

A structure-based drug-discovery program is under way in order to identify and develop small-molecule inhibitors of S100B for the treatment of malignant melanoma. X-ray crystallographic analysis of two S100B inhibitors reveals newly identified inhibitor–target interactions within site 3 of Ca2+-bound S100B. These data contribute to the structural biology knowledge base for this target, which is important for rational drug-design efforts.
Keywords: S100B, inhibitor, melanoma, X-ray crystallography, drug design
Abstract
Structure-based drug discovery is under way to identify and develop small-molecule S100B inhibitors (SBiXs). Such inhibitors have therapeutic potential for treating malignant melanoma, since high levels of S100B downregulate wild-type p53 tumor suppressor function in this cancer. Computational and X-ray crystallographic studies of two S100B–SBiX complexes are described, and both compounds (apomorphine hydrochloride and ethidium bromide) occupy an area of the S100B hydrophobic cleft which is termed site 3. These data also reveal novel protein–inhibitor interactions which can be used in future drug-design studies to improve SBiX affinity and specificity. Of particular interest, apomorphine hydrochloride showed S100B-dependent killing in melanoma cell assays, although the efficacy exceeds its affinity for S100B and implicates possible off-target contributions. Because there are no structural data available for compounds occupying site 3 alone, these studies contribute towards the structure-based approach to targeting S100B by including interactions with residues in site 3 of S100B.
1. Introduction
Highly proliferative and resistant to conventional chemotherapies, malignant melanoma (MM) is driven by mutations that activate oncogenes such as BRAF and NRAS, and inactivate cell-cycle regulators such as CDKN2A and PTEN (Bresnick et al., 2015 ▸). Unlike most cancers, MM generally maintains the wild-type p53 genotype until the late stages. However, while the p53 gene typically remains intact in MM (>50%), protein levels are downregulated aberrantly by elevated S100B (Bresnick et al., 2015 ▸). In normal melanocytes, when p53 levels are elevated p53 binds directly to the promoter site of S100B and activates its expression, and in turn S100B binding to p53 disrupts p53 oligomerization and promotes the hdm2-dependent polyubiquination of p53 and degradation while also blocking p53 acetylation and phosphorylation. However, in MM, when S100B levels are high S100B is likely to co-regulate p53 degradation, since S100B binds to both the p53 E3 ligase hdm2 and the hdm2 co-regulator hdm4 (Bresnick et al., 2015 ▸). S100B also has a measurable effect on cell viability through the modulation of MAPK signaling via direct interaction with the p90 ribosomal S6 kinase (RSK). This interaction blocks ERK-dependent phosphorylation and sequesters RSK into the cytoplasm. Systemic circulation of S100B also is a likely contributor to melanoma progression via its role in chronic inflammation (Hartman et al., 2014 ▸), so the role of S100B in several important cancer pathways makes this protein a new and relevant therapeutic target. In fact, the suppression of S100B activity via RNA interference or by small-molecule inhibitors has been demonstrated to raise p53 protein levels and its tumor suppression activities, including UV-activated apoptosis (Lin et al., 2004 ▸; Markowitz et al., 2004 ▸), and an S100B-directed inhibitor has been tested in a stage II clinical trial for the treatment of relapsed or refractory MM in patients with wild-type p53 and detectable S100B (http://www.clinicaltrials.gov, identifier NCT00729807; Smith et al., 2010 ▸). Importantly, elevated S100B is an established diagnostic tool indicating a poor prognosis for MM patients, so personalized medicine approaches for targeting S100B are a possibility.
As part of a drug-discovery program, a rational drug-design approach for developing S100B inhibitors is being used. This approach relies heavily on structural biology and computational and experimental screening techniques for the detailed characterization of small-molecule/protein interactions. Previous inhibitor-design studies have generated small molecules that target two pockets on S100B simultaneously, including the p53-binding cleft of calcium-loaded S100B (i.e. sites 1 and 2; Cavalier et al., 2016 ▸; Rustandi et al., 2000 ▸) or the pentamidine-binding sites (i.e. sites 2 and 3; Cavalier et al., 2016 ▸; Hartman et al., 2013 ▸; McKnight et al., 2012 ▸); however, a detailed examination of molecules that bind to site 3 alone has not yet been performed. The data presented here for two site 3 binders (apomorphine hydrochloride or SC0025, and ethidium bromide or SC1990) have revealed new protein–inhibitor interactions that can be considered for improving existing site 2/3 binders. As part of this drug-design approach, the novel interactions discovered here with site 3 residues, along with computational functional group mapping results, may produce inhibitors that bind all three persistent sites in S100B simultaneously using a single small-molecule inhibitor.
2. Experimental procedures
2.1. Materials
(R)-6-Methyl-5,6,6a,7-tetrahydro-4H-dibenzo[de,g]quinoline-10,11-diol hydrochloride (apomorphine hydrochloride or SC0025) was acquired from Sigma–Aldrich (catalog No. A4393) and stocks were prepared in DMSO.
3,8-Diamino-5-ethyl-6-phenylphenanthridin-5-ium bromide (ethidium bromide or SC1990) was acquired from Sigma–Aldrich (catalog No. E8751) and stocks were prepared in DMSO.
