding tag ladders were tethered to the bead interior via cleavable linkers (CL). Please see Scheme S1 for details of the library synthesis. The M13 phage display cDNA library was constructed by Spring Bioscience using a random primer method according to the manfacturer’s manual. The resulting cDNA fragments were fractionated, and the 0.2–2kb fragments were extracted to construct the library. The redundency or sparseness of the protein motif coverage in this library, however, had never been verified.
The phage library against the OBOC library (or “library-against-library”) screening procedure was adapted from the two-step enzyme-linked colorimetric OBOC library screening method developed in our lab.23 In this method, the small molecule bead library was first incubated with bovine serum albumin (BSA) in phosphate buffered saline (PBS) to block the nonspecific binding between small molecules and proteins. Before mixing the phage display cDNA proteome domain expression library with the beads, the BSA-treated beads were sequentially incubated with mouse anti-M13 IgG, rabbit anti-mouse-IgG-alkaline phosphatase, and then 5-bromo-4-chloro-3-indolyl phosphate (BCIP), the colorimetric substrate of alkaline phosphatase. The beads that turned turquoise in this first step, due to interactions with the primary or secondary antibodies, were considered as the background false positive beads. The bead library was then thorougly washed and incubated sequentially with the phage display proteome library and primary and secondary antibodies. It was then immobilized on agarose gel followed by the addition of BCIP. Beads that turned tuorquoise in this second step were the true positive beads that interacted with the phage display protein domains, and they were readily identified by image subtraction of the bead arrays before and after the addition of BCIP substrate (Scheme 1). True positive beads were then picked, stripped with acidic guanidine HCl to remove bound phages and antibodies, and treated with cyanogen bromide to cleave the CL; the coding tags were released for chemical decoding with MALDI-TOF (Figure S1) and a Matlab scripts (Supporting Information). The molecular weight difference between the three distinct coding tags released from each bead provided information needed to determine the three building blocks used for each compound. Ninety one small molecule structures were unequivacally determined. Please see Table S4 for the structure information. We show here that our library-against-library screening strategy can evaluate more than 1013 combinations of small molecule–protein interactions in a relatively short amount of time in one small tube (1.5 mL). Interrogation of such a huge number of potential molecular interactions is difficult to achieve solely by increasing the permutations of a library but can easily be accomplished by employing a proteome library as probes to screen a small molecule OBOC library. Given this number of combinations, it is likely that binding partners with high binding affinity and specificity can be discovered.
Scheme 1. Screening of the Phage Display Library against the OBOC Library.

Four Compounds out of the 19 Randomly Selected Compounds from the 91 Hits Were Cytotoxic to Both Jurkat and HepG2 Cells
We randomly picked 19 compounds from the 91 hits for resynthesis in soluble form (please see the upper panel of Figure 1A for structural examples) and evaluated their cell cytotoxic activities with an MTT (3-(4,5-dimethylthiazol-yl)-2,5-diphenyltetrazolium bromide) assay using the T-leukemia Jurkat cell line as the target indicator cells. Four out of the 19 compounds, LWW29, LWW31, LWW32, and LWW55, were found to be cytotoxic to Jurkat cells at 40 μM (Figure 1B). These results suggested that these compounds discovered from the library at midmicromolar concentrations and in their carboxyl-amide free form were able to enter the cells and interact with the intracellular proteomes to affect cell functions. Here, we demonstrate their antiproliferation effects on Jurkat cells. As we later found that LWW31 may bind to eukaryotic initiation factor 5B (EIF5B) displayed on T7 phages from a liver tumor cDNA phage display library, we also evaluated its cytotoxic activity against HepG2, a hepatocellular carcinoma cell line, and normal human fetal hepatocytes (hHF). LWW31 had a lower IC50 (∼30 μM) against HepG2 cells than against normal human fetal hepatocytes (hHF) with an IC50 of approximately 50 μM (Figure 1C). The result indicates that LWW31 may be a potential lead compound for the development of cancer therapeutics.
Figure 1.

Characterization of LWW31. (A) Structures of LWW31 (from the top) in soluble form, LWW31 on beads, and LWW31 conjugated with linker-lysine-biotin (LKB). (B) Preliminary cytotoxicity assay for the 19 randomly chosen compounds at 40 μM. (C) LWW31 inhibited HepG2 cell proliferation with a lower IC50 than that observd for hHF cells. (D) LWW31-LKB pulls down EIF5B from both Jurkat (lane 4) and HepG2 (lane 9) cell lysates. Western blot: lane 1, total Jurkat cell lysate with mouse anti-EIF5B IgG; lane 2, Jurkat cell proteins pulled down with LKB; lane 3, Jurkat cell proteins pulled down with LWW29-LKB; lanes 5 and 6, molecular weight markers; lane 7, total HepG2 cell lysate with mouse anti-EIF5B; lane 8, HepG2 proteins pulled down with LKB. (E) LWW31 induced Jurkat cell apoptosis, whereas (F) LKB did not.
Target Proteins Were Identified by Performing PDL Biopanning against the On-Bead Compounds and Subsequently Confirmed by Pull-Down Assays
The identification of proteins displayed on the phages bound to the identified small molecules enabled us to create a small molecule–protein domain binding database, which will be valuable for functional proteomics as well as drug and therapeutic target discovery. To establish the database, we performed four rounds of PDL panning against beads displaying the resynthesized compounds. In principle, one single positive bead can be used to retrieve the binding phages for identification of the displayed protein domain(s). However, in practice, to pan the phage library with a single bead is very difficult, and there is a good chance of losing the bead. This is especially true when multiple cycles of phage panning are required so that strong binding phages can be amplified and selected. To overcome this problem, we first decoded each positive bead, and based on the elucidated chemical structure, each of these compounds were resynthesized on ∼250 mg of Tentagel beads (please see the structure column of Table 1 and the middle panel of Figure 1A for structural examples).The carboxyl end of each molecule was tethered to the beads. Approximately 20,000 beads displaying the same compound were used for each round of biopanning.
Table 1. Structures and Biopanning Results of the Four Compounds Identified that Showed Cellular Toxicitya.
