

Abstract
Background
Proinflammatory immune response involves a complex series of molecular events leading to inflammatory reaction at a site, which enables host to combat plurality of infectious agents. It can be initiated by specific stimuli such as viral, bacterial, parasitic or allergenic antigens, or by non-specific stimuli such as LPS. On counter with such antigens, the complex interaction of antigen presenting cells, T cells and inflammatory mediators like IL1α, IL1β, TNFα, IL12, IL18 and IL23 lead to proinflammatory immune response and further clearance of infection. In this study, we have tried to establish a relation between amino acid sequence of antigen and induction of proinflammatory response.
Results
A total of 729 experimentally-validated proinflammatory and 171 non-proinflammatory epitopes were obtained from IEDB database. The A, F, I, L and V amino acids and AF, FA, FF, PF, IV, IN dipeptides were observed as preferred residues in proinflammatory epitopes. Using the compositional and motif-based features of proinflammatory and non-proinflammatory epitopes, we have developed machine learning-based models for prediction of proinflammatory response of peptides. The hybrid of motifs and dipeptide-based features displayed best performance with MCC = 0.58 and an accuracy of 87.6 %.
Conclusion
The amino acid sequence-based features of peptides were used to develop a machine learning-based prediction tool for the prediction of proinflammatory epitopes. This is a unique tool for the computational identification of proinflammatory peptide antigen/candidates and provides leads for experimental validations. The prediction model and tools for epitope mapping and similarity search are provided as a comprehensive web server which is freely available at http://metagenomics.iiserb.ac.in/proinflam/ and http://metabiosys.iiserb.ac.in/proinflam/.
Electronic supplementary material
The online version of this article (doi:10.1186/s12967-016-0928-3) contains supplementary material, which is available to authorized users.
Keywords: Proinflammatory, Antigens, Prediction, Vaccine, Machine-learning
Background
The role of peptides as therapeutic agents has gained considerable importance recently, and more than 7000 natural peptides have been reported which play a pivotal role in human physiology and have different applications such as, vaccines, and other immunotherapeutics [1]. However, in addition to the desired action, these peptides may show undesirable immuno-activity, for example B cell or T cell activation and other proinflammatory events [2–4]. Similarly in nature, different infectious agents also harbor immunomodulatory properties present in their proteins, which help them in initiation and progression of the disease [5, 6]. Several examples of proinflammatory reactions are known where the pathogens get advantage of inflammation caused by their antigen. A well-known example is the proinflammatory response induced by the peptide Hp(2–20) of Helicobacter pylori which induces proinflammatory activities such as, recruiting and activating various immune cells like neutrophils and monocytes, upregulation of integrins (Mac-1) and activation of the oxygen radical producing NADPH-oxidase. This leads to destruction of host mucosal tissue along with reduction in the viability and function of antineoplastic lymphocytes [7]. Similarly, the peptide gG-2p20, which corresponds to amino acids 190–205 of glycoprotein G-2 of Herpes Simplex Virus-2 (HSV-2), induces proinflammatory effects by recruiting and activating the phagocytic cells. This, in turn, leads to reduced function and viability of NK cells [8]. Since NK cells constitute early line of defense and particularly important in protection against HSV-2, such proinflammatory reaction caused by gG-2p20 peptide leads to HSV-2 infection. Furthermore, there are examples of other physiological diseases, such as transmissible spongiform encephalopathies (TSEs), where prion peptide PrP(106–126) increases the pathogenicity due to its proinflammatory nature [9]. Similarly, LL-37, a 37 amino acid proinflammatory peptide generated from hCAP18 protein, has a role in pathogenesis of rheumatoid arthritis, systemic lupus erythematosus, atherosclerosis etc. [10]. Another example of proinflammatory peptide is C-peptide, a cleavage product of proinsulin which is used in peptide-therapeutics. It has a proinflammatory response in different tissues and this property leads to inflammation in kidney and vasculature, worsening the disease in long term [11].
The above evidences of proinflammatory property of peptide sequences underscore the correlation between amino acid sequence and its proinflammatory behavior. To the best of authors knowledge, there are no computational studies reported till date where any sequence-based signature or feature has been investigated which could be responsible for proinflammatory behavior of a peptide. Although, several studies have focused on the prediction of different kind of immune epitopes, such as B cell epitopes [12–14], T cell epitopes [15–17], MHC binders [18], IL4-inducing peptides [19], IFN-gamma inducing MHC binders [20] and allergenicity [21, 22], there is no study known where the sequence-based features have been examined to determine the proinflammatory nature of peptides. In this work, we have analyzed amino acid sequence of experimentally validated proinflammatory epitopes (PiEs) in contrast to non-proinflammatory epitopes (NPiEs) and developed a machine learning-based classification method incorporating the sequence-based features, to predict the proinflammatory nature of peptides and proteins.
Results and discussion
The induction of proinflammatory immune response may be a desirable or undesirable property of peptide therapeutics. There are examples of therapeutic peptides where inflammation is a desirable property [3, 23]. However, examples like C-peptide have an undesirable proinflammatory behavior, which worsen the disease [11]. The aim of this study is to develop an in silico method for predicting PiEs. In this study, we have analyzed the sequence-based properties which may contribute to its proinflammatory nature. Although in the past, several studies have been carried out on allergenic proteins/peptides [21, 22], toxic peptides [24], MHC binders [18], CTL epitopes [17], and B cell epitopes [12]; this study focus on investigating the basic property of peptide antigens to initiate proinflammatory cascade, which involves recruiting several immune cells, activation of complement proteins and communication via different immune mediators, which are also known as cytokines. The cytokines, such as IL1α, IL1β, TNFα, IL12, IL18 and IL23, are considered as proinflammatory cytokines [25], which are established mediators measured during a proinflammatory reaction assay. In this study, the experimentally validated epitopes which are assayed positive for these cytokines were considered as PiEs. The epitopes which gave negative assay were considered as NPiEs (Fig. 1). The compositional and motif-based analysis were carried out on the main dataset, however, the prediction models were developed using both main and alternate dataset, as discussed in methods section.