2.2. Computational methods
The general procedure for carrying out site identification by ligand competitive saturation (SILCS; Guvench & MacKerell, 2009 ▸; Raman et al., 2011 ▸) calculations was similar to that used in our previously reported studies (Raman et al., 2013 ▸; Cao, 2013 ▸). Briefly, SILCS involves computationally immersing a protein in an aqueous solution simultaneously containing different types of small solutes (propane, benzene, imidazole, methanol, formamide, acetaldehyde, methylammonium and acetate) that represent different classes of chemical functional groups (Raman et al., 2013 ▸). The system is then subjected to multiple molecular-dynamics (MD) simulations allowing competitive binding of the small solutes with each other and with water to the protein. The MD simulations include protein flexibility, allowing the identification of regions of the protein that can undergo conformational changes to allow ligand binding to be identified. Three-dimensional probability distributions of the functional groups are obtained from the simulations. These are normalized by the bulk voxel occupancies and converted into free energies based on a Boltzmann distribution, yielding grid free-energy (GFE) FragMaps. The FragMaps may then be used for a number of ligand-design protocols (Yu et al., 2015 ▸; Lakkaraju et al., 2015 ▸), including the identification of regions of the protein that are suitable for ligand design, as performed in the present study. MD simulations were initiated from the structure of S100B in the presence of inhibitor (PDB entry 3gk1; Charpentier et al., 2009 ▸), with the inhibitor removed, and performed using in-house analysis scripts along with the GROMACS simulation program (Hess et al., 2008 ▸). All calculations used the CHARMM36 protein force field (MacKerell et al., 1998 ▸; Best et al., 2012 ▸) with the CHARMM TIP3P (Jorgensen, 1981 ▸) water model (Reiher, 1985 ▸) and the CHARMM General Force Field for the solute molecules (Vanommeslaeghe et al., 2010 ▸, 2012 ▸; Vanommeslaeghe & Mackerell, 2012 ▸).
2.3. Purification
15N-labelled bovine or rat S100B was expressed and purified (>99%) using methods similar to those described previously (Drohat et al., 1996 ▸; Amburgey et al., 1995 ▸). The concentrations of S100B stock solutions were determined using the Bio-Rad Protein Assay (Bio-Rad, Hercules, California, USA) using bovine serum albumin as a standard. The S100B was stored at a concentration of ∼10 mM in 0.25 mM Tris–HCl pH 7.2 with 0.5 mM DTT at −20°C until use.
2.4. Direct fluorescence
The affinities between S100B and inhibitors were determined by measuring the decreases in compound fluorescence intensity with titrated amounts of rat S100B. The assays were performed in 10 mM HEPES pH 7.2, 10 mM CaCl2, 15 mM NaCl, 100 mM KCl, 0.01%(v/v) Triton X-100. The fluorescence data for 25 µM SC0025 (λex = 273 nm, λem = 465 nm) were collected in quartz cuvettes on a Varian Cary Eclipse fluorescence spectrophotometer with the temperature maintained at 37°C using a circulating constant-temperature bath. The fluorescence data for 25 µM SC1990 in black 384-well microplates (20 µl total volume) were measured at room temperature in a BMG PHERAstar FS multimode microplate reader with excitation at 540 nm and emission at 590 nm.
2.5. Cell-based assay with WM115 malignant melanoma cells
The malignant melanoma cell line WM115 was obtained from the American Type Culture Collection (ATCC) and was cultured in Minimum Essential Medium (MEM; Invitrogen) supplemented with 10%(v/v) heat-inactivated fetal bovine serum (FBS) and 100 units ml−1 penicillin–streptomycin (PS). SC0025 was tested for its ability to inhibit the growth of WM115 human melanoma cells expressing high levels of S100B (shRNAscrambled) and low levels of S100B (shRNAS100B) (Cavalier et al., 2014 ▸, 2016 ▸; Hartman et al., 2014 ▸) using a modification of the high-throughput screening assay performed by Bachman et al. (2015 ▸). The methods used were similar to previous methods and included the use of a Biomek FX Laboratory Automation Workstation (Beckman Coulter) equipped with a 96-channel pipetting head (Bachman et al., 2005 ▸). Specifically, 20 µl of MEM (Corning) supplemented with 10%(v/v) fetal bovine serum, 0.5 µg ml−1 puromycin and 1%(v/v) PS was added to each well of a 384-well clear-bottom tissue-culture plate (Corning) containing 600 cells per well such that growing uninhibited they reached 80% confluence in 5 d. After 24 h of growth at 37°C in a 5% CO2 humidity-controlled incubator, 20 µl of cell-culture medium containing compound from DMSO stocks was added to the cells, while control cultures received medium containing an equivalent amount of DMSO. After an additional 4 d of incubation, the cells were lysed by transferring 20 µl lysis buffer consisting of 1.8%(v/v) IGEPAL with a 1:10 000 dilution of SYBR Green I (10 000×, Invitrogen) to each well. The plates were then returned to the incubator for 24 h. The fluorescence intensity was then read through the bottom of the plate using a PolarStar plate reader (BMG) using 485 nm excitation and 520 nm emission filters. The SYBR Green fluorescence was used to measure total DNA, which in turn correlates with cell number as described previously (Myers, 1998 ▸). The EC50 of each compound was determined using serial dilutions and performed as a minimum of three replicates. Hill curves for each replicate were generated using the Origin data-analysis software (OriginLab).
To analyze the changes in the total p53 protein levels upon treatment with SC0025, WM115 cells were seeded in triplicate at 70 × 104 cells ml−1 in 60 mm dishes in 1× MEM (Cellgro) supplemented with 10%(v/v) FBS, 100 units ml−1 PS, 0.5 µg ml−1 puromycin and allowed to adhere overnight. The following day, the old media were removed and new media containing 15 µM SC0025 or an equivalent volume of DMSO were added. The cells were harvested at 4 h post-treatment using cold 1× RIPA lysis buffer [0.5 M Tris–HCl pH 7.4, 1.5 M NaCl, 2.5%(w/v) deoxycholic acid, 10%(v/v) NP-40, 10 mM EDTA] and subjected to Western blotting.