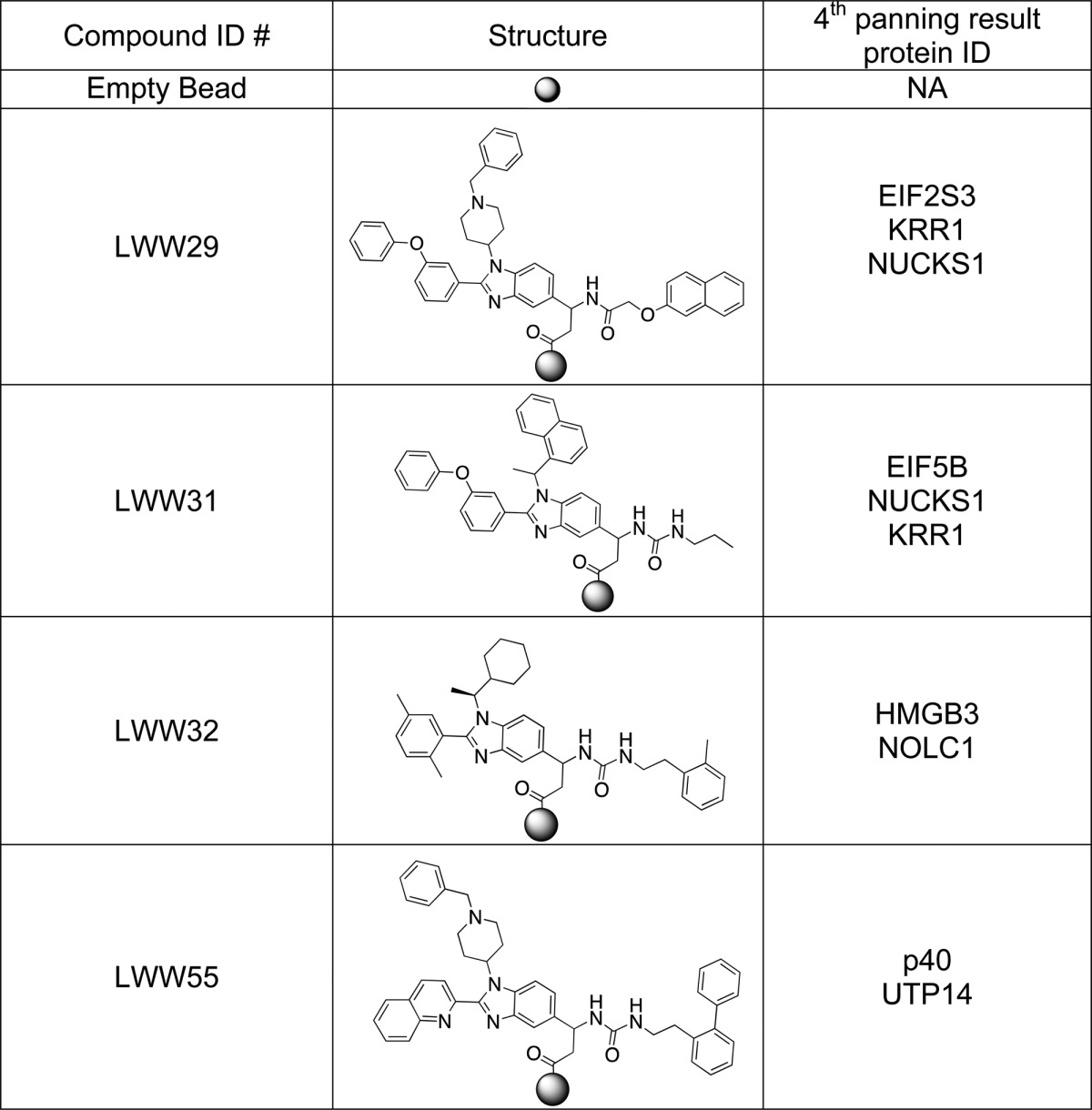
NA, peptide sequences were not available due to the failure of DNA sequencing, the failure of translation of DNA to putative peptide sequences, or the peptide sequences not matching any sequences in the database after the BLAST search; EIF2S3, eukaryotic translation initiation factor 2, subunit 3 gamma; HMGN2, high-mobility group nucleosomal binding domain 2; EIF5B, eukaryotic translation initiation factor 5B; NUCKS1, nuclear casein kinase and cyclin-dependent kinase substrate 1; HMGB3, high-mobility group box 3; NOLC1, nucleolar and coiled-body phosphoprotein 1; UTP14, U3 small nucleolar ribonucleoprotein, homologue A.
We used the T7select phage display liver tumor cDNA proteome expression library (Merck KGaA, Darmstadt, Germany) to perform the biopanning against each of the 91 identified small molecules. Beads displaying the compounds were first incubated with the phage display proteome library. After washing, the bead-bound phages were amplified by adding Escherichia coli (E. coli) BLT5403 to the panned beads. The E. coli culture became clear in 2 h due to cell lysis after phage infection, indicating that the bound phages were infectious. After centrifugation, the resulting clear broth with enriched phages that carried target proteins was used for the next round of panning. At the end of the fourth round of panning, the bound phages were incubated with E. coli, mixed with soft LB-agar, and poured onto hard LB-agar on each Petri-dish. The infected E. coli formed individual plaques on the E. coli lawn after a 3 h incubation at 37 °C. We then picked 10 phage plaques and amplified the inserted cDNA fragments by polymerase chain reaction (PCR) for DNA sequencing. We matched the identified coding sequences with the corresponding on-bead compounds and created the small molecule–proteome binding pair database. Table 1 summarizes the decoded structures and corresponding biopanning results for four compounds shown to have cytotoxic activity against cancer cell lines. For LWW31, the biopanning experiment yielded the N-terminal domain (104 amino acids, aa 248–351) of EIF5B as the putative target protein. Please see the Supporting Information for EIF5B nucleotide and peptide sequences. The underlined sequences matched the sequence databases through a BLAST28 (basic local alignment searching tools) search. To confirm the binding, we performed pull-down assays with biotinylated compounds (for example, please see the lower panel of Figure 1A) and neutravidin agarose beads against cell lysates, followed by SDS-PAGE and Western blotting with an anti-EIF5B antibody. LWW31 pulled down EIF5B from both Jurkat (Figure 1D, lane 4) and HepG2 (Figure 1D, lane 9) cell lysates; LWW29, another cell-killing compound with the same scaffold, could not pull down EIF5B from the Jurkat cell lysate (Figure 1D, lane 3). As a negative control, the linker-lysine-biotin (LKB) fragment could not pull down EIF5B from either lysate (Figure 1D, lanes 2 and 8). This result further confirmed that LWW31 bound EIF5B. Please see Table S5 for more biopanning results against other compounds.