Fig. 1.
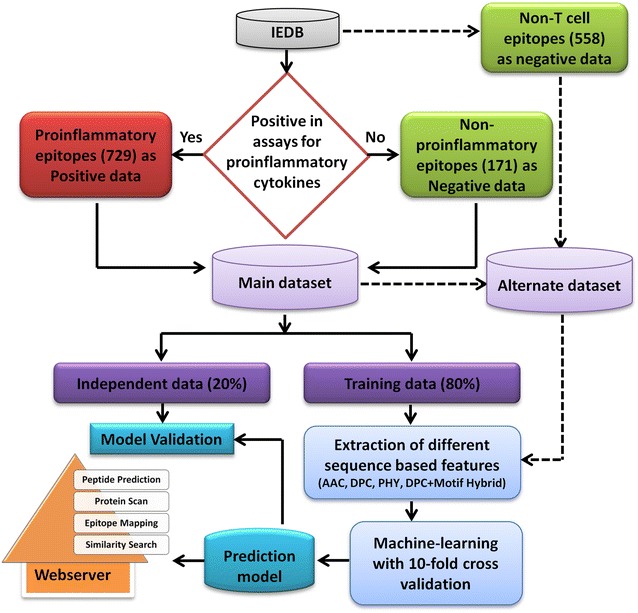
Flowchart showing steps involved in the development of prediction model and web server
Compositional analysis
In order to examine the abundance of amino acids in PiEs as compared to NPiEs, amino acid composition was computed for both the epitope classes. The compositional analysis revealed that the average composition of Ala, Phe, Iso, Leu and Val amino acids is higher in PiEs as compared to NPiEs (Fig. 2; Additional file 1: Table S1), whereas, the amino acids Cys, Gly, Pro and Thr are less abundant in PiEs. It suggests a preferential abundance of some amino acids in PiEs. Similarly, the dipeptide composition was examined in both the classes and several dipeptides were observed to be significantly abundant (Welch’s t test, p < 0.05) in PiEs. The composition of 91 dipeptides were found to differ significantly in PiEs as compared to NPiEs, of which the dipeptides AF, DA, GF, IN, KA, KD, RK, RM, TL and YA are the top ten dipeptides which showed the most significant differences in composition. Of these 91 significantly different dipeptides, AF, FA, FF, PF, IV, IN were also observed to be the most abundant in PiEs (Additional file 2: Table S2).
Fig. 2.
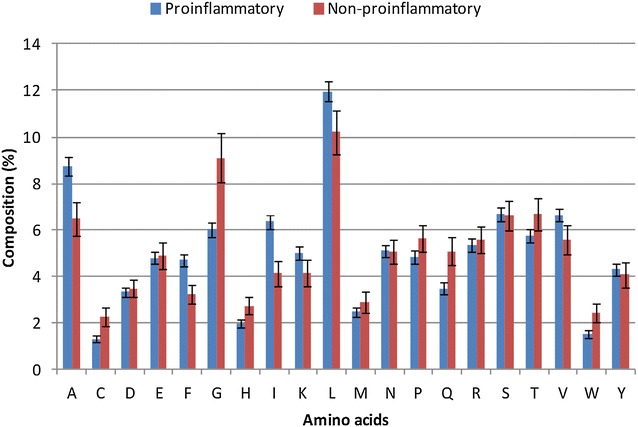
Compositional analysis of proinflammatory and non-proinflammatory epitopes
The composition-based analysis revealed that in PiEs, most of the preferred residues (Ala, Phe, Iso, Leu and Val) are aliphatic or hydrophobic amino acids. Similarly, the significant dipeptides, discussed in the above section, mostly included pairs of aliphatic or hydrophobic amino acids in different local orders.
Motif analysis
The proinflammatory immune response requires the activation of T cells by presentation of peptide bound to MHC molecule. Several key amino acids of the peptide bind at certain positions in the binding core of MHC which suggests that peptide may contain specific motif required for binding. In order to investigate if there are particular sequential motifs present in PiEs, the motifs were searched in the training datasets using MERCI software as described in the Methods section. After applying different algorithms available in MERCI software, such as none, Koolman–Rohm and Betts–Russell, several motifs were discovered which were found to be exclusively present in PiEs and NPiEs (Table 1; Additional file 3: Table S3). The overall coverage of a motif represents the total number of epitopes harboring that particular motif and same epitopes might also be covered by other motifs. Out of 583 PiEs present in positive dataset, the Betts–Russell algorithm, Koolman–Rohm algorithm and none algorithm could identify 256, 192 and 179 unique PiEs using 11, 10 and 10 motifs, respectively. Similarly, the above algorithms were used to identify motifs from NPiEs (Additional file 3: Table S3).
Table 1.