2.5.1. Western blotting
Western blotting was performed using 30 µg cell lysates loaded onto a 12% SDS–PAGE gel (NuPage). They were subsequently transferred to PVDF membranes (Millipore) and reacted with p53 mouse monoclonal antibody (DO-1, Santa Cruz), mouse anti-S100B antibody (BD Biosciences) and mouse anti-GAPDH at dilutions recommended by the reagent suppliers. The blots were then reacted with goat anti-mouse secondary antibodies (Kirkegaard & Perry Laboratories) and treated with Immobilon Western Chemiluminescent HRP Substrate (Millipore) at dilutions recommended by the reagent suppliers.
2.6. Crystallization
All crystallization experiments were conducted using vapor-diffusion methods and were set up as follows. S100B–SC0025 crystals were grown in sitting drops consisting of 0.75:0.75 µl protein solution (40 mg ml−1 bovine S100B, 10 mM cacodylate pH 7.2, 7.5 mM CaCl2, 4 mM SC0025 prepared in DMSO) and mother liquor [25%(v/v) polyethylene glycol monomethyl ether 550, 0.1 M HEPES pH 8.0, 5%(v/v) glycerol, 7.5 mM CaCl2]. S100B–SC1990 crystals were grown in sitting drops consisting of 0.75:0.75 µl protein solution (40 mg ml−1 bovine S100B, 10 mM cacodylate pH 7.2, 7.5 mM CaCl2, 4 mM SC1990 prepared in DMSO) and mother liquor [25%(w/v) PEG 3350, 0.1 M HEPES pH 7.0, 5%(v/v) glycerol, 7.5 mM CaCl2]. The crystals were grown over a period of 1–14 d at a temperature of 295 K. Crystals were not cryoprotected before being flash-cooled in liquid nitrogen.
2.7. Data collection
Diffraction data for S100B–SC0025 crystals were collected at 100 K using a MicroMax-007 X-ray generator (Rigaku, The Woodlands, Texas, USA) and an R-AXIS IV++ imaging-plate detector (Rigaku/MSC) at the University of Maryland School of Medicine X-ray Crystallography Core Facility. A 1.81 Å resolution data set was collected at a wavelength of 1.5418 Å while rotating the crystal by 1.8° each frame. The data were processed and integrated using MOSFLM (Leslie & Powell, 2007 ▸) within the CCP4 program suite (Winn et al., 2011 ▸). The space group was determined to be C2221.
Diffraction data for S100B–SC1990 crystals were collected remotely (Soltis et al., 2008 ▸; Cohen et al., 2002 ▸) on beamline 7-1 at the Stanford Synchrotron Radiation Lightsource (SSRL). Data were recorded at 100 K on an ADSC Q315 (315 × 315 mm) detector with data-collection strategies generated by Blu-Ice (McPhillips et al., 2002 ▸). Data sets were processed and integrated by AUTOXDS (Gonzalez & Tsai, 2010 ▸). A 1.26 Å resolution data set was collected at a wavelength of 1.1271 Å while rotating the crystal by 0.55° each frame. The space group was determined to be C2221.
Diffraction data statistics are summarized in Table 1 ▸.
Table 1. X-ray data-collection and refinement statistics.
Values in parentheses are for the outer shell.
| S100B ligand | SC0025 | SC1990 |
|---|---|---|
| PDB code | 5er4 | 5er5 |
| Diffraction source | MicroMax-007 X-ray generator | Beamline 7-1, SSRL |
| Wavelength (Å) | 1.5418 | 1.1271 |
| Temperature (K) | 100 | 100 |
| Detector | R-AXIS IV++ imaging-plate detector | ADSC Q315 |
| Crystal-to-detector distance (mm) | 125.0 | 120.0 |
| Rotation range per image (°) | 1.0 | 0.55 |
| Total rotation range (°) | 133 | 180 |
| Exposure time per image (s) | 600 | 2.6 |
| Space group | C2221 | C2221 |
| a, b, c (Å) | 34.91, 89.24, 60.27 | 35.42, 88.28, 59.11 |
| α, β, γ (°) | 90, 90, 90 | 90, 90, 90 |
| Mosaicity (°) | 0.80 | 0.30 |
| Resolution range (Å) | 35.86–1.81 (1.85–1.81) | 35.37–1.26 (1.28–1.26) |
| Total No. of reflections | 44341 (2477) | 173942 (6301) |
| No. of unique reflections | 8890 (521) | 25238 (1172) |
| Completeness (%) | 99.9 (98.3) | 99.1 (94.3) |
| Multiplicity | 5.0 (4.8) | 6.9 (5.4) |
| 〈I/σ(I)〉 | 14.9 (4.8) | 28.7 (1.6)† |
| R meas | 0.072 (0.150) | 0.036 (1.150) |
| R p.i.m. | 0.033 | 0.014 |
| Overall B factor from Wilson plot (Å2) | 18.5 | 15.0 |
| Resolution range (Å) | 32.51–1.81 (1.93–1.81) | 35.37–1.26 (1.29–1.26) |
| Completeness (%) | 99.7 | 98.9 |
| σ Cutoff | F > 1.35σ(F) | F > 1.34σ(F) |
| No. of reflections, working set | 7974 (1305) | 23218 (1550) |
| No. of reflections, test set | 886 (144) | 2000 (133) |
| Final R cryst | 0.181 (0.176) | 0.152 (0.237) |
| Final R free | 0.224 (0.220) | 0.187 (0.284) |
| Cruickshank DPI | 0.129 | 0.060 |
| No. of non-H atoms | ||
| Protein | 727 | 727 |
| Ligand | 23 | 26 |
| Solvent | 138 | 158 |
| Total | 888 | 911 |
| R.m.s. deviations | ||
| Bonds (Å) | 0.010 | 0.009 |
| Angles (°) | 0.954 | 1.018 |
| Average B factors (Å2) | ||
| Protein | 23.20 | 17.08 |
| Ligand | 26.66 | 20.35 |
| Ramachandran plot | ||
| Most favored (%) | 100.0 | 100.0 |
〈I/σ(I)〉 in the outer shell is <2.0 as a CC1/2 of 0.531 was used to determine the resolution cutoff.
