

Introduction of the flexible aminoalcohol substructure of ifenprodil into a more rigid ring system resulted in 2-methyl-2,3,4,5-tetrahydro-1H-3-benzazepin-1-ols, (3) and (4), showing GluN2B affinity in the low nanomolar range. The chiral pool synthesis starting with (R)-alanine led to two diastereomers. The relative configuration of the benzazepines (3) and (4), that crystallized as racemates, was determined to be (S*,R*)-3 and (R*,R*)-4.
Keywords: crystal structure, NMDA receptor antagonists, GluN2B antagonists, ifenprodil analogs, tetrahydro-3-benzazepines, relative configuration, conformational restriction
Abstract
The title compounds, C22H29NO2 (3) and C22H29NO2 (4) [systematic names: (1S*,2R*)-7-methoxy-2-methyl-3-(4-phenylbutyl)-2,3,4,5-tetrahydro-1H-3-benzazepin-1-ol and (1R*,2R*)-7-methoxy-2-methyl-3-(4-phenylbutyl)-2,3,4,5-tetrahydro-1H-3-benzazepin-1-ol, are diastereomers with the relative configuration of the adjacent hydroxyl and methyl groups at the seven-membered azepine ring being trans in (3) and cis in (4). In the crystals the orientation of these groups is −anti-periplanar (3) and +syn-clinal (4). In both cases, the crystals studied proved to be of a racemic mixture, with relative configurations (R*,S*)-3 and (R*,R*)-4. In both compounds, the seven-membered azepine ring has a chair-like conformation, and the 4-phenylbutyl side chain adopts a extended conformation in (R*,S*)-3, but a twisted conformation in (R*,R*)-4. In the crystal of (S*,R*)-3, molecules are linked via C—H⋯O hydrogen bonds, forming slabs parallel to the ac plane. In the crystal of (R*,R*)-4, molecules are linked via O—H⋯N hydrogen bonds, forming chains propagating along the c-axis direction. The chains are linked by C—H⋯O hydrogen bonds, forming slabs parallel to the ac plane.
Chemical context
(S)-Glutamate is the most important excitatory neurotransmitter in the central nervous system. It interacts with different metabotropic and ionotropic glutamate receptors. The NMDA (N-methyl-d-aspartate) receptor is one of three ionotropic receptors, which control the influx of cations, in particular Na+ and Ca2+ ions, into neurons (Bräuner-Osborne et al., 2000 ▸; Kew & Kemp, 2005 ▸). Physiological activation of the NMDA receptor is associated with processes like learning and memory. However, over-activation of the NMDA receptor is connected with damage of neuronal cells leading finally to neuronal cell death. Therefore, inhibition of the NMDA associated ion channel could be useful for the treatment of traumatic brain injury, cerebral ischemia, neuropathic pain, depression and neurodegenerative disorders like Alzheimer’s and Parkinson’s disease (Bräuner-Osborne et al., 2000 ▸; Kew & Kemp, 2005 ▸; Paoletti et al., 2013 ▸; Wu & Zhou, 2009 ▸).
The aminoalcohol ifenprodil inhibits selectively NMDA receptors containing GluN2B subunits (Williams, 2001 ▸; Borza & Domány, 2006 ▸; Layton et al., 2006 ▸; Karakas et al., 2011 ▸). In order to improve the affinity, selectivity and metabolic stability of ifenprodil, the β-aminoalcohol substructure of ifenprodil was incorporated into a ring system resulting in seven-membered 3-benzazepines with high GluN2B affinity, high selectivity over related receptors and high metabolic stability (Tewes et al., 2010a ▸,b ▸; Schepmann et al., 2010 ▸; Falck et al., 2014 ▸).
Elucidation of the relative configuration
The 3-benzazepines (3) and (4) were prepared in a chiral pool synthesis starting with (R)-alanine. In a seven-step sequence the secondary amines (S,R)-1 and (R,R)-2 were obtained. In the last step, the secondary amines (S,R)-1 and (R,R)-2 were alkylated with 1-chloro-4-phenylbutane to afford the conformationally constrained ifenprodil analogues (3) and (4) which reveal high GluN2B affinity with K
i values of 47 nM and 41 nM, respectively (Tewes et al., 2015 ▸) (Fig. 1 ▸).
Figure 1.

Reaction scheme. Reagents and reaction conditions: (a) 1-chloro-4-phenylbutane, CH3CN, Bu4NI, K2CO3, Δ, 72 h.
As a result of the flexibility of the tetrahydro-3-benzazepine system of (1)–(4), the relative configuration of the 3-benzazepines (3) and (4) could not be determined unequivocally by interpretation of NMR spectra. However, crystallization of 70:30 mixtures of (S,R)-3 and (R,S)-3, as well as (R,R)-4 and (S,S)-4, led to colourless crystals which were suitable for X-ray crystal structure analysis. In both cases, the crystals proved to be of a racemic mixture, with the compounds having relative configurations (S*,R*)-3 and (R*,R*)-4.
Structural commentary
The molecular structures of compounds (S*,R*)-3 and (R*,R*)-4 are depicted in Figs. 2 ▸ and 3 ▸, respectively. In the structure of (S*,R*)-3 (Fig. 2 ▸), a trans-configuration with −anti-periplanar conformation and a torsion angle O12—C1—C2—C13 = −175.00 (12)°, of the OH group and the methyl group at the seven-membered azepine ring is shown. In (R*,R*)-4 (Fig. 3 ▸) the same substituents are cis-configured, in +syn-clinal conformation with torsion angle O12—C1—C2—C13 = 73.2 (7)°.
Figure 2.

The molecular structure of compound (S*,R*)-3, with atom labelling. Displacement ellipsoids are drawn at the 30% probability level.
Figure 3.

The molecular structure of compound (R*,R*)-4, with atom labelling. Displacement ellipsoids are drawn at the 30% probability level.
In compound (S*,R*)-3 the 4-phenylbutyl side chain adopts an extended conformation [torsion angle C16—C17—C18—C19 = 172.13 (14)°]. The CH3 and OH groups are on opposite sides of the azepine ring adopting an almost axial orientation. The bonds between atom N3 and its adjacent C atoms (C2, C16, C4) are shorter (ca. 1.47 Å) than the C—C bonds in the azepine ring (ca 1.52–1.54 Å). There is an intramolecular O-H⋯N contact present (Table 1 ▸) involving the O12 hydroxyl group and atom N3 of the 3-benzazepine ring, enclosing an S(5) ring motif.
Table 1. Hydrogen-bond geometry (Å, °) for (R*,S*)-3 .
| D—H⋯A | D—H | H⋯A | D⋯A | D—H⋯A |
|---|---|---|---|---|
| O12—H12⋯N3 | 0.83 | 2.17 | 2.6883 (17) | 120 |
| C15—H15B⋯O12i | 0.97 | 2.59 | 3.295 (2) | 130 |
| C21—H21⋯O12ii | 0.94 | 2.55 | 3.349 (2) | 143 |
| C22—H22⋯O14iii | 0.94 | 2.59 | 3.373 (3) | 141 |
Symmetry codes: (i) 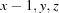 ; (ii)
; (ii) 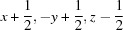 ; (iii)
; (iii) 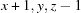 .
.
In compound (R*,R*)-4 the 4-phenylbutyl side chain exists in a twisted conformation torsion angle C16—C17—C18—C19 = 76.1 (9)°]. The CH3 group is on the opposite side of the azepine ring adopting an almost axial orientation, as for (S*,R*)-3. However, here the OH group adopts a more equatorial orientation at the seven-membered azepine ring, in contrast to the OH group of (S*,R*)-3. The angles of the aliphatic part of the 3-benzazepine ring are close to the tetrahedral angle value.