Number of exclusive proinflammatory (Np) and non-proinflammatory epitopes (Nn) covered by motifs identified using different algorithms of MERCI software
| Algorithm for motif discovery | Np | Nn |
|---|---|---|
| Betts–Russell | 256 | 29 |
| Koolman–Rohm | 192 | 15 |
| None | 179 | 9 |
For example, Betts–Russell algorithm-based proinflammatory and non-proinflammatory motifs could identify 256 proinflammatory as well as 29 non-proinflammatory unique epitopes, respectively
The “hydrophobic hydrophobic K hydrophobic hydrophobic” and “hydrophobic aliphatic polar N” were found to be the most frequent motifs and covered 54 and 44 unique proinflammatory epitopes, respectively. Among motifs given by Koolman–Rohm algorithm, “basic A aliphatic” was the most recurring motif covering 41 unique proinflammatory epitopes. Interestingly, the 10 motifs obtained in MERCI analysis with none algorithm were same as the significant dipeptides obtained in the compositional analysis. MERCI motifs, which were discovered exclusively in proinflammatory epitopes against non-proinflammatory epitopes, showed similar conservation of residues as observed in compositional analysis, for example, the motifs having highest coverage as shown in Table 2, are abundant in hydrophobic and aliphatic residues at various positions. Similar observations were also reported earlier where MBP(85–105) peptide binds to MHC allele DRB1 * 1501 with a nonaromatic, hydrophobic anchor (L, V, or I) at position i and by a bulky hydrophobic residue (F or Y) at position i + 3 as primary anchor which may contribute to its immunodominance [26].
Table 2.
Motifs discovered in proinflammatory epitopes along with the overall coverage for each motif
| Proinflammatory MERCI motifs | Overall coverage |
|---|---|
| Hydrophobic hydrophobic K hydrophobic hydrophobic | 54 |
| Hydrophobic aliphatic polar N | 48 |
| K hydrophobic aliphatic polar | 46 |
| Hydrophobic hydrophobic K small hydrophobic | 45 |
| Aliphatic R hydrophobic hydrophobic | 44 |
| Positive tiny L | 43 |
| Polar tiny hydrophobic aromatic hydrophobic | 43 |
| K hydrophobic L | 42 |
| Hydrophobic positive tiny hydrophobic polar | 42 |
| Hydrophobic hydrophobic aliphatic polar small aliphatic | 41 |
| Hydrophobic N aromatic hydrophobic | 41 |
Machine learning-based classification
The preliminary analysis unveiled that the PiEs differ from NPiEs in amino acid sequence-based features, and thus, sequence-based features can be exploited for their classification into PiEs and NPiEs epitopes using machine learning-based classification. Classification models for different features were developed using 6 different machine learning techniques (SVMlight and RandomForest, BayesNet, NaiveBayes, IBk and J48). However, the performance of RandomForest, BayesNet, NaiveBayes, IBk and J48 models was observed to be lower as compared to SVM-based models (Additional file 4: Table S4). Therefore, SVM-based models were implemented and discussed in the manuscript. Similarly, the performance of models on alternate dataset is mentioned in Additional file 5: Table S5.
Amino acid composition-based models
The two classes of epitopes differed in amino acid composition as mentioned in the compositional analysis (Fig. 2). Therefore, the amino acid composition was utilized to classify the two classes by developing machine learning models. After optimization of parameters, the best performing SVM-based model was selected with rbf kernel (t = 2), gamma parameter (g) = 0.005, trade off factor (c) = 80 and a cost factor (j) of 1. The model performed with an overall accuracy of 72.9 % and MCC was measured as 0.36. The threshold independent parameter area under curve (AUC) was found to be 0.77 (Table 3; Fig. 3).
Table 3.
Performance of different classification models developed using support vector machine as machine learning technique
| Feature | Thre | Sen | Spec | Acc | MCC | AUC | Parameters |
|---|---|---|---|---|---|---|---|
| Performance on training data | |||||||
| AAC | 0.6 | 73.58 | 70.07 | 72.92 | 0.36 | 0.77 | t:2 g:0.005 c:80 j:1 |
| DPC | 0.4 | 86.11 | 62.04 | 81.53 | 0.45 | 0.8 | t:2 g:0.001 c:10 j:1 |
| PHY | 0.7 | 91.25 | 24.82 | 78.61 | 0.20 | 0.57 | t:2 g:0.001:c:50:j:4 |
| DPCHyb_NONE | 0.4 | 87.82 | 62.04 | 82.92 | 0.48 | 0.84 | t:2 g:0.001 c:20 j:1 |
| DPCHyb_KOOL | 0.4 | 89.54 | 60.58 | 84.03 | 0.49 | 0.85 | t:2 g:0.001 c:4 j:2 |
| DPCHyb_BETTS | 0.3 | 93.65 | 62.04 | 87.64 | 0.58 | 0.88 | t:2 g:0.001 c:8 j:3 |
| Performance on validation data | |||||||
| DPCHyb_BETTS | 0.3 | 91.1 | 50 | 83.33 | 0.43 | 0.71 | |
The hybrid model prepared using Dipeptide composition based features and MERCI displayed the best performance with an accuracy of 87.6 %. The same model showed an accuracy of 83.3 % on validation dataset
Fig. 3.