2.8. Phasing and refinement
To determine the structure of S100B in complex with SBiXs, molecular replacement (MR) was performed using a previously determined S100B structure (PDB entry 1mho; Matsumura et al., 1998 ▸). MR was carried out using the AUTOMR (McCoy et al., 2007 ▸) function of the PHENIX software suite (Adams et al., 2010 ▸). The models were finished by manual building within Coot (Emsley et al., 2010 ▸). The models were refined with the phenix.refine (Afonine et al., 2012 ▸) function of the PHENIX software suite (Adams et al., 2010 ▸). Ligands and waters were incorporated into the models by referring to the |F o| − |F c| OMIT maps. As the diffraction data from S100B–SC1990 crystals were collected at such high resolutions, careful refinement while extending the phases in reasonable steps was performed. H atoms were added to the model and anisotropic B factors were used. The structure-refinement statistics are summarized in Table 1 ▸.
Coordinates and structure factors have been deposited in the Protein Data Bank as PDB entries 5er4 (S100B–SC0025) and 5er5 (S100B–SC1990).
3. Results and discussion
3.1. Overall structure of S100B
S100B is functional as a homodimer, with each subunit consisting of four helices (helix 1, Glu2–Gly19; helix 2, Lys28–Leu40; helix 3, Glu49–Asp61; helix 4, Asp69–Phe88). The S100B fold contains both typical and noncanonical EF-hand calcium-binding domains in each subunit. In the presence of calcium, each EF-hand coordinates a single Ca2+ ion. In this Ca2+-bound state the typical EF-hand of S100B is in an ‘open’ conformation and the dimer interface forming the biologically active unit is aligned as a symmetric X-type four-helix bundle comprising helices 1, 1′ and 4, 4′, respectively (Matsumura et al., 1998 ▸; McKnight et al., 2012 ▸; Charpentier et al., 2008 ▸, 2010 ▸; Wilder et al., 2010 ▸). Comparisons of S100B models derived by X-ray crystallography both in the apo form and in the presence of ligands shows a strong conservation of the global fold, with the primary deviation between models seen at the C-terminal residues His85–Glu91 (to compare the models introduced here, see Supplementary Table S1; Cavalier et al., 2014 ▸). This region of the polypeptide readily accommodates the binding of small molecules.
3.2. S100B has three sites suitable for ligand binding
Through the structural characterization of the interactions between S100B and both peptidic and small-molecule inhibitors, three persistent binding sites were revealed. Site 1 is the pocket where targeted peptides, such as the TRTK peptide or the C-terminus of p53, are bound. Accordingly, site 1 is an obvious early choice for targeting S100B with small molecules to inhibit its interaction with p53. However, until more recently (Cavalier et al., 2016 ▸) very few inhibitors had been found to occupy this site, whereas targeting of sites 2 and 3 has been well documented (Cavalier et al., 2014 ▸; Wilder et al., 2010 ▸; Charpentier et al., 2009 ▸), particularly in studies involving the binding of pentamidine and pentamidine derivatives (Charpentier et al., 2008 ▸; McKnight et al., 2012 ▸; Hartman et al., 2013 ▸). These studies include both reversible inhibitors as well as those that form covalent adducts (Cavalier et al., 2014 ▸). Altogether, the permissible chemical space and the available interactions within site 2 have been well explored. Conversely, the exploration of site 3 has been limited to pentamidine and its analog heptamidine (Charpentier et al., 2008 ▸; McKnight et al., 2012 ▸). Furthermore, no inhibitors that bind only site 3 have been characterized to date. The present study extends the characterization of site 3 and its relationship to sites 1 and 2, suggesting how the spatial relationship of these sites can be exploited for ligand design using fragment-based approaches.
To investigate the potential of the development of ‘multisite’ ligands, the three-dimensional functional group affinity patterns (GFE FragMaps) of the entire protein were mapped using the SILCS approach. Shown in Fig. 1 ▸ are the Apolar GFE FragMaps overlaid on S100B along with the three sites shown in solvent-accessible surface representation (site 1, magenta; site 2, yellow; site 3, gray). Apolar FragMaps are evident in each of the sites, with the map in site 2 being elongated. The presence of the FragMaps indicates the potential for favorable ligand binding in each of the sites, consistent with the known ligand structures from crystallographic studies discussed above. Notable is the elongated, continuous nature of the FragMaps through the three sites, indicating the potential of designing inhibitors that link all three sites, taking advantage of the binding potential of all three sites.
