

Abstract
Introduction
Meta-analyses of genetic association studies are affected by biases and quality shortcomings of the individual studies. We previously developed and validated a risk of bias tool for use in systematic reviews of genetic association studies. The present study describes a larger empirical evaluation of the Q-Genie tool.
Methods and analysis
MEDLINE, Embase, Global Health and the Human Genome Epidemiology Network will be searched for published meta-analyses of genetic association studies. Twelve reviewers in pairs will apply the Q-Genie tool to all studies in included meta-analyses. The Q-Genie will then be evaluated on its ability to (i) increase precision after exclusion of low quality studies, (ii) decrease heterogeneity after exclusion of low quality studies and (iii) good agreement with experts on quality rating by Q-Genie. A qualitative assessment of the tool will also be conducted using structured questionnaires.
Discussion
This systematic review will quantitatively and qualitatively assess the Q-Genie's ability to identify poor quality genetic association studies. This information will inform the selection of studies for inclusion in meta-analyses, conduct sensitivity analyses and perform metaregression. Results of this study will strengthen our confidence in estimates of the effect of a gene on an outcome from meta-analyses, ultimately bringing us closer to deliver on the promise of personalised medicine.
Ethics and dissemination
An updated Q-Genie tool will be made available from the Population Genomics Program website and the results will be submitted for a peer-reviewed publication.
Keywords: Risk of bias, Genetic association studies, Meta-analyses, Systematic reviews, Genomics
Strengths and limitations of this study.
Novel methodology to empirically assess and modify a risk of bias tool.
Large sample size.
Older, poorer quality studies may be missed, as the search is limited to the past 2 years.
Background
Meta-analyses from systematic reviews have gained popularity in the past few decades, in parallel with the rise of evidence-based medicine. The primary goal of a meta-analysis is to precisely estimate the effect of an exposure on an outcome by pooling data from previously conducted studies.1 In the field of genetic epidemiology, published meta-analyses have increased nearly 50-fold over the last 20 years, from 27 in 1994–19982 to 1302 in 2014 alone. A particularly attractive feature of the meta-analysis method lies in its potential to deal with a major shortcoming of most genetic association studies: a lack of power to detect associations of modest sizes.2 Conversely, a major constraint is its reliance on aggregate data from individual study reports; therefore, results are not free from biases and quality shortcomings of the individual studies. It is thus critical that a systematic review, and subsequent meta-analysis, incorporates an assessment of risk of bias of its included studies and the extent to which design and conduct of each study has minimised the impact of this bias.3 In many disciplines, tools have been constructed to examine the internal validity of systematic reviews in a standardised way. Examples include the Cochrane risk of bias tool for randomised control trials and the Newcastle-Ottawa scale for observational studies.3–5 Such tools allow for a standardised assessment of biases introduced by included studies, create criteria to facilitate exclusion of poor quality studies and lend credence to the pooled results. The tools incorporate items on bias in methodology and reporting of results, such as baseline comparability of groups, blinding and power to detect associations.6 7
While many risk of bias tools have been developed for epidemiological studies, they do not capture sources of bias pertinent to genetic association studies, including population stratification (confounding that arises from differences in genotype prevalence and disease risk between subpopulations); variations in the collection, handling and processing of DNA; classification of genotypes; and degree of relatedness/consanguinity in a population under study.8 9 Specific to population stratification, heterogeneity could also result from variable gene expression. These unique biases and their absence in other tools prompted us to develop and validate a risk of bias tool for use in systematic reviews of genetic association studies.10 The Q-Genie was developed at McMaster University's Population Genomics Program. It contains 11 items assessing the following dimensions: scientific basis for development of the research question, ascertainment of comparison groups (ie, cases and controls), technical and non-technical classification of genetic variant tested, classification of the outcome, discussion of sources of bias, appropriateness of sample size, description of planned statistical analyses, statistical methods used, test of assumptions in the genetic studies (eg, agreement with the Hardy-Weinberg equilibrium) and appropriate interpretation of results (see online supplementary table S1). Each question is rated on a 7-point Likert scale. The tool took ∼20 min per study to complete the pilot testing. The authors assessed reliability and construct validity of the tool and found excellent performance characteristics (inter-rater reliability of 0.74, internal consistency of 0.82 and overall reliability of 0.64). The Q-Genie tool was applied to a published systematic review assessing the association of a single nucleotide polymorphism (SNP) in the CDKAL1 gene with type 2 diabetes.11 Exclusion of poor quality studies, based on the tool's classification, led to a reduction in heterogeneity and an increase in precision. Specifically, I2 was reduced from 72% with 7 studies to 0% with 6 studies, and the summary effect size changed from an OR of 1.25 (95% CI 1.09 to 1.45) to an OR of 1.15 (95% CI 1.07 to 1.24). Since this was a validation study, the tool was only tested on a small meta-analysis in one population. A larger empirical evaluation of the Q-Genie tool is warranted prior to ascertaining its effectiveness and applicability.