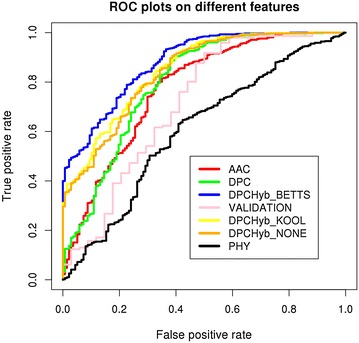
ROC plots of prediction models developed using SVMlight as machine learning technique. The DPCHyb_BETTS model (shown in blue) achieved highest area under curve (AUC = 0.88 as given in Table 3)
Dipeptide composition-based models
The models developed on AAC-based feature with vector size of 20, could not perform well both on threshold dependent as well as threshold independent parameters. Further, seeking better performance, dipeptide composition (DPC) was utilized, as input feature. The DPC-based SVM models yielded an overall accuracy of 81.5 % and MCC = 0.45 and AUC = 0.8. The optimized parameters for this model included rbf kernel (t = 2), g = 0.001, c = 10 and j = 1 (Table 3; Fig. 3).
Physiochemical properties-based models
While exploring different sequence-based features for better classification of PiEs from NPiEs, the physiochemical property-based models (explained in Methods) could only provide an accuracy of 79 % with MCC = 0.2, which was much lower as compared to AAC and DPC-based models (Table 3; Fig. 3) and hence, were not included in the tool.
Hybrid model
Among three sequence-based features mentioned above, the DPC-based model displayed the best performance. In order to further explore the possibility of better performance, the motif information was incorporated into the DPC-based models. A hybrid model of DPC and MERCI motifs were developed using the same methodology which was used to develop the DPC models and validated by tenfold cross-validation. Furthermore, three different models were developed employing three different algorithms of MERCI as described in method section. The overall accuracy of DPC-motif hybrid model with none algorithm (DPCHyb_NONE) was found to be 83 %, whereas, the DPC-motif hybrid model with Koolman–Rohm (DPCHyb_KOOL) gave an accuracy of 84 %. The DPC-motif hybrid with Betts–Russell algorithm yielded an overall accuracy of 87.6 %, MCC = 0.58 along with an AUC of 0.88 and was selected. The model was developed using rbf kernel with g = 0.001, c = 8, j = 3 (Table 3; Fig. 3).
Performance on validation dataset
Although, the tenfold cross validation technique is well accepted practice in machine learning methods, there are chances of over-fitting. In order to examine the possibility that the observed performance of the final model could be due to over-optimization, the model was tested on validation dataset. The DPCHyb_BETTS model displayed an accuracy of 83.3 % with MCC = 0.43 and AUC of 0.77 on validation dataset (Table 3).
The DPCHyb_BETTS model could achieve good accuracy and MCC on DPC-motif hybrid inputs. The strategy of giving weightage worked well and enhanced the performance of model in terms of accuracy, MCC and AUC. Since the prediction model performed well on unseen validation dataset, it attests that the performance of the model is not due to over optimization.
The performance of models developed on alternate dataset is mentioned in the Additional file 5: Table S5. In brief, among models developed on alternate dataset, RandomForest performed best using AAC as input feature. The accuracy of the AAC-based RandomForest model was 67.2 % with MCC = 0.34.
The DPCHyb_BETTS model (developed on main dataset) was incorporated in the webserver to allow the users to analyze and get predictions for their queries. It could serve as a computational substitute to the costly and time consuming experiments, as mentioned in case of HP(2–20) of H. pylori, gG2p20 of HSV-2, prion peptide PrP(106–126) and C-peptide of proinsulin. Using this web-based tool, users can sort down number of candidates responsible for proinflammatory nature of an antigen, which can further be validated by experimental studies.
Webserver and tools
The predictive modeling yielded classification tools with good accuracy for predicting the proinflammatory property of a peptide/protein and were used to develop a webserver by incorporating prediction model along with different other analysis tools. To show the translational application of “Proinflam” web server, a highly relevant example of the C-peptide from proinsulin protein is included in different modules (Peptide prediction, Protein Scan, Epitopes mapping and Similarity Search) of the web server. The C-peptide is a byproduct of proinsulin, located at position from 57 to 87 amino acids in the proinsulin protein, and was predicted as a proinflammatory epitope from the proinsulin protein using the web server. This peptide is responsible for the proinflammatory events in kidney and vasculature leading to diabetic nephropathy and atherosclerosis, respectively [11]. This example demonstrates the application and ability of the server to predict the proinflammatory epitopes in clinically relevant real proteins.
Peptide prediction
This module is designed for submission of single or batch of peptide/protein sequences in FASTA format with a length ranging from 4 to 30 amino acids. This tool runs the queries through the prediction pipeline with DPCHyb_BETTS model and classifies the queries into PiEs or NPiEs. The threshold option is provided to select the stringency of positive prediction.
As an additional function, virtual screening and designing option has been provided in the result table which allows the resubmission of the selected peptide. The virtual screening and designing involves substitution of amino acids at each position of peptide with the other 19 natural amino acids, which are further predicted in terms of PiEs or NPiEs and the results are displayed as a table. It allows the user to predict the proinflammatory nature of different variants of a given query.
Protein scan
Apart from peptide prediction module, which was meant for small length peptides, this module can be used for identification of antigenic regions in a protein, which can induce proinflammatory response in a host. The provision of window length allows user to select desired length of peptide for prediction. Similar to the peptide prediction module, this module runs query through prediction pipeline and virtual screening and provides the results as a table.
Epitope mapping
The prediction models achieved good accuracy in this study. However, user might want to investigate if there are previous reports of experimentally validated epitopes mapping to the query sequence. Therefore, ‘Epitope mapping’ module is developed to assist the user for mapping experimentally validated PiEs on the query sequence. Using this tool, the user can map the query sequence with PiEs and can also link to related assays in IEDB database.
Similarity search
In contrast to the epitope mapping module where exact match with experimental data is carried out, the ‘Similarity Search’ module performs Smith-Waterman search of query sequence in the database of experimentally validated PiEs. The top hits are shown with alignment and links to related assays in IEDB database.