According to our database shown in Table S5, phage panning by many of the small molecule ligands yielded unique repeating phage clones for each compound, indicating that the affinity and specificity between these binding pairs were relatively high. Although less frequent, we had also observed single copy phage clones retrieved by some of the small molecule ligands. Interactions between such binding pairs were likely to be weaker. All eluted phages that contained DNA inserts and/or expressed peptides that could not be found on BLAST were annotated “NA” in the database. For example, from the nucleotide sequencing and BLAST nucleotide searching result, LWW13 bound to a phage that contained an insert of Homo sapiens WW domain binding protein 5 (WBP5) cDNA, whose sequence 100% matched the BLAST database, but its putative protein sequence did not match anything in the BLAST protein search due to the coding DNA frameshift. It is conceivable that the actual expressed peptide sequence and its corresponding small molecule structure may still provide some peptide–small molecule binding information. Some compounds like LWW14 bound to a phage that had a cDNA insert whose 1–193 nucleotides did not match anything in the BLAST search and therefore generated a peptide sequence that also did not match anything through a BLAST peptide search. Some compounds like LWW45 and LWW48 bound to some phages that carried cDNAs that could not be sequenced completely for some unknown reason.
LWW31 Induced Jurkat Cell Apoptosis
LWW31 was found to be cytotoxic against Jurkat cells with an IC50 of approximately 25–30 μM. To observe the effect of LWW31 on the Jurkat cell cycle, we incubated the cells with 25 μM of LWW31 for 24 h, collected and permeabilized the cells, and then stained the DNA inside the cells with propidium iodide (PI). We found by flow cytometry that a hypodiploid sub-G1 peak was generated from treated cells, indicative of apoptotic DNA fragmentation, whereas all untreated cells in different phases of the cell cycle only had normal DNA content from the G1, S, or G2/M peaks (Figure 1E,F). On the basis of this result, we hypothesize that the binding of LWW31 to the N-terminal domain of EIF5B inhibited initiation of cellular translation, resulting in apoptosis and cell death.
LWW31 Inhibited Protein Expression in HepG2 Cells
To test whether binding of LWW31 to EIF5B inhibited protein synthesis, we conducted a [35S]-methionine incorporation assay. The cells were first starved in culture media with dialyzed fetal bovine serum (dFBS) for one hour to deplete amino acids for protein synthesis. Medium containing 10 μCi [35S]-methionine, 2 μg of unlabeled methionine, and various concentrationa of LWW31 was then added to each well of cells and incubated for another 24 h. The cells were harvested and disrupted to obtain cell lysate proteins for SDS-PAGE analysis. The SDS-PAGE gels were stained with coomassie blue and dried with a gel dryer. Kodak phosphorimaging screen-K was exposed to the dried gels for 3 days and scanned with a Biorad Molecular Imager system. After normalizing the radioisotope intensity of each lane on the SDS-PAGE gel with the coomassie blue intensity of each corresponding lane, we observed a dose-dependent reduction of incorporation of [35S]-methionine into newly synthesized proteins (Figure 2), suggesting that LWW31 bound to EIF5B and interfered with protein translation. We also found a similar inhibition against rabbit reticulocyte lysate in an in vitro expression system in which luciferase expression was inhibited and therefore generated less luminescence in the presense of LWW31 (Figure S2). However, a much higher concentration of LWW31 was needed to demonstrate a comparable inhibitory effect. This could be due to (i) rabbit instead of human reticulocyte lysate or (ii) a highly optimized and concentrated in vitro translation system.
Figure 2.

LWW31 inhibited the incorporation of [35S]-methionine. (A) Phosphorimaging of SDS-PAGE resolved [35S]-methionine labeled proteins from cells treated with various concentrations of LWW31. (B) Coomassie blue staining of the SDS-PAGE used for phosphorimaging. (C) The ratio of [35S]-methionine-labeled protein radioactivity (CNT*mm2) to the amount of total protein (band density) of each lane on the same SDS-PAGE gel.
Therefore, this proof of concept study found that the small molecule LWW31 may bind to the N-terminal domain (aa 248–351) of EIF5B, perturb protein translation, and induce apoptosis of target cells. EIF5B is a eukaryotic translational initiation factor, which is a homologue to prokaryotic IF2 proteins. It plays an essential role in protein translation initiation by joining the 40s and 60s ribosome subunits to form the translation complex with mRNA and Met-tRNAimet.29 The structure and function of the protein have been widely reported and reviewed.30−33 Little is known about the functional significance of the region between the N-terminal end and the GTP binding site. Although it has been reported that the entire N-terminal region (aa 1–396 in M. therms) is nonessential;32 in our study, we found that our small molecule LWW31 could bind to amino acids 248–351 of EIF5B, resulting in Jurkat and HepG2 cell apoptosis and translational inhibition, which has not previously been reported. More experiments are needed to confirm the structure and function of this protein domain.
Compared with conventional approaches, our library-against-library approach can greatly reduce the time required to identify the proteins or protein domains to which the small molecule binds. With a conventional method of using cell lysates to screen the OBOC libraries together with pull-down assays followed by SDS-PAGE and MS analysis to determine the identity of the putative target proteins,22 it can take weeks or months to determine the identity of just one capturing protein. To determine the binding domain within a full length protein, one will need to produce protein domains that could then be evaluated for their binding to the affinity compound. Furthermore, in the conventional approach, the relative abundance of proteins in the lysate could greatly affect the screening result, such that rare proteins may be missed. In contrast to the conventional approach, our library-against-library approach employs a randomly primed cDNA library, so that truncated rather than full-length proteins are displayed on the phage, resulting in immediate identification of a candidate binding domain for the affinity small molecule. With the phage-display approach, dozens of ligands can be analyzed and putative target proteins determined in less than a week. Furthermore, critical proteins with low cellular expression levels can also be discovered as new targets by using our screening strategy.