Figure 1.

SILCS Apolar GFE FragMaps. FragMaps (green wire mesh) are shown at a cutoff of −1.0 kcal mol−1 overlaid on the crystal structure of S100B (PDB entry 3gk1). The full protein is represented as a ribbon diagram (blue) with site 1 (magenta), site 2 (yellow) and site 3 (gray) highlighted as solvent-accessible protein surfaces. The presence of Apolar FragMaps in each site indicates the potential for the favorable binding of drug-like molecules, with the extended nature of the site 2 FragMap indicating the potential to link sites 1 and 3 via site 2. Apolar FragMaps were calculated based on the benzene and propane carbon three-dimensional probability distributions.
3.3. In vitro characterization of S100B inhibitors
SC0025 and SC1990 were first identified as inhibitors of S100B during the development of a fluorescence polarization competition assay (FPCA; Table 2 ▸; Wilder et al., 2010 ▸). Using FPCA-derived IC50 data and the Nikolovska–Coleska equation, the dissociation constant (K d) of SC1990 was determined to be 422.9 ± 219.0 µM. The same could not be applied to SC0025 as the compound produced fluorescent interference in the assay. To better characterize the affinity of these compounds for S100B, direct fluorescence of the compounds was measured while S100B protein was titrated. Direct fluorescence measurements determined the K d values for SC0025 and SC1990 to be 1752 ± 176.5 and 215.4 ± 9.0 µM, respectively (Table 2 ▸ and Fig. 2 ▸).
Table 2. Cellular assays and binding data for the investigated compounds.
| SC0025 | SC1990 | |
|---|---|---|
| Structure |
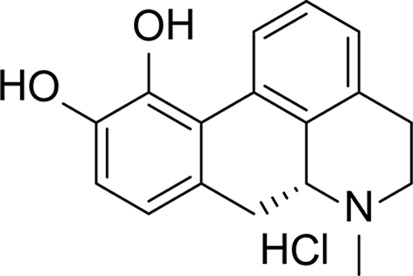
|
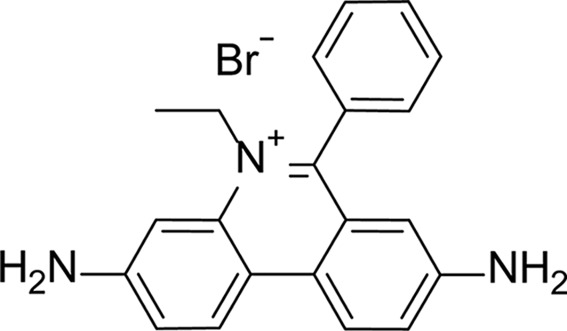
|
| K d, FPCA† (µM) (Wilder et al., 2010 ▸) | FI‡ | 423 ± 219 |
| K d, direct fluorescence§ (µM) | 1752 ± 177 | 215.4 ± 9.0 |
| Cell assay (high S100B)† ¶ (µM) | 10.8 ± 3.3 | ND†† |
| Cell assay (low S100B)† ¶ (µM) | 19.2 ± 2.3 | ND†† |
Error represented as the standard deviation.
Fluorescent interference.
Error represented as the standard error.
High S100B (shRNAscrambled), low S100B (shRNAS100B), n = 8, t-test < 0.0001.
Not determined.
Figure 2.

The affinity of inhibitors for S100B as measured by direct fluorescence. SC0025 or SC1990 was added to increasing concentrations of S100B protein and the binding is measured as the change in fluorescence intensity. The change in fluorescence intensity is plotted versus S100B concentration and fitted to a single ligand-binding model.
The in vivo efficacy (LD50) of SC0025 was determined in WM115 melanoma cell lines containing lentiviral infections (Table 2 ▸). These cell lines allow testing of S100B-dependent killing through stable or impotent knockdowns of S100B expression by S100B-directed (shRNAS100B) or scrambled shRNA (shRNAscrambled), respectively. Indeed, SC0025 demonstrated S100B-dependent killing with an in vivo efficacy of 10.75 µM in shRNAscrambled cells and an in vivo efficacy of 19.19 µM in shRNAS100B cells. However, SC0025 did not demonstrate an S100B-dependent increase in p53 levels, suggesting that binding this site alone may not be sufficient to completely block the S100B–p53 interaction (see Supplementary Fig. S1). SC1990 was not tested in cellular studies owing to its low binding affinity and its well documented toxicity (National Toxicity Program, 2015 ▸).
3.4. Crystallographic structure of the S100B–SC1990 complex
Despite its poor binding affinity and its potential toxicity (National Toxicity Program, 2015 ▸), SC1990 was included in co-crystallization trials as it may produce additional insights into ligand binding to S100B. The X-ray structure of SC1990-bound, calcium-loaded S100B (S100B–SC1990) was solved at 1.26 Å resolution (Fig. 3 ▸ a, Table 1 ▸). The asymmetric unit contained one S100B molecule, which is one-half of the biologically active dimeric form of two S100B molecules in the crystal. Residues Met0–Glu89 were built into the electron-density map. Each subunit in the S100B–SC1990 complex bound two calcium ions and a single SC1990 molecule. In the final refined model (R cryst and R free of 0.152 and 0.187, respectively), all residues were in the favored region of the Ramachandran plot, with no outliers (see Table 1 ▸). SC1990 is situated in an area defined by helix 4 and helix 1′ (Fig. 2 ▸ b) occupying the pocket formed by residues Ile11, Asp12, His15, His25, Phe88′ and Glu89′. It makes interactions with helix 1′, including hydrogen bonds with Asp12 and a water-bridged interaction with the backbone carbonyl of Val8. The tertiary amine of SC1990 is also 3.0 Å from the side chain of Glu89 (helix 4). Only a single molecule of SC1990 was modeled, but owing to the ligand density sitting on a rotation axis in the crystal symmetry, a second symmetry-generated copy is represented within Fig. 2 ▸(a) so that the entirety of the observed electron density could be interpreted. For the same reason, the SC1990 molecule was modeled at one-half occupancy.