Conclusion
The tendency of an antigen to initiate proinflammatory cascade, such as recruiting neutrophils, monocytes, and activate complement proteins, is of great importance in immunology and peptide therapeutics. Therefore, we have investigated sequence-based signatures which could be responsible for the proinflammatory nature of a peptide and developed a machine learning-based prediction tool for the prediction of proinflammatory epitopes. The computational identification of proinflammatory antigenic candidates before going for expensive and time-consuming experiments would be of great help to the scientific community. The developed computational tools are available freely for academic usage as a web server.
Methods
Dataset
The availability of clean experimental data is very crucial for any predictive modeling. Therefore, the Immune Epitope Database (IEDB) [27] for immune assays carried out for different peptide antigen was used to retrieve a clean dataset of 729 epitopes which were reported positive in assays in which any one of the proinflammatory cytokine [IL1α, IL1β, TNFα, IL12, IL18 and IL23] [25] was measured. Since these 729 epitopes were showing proinflammatory response in assays, these epitopes were considered as proinflammatory epitopes (PiEs) in this study. All those peptides which were assayed negative for proinflammatory cytokines were considered as non-proinflammatory epitopes (NPiEs). The total retrieved dataset contained 729 PiEs and 171 NPiEs ranging in length between 4 and 30 amino acids and was termed as ‘Main Dataset’. From the main dataset, 80 % of the data was assigned as training data and 20 % of the data was picked up randomly and kept as validation dataset (Fig. 1). The final training dataset contained 583 PiEs (positive data) and 137 NPiEs (negative data). The validation dataset contained 146 PiEs and 34 NPiEs.
Since the NPiEs (negative dataset) in main dataset was lesser in numbers as compare to PiEs (positive dataset), an additional dataset was constructed where the number of negative examples were kept equal to the number of positive examples. For this, 558 randomly picked non-T cell epitopes (NTCEs) data of 4–31 amino acid length were added to NPiEs of main dataset. The finalized alternate dataset contained 729 PiEs and 729 NPiEs (171 NPiEs + 558 NTCEs). The models were trained and tested in the same manner as done for the main dataset.
The main dataset was used for compositional and motif-based analysis, whereas, both main dataset and alternate dataset were used for the development of machine learning-based models. The models developed using the main dataset were incorporated in web server as prediction tool.
Input features for machine learning
Composition-based features
Amino acid composition
Amino acid composition (AAC) is the percentages of each amino acid in the given length of amino acid sequence. AAC has widely been applied in different peptide and protein composition based classification method [28, 29]. Since there are 20 amino acids, each peptide/protein is represented by 20 types of compositions or a vector size of 20.
where, AAC(i) is the amino acid composition of the amino acid (i) and amino acid (i) is one of the 20 amino acids.
Di-peptide composition
Similar to the AAC, dipeptide composition (DPC) has also been extensively applied in sequence-based classifications, particularly in the immune epitope prediction algorithms [30, 31]. DPC differs from AAC in having pair of amino acids and thus also provides information on local arrangement. The percentage of every possible pair (dipeptide) of amino acids was calculated. The following equation has been used for this calculation:
where, DPC(i) is the dipeptide frequency of dipeptide (i) and the dipeptide (i) is one out of 400 dipeptides.
Physiochemical properties
In earlier studies, different physiochemical properties of amino acids have been used in several classification methods for predicting immune epitopes [24, 32] and these features are also implemented in this study for developing the prediction models. 10 different physico-chemical properties were computed, namely amphipathicity, hydrophobicity, pI value, bulky side chain, hydrophilicity, net-hydrogen, steric hindrance, charge, hydropathy, molecular weight [32, 33].
Motif-based features
Identification of functional motifs in amino acid sequences has widely been exploited in functional annotation of protein/peptide sequences. Several authors have discovered immunologically relevant motifs in immunoinformatics studies [19, 34]. In this analysis, motifs specific to PiEs were identified using MERCI software (http://dtai.cs.kuleuven.be/ml/systems/merci) [35]. MERCI software is a tool to identify exclusive motifs present in positive data by comparing it with negative data. The exclusive motif discovery using MERCI was carried out using two steps. In the first step, PiEs were taken as positive and NPiEs were taken as negative data, which yielded exclusive motifs present in PiEs. In the second step, in order to get the exclusive motifs present in NPiEs, the datasets were reversed, i.e. NPiEs as positive data and PiEs as negative data.
While discovering the motifs with MERCI software, three algorithms were adopted: (a) none (b) Koolman–Rohm, and (c) Betts–Russell. The length of motif was set as maximum of 9 amino acids because the size of binding core in both MHC I and II is 9 amino acids [36, 37]. The gap length was set to default (1).
Hybrid feature
The hybrid of compositional features and motif-based feature has already been used in various prediction tools by different authors [19, 20]. In this study, a hybrid of DPC and MERCI-based motifs was used to improve the classification performance. As described above, two sets of motifs were identified as exclusively found in PiEs and NPiEs, respectively. In order to make a hybrid feature, the presence of proinflammatory and non-proinflammatory motifs was searched in the peptide. If the peptide is positive for proinflammatory motif, the weight of +1 was assigned to the DPC based SVM score. Similarly, if the peptide is positive for non-proinflammatory motif, a weight of −1 was assigned to the SVM score.