bmjopen-2015-010403supp_table.pdf (57.3KB, pdf)
The objectives of the present study are (1) to confirm the validity of the Q-Genie by applying the tool to a large sample of published meta-analyses of genetic association studies, (2) examine whether estimates of pooled effects and heterogeneity vary according to quality and (3) modify the tool based on findings of the present study.
Methods
The present study will be conducted in two parts. First, a systematic review of the literature will be undertaken to identify a sample of meta-analyses to which the Q-Genie tool can be applied. The first section describes methods for this systematic review. The following section outlines our methodology for application of the Q-Genie tool and subsequent modifications based on the findings.
Systematic review of literature to identify meta-analyses of genetic variants
This section describes the methods for a systematic review to identify a sample of meta-analyses to which the Q-Genie tool will be applied.
Sources of studies
We will conduct a thorough search for published systematic reviews investigating the impact of a genetic variant on an outcome. No restrictions will be placed on the type of outcome, the population studied or the type of genetic variant studied. However, only studies published in English and those conducted in human samples will be included. We will limit the search to only systematic reviews with meta-analyses conducted between 1 January 2014 and 31 December 2014 to obtain an appropriate sample size (see below). MeSH and free text terms will be used to identify relevant articles. The search strategy was created in consultation with a librarian and is presented in box 1. The following databases will be searched using the OVID interface: MEDLINE, Embase and Global Health. Additionally, we will perform a search of the Human Genome Epidemiology (HuGE) Network for published meta-analyses.
Box 1. Search strategy.
- OVID Search (MEDLINE, Embase, Global Health):
- ((genetic association study or gene association study) and meta-analysis).af.;
- limit 1 to english language;
- limit 2 to humans;
- limit 3 to yr=‘2014’.
- Search using HuGE Literature finder:
- ‘meta-analysis’ OR ‘systematic review’ OR ‘HuGE review’; filtered by Year ‘2014’*.
- *In searching HuGE, we did not restrict the search to ‘humans’ as this database is designed to capture only human studies. Furthermore, any non-English studies were manually removed during Title and Abstract Screening.
The goal of this search is to provide a sampling frame to select studies for our evaluation. To ensure that two meta-analyses published on the same topic in independent Journals with the same primary studies will not be included in our sample, we will sort by Authors and Year of the primary studies to identify duplicate publications. If duplicate meta-analyses are found, we will include the earlier publication. For meta-analysis with some, but not all, overlapping studies, the more comprehensive of the two will be used.