Supramolecular features
In the crystal of (S*,R*)-3, molecules are linked via C—H⋯O hydrogen bonds, forming slabs parallel to the ac plane (Table 1 ▸ and Fig. 4 ▸). In the crystal of (R*,R*)-4, molecules, are linked via O—H⋯N hydrogen bonds, forming chains propagating along the c-axis direction. The chains are linked by C—H⋯O hydrogen bonds, forming slabs parallel to the ac plane (Table 2 ▸ and Fig. 5 ▸).
Figure 4.

A view along the b axis of the crystal packing of compound (S*,R*)-3. The hydrogen bonds are shown as dashed lines (see Table 1 ▸); for clarity, only the H atoms involved in these interactions are included.
Table 2. Hydrogen-bond geometry (Å, °) for (R*,R*)-4 .
| D—H⋯A | D—H | H⋯A | D⋯A | D—H⋯A |
|---|---|---|---|---|
| O12—H12⋯N3i | 0.83 | 1.97 | 2.796 (6) | 172 |
| C15—H15C⋯O14ii | 0.97 | 2.58 | 3.365 (9) | 138 |
Symmetry codes: (i) 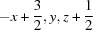 ; (ii)
; (ii) 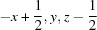 .
.
Figure 5.

A view along the b axis of the crystal packing of compound (R*,R*)-4. The hydrogen bonds are shown as dashed lines (see Table 2 ▸; for clarity, only the H atoms involved in these interactions are included.
Synthesis and crystallization
(1 S* ,2 R* )-7-Methoxy-2-methyl-3-(4-phenylbutyl)-2,3,4,5-tetrahydro-1 H -3-benzazepin-1-ol: ( S *,R *)-3
As described for the synthesis of (R,S)-3 (Tewes et al., 2015 ▸), the enantiomer (S,R)-3 was prepared in the same manner by alkylation of secondary amine (S,R)-1 [(S,R)-1:(R,S)-1 = 70:30] with 1-chloro-4-phenylbutane. Purification by flash chromatography (2 cm, n-hexane:ethyl acetate 95:5 and 1% N,N-dimethylethanamine, 10 ml, R f = 0.10) resulted in colourless crystals. The sample, contained the enantiomers (S,R)-3 and (R,S)-3 in the ratio 70:30. Spectroscopic data are given in Tewes et al. (2015 ▸).
(1 R* ,2 R* )-7-Methoxy-2-methyl-3-(4-phenylbutyl)-2,3,4,5-tetrahydro-1 H -3-benzazepin-1-ol: ( R *,R *)-4
As described for the synthesis of (S,S)-4 (Tewes et al., 2015 ▸), the enantiomer (R,R)-4 was prepared in the same manner by alkylation of secondary amine (R,R)-2 [(R,R)-1:(S,S)-1 = 70:30] with 1-chloro-4-phenylbutane. Purification by flash chromatography (2 cm, n-hexane:ethyl acetate 70: 30 and 1% N,N-dimethylethanamine, 10 ml, R f = 0.29) resulted in colourless crystals. The sample contained the enantiomers (R,R)-4 and (S,S)-4 in the ratio 70:30. Spectroscopic data are given in Tewes et al. (2015 ▸).
In both cases, the compounds were used for recrystallization with ethyl acetate and the crystals obtained were used for the subsequent X-ray crystal structure analyses. The crystals thus obtained proved to be racemic mixtures, with the compounds having relative configurations (R*,S*)-3 and (R*,R*)-4.
Refinement details
Crystal data, data collection and structure refinement details are summarized in Table 3 ▸. For both compounds the OH and C-bound H atoms were included in calculated positions and treated as riding atoms: O—H = 0.83 Å, C—H = 0.94–0.99 Å with U iso(H) = 1.5U eq(O or C-methyl) and 1.2U eq(C) for other H atoms.
Table 3. Experimental details.
| (R*,S*)-3 | (R*,R*)-4 | |
|---|---|---|
| Crystal data | ||
| Chemical formula | C22H29NO2 | C22H29NO2 |
| M r | 339.46 | 339.46 |
| Crystal system, space group | Monoclinic, P21/n | Orthorhombic, P c a21 |
| Temperature (K) | 223 | 223 |
| a, b, c (Å) | 10.3594 (2), 18.8246 (4), 10.9981 (3) | 9.2049 (5), 25.4468 (17), 8.2451 (6) |
| α, β, γ (°) | 90, 117.889 (1), 90 | 90, 90, 90 |
| V (Å3) | 1895.65 (8) | 1931.3 (2) |
| Z | 4 | 4 |
| Radiation type | Cu Kα | Cu Kα |
| μ (mm−1) | 0.59 | 0.58 |
| Crystal size (mm) | 0.40 × 0.25 × 0.10 | 0.35 × 0.05 × 0.03 |
| Data collection | ||
| Diffractometer | Nonius KappaCCD APEXII | Nonius KappaCCD APEXII |
| Absorption correction | Multi-scan (DENZO; Otwinowski et al., 2003 ▸) | Multi-scan (DENZO; Otwinowski et al., 2003 ▸) |
| T min, T max | 0.799, 0.944 | 0.824, 0.983 |
| No. of measured, independent and observed [I > 2σ(I)] reflections | 8812, 3077, 2864 | 8312, 2885, 2164 |
| R int | 0.034 | 0.082 |
| (sin θ/λ)max (Å−1) | 0.600 | 0.599 |
| Refinement | ||
| R[F 2 > 2σ(F 2)], wR(F 2), S | 0.045, 0.118, 1.04 | 0.087, 0.231, 1.25 |
| No. of reflections | 3077 | 2885 |
| No. of parameters | 229 | 229 |
| No. of restraints | 0 | 1 |
| H-atom treatment | H-atom parameters constrained | H-atom parameters constrained |
| Δρmax, Δρmin (e Å−3) | 0.16, −0.13 | 0.26, −0.25 |
Supplementary Material
Crystal structure: contains datablock(s) SR-3, RR-4, global. DOI: 10.1107/S2056989016005843/su5285sup1.cif
Structure factors: contains datablock(s) S,R-3. DOI: 10.1107/S2056989016005843/su5285SR-3sup3.hkl
Structure factors: contains datablock(s) R,R-4. DOI: 10.1107/S2056989016005843/su5285RR-4sup2.hkl
Supporting information file. DOI: 10.1107/S2056989016005843/su5285SR-3sup4.cml
Supporting information file. DOI: 10.1107/S2056989016005843/su5285RR-4sup5.cml
Additional supporting information: crystallographic information; 3D view; checkCIF report
Acknowledgments
Financial support by the Deutsche Forschungsgemeinschaft is gratefully acknowledged.