Machine-learning-based prediction models
Support vector machine
In this study, support vector machine is used as the machine learning algorithm implemented using SVMlight package available at http://svmlight.joachims.org/. SVM-based models are trained with a learning algorithm where it draws an optimal hyperplane in a multi-dimensional feature space that creating a boundary dividing the datasets in two classes. Among different machine learning techniques, SVM performs well because it is effective in controlling the classifier’s capacity and associated potential for overfitting [38]. In particular, SVM has been hugely implemented in various immune epitopes prediction tools [31, 39], protein structure prediction [40] and genomic data [41] because of its ability to handle noise and large dataset [42].
RandomForest
In this study, RandomForest (RF) has been implemented using randomForest package in R (http://cran.r-project.org//). RF has been widely used for the binary as well as multiclass classification using nucleotide or amino acid compositions as feature inputs [43]. RF classification model, at optimized parameters with lowest out-of-bag (OOB) error, was selected for the classification purpose. The overall performance of the selected model was evaluated in terms of sensitivity, specificity, accuracy and MCC from the confusion matrix.
WEKA-based techniques
In addition to the SVM and RF, BayesNet, NaiveBayes, IBk (kNN) and J48 machine learning algorithms were also evaluated through WEKA package. These techniques have already been implemented in immune epitope prediction studies in earlier studies [31, 44]. Similar to SVM and RF, the performances were evaluated using sensitivity, specificity, accuracy, MCC and AUC.
Evaluating performance of models
Evaluation and comparison of learning methods is essential part of predictive modeling. Cross-validation technique is among most practiced techniques which involve dividing the data into two segments; the first part is used to train the model and the other holdout or test data is used to test the model. Tenfold cross validation technique has been adopted where at a given instance, nine segments were used in training the model and the rest one segment was used to test the model. The process is repeated ten times such that each segment can be tested. The performance of model was calculated by including results from all the ten predictions taken together. The performance of models can be measured by both threshold dependent as well as threshold independent parameters. For threshold independent parameter, AUC was measured which was calculated by PERF software. For threshold dependent parameters, the parameters like sensitivity (Sen), specificity (Spec), accuracy (Acc) and Matthews’s correlation coefficient (MCC) were calculated. The following equations were used for the calculation of these parameters:
where, TP = True Positive, FP = False Positive, FN = False Negative, TN = True Negative.
Authors’ contributions
SG developed SVM and WEKA-based models and drafted the manuscript. AKS and MKM developed RandomForest models and revised the manuscript critically. SG and MKM developed web server. SG and VKS conceived the work, participated in the design of the study, and drafted the manuscript. All authors read and approved the final manuscript.
Acknowledgements
We thank MHRD, Govt of India, funded Centre for Research on Environment and Sustainable Technologies (CREST) at IISER Bhopal for its support. The views expressed in this manuscript are that of the authors alone and no approval of the same, explicit or implicit, by MHRD should be assumed.
Availability of data and materials
We have provided prediction models and other tools freely in the public domain at http://metagenomics.iiserb.ac.in/proinflam/ and http://metabiosys.iiserb.ac.in/proinflam/. In order to ensure reproducibility of our study and method, we have also provided the training datasets as well as validation datasets on the webserver.
Competing interests
The authors declare that they have no competing interests.
Funding
This work was supported by the intramural funding received from IISER Bhopal.
Abbreviations
- AAC
amino acid composition
- Acc
accuracy
- AUC
area under curve
- DPC
dipeptide composition
- IEDB
immune epitope database
- MCC
Matthews correlation coefficient
- MHC
major histocompatibility complex
- NPiEs
non-proinflammatory epitopes
- NTCEs
non T Cell epitopes
- OOB
out-of-bag
- PHY
physiochemical properties
- PiEs
proinflammatory epitopes
- RF
RandomForest
- Sen
sensitivity
- Spec
specificity
- SVM
support vector machine
Additional files
10.1186/s12967-016-0928-3 Amino acid composition distribution between proinflammatory and non-proinflammatory data.
10.1186/s12967-016-0928-3 Dipeptide composition distribution between proinflammatory and non proinflammatory data.
10.1186/s12967-016-0928-3 MERCI motifs extracted form proinflammatory and non-proinflammatory data.
10.1186/s12967-016-0928-3 Performance of models developed on main dataset, with RandomForest, BayesNet, NaiveBayes, IBk and J48.
10.1186/s12967-016-0928-3. Performance of different machine learning models developed on alternate dataset.
Contributor Information
Sudheer Gupta, Email: sudheer@iiserb.ac.in.