Selection of studies
Two reviewers (ZNS and SS) will independently assess each study for eligibility based on the title and abstract using the inclusion and exclusion criteria described below. Disagreements will be resolved by consensus. Articles that pass the screening phase will be carried forward to the data extraction phase. Agreement between reviewers will be assessed using Cohen's κ. A flow diagram displaying the screening process and a detailed table of the studies selected in the systematic review will be included in the publication, as per guidelines set by the Meta-analysis of Observational Studies in Epidemiology (MOOSE)12 and the Preferred Reporting Items for Systematic Reviews and Meta-Analyses (PRISMA).13
Inclusion and exclusion criteria
Any systematic review with at least one meta-analysis of studies assessing the impact of a genetic variant on any outcome will be considered for inclusion. If several SNPs and/or several outcomes are tested in one published article, we will include all meta-analyses. Meta-analyses published in English between 1 January 2013 and 31 December 2014 will be included. Meta-analyses published prior to January 2013 will be excluded in order to provide a sufficient but feasible sample pool. Second, meta-analyses with fewer than four studies will be excluded to ensure that the pool of studies has some degree of variability. This degree of variability is needed for the Q-Genie tool to assess whether study quality affects heterogeneity and precision of summary estimates. Lastly, we will also exclude primary data meta-analyses, such as consortium meta-analyses, as these do not include any individual studies to evaluate. Similarly, meta-analyses of genome-wide association study will be systematically excluded.
Sample size
A method for conducting sample size calculations for this type of endeavour has not yet been established. While simulations can be used to determine appropriate sample sizes, a number of factors render this difficult. These include lack of estimates for classic meta-analysis—parameters such as the number of studies, effect sizes, error variance and between-study heterogeneity. Additionally, specific to this study, information on overall study quality, variability of between-study quality, extent of bias introduced due to poor study quality and sensitivity of the Q-Genie tool would be needed.14 15 As a result of difficulty in estimating power, we used a pragmatic approach that considered feasibility, similar studies of tools conducted in the past,3 16 17 and input from methodology experts. Based on these considerations, we aim to include at least 50 meta-analyses (with at least 4 studies in each, therefore >200 independent studies). This will be accomplished by using all meta-analyses meeting our inclusion and exclusion criteria acquired from our search of literature as a sampling frame from which 50 will be randomly selected.
Data extraction
A total of 12 reviewers familiar with the conduct of genetic association studies will be recruited to appropriately represent users of the tool.10 In pairs, reviewers will conduct a full-text assessment and data extraction from the assigned meta-analyses. Standardised forms with the scoring criteria will be given to each reviewer. Data extraction forms will be pilot tested to ensure feasibility and consistency in use among reviewers in a first round of review. A second round of review will be conducted with the same group of reviewers after addressing any concerns that may arise from the first round. The following will be extracted from all included systematic reviews: title of the journal, outcome studied, genetic variant studied, list of included studies with the associated risk estimate, sample size, SE, CIs and p values, as well as the statistical methods used for meta-analysis and information on design of included studies (ie, case–control, cohort, etc). Data will be extracted for the model of inheritance investigated by the authors of the meta-analysis. Specifically, if the meta-analysis was conducted based on an additive genetic model, we will also use this model to pool our results. If multiple models are investigated, data for all models will be extracted. However, if a formal assessment of inheritance model fit has been conducted, preference will be given to the best fitted model.
Assessment of the Q-Genie tool
This section describes the quantitative and qualitative assessment of our confidence in the reported association estimate provided by meta-analyses based on information from the Q-Genie tool.
Criteria for evaluation of effectiveness
The Q-Genie tool will be applied to all meta-analyses included from our systematic review, described above. The main objective of the present study is to examine our level of confidence in the findings from the meta-analyses in light of the study ratings by Q-Genie. If the Q-Genie tool serves its purpose to correctly identify poor quality studies, the score will inform confidence (or ‘certainty’) in the estimates derived from the meta-analysis on the basis of quality of its included studies. To this end, Q-Genie will be evaluated on the following criteria.