supplementary crystallographic information
(SR-3) (1S*,2R*)-7-Methoxy-2-methyl-3-(4-phenylbutyl)-2,3,4,5-tetrahydro-1H-3-benzazepin-1-ol . Crystal data
| C22H29NO2 | F(000) = 736 |
| Mr = 339.46 | Dx = 1.189 Mg m−3 |
| Monoclinic, P21/n | Cu Kα radiation, λ = 1.54178 Å |
| Hall symbol: -P 2yn | Cell parameters from 1877 reflections |
| a = 10.3594 (2) Å | θ = 0.9–68.3° |
| b = 18.8246 (4) Å | µ = 0.59 mm−1 |
| c = 10.9981 (3) Å | T = 223 K |
| β = 117.889 (1)° | Plate, colourless |
| V = 1895.65 (8) Å3 | 0.40 × 0.25 × 0.10 mm |
| Z = 4 |
(SR-3) (1S*,2R*)-7-Methoxy-2-methyl-3-(4-phenylbutyl)-2,3,4,5-tetrahydro-1H-3-benzazepin-1-ol . Data collection
| Nonius KappaCCD APEXII diffractometer | 3077 independent reflections |
| Radiation source: fine-focus sealed tube | 2864 reflections with I > 2σ(I) |
| Graphite monochromator | Rint = 0.034 |
| ω and \ scans | θmax = 67.6°, θmin = 5.1° |
| Absorption correction: multi-scan (DENZO; Otwinowski et al., 2003) | h = 0→12 |
| Tmin = 0.799, Tmax = 0.944 | k = 0→21 |
| 8812 measured reflections | l = −13→11 |
(SR-3) (1S*,2R*)-7-Methoxy-2-methyl-3-(4-phenylbutyl)-2,3,4,5-tetrahydro-1H-3-benzazepin-1-ol . Refinement
| Refinement on F2 | Primary atom site location: structure-invariant direct methods |
| Least-squares matrix: full | Secondary atom site location: difference Fourier map |
| R[F2 > 2σ(F2)] = 0.045 | Hydrogen site location: inferred from neighbouring sites |
| wR(F2) = 0.118 | H-atom parameters constrained |
| S = 1.04 | w = 1/[σ2(Fo2) + (0.056P)2 + 0.5327P] where P = (Fo2 + 2Fc2)/3 |
| 3077 reflections | (Δ/σ)max < 0.001 |
| 229 parameters | Δρmax = 0.16 e Å−3 |
| 0 restraints | Δρmin = −0.13 e Å−3 |
(SR-3) (1S*,2R*)-7-Methoxy-2-methyl-3-(4-phenylbutyl)-2,3,4,5-tetrahydro-1H-3-benzazepin-1-ol . Special details
| Geometry. All esds (except the esd in the dihedral angle between two l.s. planes) are estimated using the full covariance matrix. The cell esds are taken into account individually in the estimation of esds in distances, angles and torsion angles; correlations between esds in cell parameters are only used when they are defined by crystal symmetry. An approximate (isotropic) treatment of cell esds is used for estimating esds involving l.s. planes. |
| Refinement. Refinement of F2 against ALL reflections. The weighted R-factor wR and goodness of fit S are based on F2, conventional R-factors R are based on F, with F set to zero for negative F2. The threshold expression of F2 > 2sigma(F2) is used only for calculating R-factors(gt) etc. and is not relevant to the choice of reflections for refinement. R-factors based on F2 are statistically about twice as large as those based on F, and R- factors based on ALL data will be even larger. |
(SR-3) (1S*,2R*)-7-Methoxy-2-methyl-3-(4-phenylbutyl)-2,3,4,5-tetrahydro-1H-3-benzazepin-1-ol . Fractional atomic coordinates and isotropic or equivalent isotropic displacement parameters (Å2)
| x | y | z | Uiso*/Ueq | ||
| C1 | 0.66460 (15) | 0.15839 (8) | 0.42463 (15) | 0.0438 (4) | |
| H1 | 0.7477 | 0.1434 | 0.5128 | 0.053* | |
| C2 | 0.64344 (16) | 0.10045 (8) | 0.31824 (16) | 0.0461 (4) | |
| H2 | 0.7422 | 0.0894 | 0.3305 | 0.055* | |
| N3 | 0.56166 (13) | 0.13021 (7) | 0.17856 (13) | 0.0450 (3) | |
| C4 | 0.40433 (16) | 0.13908 (9) | 0.12903 (16) | 0.0489 (4) | |
| H4A | 0.3643 | 0.0943 | 0.1422 | 0.059* | |
| H4B | 0.3575 | 0.1489 | 0.0301 | 0.059* | |
| C5 | 0.36506 (16) | 0.19818 (9) | 0.20015 (16) | 0.0470 (4) | |
| H5A | 0.4188 | 0.2409 | 0.1996 | 0.056* | |
| H5B | 0.2608 | 0.2086 | 0.1447 | 0.056* | |
| C6 | 0.39431 (15) | 0.18510 (7) | 0.34623 (15) | 0.0417 (3) | |
| C7 | 0.27908 (16) | 0.19112 (8) | 0.37801 (16) | 0.0448 (4) | |
| H7 | 0.1850 | 0.2020 | 0.3078 | 0.054* | |
| C8 | 0.30111 (17) | 0.18140 (8) | 0.51121 (17) | 0.0456 (4) | |
| C9 | 0.43943 (18) | 0.16572 (9) | 0.61484 (17) | 0.0500 (4) | |
| H9 | 0.4556 | 0.1595 | 0.7057 | 0.060* | |
| C10 | 0.55380 (17) | 0.15931 (8) | 0.58400 (16) | 0.0475 (4) | |
| H10 | 0.6474 | 0.1483 | 0.6549 | 0.057* | |
| C11 | 0.53452 (15) | 0.16869 (7) | 0.45093 (15) | 0.0421 (3) | |
| O12 | 0.70726 (11) | 0.22260 (6) | 0.38429 (11) | 0.0492 (3) | |
| H12 | 0.6794 | 0.2218 | 0.3002 | 0.074* | |
| C13 | 0.5846 (2) | 0.03100 (9) | 0.3454 (2) | 0.0621 (5) | |
| H13A | 0.4891 | 0.0393 | 0.3389 | 0.093* | |
| H13B | 0.6509 | 0.0138 | 0.4368 | 0.093* | |
| H13C | 0.5763 | −0.0042 | 0.2777 | 0.093* | |
| O14 | 0.19492 (12) | 0.18687 (7) | 0.55197 (13) | 0.0607 (3) | |
| C15 | 0.04982 (18) | 0.20161 (11) | 0.4508 (2) | 0.0653 (5) | |
| H15A | 0.0473 | 0.2467 | 0.4069 | 0.098* | |
| H15B | −0.0131 | 0.2041 | 0.4939 | 0.098* | |
| H15C | 0.0159 | 0.1642 | 0.3822 | 0.098* | |