Midhun K. Madhu, Email: midhunk@iiserb.ac.in
Ashok K. Sharma, Email: ashok@iiserb.ac.in
Vineet K. Sharma, Email: vineetks@iiserb.ac.in
References
- 1.Fosgerau K, Hoffmann T. Peptide therapeutics: current status and future directions. Drug Discov Today. 2015;20:122–128. doi: 10.1016/j.drudis.2014.10.003. [DOI] [PubMed] [Google Scholar]
- 2.Shi J, Liu Y, Wang Y, Zhang J, Zhao S, Yang G. Biological and immunotoxicity evaluation of antimicrobial peptide-loaded coatings using a layer-by-layer process on titanium. Sci Rep. 2015;5:16336. doi: 10.1038/srep16336. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Gustafsson A, Sigel S, Ljunggren L. The antimicrobial peptide LL37 and its truncated derivatives potentiates proinflammatory cytokine induction by lipoteichoic acid in whole blood. Scand J Clin Lab Invest. 2010;70:512–518. doi: 10.3109/00365513.2010.521255. [DOI] [PubMed] [Google Scholar]
- 4.Gordon YJ, Romanowski EG, McDermott AM. A review of antimicrobial peptides and their therapeutic potential as anti-infective drugs. Curr Eye Res. 2005;30:505–515. doi: 10.1080/02713680590968637. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Hsu HY, Chang MH, Ni YH, Huang SF. Cytomegalovirus infection and proinflammatory cytokine activation modulate the surface immune determinant expression and immunogenicity of cultured murine extrahepatic bile duct epithelial cells. Clin Exp Immunol. 2001;126:84–91. doi: 10.1046/j.1365-2249.2001.01558.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Desmet VJ. Cholangiopathies: past, present, and future. Semin Liver Dis. 1987;7:67–76. doi: 10.1055/s-2008-1040566. [DOI] [PubMed] [Google Scholar]
- 7.Bylund J, Christophe T, Boulay F, Nystrom T, Karlsson A, Dahlgren C. Proinflammatory activity of a cecropin-like antibacterial peptide from Helicobacter pylori. Antimicrob Agents Chemother. 2001;45:1700–1704. doi: 10.1128/AAC.45.6.1700-1704.2001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Bellner L, Thoren F, Nygren E, Liljeqvist JA, Karlsson A, Eriksson K. A proinflammatory peptide from herpes simplex virus type 2 glycoprotein G affects neutrophil, monocyte, and NK cell functions. J Immunol. 2005;174:2235–2241. doi: 10.4049/jimmunol.174.4.2235. [DOI] [PubMed] [Google Scholar]
- 9.Bacot SM, Lenz P, Frazier-Jessen MR, Feldman GM. Activation by prion peptide PrP106-126 induces a NF-kappaB-driven proinflammatory response in human monocyte-derived dendritic cells. J Leukoc Biol. 2003;74:118–125. doi: 10.1189/jlb.1102521. [DOI] [PubMed] [Google Scholar]
- 10.Kahlenberg JM, Kaplan MJ. Little peptide, big effects: the role of LL-37 in inflammation and autoimmune disease. J Immunol. 2013;191:4895–4901. doi: 10.4049/jimmunol.1302005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Vasic D, Walcher D. Proinflammatory effects of C-Peptide in different tissues. Int J Inflamm. 2012;2012:932725. doi: 10.1155/2012/932725. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Saha S, Raghava GP. Prediction methods for B-cell epitopes. Methods Mol Biol. 2007;409:387–394. doi: 10.1007/978-1-60327-118-9_29. [DOI] [PubMed] [Google Scholar]
- 13.Singh H, Ansari HR, Raghava GP. Improved method for linear B-cell epitope prediction using antigen’s primary sequence. PLoS One. 2013;8:e62216. doi: 10.1371/journal.pone.0062216. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Y EL-Manzalawy Y, Honavar V. Building classifier ensembles for B-cell epitope prediction. Methods Mol Biol. 2014;1184:285–294. doi: 10.1007/978-1-4939-1115-8_15. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Desai DV, Kulkarni-Kale U. T-cell epitope prediction methods: an overview. Methods Mol Biol. 2014;1184:333–364. doi: 10.1007/978-1-4939-1115-8_19. [DOI] [PubMed] [Google Scholar]
- 16.Nielsen M, Lundegaard C, Worning P, Lauemoller SL, Lamberth K, Buus S, Brunak S, Lund O. Reliable prediction of T-cell epitopes using neural networks with novel sequence representations. Protein Sci. 2003;12:1007–1017. doi: 10.1110/ps.0239403. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Bhasin M, Raghava GP. Prediction of CTL epitopes using QM, SVM and ANN techniques. Vaccine. 2004;22:3195–3204. doi: 10.1016/j.vaccine.2004.02.005. [DOI] [PubMed] [Google Scholar]
- 18.Bhasin M, Raghava GP. A hybrid approach for predicting promiscuous MHC class I restricted T cell epitopes. J Biosci. 2007;32:31–42. doi: 10.1007/s12038-007-0004-5. [DOI] [PubMed] [Google Scholar]
- 19.Dhanda SK, Gupta S, Vir P, Raghava GP. Prediction of IL4 inducing peptides. Clin Dev Immunol. 2013;2013:263952. doi: 10.1155/2013/263952. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Dhanda SK, Vir P, Raghava GP. Designing of interferon-gamma inducing MHC class-II binders. Biol Direct. 2013;8:30. doi: 10.1186/1745-6150-8-30. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Saha S, Raghava GP. AlgPred: prediction of allergenic proteins and mapping of IgE epitopes. Nucleic Acids Res. 2006;34:W202–W209. doi: 10.1093/nar/gkl343. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Dimitrov I, Flower DR, Doytchinova I. AllerTOP—a server for in silico prediction of allergens. BMC Bioinform. 2013;14(Suppl 6):S4. doi: 10.1186/1471-2105-14-S6-S4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Wan M, Godson C, Guiry PJ, Agerberth B, Haeggstrom JZ. Leukotriene B4/antimicrobial peptide LL-37 proinflammatory circuits are mediated by BLT1 and FPR2/ALX and are counterregulated by lipoxin A4 and resolvin E1. FASEB J. 2011;25:1697–1705. doi: 10.1096/fj.10-175687. [DOI] [PubMed] [Google Scholar]