Decreased heterogeneity after exclusion of low quality studies. It is expected that variation in the methodological rigour of conduct will be a major source of between-studies heterogeneity. Thus, elimination of studies assessed to be of poor design/conduct as measured by Q-Genie should reduce variability in the meta-analysis, thus increasing our certainty in the synthesis of the remaining studies.
Increased precision after exclusion of low quality studies. It is expected that pooled estimate with narrow CIs is more trustworthy than those with wider CIs. Elimination of studies that are of poor quality, such as those that are underpowered, will result in more precise estimates, thus increasing our certainty in the synthesis of the remaining studies.
The following criteria evaluate external validity.
Good agreement with experts on quality rating by Q-Genie.
Agreement with the Venice criteria.
Methods for evaluation of the above criteria are detailed below.
Application of the Q-Genie tool
The Q-Genie tool is available from the Population Genomics Program website http://fhs.mcmaster.ca/pgp/links.html. In the same pairs as data extraction, reviewers will independently rate studies from included meta-analyses using the Q-Genie tool. Their average score will be used. Reviewers will pilot test the Q-Genie on two or three studies in pairs and will meet to discuss disagreements in interpretation, if necessary. Agreement between pairs of reviewers will be reported with inter-rater G-coefficients, as this measure is able to produce a single estimate of reliability from all six rater-pairs.18 As an adjunct, we will also report Cohen's κ for each pair as this is commonly used in systematic reviews to assess concordance. Lastly, an overall G-coefficient will be used to report the overall agreement, using data from all reviewers.
Statistical analysis plan
The following analyses will be conducted to evaluate the Q-Genie tool based on the above-described criteria.
Association analysis
Random-effect metaregression models assuming normality will be used to assess the relationship between effect size estimates and study quality as the moderator variable measured by the Q-Genie tool.19
The random-effect metaregression model will be fitted as follows:
Let i=1, …, n, where n is the number of studies to be included in a meta-analysis. Then,
where yi are effect size estimates, xi are Q-Genie scores and 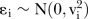 as the residual random errors with vi as the SE of effect size estimates from each genetic association study included in the meta-analysis, respectively.
as the residual random errors with vi as the SE of effect size estimates from each genetic association study included in the meta-analysis, respectively.
Between-study variability is incorporated as ηi∼N(0,τ2), where τ2 is the residual heterogeneity in the true effect size between studies that is not accounted for by quality scores incorporated in this model.
The overall pooled random-effect meta-analysis estimate is given by μ adjusted for study quality. The overall effect of study quality per 1 unit of change in the Q-Genie score on this pooled estimate is given by the regression estimate, β, which could be regarded as the bias introduced due to study quality. This bias is due to systematic error and not random error as assumed in classic random-effect meta-analysis with unobserved study qualities. A test for β under the null hypothesis, H0: β=0, will shed light on the impact of study quality on the corresponding meta-analysis with respect to the bias introduced due to poor quality studies. In this case, the Q-Genie tool will increase the accuracy of the pooled meta-analysis estimate, and decrease between-study heterogeneity. Similar analysis will be conducted on individual scores for each of the Q-Genie questions to assess their impact on meta-analysis. Model averaging using all sample meta-analyses will be utilised for constructing a robust Q-Genie score by weighing the stable relevance of individual Q-Genie questions for meta-analysis of genetic association studies. Unweighted and weighted Q-Genie score will be compared using goodness-of-fit measures such as the residual sum of squares applied in our pool of meta-analyses.