| C16 | 0.59495 (18) | 0.09299 (9) | 0.07866 (17) | 0.0528 (4) | |
| H16A | 0.5150 | 0.1010 | −0.0142 | 0.063* | |
| H16B | 0.6004 | 0.0418 | 0.0970 | 0.063* | |
| C17 | 0.73646 (17) | 0.11704 (9) | 0.08386 (17) | 0.0497 (4) | |
| H17A | 0.8169 | 0.1068 | 0.1754 | 0.060* | |
| H17B | 0.7329 | 0.1686 | 0.0703 | 0.060* | |
| C18 | 0.76778 (17) | 0.08148 (9) | −0.02367 (17) | 0.0492 (4) | |
| H18A | 0.7855 | 0.0307 | −0.0025 | 0.059* | |
| H18B | 0.6819 | 0.0861 | −0.1141 | 0.059* | |
| C19 | 0.89835 (18) | 0.11362 (8) | −0.02919 (18) | 0.0511 (4) | |
| H19A | 0.9830 | 0.1083 | 0.0619 | 0.061* | |
| H19B | 0.8804 | 0.1647 | −0.0464 | 0.061* | |
| C20 | 0.93912 (16) | 0.08411 (8) | −0.13445 (15) | 0.0439 (4) | |
| C21 | 1.04410 (18) | 0.11928 (9) | −0.15571 (17) | 0.0521 (4) | |
| H21 | 1.0858 | 0.1611 | −0.1062 | 0.063* | |
| C22 | 1.0888 (2) | 0.09450 (12) | −0.24730 (19) | 0.0661 (5) | |
| H22 | 1.1605 | 0.1193 | −0.2596 | 0.079* | |
| C23 | 1.0292 (2) | 0.03362 (12) | −0.32099 (19) | 0.0683 (5) | |
| H23 | 1.0608 | 0.0162 | −0.3826 | 0.082* | |
| C24 | 0.9229 (2) | −0.00145 (10) | −0.30370 (19) | 0.0638 (5) | |
| H24 | 0.8802 | −0.0426 | −0.3554 | 0.077* | |
| C25 | 0.87799 (18) | 0.02311 (8) | −0.21089 (18) | 0.0533 (4) | |
| H25 | 0.8056 | −0.0017 | −0.1996 | 0.064* |
(SR-3) (1S*,2R*)-7-Methoxy-2-methyl-3-(4-phenylbutyl)-2,3,4,5-tetrahydro-1H-3-benzazepin-1-ol . Atomic displacement parameters (Å2)
| U11 | U22 | U33 | U12 | U13 | U23 | |
| C1 | 0.0332 (7) | 0.0498 (8) | 0.0415 (8) | 0.0023 (6) | 0.0116 (7) | −0.0005 (6) |
| C2 | 0.0356 (7) | 0.0512 (8) | 0.0452 (9) | 0.0044 (6) | 0.0135 (7) | −0.0032 (6) |
| N3 | 0.0365 (6) | 0.0552 (7) | 0.0404 (7) | 0.0007 (5) | 0.0155 (6) | −0.0060 (5) |
| C4 | 0.0364 (8) | 0.0644 (10) | 0.0401 (8) | 0.0007 (6) | 0.0130 (7) | −0.0025 (7) |
| C5 | 0.0350 (7) | 0.0598 (9) | 0.0402 (9) | 0.0078 (6) | 0.0127 (7) | 0.0047 (6) |
| C6 | 0.0362 (7) | 0.0444 (7) | 0.0416 (8) | 0.0008 (6) | 0.0157 (7) | −0.0010 (6) |
| C7 | 0.0342 (7) | 0.0486 (8) | 0.0465 (9) | 0.0010 (6) | 0.0145 (7) | 0.0003 (6) |
| C8 | 0.0427 (8) | 0.0463 (8) | 0.0524 (9) | −0.0032 (6) | 0.0259 (8) | 0.0006 (6) |
| C9 | 0.0482 (9) | 0.0588 (9) | 0.0422 (9) | 0.0015 (7) | 0.0205 (8) | 0.0050 (7) |
| C10 | 0.0393 (8) | 0.0554 (9) | 0.0404 (8) | 0.0039 (6) | 0.0125 (7) | 0.0047 (6) |
| C11 | 0.0362 (7) | 0.0444 (8) | 0.0413 (8) | 0.0005 (6) | 0.0145 (7) | −0.0007 (6) |
| O12 | 0.0407 (6) | 0.0532 (6) | 0.0529 (7) | −0.0060 (4) | 0.0212 (6) | −0.0065 (5) |
| C13 | 0.0674 (11) | 0.0498 (9) | 0.0614 (11) | 0.0020 (8) | 0.0236 (10) | 0.0011 (8) |
| O14 | 0.0451 (6) | 0.0837 (8) | 0.0618 (8) | 0.0015 (5) | 0.0321 (6) | 0.0090 (6) |
| C15 | 0.0398 (9) | 0.0861 (13) | 0.0710 (13) | −0.0001 (8) | 0.0267 (9) | −0.0064 (9) |
| C16 | 0.0457 (9) | 0.0631 (10) | 0.0487 (9) | −0.0033 (7) | 0.0213 (8) | −0.0118 (7) |
| C17 | 0.0466 (9) | 0.0521 (9) | 0.0500 (9) | 0.0022 (6) | 0.0225 (8) | −0.0040 (7) |
| C18 | 0.0442 (8) | 0.0551 (9) | 0.0473 (9) | 0.0006 (6) | 0.0206 (8) | −0.0021 (7) |
| C19 | 0.0508 (9) | 0.0485 (8) | 0.0558 (10) | −0.0009 (7) | 0.0264 (8) | −0.0040 (7) |
| C20 | 0.0388 (8) | 0.0473 (8) | 0.0413 (8) | 0.0073 (6) | 0.0152 (7) | 0.0063 (6) |
| C21 | 0.0472 (9) | 0.0595 (9) | 0.0452 (9) | −0.0030 (7) | 0.0179 (8) | 0.0050 (7) |
| C22 | 0.0559 (10) | 0.0947 (14) | 0.0531 (11) | −0.0001 (9) | 0.0299 (9) | 0.0124 (10) |
| C23 | 0.0643 (11) | 0.0977 (15) | 0.0456 (10) | 0.0195 (10) | 0.0281 (9) | 0.0025 (9) |
| C24 | 0.0610 (11) | 0.0631 (11) | 0.0583 (11) | 0.0095 (8) | 0.0205 (9) | −0.0109 (8) |
| C25 | 0.0485 (9) | 0.0517 (9) | 0.0611 (10) | 0.0016 (7) | 0.0267 (8) | −0.0014 (7) |
(SR-3) (1S*,2R*)-7-Methoxy-2-methyl-3-(4-phenylbutyl)-2,3,4,5-tetrahydro-1H-3-benzazepin-1-ol . Geometric parameters (Å, º)
| C1—O12 | 1.4271 (18) | C9—C10 | 1.382 (2) |
| C1—C11 | 1.517 (2) | C10—C11 | 1.393 (2) |
| C1—C2 | 1.539 (2) | O14—C15 | 1.418 (2) |
| C2—N3 | 1.474 (2) | C16—C17 | 1.510 (2) |
| C2—C13 | 1.530 (2) | C17—C18 | 1.520 (2) |
| N3—C4 | 1.4664 (19) | C18—C19 | 1.509 (2) |
| N3—C16 | 1.4736 (19) | C19—C20 | 1.511 (2) |
| C4—C5 | 1.521 (2) | C20—C21 | 1.383 (2) |
| C5—C6 | 1.509 (2) | C20—C25 | 1.389 (2) |
| C6—C7 | 1.397 (2) | C21—C22 | 1.372 (3) |
| C6—C11 | 1.402 (2) | C22—C23 | 1.372 (3) |
| C7—C8 | 1.385 (2) | C23—C24 | 1.371 (3) |
| C8—O14 | 1.3721 (18) | C24—C25 | 1.385 (2) |
| C8—C9 | 1.383 (2) | ||
| O12—C1—C11 | 112.51 (12) | C10—C9—C8 | 119.56 (14) |
| O12—C1—C2 | 108.58 (12) | C9—C10—C11 | 121.94 (14) |
| C11—C1—C2 | 114.35 (12) | C10—C11—C6 | 118.37 (13) |
| N3—C2—C13 | 116.16 (13) | C10—C11—C1 | 118.72 (13) |
| N3—C2—C1 | 109.33 (12) | C6—C11—C1 | 122.88 (13) |
| C13—C2—C1 | 112.66 (13) | C8—O14—C15 | 118.41 (13) |