Finding hits and identifying new druggable targets are the two major tasks of drug discovery in the pharmaceutical industry.34 The development of HTS screening methodology is critical to enhance the efficiency of affinity-based drug screening. Conventional screening methods are often performed in 384-well format with soluble compound libraries and require costly equipment (e.g., robotics) and supplies. An affinity-based screening strategy using NMR and mass spectrometry has also been reported, but they are expensive and relatively slow.35 Our library-against-library screening approach not only accomplishes these two major tasks simultaneously but also requires less instrumentation, making it more efficient and affordable. Library generation with the OBOC technology is highly efficient and economical. The enzyme-linked colorimetric screening method enables us to visualize the binding between small molecules and proteomes using only a simple table-top scanner. Chemical decoding can be achieved with MALDI-TOF, which is readily available in many facilitates. Phage panning and DNA sequencing for protein identificaiton are easy and inexpensive.
Thus far, of the 91 small molecule ligands identified in this research, only 19 have been resynthesized and evaluated for their antitumor properties. Of the four antitumor compounds identified, one has been explored for its antitumor mechanism. The data outlined in Table S5 on the small molecule–proteome binding pairs provide a rich source of information that will need to be explored for the discovery of potential anticancer agents and small molecules that may perturb other specific biochemical or cellular pathways, leading to unique cellular phenotypes. In addition, some of them may serve as specific capturing or affinity agents for unique proteins or protein complexes. This innovative library-against-library screening approach holds great promise for finding new targets and hits that are invaluable for basic research and drug discovery.
Experimental Procedures
Synthesis of the OBOC Benzimidazole Small Molecule Library
The topological segregation method used to construct multiple-layer Tentagel beads for coupling coding tag ladders, which encode the small molecule structures displayed on the surface of the beads, has been previously reported.36 Briefly, Tentagel beads were first swollen in water overnight, and 0.1 equiv of Alloc-Osu with dichloromethane/ether (45:55) and N,N-diisopropylethylamine (DIEA) were then added to the beads after the water was drained. Because the amount of Alloc-Osu was only enough to conjugate to the outer layer of the beads (10% of the overall bead substitution), the free amine groups inside the beads were used to couple the Fmoc-protected cleavable linker (Fmoc-CL) containing methionine (Met), arginine (Arg), 3-(4-bromophenyl)-β-alanine (A(Br)), and 2,2′-ethylenedioxybis(ethylamine)monosuccinamide (linker).36 After removal of Fmoc and Alloc protective groups, exposed amine groups, 10% of the surface, and 10% of the inner CL layer were coupled with 0.2 equiv of Fmoc-Osu. The resulting inner 80% of the CL was protected with 3 equiv of Alloc-Osu. After the Fmoc deprotection, the outer 20% of the exposed amines were conjugated to the scaffold Dde-3-amino-3-(4-fluoro-3-nitrophenyl)propanoic acid, and the 80% Alloc inside the beads was then removed to conjugate the other two truncated scaffold mixtures, 4-fluoro-3-nitrobenzoic acid (R1 and R2 groups) and 2-bromoacetic acid (R1). The resulting beads were split in 24 equal portions and reacted individually with 24 different R1 groups, which were amine building blocks, so that all of the scaffolds all over the beads could be coupled to the amines by substitution with its fluorine groups (and bromine group on the 2-bromoacetic acid). After the R1 coupling, the beads were pooled together and the nitro groups (NO2) on the scaffolds were reduced to amine (NH2) groups with SnCl2. The reduced beads were split again into 22 equal portions and reacted with 22 R2 aldehydes individually to form the benzimidazole structure. After the reaction, the beads were pooled together again. The secondary amines, which resulted from the bromine replacement on 2-bromoacetic acid with R1s in the 80% inner layer, were then protected with Boc groups. Finally, Dde protective groups of the 10% surface scaffold were removed to expose the amine groups, and the resulting beads were split into 32 portions and reacted individually with 32 R3 building blocks (Fmoc-amino acids, acids, and isocyanates). After coupling, all of the Fmoc groups were removed with 20% 4-methylpiperidine in DMF. All of the other side chain protective groups were removed with a mixture of TFA/TIS/H2O. After each coupling, beads were washed 3 times with DMF. After each Fmoc deprotection, beads were washed 3 times with each solvent in the following order: DMF, MeOH, DCM, and DMF. The library was stored in 70% ethanol solution at 4 °C. Please see Scheme S1 for the library synthesis and Tables S1–3 for the list of the three building blocks.
Screening of the M13 Phage Display cDNA Library Binding Small Molecules
Approximately 200,000 beads (∼500 μL of 50% (v/v) bead slurry) of the OBOC library were washed 3 times with PBSTB (PBS, pH 7.4, with 0.5% of Tween and 0.5% of BSA), swollen in 10.0 mL of the same buffer in polypropylene columns (Thermo Fisher Scientific, Rockford, IL), and spun at room temperature overnight prior to screening. On the day of screening, PBSTB was drained, and the beads were incubated with a 1:5000 dilution of mouse anti-M13 monoclonal antibody in PBSTB for 1 h. The unbound antibodies were drained and washed 3 times with PBSTB. The beads were then incubated with a 1:5000 dilution of rabbit ant-imouse IgG (H+L)-AP for another 1 h, and the unbound antibodies were again washed away from the beads with 3 PBSTB washes. Further washing with 15 mL of TBS buffer (2.5 mM Tris-HCl, 13.7 mM NaCl, and 0.27 mM KCl, pH 8) and 3 times with 15 mL of BCIP buffer (0.1 M Tris-HCl, 0.1 M NaCl, and 2.34 mM MgCl2, pH 8.5–9.0) was performed. The beads were equilibrated with BCIP buffer and subjected to incubation with BCIP solution for another 2 h. These beads that turned turquoise within these 2 h, due to the removal of the phosphate groups from the BCIP catalyzed by alkaline phosphatase (AP) on the rabbit antimouse-AP, were considered to be the false positive beads that bound to either the anti-M13 antibody or rabbit anti-mouse antibody. The false positive beads were eventually ruled out using the image subtraction method,23 which will be described briefly below. The beads, including the stained beads, were then incubated with approximately 109 colony forming units (cfu) of M13 phage display Jurkat cell cDNA library for 1 h, and the unbound phages were washed away with 3 PBSTB flushes. The beads were again incubated with mouse anti-M13 and followed by incubation with rabbit anti-mouse-AP. The incubation and washing conditions are described above. The beads were mixed with 10 mL of 1% low melting point agarose melted in BCIP buffer. Red landmark beads were also included. One milliliter of the mixture was dispensed into a 35 mm falcon Petri-dish lid. The mixture was allowed to cool for 15 min or until totally solidified. One milliliter of BCIP buffer was added to the surface of each solidified agarose-bead mixture and incubated for another 15 min for equilibrium. The BCIP buffer was then removed by vacuum. The plates were transferred to the Umax (Taipei, Taiwan) Astra 2400 image scanner equipped with the transparency adaptor UTA2400, and the BCIP solution was added to the agarose-bead mixture. The 0 h image was taken immediately after the BCIP solution was added. After 2 h, the beads that turned turquoise “in situ” were considered to be positive, which bound to the phages. Without changing the initial plate setting on the scanner, the location of the positive beads could be easily visualized and indicated with arrowheads. After subtracting the 0 h image from the second hour image using computer software,23 false positive beads were readily ruled out. The subtracted image was printed out, put under the agarose-bead mixtures, and aligned with the help of landmark beads under the dissection microscope so that the locations where the arrowheads indicated on the subtracted image could be perfectly superimposed with the corresponding beads in the agarose mixture. The positive beads were then picked up with watchmaker forceps and stored in H2O for structure decoding. Please see Scheme 1 for an illustration of the screening procedure.