Figure 3.

Crystallographic structure of the S100B–SC1990 complex. (a) SC1990 (ball-and-stick representation) is shown within site 3 of the S100B dimer (surface rendering with site 3 highlighted in gray). An |F o| − |F c| electron-density OMIT map (orange) of SC1990 is also shown with the map contoured at the 2.5σ level. Only a single molecule of SC1990 was modeled, but owing to the ligand density sitting on the rotation axis in the crystal symmetry a second symmetry-generated copy (green sticks) is represented so that the entirety of the observed electron density could be interpreted. (b) Dimeric S100B is rendered in a ribbon diagram with residues (sticks) within 4 Å of the SC1990 (ball-and-stick representation) highlighted in gray. SC1990 is situated within the pocket formed by Ile11, Asp12, His15 and His25 of helix 1′ and Phe88 and Glu89 of helix 4. Polar interactions are available with SC1990 at the side chains of Asp12 and Glu89.
3.5. Crystallographic structure of the S100B–SC0025 complex
Co-crystallization trials were also attempted with S100B and SC0025. The X-ray structure of SC0025-bound, calcium-loaded S100B (S100B–SC0025) was solved at 1.81 Å resolution (Fig. 4 ▸ a, Table 1 ▸). The asymmetric unit contained one S100B molecule, which is one-half of the biologically active dimeric form of two S100B molecules in the crystal. Residues Met0–Glu89 were built into the electron-density map. Each subunit in the S100B–SC0025 complex bound two calcium ions and a SC0025 molecule. In the final refined model (R cryst and R free of 0.181 and 0.224, respectively), all residues were in the favored region of the Ramachandran plot, with no outliers (see Table 1 ▸). SC0025 is situated in an area defined by helix 4 and helix 1′ (Fig. 3 ▸ b). It makes interactions with helix 4, including hydrogen bonds to the backbone carbonyls of Cys84 and His85, a water-bridged interaction with Glu89 and hydrophobic interactions with Phe88. The amine of SC0025 is also within hydrogen-bonding distance of the side chain of Asp12 (helix 1′).
Figure 4.

Crystallographic structure of the S100B–SC0025 complex. (a) SC0025 (ball-and-stick representation) is shown within site 3 of the S100B dimer (surface rendering with site 3 highlighted in gray). An |F o| − |F c| electron-density OMIT map (orange) of SC0025 is also shown with the map contoured at the 2.5σ level. (b) Dimeric S100B is rendered in a ribbon diagram with residues (sticks) within 4 Å of the SC0025 (ball-and-stick representation) highlighted in gray. SC0025 is situated within the pocket formed by Ile11 and Asp12 of helix 1′ and Cys84, His85 and Phe88 of helix 4. Polar interactions are available to SC0025 at the side chain of Asp12 and the backbones of Cys84 and His85.
As observed in the crystal structures, both compounds aid in the characterization of the poorly explored site 3 pocket in the S100B binding cleft (Figs. 2 ▸ a and 3 ▸ a). The contribution of SC1990 to drug-discovery efforts will likely be gained solely from these structure–function details, as the health risks of SC1990 (ethidium bromide) as a potent mutagen have been well documented (National Toxicity Program, 2015 ▸). Conversely, SC0025 is a significantly more interesting lead and may have value outside of the knowledge gained from its occupancy of site 3. SC0025 (apomorphine) is already an FDA-approved drug for the treatment of Parkinson’s disease, functioning as a strong nonergoline D1 and D2 receptor agonist with a dopaminergic effect, and is well tolerated by patients (Henriksen, 2014 ▸; Deleu et al., 2004 ▸). In the case of MM, we have demonstrated S100B-dependent killing via cellular assays (Table 2 ▸). Together, the on-target effects demonstrated in MM and the known tolerance in Parkinson’s patients warrant further investigation of SC0025 in our structure-based approach for developing SBiXs.
4. Conclusions
The characterization of two inhibitors, SC0025 and SC1990, using X-ray crystallography reveals that they occupy site 3 of the S100B binding pocket. Both structures, in combination with SILCS computational analysis, support the feasibility of polycyclic aromatics as S100B inhibitors at site 3, which can be tethered to or applied to the scaffolds of pentamidine or other site 2 inhibitor scaffolds. These structures also highlight important protein interactions that facilitate compound binding within the hydrophobic cleft. Specifically, there are important hydrogen-bonding opportunities with the side chains of Asp12 and Glu89 as well as the backbone at residues Cys84 and His85. Hydrophobic interactions with Phe88 and Ile11 are also important for compounds binding to site 3. Despite favorable initial results in cellular assays, nonspecific cellular effects make it clear that SC0025 is not a pharmaceutical endpoint, but that it may be useful for engineering novel SBiXs via a structure-based approach for targeting S100B in MM.