| C4—N3—C16 | 112.60 (12) | N3—C16—C17 | 113.02 (13) |
| C4—N3—C2 | 115.30 (12) | C16—C17—C18 | 113.29 (13) |
| C16—N3—C2 | 112.04 (12) | C19—C18—C17 | 112.11 (13) |
| N3—C4—C5 | 114.23 (13) | C18—C19—C20 | 117.42 (14) |
| C6—C5—C4 | 117.34 (13) | C21—C20—C25 | 117.73 (15) |
| C7—C6—C11 | 119.38 (14) | C21—C20—C19 | 118.49 (14) |
| C7—C6—C5 | 118.92 (13) | C25—C20—C19 | 123.78 (14) |
| C11—C6—C5 | 121.68 (13) | C22—C21—C20 | 121.55 (17) |
| C8—C7—C6 | 121.11 (14) | C21—C22—C23 | 120.28 (17) |
| O14—C8—C9 | 115.27 (14) | C24—C23—C22 | 119.27 (17) |
| O14—C8—C7 | 125.09 (14) | C23—C24—C25 | 120.67 (18) |
| C9—C8—C7 | 119.63 (14) | C24—C25—C20 | 120.47 (16) |
| O12—C1—C2—N3 | −44.25 (15) | C7—C6—C11—C1 | 177.61 (13) |
| C11—C1—C2—N3 | 82.30 (15) | C5—C6—C11—C1 | −3.9 (2) |
| O12—C1—C2—C13 | −175.00 (12) | O12—C1—C11—C10 | −114.59 (15) |
| C11—C1—C2—C13 | −48.45 (18) | C2—C1—C11—C10 | 120.93 (15) |
| C13—C2—N3—C4 | 52.51 (18) | O12—C1—C11—C6 | 67.47 (18) |
| C1—C2—N3—C4 | −76.33 (15) | C2—C1—C11—C6 | −57.01 (19) |
| C13—C2—N3—C16 | −78.04 (16) | C9—C8—O14—C15 | −178.90 (15) |
| C1—C2—N3—C16 | 153.12 (12) | C7—C8—O14—C15 | 1.9 (2) |
| C16—N3—C4—C5 | −158.97 (13) | C4—N3—C16—C17 | 147.99 (14) |
| C2—N3—C4—C5 | 70.75 (17) | C2—N3—C16—C17 | −80.09 (17) |
| N3—C4—C5—C6 | −72.96 (18) | N3—C16—C17—C18 | −177.17 (14) |
| C4—C5—C6—C7 | −123.71 (15) | C16—C17—C18—C19 | 172.13 (14) |
| C4—C5—C6—C11 | 57.74 (19) | C17—C18—C19—C20 | −178.61 (13) |
| C11—C6—C7—C8 | 0.2 (2) | C18—C19—C20—C21 | 170.56 (14) |
| C5—C6—C7—C8 | −178.36 (13) | C18—C19—C20—C25 | −10.0 (2) |
| C6—C7—C8—O14 | 179.40 (14) | C25—C20—C21—C22 | −1.1 (2) |
| C6—C7—C8—C9 | 0.2 (2) | C19—C20—C21—C22 | 178.45 (15) |
| O14—C8—C9—C10 | −179.82 (14) | C20—C21—C22—C23 | 0.2 (3) |
| C7—C8—C9—C10 | −0.6 (2) | C21—C22—C23—C24 | 1.0 (3) |
| C8—C9—C10—C11 | 0.5 (2) | C22—C23—C24—C25 | −1.4 (3) |
| C9—C10—C11—C6 | 0.0 (2) | C23—C24—C25—C20 | 0.5 (3) |
| C9—C10—C11—C1 | −178.04 (14) | C21—C20—C25—C24 | 0.7 (2) |
| C7—C6—C11—C10 | −0.3 (2) | C19—C20—C25—C24 | −178.77 (16) |
| C5—C6—C11—C10 | 178.20 (14) |
(SR-3) (1S*,2R*)-7-Methoxy-2-methyl-3-(4-phenylbutyl)-2,3,4,5-tetrahydro-1H-3-benzazepin-1-ol . Hydrogen-bond geometry (Å, º)
| D—H···A | D—H | H···A | D···A | D—H···A |
| O12—H12···N3 | 0.83 | 2.17 | 2.6883 (17) | 120 |
| C15—H15B···O12i | 0.97 | 2.59 | 3.295 (2) | 130 |
| C21—H21···O12ii | 0.94 | 2.55 | 3.349 (2) | 143 |
| C22—H22···O14iii | 0.94 | 2.59 | 3.373 (3) | 141 |
Symmetry codes: (i) x−1, y, z; (ii) x+1/2, −y+1/2, z−1/2; (iii) x+1, y, z−1.
(RR-4) (1R*,2R*)-7-Methoxy-2-methyl-3-(4-phenylbutyl)-2,3,4,5-tetrahydro-1H-3-benzazepin-1-ol . Crystal data
| C22H29NO2 | F(000) = 736 |
| Mr = 339.46 | Dx = 1.167 Mg m−3 |
| Orthorhombic, Pca21 | Cu Kα radiation, λ = 1.54178 Å |
| Hall symbol: P 2c -2ac | Cell parameters from 2216 reflections |
| a = 9.2049 (5) Å | θ = 0.9–70.1° |
| b = 25.4468 (17) Å | µ = 0.57 mm−1 |
| c = 8.2451 (6) Å | T = 223 K |
| V = 1931.3 (2) Å3 | Needle, colourless |
| Z = 4 | 0.35 × 0.05 × 0.03 mm |
(RR-4) (1R*,2R*)-7-Methoxy-2-methyl-3-(4-phenylbutyl)-2,3,4,5-tetrahydro-1H-3-benzazepin-1-ol . Data collection
| Nonius KappaCCD APEXII diffractometer | 2885 independent reflections |
| Radiation source: fine-focus sealed tube | 2164 reflections with I > 2σ(I) |
| Graphite monochromator | Rint = 0.082 |
| ω and φ scans | θmax = 67.5°, θmin = 3.5° |
| Absorption correction: multi-scan (DENZO; Otwinowski et al., 2003) | h = −10→10 |
| Tmin = 0.824, Tmax = 0.983 | k = −30→30 |
| 8312 measured reflections | l = −9→9 |
(RR-4) (1R*,2R*)-7-Methoxy-2-methyl-3-(4-phenylbutyl)-2,3,4,5-tetrahydro-1H-3-benzazepin-1-ol . Refinement
| Refinement on F2 | Primary atom site location: structure-invariant direct methods |
| Least-squares matrix: full | Secondary atom site location: difference Fourier map |
| R[F2 > 2σ(F2)] = 0.087 | Hydrogen site location: inferred from neighbouring sites |
| wR(F2) = 0.231 | H-atom parameters constrained |
| S = 1.25 | w = 1/[σ2(Fo2) + (0.0636P)2 + 2.9552P] where P = (Fo2 + 2Fc2)/3 |
| 2885 reflections | (Δ/σ)max < 0.001 |
| 229 parameters | Δρmax = 0.26 e Å−3 |
| 1 restraint | Δρmin = −0.24 e Å−3 |
(RR-4) (1R*,2R*)-7-Methoxy-2-methyl-3-(4-phenylbutyl)-2,3,4,5-tetrahydro-1H-3-benzazepin-1-ol . Special details
| Geometry. All esds (except the esd in the dihedral angle between two l.s. planes) are estimated using the full covariance matrix. The cell esds are taken into account individually in the estimation of esds in distances, angles and torsion angles; correlations between esds in cell parameters are only used when they are defined by crystal symmetry. An approximate (isotropic) treatment of cell esds is used for estimating esds involving l.s. planes. |