On-Bead Small Molecule Structure Decoding
The retrieved positive beads that bound to phages were first washed with 8 M guanidine-HCl (pH 1) to strip off the bound phages. These beads were then rinsed 3 times with H2O to totally remove the guanidine. For easier handling of the tiny 90 μm beads, all of the washing steps were performed in Millipore (Billerica, MA) MultiScreen Solvinert hydrophilic PTFE 0.45 μm 96-well filter plates. The washed beads were transferred to individual 200 μL polypropylene PCR tubes that contained approximately 50 μL of ethanol. After removal of the ethanol by natural evaporation under a fume hood, 20 μL of cyanogen bromide (CNBr) solution (20 mg/mL) in 70% TFA was added to each tube to release the coding tags from the bead.36 The solution containing the cleaved coding tags was lyophilized and then analyzed on an Applied Biosystem (Carlsbad, CA) 4700 proteomic analyzer MALDI-TOF-MS. The molecular weights (MWs) of the three coding tags for one compound were M1, M2, and M3. The molecular weights were easily located on the mass spectra. The molecular weight of the R1 building block was M1 = 753; the MW of R2 was M2 – M1 = 57, and the MW of the R3 building block was M3 – M2 = 42. The identity of each building block can be retrieved from the building block lists in Tables S1–3 based upon the MWs obtained from the corresponding MS spectrum. Please see Figure S1 for details of decoding a small molecule structure.
Resynthesis of the Decoded Small Molecules
Decoded small molecules, which bound to phages, were resynthesized individually in soluble form, soluble form conjugated with Linker-Lysine-Biotin (LKB), and on-bead form. The synthesis procedures were identical to the library synthesis procedures excluding the topological segregation and the conjugation of the CL. For making the soluble small molecules conjugate with LKB, the LKB was first coupled to rink resin. The small molecule was then conjugated to the α-amine of the lysine. Each small molecule on the rink resin was cleaved off with 9 mL of TFA/TIS/H2O (9.5:0.25:0.25). The solution was then reduced to less than 1 mL, and 10 mL of cold ether was added to the reduced solution to precipitate the small molecule product. The precipitate was pelleted and resuspended in cold ether 3 times to wash the compound and then dried under a fume hood for a few hours. The crude product was purified with preparative RP-HPLC.37 Please see Figure 1A for the molecule structures of LWW31, LWW31 conjugated with LKB, and LWW31 attached to beads. For making the on-bead small molecules, 75% of the TentaGel resin reaction sites were capped so that only 25% of the sites would react for the following small molecule synthesis. Again, neither topological segregation nor CL coupling was performed. Following the above procedure, the small molecule was coupled to the TentaGel resin, and the side chain protective groups were removed with TFA/TIS/H2O. The beads were then washed 3 or more times with DMF, DCM, and methanol and stored in 70% ethanol.
Biopanning of the T7select Phage Display Liver Tumor (Hepatocellular Carcinoma) cDNA Library against On-Bead Small Molecules
For identifying small molecule binding pairs, each identified small molecule was synthesized on thousands of Tentagel beads for easier handling during biopanning. The biopanning procedure was adapted from the phage display library manufacturer’s product manuals. Briefly, approximately 109 pfu of T7 phage display library was incubated with approximately 20,000 beads that conjugated to a single small molecule structure in each 1.5 mL polypropylene column. After 1–3 h of incubation, the beads were washed 3 times with PBSTB to wash out the unbound phages. The beads and the bound phages were subjected to incubation with 50 μL of log-phase E. coli BLT 5403 on the 280 rpm shaker at 37 °C for 30 min to elute bound phages from the beads by infecting E. coli. The beads were allowed to settle by gravity. Ten microliters of the supernatant was transferred to 1 mL of the log-phase E. coli and incubated on the 280 rpm shaker at 37 °C for 2 h or until the E. coli cells were totally lysed to amplify the bound phage population. After the cell debris was removed by centrifugation, one microliter of the supernatant containing amplified phages were diluted (1:1000) with 1 mL of PBSTB. The phages were incubated with another 20,000 beads, which carried the same small molecule as the previous round. After 4 rounds of biopanning with 3 rounds of amplification in between, the bound phages were eluted from beads after the fourth round of biopanning and diluted in 100 μL of LB, mixed with 250 μL of log-phase E. coli before immediately being mixed with 3 mL of 0.6% LB top agar. Prior to the top agar solidification, the entire mixture was poured onto the LB agar plate with 50 μg/mL of carbenicillin, cooled at room temperature for 5 min, and incubated at 37 °C for approximately 3 h or until plaques with reasonable sizes were visualized. Each plaque was picked up and inoculated in 1 mL of log phase E. coli to amplify the number of phage particles for storage, PCR amplification, and DNA sequencing. The PCR reagent composition and thermocycles were adapted from the manufacturer’s manual. After PCR amplification, 5 μL of the PCR product was mixed with 2 μL of ExoSAP-IT to remove excess primers according to the manufacturer’s manual. The total 7 μL solution was sent to Davis Sequencing, Inc. for DNA sequencing. The results were matched with sequence IDs in databases by using BLAST28 (basic local alignment searching tools). The DNA sequences were also submitted to Transeq to translate the DNA sequences into peptide sequences. These peptide sequences were identified with protein BLAST to make sure the cDNA, which encoded the corresponding protein, did not incur a frame-shift or mutation. The bound phage display protein ID and its small molecule partners could be paired up to create a database for further use to discover novel inhibitors and targets.