Supplementary Material
PDB reference: calcium-loaded S100B bound to SC0025, 5er4
PDB reference: calcium-loaded S100B bound to SC1990, 5er5
Supporting Information.. DOI: 10.1107/S2059798316005532/dw5162sup1.pdf
Acknowledgments
The Department of Energy (DOE) Office of Science and Office of Basic Energy support the Stanford Synchrotron Radiation Lightsource, SLAC National Accelerator Laboratory. The SSRL Structural Molecular Biology Program is supported by the DOE Office of Biological and Environmental Research and by the National Institutes of Health, National Institute of General Medical Sciences. This work was also supported by R01 NIH grants (CA107331 and GM58888 to DJW). The training of MCC and ZM was also supported by T32 NIH grants (CA154274 and AR007592).
References
- Adams, P. D. et al. (2010). Acta Cryst. D66, 213–221.
- Afonine, P. V., Grosse-Kunstleve, R. W., Echols, N., Headd, J. J., Moriarty, N. W., Mustyakimov, M., Terwilliger, T. C., Urzhumtsev, A., Zwart, P. H. & Adams, P. D. (2012). Acta Cryst. D68, 352–367. [DOI] [PMC free article] [PubMed]
- Amburgey, J. C., Abildgaard, F., Starich, M. R., Shah, S., Hilt, D. C. & Weber, D. J. (1995). J. Biomol. NMR, 6, 171–179. [DOI] [PubMed]
- Bachman, K. E., Sager, J., Cheong, I., Catto, M., Bardelli, A., Park, B. H., Vogelstein, B., Carotti, A., Kinzler, K. W. & Lengauer, C. (2005). Mol. Cancer Ther. 4, 1026–1030. [DOI] [PubMed]
- Best, R. B., Zhu, X., Shim, J., Lopes, P. E. M., Mittal, J., Feig, M. & MacKerell, A. D. Jr (2012). J. Chem. Theory Comput. 8, 3257–3273. [DOI] [PMC free article] [PubMed]
- Bresnick, A. R., Weber, D. J. & Zimmer, D. B. (2015). Nature Rev. Cancer, 15, 96–109. [DOI] [PMC free article] [PubMed]
- Cao, X. et al. (2013). Mol. Cancer, 12, 42. [DOI] [PMC free article] [PubMed]
- Cavalier, M. C., Ansari, M. I., Pierce, A. D., Wilder, P. T., McKnight, L. E., Raman, E. P., Neau, D. B., Bezawada, P., Alasady, M. J., Charpentier, T. H., Varney, K. M., Toth, E. A., MacKerell, A. D. Jr, Coop, A. & Weber, D. J. (2016). J. Med. Chem. 59, 592–608. [DOI] [PMC free article] [PubMed]
- Cavalier, M. C., Pierce, A. D., Wilder, P. T., Alasady, M. J., Hartman, K. G., Neau, D. B., Foley, T. L., Jadhav, A., Maloney, D. J., Simeonov, A., Toth, E. A. & Weber, D. J. (2014). Biochemistry, 53, 6628–6640. [DOI] [PMC free article] [PubMed]
- Charpentier, T. H., Thompson, L. E., Liriano, M. A., Varney, K. M., Wilder, P. T., Pozharski, E., Toth, E. A. & Weber, D. J. (2010). J. Mol. Biol. 396, 1227–1243. [DOI] [PMC free article] [PubMed]
- Charpentier, T. H., Wilder, P. T., Liriano, M. A., Varney, K. M., Pozharski, E., MacKerell, A. D. Jr, Coop, A., Toth, E. A. & Weber, D. J. (2008). J. Mol. Biol. 382, 56–73. [DOI] [PMC free article] [PubMed]
- Charpentier, T. H., Wilder, P. T., Liriano, M. A., Varney, K. M., Zhong, S., Coop, A., Pozharski, E., MacKerell, A. D. Jr, Toth, E. A. & Weber, D. J. (2009). Biochemistry, 48, 6202–6212. [DOI] [PMC free article] [PubMed]
- Cohen, A. E., Ellis, P. J., Miller, M. D., Deacon, A. M. & Phizackerley, R. P. (2002). J. Appl. Cryst. 35, 720–726. [DOI] [PMC free article] [PubMed]
- Deleu, D., Hanssens, Y. & Northway, M. G. (2004). Drugs Aging, 21, 687–709. [DOI] [PubMed]
- Drohat, A. C., Amburgey, J. C., Abildgaard, F., Starich, M. R., Baldisseri, D. & Weber, D. J. (1996). Biochemistry, 35, 11577–11588. [DOI] [PubMed]
- Emsley, P., Lohkamp, B., Scott, W. G. & Cowtan, K. (2010). Acta Cryst. D66, 486–501. [DOI] [PMC free article] [PubMed]
- Gonzalez, A. & Tsai, Y. (2010). AUTOXDS. http://smb.slac.stanford.edu/facilities/software/xds/#autoxds_script.
- Guvench, O. & MacKerell, A. D. Jr (2009). PLoS Comput. Biol. 5, e1000435. [DOI] [PMC free article] [PubMed]
- Hartman, K. G., McKnight, L. E., Liriano, M. A. & Weber, D. J. (2013). Future Med. Chem. 5, 97–109. [DOI] [PMC free article] [PubMed]
- Hartman, K. G., Vitolo, M. I., Pierce, A. D., Fox, J. M., Shapiro, P., Martin, S. S., Wilder, P. T. & Weber, D. J. (2014). J. Biol. Chem. 289, 12886–12895. [DOI] [PMC free article] [PubMed]
- Henriksen, T. (2014). Neurodegener. Dis. Manag. 4, 271–282. [DOI] [PubMed]
- Hess, B., Kutzner, C., van der Spoel, D. & Lindahl, E. (2008). J. Chem. Theory Comput. 4, 435–447. [DOI] [PubMed]
- Jorgensen, W. L. (1981). J. Am. Chem. Soc. 103, 335–340.