| Refinement. Refinement of F2 against ALL reflections. The weighted R-factor wR and goodness of fit S are based on F2, conventional R-factors R are based on F, with F set to zero for negative F2. The threshold expression of F2 > 2sigma(F2) is used only for calculating R-factors(gt) etc. and is not relevant to the choice of reflections for refinement. R-factors based on F2 are statistically about twice as large as those based on F, and R- factors based on ALL data will be even larger. |
(RR-4) (1R*,2R*)-7-Methoxy-2-methyl-3-(4-phenylbutyl)-2,3,4,5-tetrahydro-1H-3-benzazepin-1-ol . Fractional atomic coordinates and isotropic or equivalent isotropic displacement parameters (Å2)
| x | y | z | Uiso*/Ueq | ||
| C1 | 0.6240 (7) | 0.2962 (3) | −0.1209 (7) | 0.0427 (15) | |
| H1 | 0.7154 | 0.3049 | −0.1780 | 0.051* | |
| C2 | 0.5358 (6) | 0.2577 (3) | −0.2273 (7) | 0.0415 (16) | |
| H2 | 0.5846 | 0.2233 | −0.2157 | 0.050* | |
| N3 | 0.5442 (5) | 0.2708 (2) | −0.4043 (6) | 0.0447 (13) | |
| C4 | 0.4711 (8) | 0.3205 (3) | −0.4472 (7) | 0.0514 (18) | |
| H4A | 0.3709 | 0.3191 | −0.4074 | 0.062* | |
| H4B | 0.4670 | 0.3233 | −0.5656 | 0.062* | |
| C5 | 0.5428 (8) | 0.3701 (3) | −0.3805 (7) | 0.0524 (18) | |
| H5A | 0.5133 | 0.4002 | −0.4472 | 0.063* | |
| H5B | 0.6484 | 0.3665 | −0.3898 | 0.063* | |
| C6 | 0.5040 (7) | 0.3814 (3) | −0.2034 (7) | 0.0416 (16) | |
| C7 | 0.4270 (7) | 0.4272 (3) | −0.1680 (7) | 0.0471 (16) | |
| H7 | 0.3990 | 0.4500 | −0.2520 | 0.057* | |
| C8 | 0.3912 (8) | 0.4391 (3) | −0.0060 (8) | 0.0554 (18) | |
| C9 | 0.4298 (8) | 0.4036 (3) | 0.1137 (8) | 0.062 (2) | |
| H9 | 0.4046 | 0.4105 | 0.2221 | 0.075* | |
| C10 | 0.5042 (8) | 0.3586 (3) | 0.0767 (7) | 0.0537 (19) | |
| H10 | 0.5286 | 0.3352 | 0.1605 | 0.064* | |
| C11 | 0.5447 (6) | 0.3465 (3) | −0.0823 (7) | 0.0427 (16) | |
| O12 | 0.6590 (5) | 0.26820 (19) | 0.0243 (5) | 0.0512 (12) | |
| H12 | 0.7485 | 0.2676 | 0.0365 | 0.077* | |
| C13 | 0.3828 (7) | 0.2497 (3) | −0.1623 (9) | 0.0564 (18) | |
| H13A | 0.3254 | 0.2810 | −0.1823 | 0.085* | |
| H13B | 0.3870 | 0.2430 | −0.0466 | 0.085* | |
| H13C | 0.3383 | 0.2199 | −0.2165 | 0.085* | |
| O14 | 0.3170 (6) | 0.4827 (2) | 0.0418 (6) | 0.0726 (16) | |
| C15 | 0.2708 (10) | 0.5186 (3) | −0.0778 (9) | 0.071 (2) | |
| H15A | 0.3543 | 0.5312 | −0.1380 | 0.106* | |
| H15B | 0.2221 | 0.5480 | −0.0264 | 0.106* | |
| H15C | 0.2040 | 0.5012 | −0.1514 | 0.106* | |
| C16 | 0.4847 (7) | 0.2273 (3) | −0.5048 (9) | 0.0536 (18) | |
| H16A | 0.4947 | 0.2372 | −0.6192 | 0.064* | |
| H16B | 0.3806 | 0.2242 | −0.4819 | 0.064* | |
| C17 | 0.5525 (7) | 0.1746 (3) | −0.4821 (8) | 0.0526 (17) | |
| H17A | 0.5222 | 0.1603 | −0.3771 | 0.063* | |
| H17B | 0.6584 | 0.1785 | −0.4801 | 0.063* | |
| C18 | 0.5110 (8) | 0.1357 (3) | −0.6165 (8) | 0.0556 (18) | |
| H18A | 0.5324 | 0.0998 | −0.5800 | 0.067* | |
| H18B | 0.4063 | 0.1380 | −0.6369 | 0.067* | |
| C19 | 0.5930 (12) | 0.1465 (4) | −0.7735 (10) | 0.090 (3) | |
| H19A | 0.5682 | 0.1819 | −0.8114 | 0.108* | |
| H19B | 0.6975 | 0.1459 | −0.7510 | 0.108* | |
| C20 | 0.5606 (9) | 0.1078 (3) | −0.9069 (9) | 0.062 (2) | |
| C21 | 0.6546 (10) | 0.0668 (4) | −0.9347 (10) | 0.077 (2) | |
| H21 | 0.7380 | 0.0627 | −0.8702 | 0.092* | |
| C22 | 0.6251 (13) | 0.0308 (4) | −1.0606 (13) | 0.090 (3) | |
| H22 | 0.6893 | 0.0027 | −1.0791 | 0.108* | |
| C23 | 0.5073 (14) | 0.0361 (4) | −1.1541 (12) | 0.094 (3) | |
| H23 | 0.4897 | 0.0119 | −1.2379 | 0.112* | |
| C24 | 0.4124 (12) | 0.0765 (4) | −1.1288 (12) | 0.092 (3) | |
| H24 | 0.3301 | 0.0805 | −1.1952 | 0.110* | |
| C25 | 0.4391 (10) | 0.1116 (4) | −1.0039 (12) | 0.080 (3) | |
| H25 | 0.3722 | 0.1388 | −0.9847 | 0.095* |
(RR-4) (1R*,2R*)-7-Methoxy-2-methyl-3-(4-phenylbutyl)-2,3,4,5-tetrahydro-1H-3-benzazepin-1-ol . Atomic displacement parameters (Å2)
| U11 | U22 | U33 | U12 | U13 | U23 | |
| C1 | 0.042 (3) | 0.055 (4) | 0.030 (3) | 0.004 (3) | −0.002 (3) | 0.008 (3) |
| C2 | 0.033 (3) | 0.061 (4) | 0.030 (3) | −0.007 (3) | 0.001 (3) | 0.007 (3) |
| N3 | 0.050 (3) | 0.060 (4) | 0.023 (2) | 0.004 (3) | 0.004 (2) | 0.003 (2) |
| C4 | 0.061 (5) | 0.063 (5) | 0.030 (3) | 0.007 (4) | −0.007 (3) | −0.001 (3) |
| C5 | 0.073 (5) | 0.063 (5) | 0.022 (3) | 0.005 (4) | 0.009 (3) | 0.004 (3) |
| C6 | 0.047 (4) | 0.051 (4) | 0.027 (3) | −0.003 (3) | 0.001 (3) | 0.000 (3) |
| C7 | 0.057 (4) | 0.060 (4) | 0.025 (3) | 0.001 (3) | 0.000 (3) | 0.001 (3) |
| C8 | 0.059 (4) | 0.067 (5) | 0.039 (4) | 0.010 (4) | 0.001 (3) | 0.001 (4) |
| C9 | 0.073 (5) | 0.080 (6) | 0.033 (4) | 0.014 (4) | 0.001 (4) | 0.000 (4) |
| C10 | 0.059 (4) | 0.074 (5) | 0.028 (4) | 0.014 (4) | 0.002 (3) | 0.004 (3) |