In Vitro Transcription/Translation Inhibition Assay
A Promega TnT-coupled reticulocyte lysate cell-free protein expression system was used to evaluate the inhibition effect of small molecule LWW31 against EIF5B, a eukaryotic translation initiation factor. For making the reaction mixture, 20 μL of the TnT Mix, 0.1 μL of 10 mM methionine, 3.9 μL of H2O, and 0.5 μL of small molecules in various concentrations in 100% DMSO were mixed in one well of a 96-well plate. The transcription and translation reactions were initiated by adding 0.5 μL of T7 luciferase control DNA also provided by Promega to the reaction mixture in each well. The luciferase expression level was monitored by adding luciferase substrate to the reaction, and the brightness of the resulting chemiluminescence was then read by a luminometer.
Pulldown Assay
Jurkat and HepG2 cells were collected and washed 3 times with PBS and then resuspended with cell lysis buffer (1% of Triton X-100, 0.1% of Tween 20, and a tablet of protease inhibitor without EDTA in 10 mL of PBS) at 200 μL per T75 flask to disrupt the cells. For ensuring the maximum rate of cell disruption, the resuspended cells were passed through a 26G needle 3 times. The cell debris was pelleted and removed by centrifugation at 15,000g. The resulting cell lysate was used for direct pulldown, DNase and RNase treatment, and/or ultra high speed (∼199,000g) air-driven centrifuge exposure (Airfuge by Beckman Coulter, Brea, CA) to remove nucleotides and organelles. Three microliters of small molecule stock solution (40 mM) in 100% DMSO was added to 150 μL of cell lysate and then incubated at 4 °C for 3 h with 120 μL of neutravidin agarose beads pre-equilibrated with PBS. The lysate–small molecule mixture was discarded after incubation, and the agarose beads were washed 4 times with cold PBS. The beads were resuspended with equal bead volume of SDS loading buffer and subjected to heat at 95 °C for 10 min and then cooled to room temperature. Thirty microliters of the heated sample were loaded onto each well of the SDS-PAGE 4–20% gradient gel and resolved at 200 V for 1 h. The resolved proteins were visualized by using silver staining. To confirm the protein ID and database match, the resolved proteins on gel were blotted on PVDF membranes, and the proteins of interest were detected with corresponding primary antibody and secondary antibodies conjugated with horseradish peroxidase (HRP). Western blot chemiluminescence images were visualized and recorded using a Kodak Image station 2000.
Cell Cycle Analysis
Jurkat cells treated with 32 μM of small molecule LWW31 for 24 h were pelleted (1500 rpm, 10 min) and resuspended in PBS. Ethanol (100%) was added to the suspension to make a 70% ethanol–cell suspension mixture. The mixture was incubated on ice for 15 min, and the cells were then pelleted again (1500 rpm for 5 min). The cell pellet was resuspended in 500 μL of 50 μg/mL propidium iodide (PI) containing 0.1 mg/mL of RNase and 0.05% of Triton X-100. The PI staining mixture was incubated at 37 °C for 40 min. Three milliliters of PBS was added to the PI staining mixture, and the stained cells were pelleted and resuspended in 500 μL of PBS for flow cytometry analysis. Data from 10,000 events were acquired using a Becton Dickenson FACstar flow cytometer (San Jose, CA), and the histograms were plotted and analyzed using a WinMDI version 2.9.
[35S]-Methionine Incorporation Assay
Approximately 105 HepG2 cells were seeded to each well of a 6-well plate with 1 mL of EMEM (Eagle’s minimum essential medium) containing 10% FBS and 5% penicillin/streptomycin. The cells were incubated in a 37 °C, 5% CO2 incubator for 1 day; the media was then removed, and the attached cells were washed with PBS 3 times. One milliliter of methionine-free RPMI 1640 (Invitrogen, Carlsbad, CA) containing 10% of dialyzed FBS (dFBS) was then added to the attached cells in each well, and the cells were incubated in a 37 °C, 5% CO2 incubator for another 1 h. After the incubation, another 1 mL of methionine-free RPMI 1640 containing 10% dFBS, 5–80 μM of LWW31, 10 μCi of [35S]-methionine, and 2 μg of cold methionine were added to each well, and the cells were incubated for another 24 h. A total of 2 mL of the media was removed after the 24 h incubation; the cells were washed with cold PBS 3 times, and 300 μL of RIPA buffer (Upstate, Temecula, CA) was added to each well to disrupt the cells. The cell lysate was centrifuged at 18,000g for 20 min to obtain a clear supernatant. Twenty micrograms of protein in each lysate was resolved in 4–20% gradient SDS-PAGE gels (Bio Rad, Hercules, CA) stained with coomassie blue and then dried in a gel drier. Kodak phosphorimaging screen-K was exposed to the dried gel in an autoradiography cassette for 3 days and scanned with a BioRad Personal Molecular Imager system to visualize and quantify the amount of [35S]-methionine-labeled protein. The dried gel was also scanned with a scanner, and the amount of total protein in each lane was quantified using ImageJ software. The amount of [35S]-methionine-labeled proteins was then normalized to the amount of total protein.
MS-Decoding MATLAB Scripts
Please see the Supporting Information.