Sensitivity analyses
The following sensitivity analyses will be conducted. First, we will exclude studies of low quality as determined by a score of ≤35 on the Q-Genie tool. Thresholds designating low, moderate and high quality studies were established in the pilot study.10 Second, we will exclude low and moderate quality studies, leaving only studies of high quality (those with a score >45 on the Q-Genie). We will compare the summary estimates and corresponding CIs before and after exclusion of studies. Heterogeneity will also be compared before and after exclusion of studies. Heterogeneity will be estimated using I2 statistic, which indicates the proportion of total variation in estimates attributed to heterogeneity compared to the expected variability, as well as the Q statistic. A cut-off of 25% for I2 will be used to represent minimal heterogeneity, 50% to represent moderate and 75% to represent high heterogeneity.20 A random-effects meta-analysis with inverse-variance weighting will be used to synthesise estimates from individual studies.21
Lastly, several items included in the Q-Genie are not traditional sources of bias, notably items 1, 8 and 11. A sensitivity analysis excluding these items will be undertaken to assess the classification of studies as ‘poor’, ‘moderate’ or ‘good’ changes.
Resampling
To assess whether excluding studies based on quality scores is more useful than excluding the same number of studies (k) by chance, we will randomly remove k studies, without regard to quality, and recalculate pooled estimates. This will allow us to determine whether Q-Genie-derived pooled estimates and reduction in heterogeneity is sufficiently different from a randomly drawn sample of a meta-analysis.
Discrimination
We will calculate the c-statistic of our tool's ability to discriminate between studies that reduce heterogeneity when excluded and those that do not. Using a leave-one-out approach, we will exclude each study in a meta-analysis and note whether heterogeneity was significantly attenuated by this procedure. We will then use the scores from Q-Genie as the predictor and a binary variable designating the presence of heterogeneity as the outcome in creating a receiver operating characteristic (ROC) curve, the area under which is understood to be the c-statistic. If our tool has perfect discrimination, that is, perfect ability to separate those studies that contribute to heterogeneity from those that do not, then the c-statistic will be 1; a value of 0.5 indicates no predictive discrimination.22 23 The ROC curve, which plots sensitivity (true positive rate) against 1−specificity (false positive rate) for consecutive cut-offs of scores on Q-Genie, will be displayed.
All statistical analyses will be conducted in R (V.3.0.2).
Agreement with experts
We will ask experts to rate a randomly selected subsample of 30 studies from all included studies. This rating of ‘poor’, ‘moderate’ or ‘good’ will be compared to the ratings based on Q-Genie. Agreement will be reported using an intraclass correlation coefficient, with a value >0.80 considered good agreement.
Agreement with the Venice criteria
In 2008, Ioannidis et al24 published criteria for evaluating credibility of evidence from genetic association studies. The evaluation is based on considering the amount of evidence, extent of replication and protection from bias. The index generates a composite assessment of ‘strong’, ‘moderate’ or ‘weak’ epidemiological credibility. We will compare ratings on the Q-Genie tool with those from the Venice criteria using an intraclass correlation coefficient; ideally, a higher risk of bias on Q-Genie will correspond to weaker ratings on the Venice criteria.
Qualitative assessment of the tool
In addition to quantitative evaluation, we will conduct a qualitative assessment to explore (1) feasibility of the Q-Genie tool, (2) ease of use and comprehension and (3) relevance of the individual questions to the overall tool. The assessment will be used to compile changes to overcome proposed challenges in the presentation and administration of the tool. To accomplish this, a structured questionnaire will be administered to the 12 reviewers who participated in this study. This survey will consist of multiple-choice and open-ended questions on ease of use, comprehension and relevance of the content (table 1). This exercise will be valuable for gathering rich data on the numerous interpretations that may exist about the effectiveness of the Q-Genie tool. The Q-Genie tool will be amended based on the results of the quantitative and qualitative evaluations.
Table 1.