| C11 | 0.043 (3) | 0.061 (5) | 0.024 (3) | −0.004 (3) | 0.005 (3) | 0.004 (3) |
| O12 | 0.050 (2) | 0.072 (3) | 0.032 (2) | 0.007 (2) | −0.0035 (19) | 0.013 (2) |
| C13 | 0.044 (4) | 0.079 (5) | 0.046 (4) | −0.005 (4) | 0.004 (3) | 0.001 (4) |
| O14 | 0.096 (4) | 0.078 (4) | 0.044 (3) | 0.030 (3) | 0.002 (3) | −0.007 (3) |
| C15 | 0.090 (6) | 0.063 (5) | 0.059 (5) | 0.020 (5) | −0.014 (4) | −0.009 (4) |
| C16 | 0.049 (4) | 0.069 (5) | 0.043 (4) | 0.004 (4) | −0.005 (3) | −0.012 (4) |
| C17 | 0.051 (4) | 0.061 (5) | 0.046 (4) | 0.002 (3) | 0.000 (3) | −0.002 (4) |
| C18 | 0.065 (4) | 0.058 (5) | 0.044 (4) | −0.011 (4) | 0.003 (3) | 0.003 (3) |
| C19 | 0.126 (9) | 0.088 (7) | 0.055 (5) | −0.024 (6) | 0.027 (5) | −0.016 (5) |
| C20 | 0.082 (6) | 0.063 (5) | 0.040 (4) | −0.012 (4) | 0.006 (4) | 0.000 (4) |
| C21 | 0.073 (5) | 0.096 (7) | 0.062 (5) | 0.001 (5) | 0.006 (4) | 0.004 (5) |
| C22 | 0.111 (8) | 0.065 (6) | 0.095 (7) | 0.006 (6) | 0.033 (6) | −0.016 (5) |
| C23 | 0.125 (9) | 0.091 (7) | 0.065 (6) | −0.030 (7) | 0.012 (6) | −0.024 (6) |
| C24 | 0.097 (7) | 0.114 (9) | 0.065 (6) | −0.021 (7) | −0.017 (5) | 0.002 (6) |
| C25 | 0.083 (6) | 0.075 (6) | 0.080 (6) | 0.012 (5) | 0.008 (5) | 0.006 (5) |
(RR-4) (1R*,2R*)-7-Methoxy-2-methyl-3-(4-phenylbutyl)-2,3,4,5-tetrahydro-1H-3-benzazepin-1-ol . Geometric parameters (Å, º)
| C1—O12 | 1.429 (7) | C13—H13C | 0.9700 |
| C1—C11 | 1.509 (9) | O14—C15 | 1.410 (9) |
| C1—C2 | 1.545 (9) | C15—H15A | 0.9700 |
| C1—H1 | 0.9900 | C15—H15B | 0.9700 |
| C2—N3 | 1.498 (7) | C15—H15C | 0.9700 |
| C2—C13 | 1.521 (9) | C16—C17 | 1.492 (9) |
| C2—H2 | 0.9900 | C16—H16A | 0.9800 |
| N3—C4 | 1.477 (8) | C16—H16B | 0.9800 |
| N3—C16 | 1.486 (8) | C17—C18 | 1.535 (10) |
| C4—C5 | 1.525 (9) | C17—H17A | 0.9800 |
| C4—H4A | 0.9800 | C17—H17B | 0.9800 |
| C4—H4B | 0.9800 | C18—C19 | 1.524 (10) |
| C5—C6 | 1.531 (8) | C18—H18A | 0.9800 |
| C5—H5A | 0.9800 | C18—H18B | 0.9800 |
| C5—H5B | 0.9800 | C19—C20 | 1.507 (11) |
| C6—C11 | 1.387 (9) | C19—H19A | 0.9800 |
| C6—C7 | 1.395 (9) | C19—H19B | 0.9800 |
| C7—C8 | 1.408 (9) | C20—C21 | 1.374 (11) |
| C7—H7 | 0.9400 | C20—C25 | 1.378 (11) |
| C8—O14 | 1.361 (8) | C21—C22 | 1.410 (13) |
| C8—C9 | 1.384 (9) | C21—H21 | 0.9400 |
| C9—C10 | 1.369 (9) | C22—C23 | 1.338 (14) |
| C9—H9 | 0.9400 | C22—H22 | 0.9400 |
| C10—C11 | 1.397 (8) | C23—C24 | 1.365 (14) |
| C10—H10 | 0.9400 | C23—H23 | 0.9400 |
| O12—H12 | 0.8300 | C24—C25 | 1.383 (13) |
| C13—H13A | 0.9700 | C24—H24 | 0.9400 |
| C13—H13B | 0.9700 | C25—H25 | 0.9400 |
| O12—C1—C11 | 110.8 (5) | H13B—C13—H13C | 109.5 |
| O12—C1—C2 | 106.2 (5) | C8—O14—C15 | 118.5 (6) |
| C11—C1—C2 | 113.8 (5) | O14—C15—H15A | 109.5 |
| O12—C1—H1 | 108.6 | O14—C15—H15B | 109.5 |
| C11—C1—H1 | 108.6 | H15A—C15—H15B | 109.5 |
| C2—C1—H1 | 108.6 | O14—C15—H15C | 109.5 |
| N3—C2—C13 | 114.9 (5) | H15A—C15—H15C | 109.5 |
| N3—C2—C1 | 112.7 (5) | H15B—C15—H15C | 109.5 |
| C13—C2—C1 | 111.8 (5) | N3—C16—C17 | 116.4 (5) |
| N3—C2—H2 | 105.5 | N3—C16—H16A | 108.2 |
| C13—C2—H2 | 105.5 | C17—C16—H16A | 108.2 |
| C1—C2—H2 | 105.5 | N3—C16—H16B | 108.2 |
| C4—N3—C16 | 109.7 (5) | C17—C16—H16B | 108.2 |
| C4—N3—C2 | 113.6 (5) | H16A—C16—H16B | 107.3 |
| C16—N3—C2 | 111.1 (5) | C16—C17—C18 | 112.7 (6) |
| N3—C4—C5 | 115.2 (5) | C16—C17—H17A | 109.0 |
| N3—C4—H4A | 108.5 | C18—C17—H17A | 109.0 |
| C5—C4—H4A | 108.5 | C16—C17—H17B | 109.0 |
| N3—C4—H4B | 108.5 | C18—C17—H17B | 109.0 |
| C5—C4—H4B | 108.5 | H17A—C17—H17B | 107.8 |
| H4A—C4—H4B | 107.5 | C19—C18—C17 | 111.9 (6) |
| C4—C5—C6 | 113.5 (5) | C19—C18—H18A | 109.2 |
| C4—C5—H5A | 108.9 | C17—C18—H18A | 109.2 |
| C6—C5—H5A | 108.9 | C19—C18—H18B | 109.2 |
| C4—C5—H5B | 108.9 | C17—C18—H18B | 109.2 |
| C6—C5—H5B | 108.9 | H18A—C18—H18B | 107.9 |
| H5A—C5—H5B | 107.7 | C20—C19—C18 | 113.8 (7) |
| C11—C6—C7 | 121.4 (5) | C20—C19—H19A | 108.8 |
| C11—C6—C5 | 120.2 (6) | C18—C19—H19A | 108.8 |
| C7—C6—C5 | 118.4 (5) | C20—C19—H19B | 108.8 |
| C6—C7—C8 | 119.8 (6) | C18—C19—H19B | 108.8 |
| C6—C7—H7 | 120.1 | H19A—C19—H19B | 107.7 |
| C8—C7—H7 | 120.1 | C21—C20—C25 | 117.9 (8) |
| O14—C8—C9 | 117.0 (6) | C21—C20—C19 | 119.6 (9) |
| O14—C8—C7 | 124.5 (6) | C25—C20—C19 | 122.6 (8) |
| C9—C8—C7 | 118.4 (7) | C20—C21—C22 | 119.6 (9) |
| C10—C9—C8 | 121.0 (6) | C20—C21—H21 | 120.2 |
| C10—C9—H9 | 119.5 | C22—C21—H21 | 120.2 |
| C8—C9—H9 | 119.5 | C23—C22—C21 | 121.0 (9) |
| C9—C10—C11 | 121.8 (6) | C23—C22—H22 | 119.5 |
| C9—C10—H10 | 119.1 | C21—C22—H22 | 119.5 |
| C11—C10—H10 | 119.1 | C22—C23—C24 | 120.4 (9) |
| C6—C11—C10 | 117.5 (6) | C22—C23—H23 | 119.8 |
| C6—C11—C1 | 121.5 (5) | C24—C23—H23 | 119.8 |
| C10—C11—C1 | 120.9 (6) | C23—C24—C25 | 119.1 (10) |
| C1—O12—H12 | 109.5 | C23—C24—H24 | 120.5 |