Acknowledgments
This work was supported in part by NIH R33CA160132 grant awarded to K.S.L. We thank Dr. Jian Wu for his generous gift of the hHF cells. We also thank Dr. Alan Buckpitt, Dr. Daniel Feldman, Dr. David Olivos, and Dr. Mary Saunders for their valuable comments on this manuscript.
Supporting Information Available
The Supporting Information is available free of charge on the ACS Publications website at DOI: 10.1021/acscombsci.5b00194.
Materials, NMR data, MTT method, EIF5B nucleotide and protein sequences, MS decoding Matlab scripts, library building blocks, library synthetic scheme, hit structures, detailed biopanning results, MS decoding illustration, and LWW31 inhibition effect on rabbit reticulocyte lysate protein expression system (PDF)
Author Contributions
∇ C.-Y.W. and D.-H.W. contributed equally to this work. C.-Y.W, D.-H.W., and K.S.L. designed the research, analyzed the data, and prepared the manuscript. X.W. designed and synthesized the OBOC small molecule library. S.M.D., L.M., S.A., J.K., and L.J.L. synthesized the soluble and bead-tethered compounds. L.M. purified LWW31 and also performed NMR analysis. D.H.E. wrote the Matlab scripts. C.-Y.C. performed the [35S]-methionine incorporation experiment. Y.M. performed flow cytometry analysis. C.F.R. performed the MTT assay. L.M.R., B.R., and B.W. performed biopanning. H.-J.K. commented on the manuscript. K.S.L. supervised the overall project.
The authors declare no competing financial interest.
Supplementary Material
References
- al-Obeidi F. A.; Wu J. J.; Lam K. S. Protein tyrosine kinases: structure, substrate specificity, and drug discovery. Biopolymers 1998, 47, 197–223. . [DOI] [PubMed] [Google Scholar]
- Gao Y.; Amar S.; Pahwa S.; Fields G.; Kodadek T. Rapid lead discovery through iterative screening of one bead one compound libraries. ACS Comb. Sci. 2015, 17 (1), 49–59. 10.1021/co500154e. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kumaresan P. R.; Natarajan A.; Song A.; Wang X.; Liu R.; DeNardo G.; DeNardo S.; Lam K. S. Development of tissue plasminogen activator specific ″on demand cleavable″ (odc) linkers for radioimmunotherapy by screening one-bead-one-compound combinatorial peptide libraries. Bioconjugate Chem. 2007, 18 (1), 175–82. 10.1021/bc0602681. [DOI] [PubMed] [Google Scholar]
- Lam K. S.; Wu J.; Lou Q. Identification and characterization of a novel synthetic peptide substrate specific for Src-family protein tyrosine kinases. Int. J. Pept. Protein Res. 1995, 45 (6), 587–92. 10.1111/j.1399-3011.1995.tb01323.x. [DOI] [PubMed] [Google Scholar]
- Liu T.; Qian Z.; Xiao Q.; Pei D. High-throughput screening of one-bead-one-compound libraries: identification of cyclic peptidyl inhibitors against calcineurin/NFAT interaction. ACS Comb. Sci. 2011, 13 (5), 537–46. 10.1021/co200101w. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lou Q.; Leftwich M. E.; Lam K. S. Identification of GIYWHHY as a novel peptide substrate for human p60c-src protein tyrosine kinase. Bioorg. Med. Chem. 1996, 4 (5), 677–82. 10.1016/0968-0896(96)00063-6. [DOI] [PubMed] [Google Scholar]
- Lou Q.; Leftwich M. E.; McKay R. T.; Salmon S. E.; Rychetsky L.; Lam K. S. Potent pseudosubstrate-based peptide inhibitors for p60(c-src) protein tyrosine kinase. Cancer Res. 1997, 57 (10), 1877–81. [PubMed] [Google Scholar]
- Meldal M. The one-bead two-compound assay for solid phase screening of combinatorial libraries. Biopolymers 2002, 66 (2), 93–100. 10.1002/bip.10229. [DOI] [PubMed] [Google Scholar]
- Meldal M.; Svendsen I.; Breddam K.; Auzanneau F. I. Portion-mixing peptide libraries of quenched fluorogenic substrates for complete subsite mapping of endoprotease specificity. Proc. Natl. Acad. Sci. U. S. A. 1994, 91 (8), 3314–8. 10.1073/pnas.91.8.3314. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Qian Z.; Upadhyaya P.; Pei D. Synthesis and screening of one-bead-one-compound cyclic peptide libraries. Methods Mol. Biol. 2015, 1248, 39–53. 10.1007/978-1-4939-2020-4_3. [DOI] [PubMed] [Google Scholar]
- Wu J.; Ma Q. N.; Lam K. S. Identifying substrate motifs of protein kinases by a random library approach. Biochemistry 1994, 33 (49), 14825–33. 10.1021/bi00253a022. [DOI] [PubMed] [Google Scholar]
- Barbas C. F. 3rd; Kang A. S.; Lerner R. A.; Benkovic S. J. Assembly of combinatorial antibody libraries on phage surfaces: the gene III site. Proc. Natl. Acad. Sci. U. S. A. 1991, 88 (18), 7978–82. 10.1073/pnas.88.18.7978. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hertveldt K.; Belien T.; Volckaert G. General M13 phage display: M13 phage display in identification and characterization of protein-protein interactions. Methods Mol. Biol. 2009, 502, 321–39. 10.1007/978-1-60327-565-1_19. [DOI] [PubMed] [Google Scholar]
- Huang L.; Sato A. K.; Sachdeva M.; Fleming T.; Townsend S.; Dransfield D. T. Discovery of human antibodies against the C5aR target using phage display technology. J. Mol. Recognit. 2005, 18 (4), 327–33. 10.1002/jmr.735. [DOI] [PubMed] [Google Scholar]
- Hufton S. E.; Moerkerk P. T.; Meulemans E. V.; de Bruine A.; Arends J. W.; Hoogenboom H. R. Phage display of cDNA repertoires: the pVI display system and its applications for the selection of immunogenic ligands. J. Immunol. Methods 1999, 231 (1–2), 39–51. 10.1016/S0022-1759(99)00139-8. [DOI] [PubMed] [Google Scholar]