Survey for Q-Genie reviewers
| Theme | Question | Response type |
|---|---|---|
| Feasibility of use | Please rate the tool on ease of use | 5-Point likert: 1 (very difficult to use) to 5 (very easy to use) |
| What made the tool difficult to use? | Free text | |
| How long did it take you (on average) to apply the tool to each study? | Free text | |
| Would you recommend the format of the tool to be changed? | Yes/no | |
| If yes, please elaborate on how the format should be changed | Select one or more of the following:
|
|
| If you suggested: modify/re-label categories (option 3), please elaborate below | Free text | |
| Would you recommend the scale of the tool be modified? | Yes/no | |
| If yes, how should the scale be modified? | Select one or more of the following:
|
|
| Was the order of the questions cohesive and easy to follow? | Yes/no | |
| If no, please recommend how the order of the questions should be changed | Free text | |
| Relevance | Were the questions relevant to estimating risk of bias in genetic association studies? | Yes/no |
| If no, which questions were not relevant? | Free text | |
| Would you recommend the content of the tool be modified? | Yes/no | |
| If yes, what question(s) should be added? | Free text | |
| If yes, what question(s) should be removed? | Free text | |
| Comprehension | Please rate the comprehensibility of the tool | 5-Point likert: 1 (very difficult to understand) to 5 (very easy to understand) |
| What made the tool difficult to understand? | Free text | |
| What made the tool easy to understand? | Free text | |
| Did you have any other difficulties/have any additional recommendations? | Free text |
Conclusions
The past few decades have seen a special interest in the potential of genomics to uncover the aetiology of disease with the promise of better prediction, classification, prevention and treatment of diseases.25 26 However, the field of genetic epidemiology has been plagued by irreproducible findings, which is largely attributable to improperly conducted studies with poor methodological designs and inappropriate reporting. When combined in a meta-analysis, these same studies introduce bias into pooled summary estimates and deter an effort to estimate the disease-causing effects of genetic variants. To facilitate quality assessment of published studies, we developed and validated the Q-Genie tool. Though the tool demonstrated excellent reliability and construct validity in the original investigation, its use in meta-analyses was not methodically evaluated. The main objective of this present study is to quantitatively and qualitatively assess the Q-Genie's ability to identify poor quality genetic association studies, and thereby facilitate exclusion of such studies from meta-analyses in the future. This process could improve precision of pooled estimates and help explain and also decrease between-study heterogeneity. By methodically identifying and excluding poor quality studies, we may be able to strengthen our confidence in estimates of the effect of a gene on an outcome from meta-analyses, ultimately bringing us closer to deliver on the promise of personalised medicine.27
Acknowledgments
The authors acknowledge Sujane Kandasamy for her assistance with developing and conducting the focus groups and Andrea McLellan for her assistance with constructing the search strategy.
Footnotes
Contributors: ZNS designed the study and drafted this protocol. SS made substantial contributions to the draft of the protocol and acquisition of data. AA and RJdS made substantial contributions to the methodology, statistical analysis plan and revision of the manuscript. SSA and DM contributed to conception and design of the study and critically revised the protocol for important intellectual content. SR-d-P, AL, AM, FY, HR, ALa, CS, AI, YKL and NV contributed to the design of the study, provided feedback on the protocol and contributed to the acquisition of data. ZNS is the guarantor of this work. All authors provided final approval of the version to be published.
Funding: This research received no specific grant from any funding agency in the public, commercial or not-for-profit sectors. ZNS is supported by the Ontario Graduate Scholarship and the Canadian Diabetes Association Doctoral Award. SSA holds the Heart and Stroke Foundation of Ontario Michael G. DeGroote endowed Chair in Population Health and a Canada Research Chair in Ethnicity and Cardiovascular Disease. DM holds a Canada Research Chair in Genetics of Epidemiology.
Competing interests: None declared.
Ethics approval: A formal ethical approval is not required for this study, as primary data of patients will not be collected. Our findings will be disseminated through presentation at national and international conferences. An updated Q-Genie tool will be made available from the Population Genomics Program website <http://fhs.mcmaster.ca/pgp/links.html>. The results will also be submitted for a peer-reviewed publication.
Provenance and peer review: Not commissioned; externally peer reviewed.