| C2—C13—H13A | 109.5 | C25—C24—H24 | 120.5 |
| C2—C13—H13B | 109.5 | C20—C25—C24 | 122.0 (9) |
| H13A—C13—H13B | 109.5 | C20—C25—H25 | 119.0 |
| C2—C13—H13C | 109.5 | C24—C25—H25 | 119.0 |
| H13A—C13—H13C | 109.5 | ||
| O12—C1—C2—N3 | −155.6 (5) | C9—C10—C11—C6 | −1.4 (10) |
| C11—C1—C2—N3 | 82.3 (6) | C9—C10—C11—C1 | −178.8 (7) |
| O12—C1—C2—C13 | 73.2 (7) | O12—C1—C11—C6 | 177.6 (6) |
| C11—C1—C2—C13 | −49.0 (7) | C2—C1—C11—C6 | −62.8 (7) |
| C13—C2—N3—C4 | 62.0 (7) | O12—C1—C11—C10 | −5.1 (8) |
| C1—C2—N3—C4 | −67.7 (7) | C2—C1—C11—C10 | 114.5 (7) |
| C13—C2—N3—C16 | −62.1 (7) | C9—C8—O14—C15 | −177.4 (7) |
| C1—C2—N3—C16 | 168.2 (5) | C7—C8—O14—C15 | 0.6 (11) |
| C16—N3—C4—C5 | −167.4 (5) | C4—N3—C16—C17 | 177.1 (6) |
| C2—N3—C4—C5 | 67.6 (7) | C2—N3—C16—C17 | −56.6 (7) |
| N3—C4—C5—C6 | −80.9 (7) | N3—C16—C17—C18 | −166.4 (6) |
| C4—C5—C6—C11 | 63.5 (8) | C16—C17—C18—C19 | 76.1 (9) |
| C4—C5—C6—C7 | −116.2 (7) | C17—C18—C19—C20 | 177.5 (7) |
| C11—C6—C7—C8 | 1.0 (10) | C18—C19—C20—C21 | −98.4 (10) |
| C5—C6—C7—C8 | −179.2 (6) | C18—C19—C20—C25 | 81.5 (11) |
| C6—C7—C8—O14 | 179.8 (7) | C25—C20—C21—C22 | 0.7 (12) |
| C6—C7—C8—C9 | −2.2 (10) | C19—C20—C21—C22 | −179.3 (8) |
| O14—C8—C9—C10 | 179.8 (7) | C20—C21—C22—C23 | 0.3 (14) |
| C7—C8—C9—C10 | 1.6 (11) | C21—C22—C23—C24 | −0.3 (16) |
| C8—C9—C10—C11 | 0.2 (12) | C22—C23—C24—C25 | −0.7 (16) |
| C7—C6—C11—C10 | 0.7 (9) | C21—C20—C25—C24 | −1.7 (13) |
| C5—C6—C11—C10 | −179.0 (6) | C19—C20—C25—C24 | 178.3 (8) |
| C7—C6—C11—C1 | 178.1 (6) | C23—C24—C25—C20 | 1.8 (15) |
| C5—C6—C11—C1 | −1.7 (9) |
(RR-4) (1R*,2R*)-7-Methoxy-2-methyl-3-(4-phenylbutyl)-2,3,4,5-tetrahydro-1H-3-benzazepin-1-ol . Hydrogen-bond geometry (Å, º)
| D—H···A | D—H | H···A | D···A | D—H···A |
| O12—H12···N3i | 0.83 | 1.97 | 2.796 (6) | 172 |
| C15—H15C···O14ii | 0.97 | 2.58 | 3.365 (9) | 138 |
Symmetry codes: (i) −x+3/2, y, z+1/2; (ii) −x+1/2, y, z−1/2.
References
- Borza, I. & Domány, G. (2006). Curr. Top. Med. Chem. 6, 687–695. [DOI] [PubMed]
- Bräuner-Osborne, H., Egebjerg, J., Nielsen, E. Ø., Madsen, U. & Krogsgaard-Larsen, P. (2000). J. Med. Chem. 43, 2609–2645. [DOI] [PubMed]
- Falck, E., Begrow, F., Verspohl, E. & Wünsch, B. (2014). J. Pharm. Biomed. Anal. 88, 96–105. [DOI] [PubMed]
- Karakas, E., Simorowski, N. & Furukawa, H. (2011). Nature, 475, 249–253. [DOI] [PMC free article] [PubMed]
- Kew, J. N. C. & Kemp, J. A. (2005). Psychopharmacology, 179, 4–29. [DOI] [PubMed]
- Layton, M. E., Kelly, M. J. III & Rodzinak, K. J. (2006). Curr. Top. Med. Chem. 6, 697–709. [DOI] [PubMed]
- Macrae, C. F., Bruno, I. J., Chisholm, J. A., Edgington, P. R., McCabe, P., Pidcock, E., Rodriguez-Monge, L., Taylor, R., van de Streek, J. & Wood, P. A. (2008). J. Appl. Cryst. 41, 466–470.
- Nonius (1998). COLLECT. Nonius BV, Delft, The Netherlands.
- Otwinowski, Z., Borek, D., Majewski, W. & Minor, W. (2003). Acta Cryst. A59, 228–234. [DOI] [PubMed]
- Otwinowski, Z. & Minor, W. (1997). Methods in Enzymology, Vol. 276, Macromolecular Crystallography, Part A, edited by C. W. Carter Jr & R. M. Sweet, pp. 307–326. New York: Academic Press.
- Paoletti, P., Bellone, C. & Zhou, Q. (2013). Nat. Rev. Neurosci. 14, 383–400. [DOI] [PubMed]
- Schepmann, D., Frehland, B., Lehmkuhl, K., Tewes, B. & Wünsch, B. (2010). J. Pharm. Biomed. Anal. 53, 603–608. [DOI] [PubMed]
- Sheldrick, G. M. (2008). Acta Cryst. A64, 112–122. [DOI] [PubMed]
- Spek, A. L. (2009). Acta Cryst. D65, 148–155. [DOI] [PMC free article] [PubMed]
- Tewes, B., Frehland, B., Schepmann, D., Robaa, D., Uengwetwanit, T., Gaube, F., Winckler, T., Sippl, W. & Wünsch, B. (2015). J. Med. Chem. 58, 6293–6305 [DOI] [PubMed]
- Tewes, B., Frehland, B., Schepmann, D., Schmidtke, K.-U., Winckler, T. & Wünsch, B. (2010a). Chem. Med. Chem. 5, 687–695. [DOI] [PubMed]
- Tewes, B., Frehland, B., Schepmann, D., Schmidtke, K.-U., Winckler, T. & Wünsch, B. (2010b). Bioorg. Med. Chem. 18, 8005–8015. [DOI] [PubMed]
- Williams, K. (2001). Curr. Drug Targets, 2, 285–298. [DOI] [PubMed]
- Wu, L.-J. & Zhou, M. (2009). Neurotherapeutics, 6, 693–702. [DOI] [PMC free article] [PubMed]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Crystal structure: contains datablock(s) SR-3, RR-4, global. DOI: 10.1107/S2056989016005843/su5285sup1.cif
Structure factors: contains datablock(s) S,R-3. DOI: 10.1107/S2056989016005843/su5285SR-3sup3.hkl
Structure factors: contains datablock(s) R,R-4. DOI: 10.1107/S2056989016005843/su5285RR-4sup2.hkl
Supporting information file. DOI: 10.1107/S2056989016005843/su5285SR-3sup4.cml
Supporting information file. DOI: 10.1107/S2056989016005843/su5285RR-4sup5.cml
Additional supporting information: crystallographic information; 3D view; checkCIF report