- 24.Gupta S, Kapoor P, Chaudhary K, Gautam A, Kumar R, Raghava GP. Peptide toxicity prediction. Methods Mol Biol. 2015;1268:143–157. doi: 10.1007/978-1-4939-2285-7_7. [DOI] [PubMed] [Google Scholar]
- 25.Dinarello CA. Historical insights into cytokines. Eur J Immunol. 2007;37(Suppl 1):S34–S45. doi: 10.1002/eji.200737772. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Vogt AB, Kropshofer H, Kalbacher H, Kalbus M, Rammensee HG, Coligan JE, Martin R. Ligand motifs of HLA-DRB5*0101 and DRB1*1501 molecules delineated from self-peptides. J Immunol. 1994;153:1665–1673. [PubMed] [Google Scholar]
- 27.Zhang Q, Wang P, Kim Y, Haste-Andersen P, Beaver J, Bourne PE, Bui HH, Buus S, Frankild S, Greenbaum J, et al. Immune epitope database analysis resource (IEDB-AR) Nucleic Acids Res. 2008;36:W513–W518. doi: 10.1093/nar/gkn254. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Gupta A, Kapil R, Dhakan DB, Sharma VK. MP3: a software tool for the prediction of pathogenic proteins in genomic and metagenomic data. PLoS One. 2014;9:e93907. doi: 10.1371/journal.pone.0093907. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Sharma AK, Gupta A, Kumar S, Dhakan DB, Sharma VK. Woods: a fast and accurate functional annotator and classifier of genomic and metagenomic sequences. Genomics. 2015;106:1–6. doi: 10.1016/j.ygeno.2015.04.001. [DOI] [PubMed] [Google Scholar]
- 30.Zhang J, Zhao X, Sun P, Gao B, Ma Z. Conformational B-cell epitopes prediction from sequences using cost-sensitive ensemble classifiers and spatial clustering. Biomed Res Int. 2014;2014:689219. doi: 10.1155/2014/689219. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Gupta S, Ansari HR, Gautam A, Raghava GP. Identification of B-cell epitopes in an antigen for inducing specific class of antibodies. Biol Direct. 2013;8:27. doi: 10.1186/1745-6150-8-27. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Melo MN, Ferre R, Feliu L, Bardaji E, Planas M, Castanho MA. Prediction of antibacterial activity from physicochemical properties of antimicrobial peptides. PLoS One. 2011;6:e28549. doi: 10.1371/journal.pone.0028549. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Sharma A, Singla D, Rashid M, Raghava GP. Designing of peptides with desired half-life in intestine-like environment. BMC Bioinform. 2014;15:282. doi: 10.1186/1471-2105-15-282. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Gupta S, Kapoor P, Chaudhary K, Gautam A, Kumar R, Raghava GP. In silico approach for predicting toxicity of peptides and proteins. PLoS One. 2013;8:e73957. doi: 10.1371/journal.pone.0073957. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Vens C, Rosso MN, Danchin EG. Identifying discriminative classification-based motifs in biological sequences. Bioinformatics. 2011;27:1231–1238. doi: 10.1093/bioinformatics/btr110. [DOI] [PubMed] [Google Scholar]
- 36.Rammensee HG, Friede T, Stevanoviic S. MHC ligands and peptide motifs: first listing. Immunogenetics. 1995;41:178–228. doi: 10.1007/BF00172063. [DOI] [PubMed] [Google Scholar]
- 37.Nielsen M, Lund O, Buus S, Lundegaard C. MHC class II epitope predictive algorithms. Immunology. 2010;130:319–328. doi: 10.1111/j.1365-2567.2010.03268.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Ward JJ, McGuffin LJ, Buxton BF, Jones DT. Secondary structure prediction with support vector machines. Bioinformatics. 2003;19:1650–1655. doi: 10.1093/bioinformatics/btg223. [DOI] [PubMed] [Google Scholar]
- 39.Wang HW, Pai TW. Machine learning-based methods for prediction of linear B-cell epitopes. Methods Mol Biol. 2014;1184:217–236. doi: 10.1007/978-1-4939-1115-8_12. [DOI] [PubMed] [Google Scholar]
- 40.Mohammad TA, Nagarajaram HA. SVM-based method for protein structural class prediction using secondary structural content and structural information of amino acids. J Bioinform Comput Biol. 2011;9:489–502. doi: 10.1142/S0219720011005422. [DOI] [PubMed] [Google Scholar]
- 41.Liu Y. Active learning with support vector machine applied to gene expression data for cancer classification. J Chem Inf Comput Sci. 2004;44:1936–1941. doi: 10.1021/ci049810a. [DOI] [PubMed] [Google Scholar]
- 42.Zavaljevski N, Stevens FJ, Reifman J. Support vector machines with selective kernel scaling for protein classification and identification of key amino acid positions. Bioinformatics. 2002;18:689–696. doi: 10.1093/bioinformatics/18.5.689. [DOI] [PubMed] [Google Scholar]
- 43.Chaudhary N, Sharma AK, Agarwal P, Gupta A, Sharma VK. 16S classifier: a tool for fast and accurate taxonomic classification of 16S rRNA hypervariable regions in metagenomic datasets. PLoS One. 2015;10:e0116106. doi: 10.1371/journal.pone.0116106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Bhasin M, Raghava GP. Pcleavage: an SVM based method for prediction of constitutive proteasome and immunoproteasome cleavage sites in antigenic sequences. Nucleic Acids Res. 2005;33:W202–W207. doi: 10.1093/nar/gki587. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Data Availability Statement
We have provided prediction models and other tools freely in the public domain at http://metagenomics.iiserb.ac.in/proinflam/ and http://metabiosys.iiserb.ac.in/proinflam/. In order to ensure reproducibility of our study and method, we have also provided the training datasets as well as validation datasets on the webserver.