- Marks J. D.; Hoogenboom H. R.; Bonnert T. P.; McCafferty J.; Griffiths A. D.; Winter G. By-passing immunization. Human antibodies from V-gene libraries displayed on phage. J. Mol. Biol. 1991, 222 (3), 581–97. 10.1016/0022-2836(91)90498-U. [DOI] [PubMed] [Google Scholar]
- McCafferty J.; Griffiths A. D.; Winter G.; Chiswell D. J. Phage antibodies: filamentous phage displaying antibody variable domains. Nature 1990, 348 (6301), 552–4. 10.1038/348552a0. [DOI] [PubMed] [Google Scholar]
- Parmley S. F.; Smith G. P. Antibody-selectable filamentous fd phage vectors: affinity purification of target genes. Gene 1988, 73 (2), 305–18. 10.1016/0378-1119(88)90495-7. [DOI] [PubMed] [Google Scholar]
- Scott J. K.; Smith G. P. Searching for peptide ligands with an epitope library. Science 1990, 249 (4967), 386–90. 10.1126/science.1696028. [DOI] [PubMed] [Google Scholar]
- Smith G. P. Filamentous fusion phage: novel expression vectors that display cloned antigens on the virion surface. Science 1985, 228 (4705), 1315–7. 10.1126/science.4001944. [DOI] [PubMed] [Google Scholar]
- Smith G. P.; Petrenko V. A. Phage Display. Chem. Rev. 1997, 97 (2), 391–410. 10.1021/cr960065d. [DOI] [PubMed] [Google Scholar]
- Miyamoto S.; Liu R.; Hung S.; Wang X.; Lam K. S. Screening of a one bead-one compound combinatorial library for beta-actin identifies molecules active toward Ramos B-lymphoma cells. Anal. Biochem. 2008, 374 (1), 112–20. 10.1016/j.ab.2007.10.028. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lehman A.; Gholami S.; Hahn M.; Lam K. S. Image subtraction approach to screening one-bead-one-compound combinatorial libraries with complex protein mixtures. J. Comb. Chem. 2006, 8 (4), 562–70. 10.1021/cc0600268. [DOI] [PubMed] [Google Scholar]
- Gray B. P.; Brown K. C. Combinatorial peptide libraries: mining for cell-binding peptides. Chem. Rev. 2014, 114 (2), 1020–81. 10.1021/cr400166n. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wang X.; Peng L.; Liu R.; Xu B.; Lam K. S. Applications of topologically segregated bilayer beads in ’one-bead one-compound’ combinatorial libraries. J. Pept. Res. 2005, 65 (1), 130–8. 10.1111/j.1399-3011.2005.00192.x. [DOI] [PubMed] [Google Scholar]
- Furka A.; Sebestyen F.; Asgedom M.; Dibo G. General method for rapid synthesis of multicomponent peptide mixtures. Int. J. Pept. Protein Res. 1991, 37 (6), 487–93. 10.1111/j.1399-3011.1991.tb00765.x. [DOI] [PubMed] [Google Scholar]
- Lam K. S.; Salmon S. E.; Hersh E. M.; Hruby V. J.; Kazmierski W. M.; Knapp R. J. A new type of synthetic peptide library for identifying ligand-binding activity. Nature 1991, 354 (6348), 82–4. 10.1038/354082a0. [DOI] [PubMed] [Google Scholar]
- Altschul S. F.; Gish W.; Miller W.; Myers E. W.; Lipman D. J. Basic local alignment search tool. J. Mol. Biol. 1990, 215 (3), 403–10. 10.1016/S0022-2836(05)80360-2. [DOI] [PubMed] [Google Scholar]
- Pestova T. V.; Lomakin I. B.; Lee J. H.; Choi S. K.; Dever T. E.; Hellen C. U. The joining of ribosomal subunits in eukaryotes requires eIF5B. Nature 2000, 403 (6767), 332–5. 10.1038/35002118. [DOI] [PubMed] [Google Scholar]
- Laursen B. S.; Mortensen K. K.; Sperling-Petersen H. U.; Hoffman D. W. A conserved structural motif at the N terminus of bacterial translation initiation factor IF2. J. Biol. Chem. 2003, 278 (18), 16320–8. 10.1074/jbc.M212960200. [DOI] [PubMed] [Google Scholar]
- Roll-Mecak A.; Cao C.; Dever T. E.; Burley S. K. X-Ray structures of the universal translation initiation factor IF2/eIF5B: conformational changes on GDP and GTP binding. Cell 2000, 103 (5), 781–92. 10.1016/S0092-8674(00)00181-1. [DOI] [PubMed] [Google Scholar]
- Shin B. S.; Maag D.; Roll-Mecak A.; Arefin M. S.; Burley S. K.; Lorsch J. R.; Dever T. E. Uncoupling of initiation factor eIF5B/IF2 GTPase and translational activities by mutations that lower ribosome affinity. Cell 2002, 111 (7), 1015–25. 10.1016/S0092-8674(02)01171-6. [DOI] [PubMed] [Google Scholar]
- Unbehaun A.; Marintchev A.; Lomakin I. B.; Didenko T.; Wagner G.; Hellen C. U.; Pestova T. V. Position of eukaryotic initiation factor eIF5B on the 80S ribosome mapped by directed hydroxyl radical probing. EMBO J. 2007, 26 (13), 3109–23. 10.1038/sj.emboj.7601751. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Comess K. M.; Schurdak M. E. Affinity-based screening techniques for enhancing lead discovery. Curr. Opin Drug Discov Devel 2004, 7 (4), 411–6. [PubMed] [Google Scholar]
- Keseru G. M.; Makara G. M. Hit discovery and hit-to-lead approaches. Drug Discovery Today 2006, 11 (15–16), 741–8. 10.1016/j.drudis.2006.06.016. [DOI] [PubMed] [Google Scholar]
- Wang X.; Zhang J.; Song A.; Lebrilla C. B.; Lam K. S. Encoding method for OBOC small molecule libraries using a biphasic approach for ladder-synthesis of coding tags. J. Am. Chem. Soc. 2004, 126 (18), 5740–9. 10.1021/ja049322j. [DOI] [PubMed] [Google Scholar]
- Yao N.; Wu C. Y.; Xiao W.; Lam K. S. Discovery of high-affinity peptide ligands for vancomycin. Biopolymers 2008, 90 (3), 421–32. 10.1002/bip.20949. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.