- Lakkaraju, S. K., Yu, W., Raman, E. P., Hershfeld, A. V., Fang, L., Deshpande, D. A. & MacKerell, A. D. Jr (2015). J. Chem. Inf. Model. 55, 700–708. [DOI] [PMC free article] [PubMed]
- Leslie, A. G. W. & Powell, H. R. (2007). Evolving Methods for Macromolecular Crystallography, edited by R. J. Read & J. L. Sussman, pp. 41–51. Dordrecht: Springer.
- Lin, J., Yang, Q., Yan, Z., Markowitz, J., Wilder, P. T., Carrier, F. & Weber, D. J. (2004). J. Biol. Chem. 279, 34071–34077. [DOI] [PubMed]
- MacKerell, A. D. Jr et al. (1998). J. Phys. Chem. B, 102, 3586–3616. [DOI] [PubMed]
- Markowitz, J., Chen, I., Gitti, R., Baldisseri, D. M., Pan, Y., Udan, R., Carrier, F., MacKerell, A. D. Jr & Weber, D. J. (2004). J. Med. Chem. 47, 5085–5093. [DOI] [PubMed]
- Matsumura, H., Shiba, T., Inoue, T., Harada, S. & Kai, Y. (1998). Structure, 6, 233–241. [DOI] [PubMed]
- McCoy, A. J., Grosse-Kunstleve, R. W., Adams, P. D., Winn, M. D., Storoni, L. C. & Read, R. J. (2007). J. Appl. Cryst. 40, 658–674. [DOI] [PMC free article] [PubMed]
- McKnight, L. E., Raman, E. P., Bezawada, P., Kudrimoti, S., Wilder, P. T., Hartman, K. G., Godoy-Ruiz, R., Toth, E. A., Coop, A., MacKerell, A. D. Jr & Weber, D. J. (2012). ACS Med. Chem. Lett. 3, 975–979. [DOI] [PMC free article] [PubMed]
- McPhillips, T. M., McPhillips, S. E., Chiu, H.-J., Cohen, A. E., Deacon, A. M., Ellis, P. J., Garman, E., Gonzalez, A., Sauter, N. K., Phizackerley, R. P., Soltis, S. M. & Kuhn, P. (2002). J. Synchrotron Rad. 9, 401–406. [DOI] [PubMed]
- Myers, M. A. (1998). J. Immunol. Methods, 212, 99–103. [DOI] [PubMed]
- National Toxicity Program (2015). Ethidium Bromide – M940107. http://ntp.niehs.nih.gov/testing/Status/agents/ts-m940107.html.
- Raman, E. P., Yu, W., Guvench, O. & MacKerell, A. D. (2011). J. Chem. Inf. Model. 51, 877–896. [DOI] [PMC free article] [PubMed]
- Raman, E. P., Yu, W., Lakkaraju, S. K. & MacKerell, A. D. Jr (2013). J. Chem. Inf. Model. 53, 3384–3398. [DOI] [PMC free article] [PubMed]
- Reiher, W. E. (1985). PhD thesis. Harvard University.
- Rustandi, R. R., Baldisseri, D. M. & Weber D. J. (2000). Nature Struct. Biol. 7, 570–574. [DOI] [PubMed]
- Smith, J., Stewart, B. J., Glaysher, S., Peregrin, K., Knight, L. A., Weber, D. J. & Cree, I. A. (2010). Anticancer Drugs, 21, 181–185. [DOI] [PMC free article] [PubMed]
- Soltis, S. M. et al. (2008). Acta Cryst. D64, 1210–1221. [DOI] [PMC free article] [PubMed]
- Vanommeslaeghe, K., Hatcher, E., Acharya, C., Kundu, S., Zhong, S., Shim, J., Darian, E., Guvench, O., Lopes, P., Vorobyov, I. & Mackerell, A. D. Jr (2010). J. Comput. Chem. 31, 671–690. [DOI] [PMC free article] [PubMed]
- Vanommeslaeghe, K. & MacKerell, A. D. Jr (2012). J. Chem. Inf. Model. 52, 3144–3154. [DOI] [PMC free article] [PubMed]
- Vanommeslaeghe, K., Raman, E. P. & MacKerell, A. D. Jr (2012). J. Chem. Inf. Model. 52, 3155–3168. [DOI] [PMC free article] [PubMed]
- Wilder, P. T., Charpentier, T. H., Liriano, M. A., Gianni, K., Varney, K. M., Pozharski, E., Coop, A., Toth, E. A., MacKerell, A. D. & Weber, D. J. (2010). Int. J. High Throughput Screen. 2010, 109–126. [DOI] [PMC free article] [PubMed]
- Winn, M. D. et al. (2011). Acta Cryst. D67, 235–242.
- Yu, W., Lakkaraju, S. K., Raman, E. P., Fang, L. & MacKerell, A. D. Jr (2015). J. Chem. Inf. Model. 55, 407–420. [DOI] [PMC free article] [PubMed]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
PDB reference: calcium-loaded S100B bound to SC0025, 5er4
PDB reference: calcium-loaded S100B bound to SC1990, 5er5
Supporting Information.. DOI: 10.1107/S2059798316005532/dw5162sup1.pdf