References
- 1.Audigé L, Ayeni OR, Bhandari M et al. A practical guide to research: design, execution, and publication. Arthroscopy 2011;27:S1–112. 10.1016/j.arthro.2011.02.001 [DOI] [PubMed] [Google Scholar]
- 2.Munafò MR, Flint J. Meta-analysis of genetic association studies. Trends Genet 2004;20:439–44. 10.1016/j.tig.2004.06.014 [DOI] [PubMed] [Google Scholar]
- 3.Hartling L, Milne A, Hamm MP et al. Testing the Newcastle Ottawa Scale showed low reliability between individual reviewers. J Clin Epidemiol 2013;66:982–93. 10.1016/j.jclinepi.2013.03.003 [DOI] [PubMed] [Google Scholar]
- 4.Wells G, Shea B, O'Connell D et al. The Newcastle-Ottawa Scale (NOS) for assessing the quality of nonrandomised studies in meta-analyses. Ottawa Hospital Research Institute, 2014. http://www.ohri.ca/programs/clinical_epidemiology/oxford.asp (accessed 3 Sep 2014). [Google Scholar]
- 5.Higgins JPT, Altman DG, Gøtzsche PC et al. The Cochrane Collaboration's tool for assessing risk of bias in randomised trials. BMJ 2011;343:d5928 10.1136/bmj.d5928 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Katrak P, Bialocerkowski AE, Massy-Westropp N et al. A systematic review of the content of critical appraisal tools. BMC Med Res Methodol 2004;4:22 10.1186/1471-2288-4-22 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Sanderson S, Tatt ID, Higgins JPT. Tools for assessing quality and susceptibility to bias in observational studies in epidemiology: a systematic review and annotated bibliography. Int J Epidemiol 2007;36:666–76. 10.1093/ije/dym018 [DOI] [PubMed] [Google Scholar]
- 8.Hattersley AT, McCarthy MI. What makes a good genetic association study? Lancet 2005;366:1315–23. 10.1016/S0140-6736(05)67531-9 [DOI] [PubMed] [Google Scholar]
- 9.Sagoo GS, Little J, Higgins JPT. Systematic reviews of genetic association studies. Human Genome Epidemiology Network. PLoS Med 2009;6:e28 10.1371/journal.pmed.1000028 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Sohani ZN, Meyre D, de Souza RJ et al. Assessing the quality of published genetic association studies in meta-analyses: the quality of genetic studies (Q-Genie) tool. BMC Genet 2015;16:50 10.1186/s12863-015-0211-2 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Sohani ZN, Deng WQ, Pare G et al. Does genetic heterogeneity account for the divergent risk of type 2 diabetes in South Asian and White European populations? Diabetologia 2014;57:2270–81. 10.1007/s00125-014-3354-1 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Stroup DF, Berlin JA, Morton SC et al. Meta-analysis of observational studies in epidemiology: a proposal for reporting. Meta-analysis Of Observational Studies in Epidemiology (MOOSE) group. JAMA 2000;283:2008–12. http://www.ncbi.nlm.nih.gov/pubmed/10789670 (accessed 19 Mar 2014). [DOI] [PubMed] [Google Scholar]
- 13.Moher D, Liberati A, Tetzlaff J et al. Preferred reporting items for systematic reviews and meta-analyses: the PRISMA statement. PLoS Med 2009;6:e1000097 10.1371/journal.pmed.1000097 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Hedges LV, Pigott TD. The power of statistical tests for moderators in meta-analysis. Psychol Methods 2004;9:426–45. 10.1037/1082-989X.9.4.426 [DOI] [PubMed] [Google Scholar]
- 15.Hedges LV, Pigott TD. The power of statistical tests in meta-analysis. Psychol Methods 2001;6:203–17. http://www.ncbi.nlm.nih.gov/pubmed/11570228 (accessed 10 Aug 2015). [PubMed] [Google Scholar]
- 16.Armijo-Olivo S, Stiles CR, Hagen NA et al. Assessment of study quality for systematic reviews: a comparison of the Cochrane Collaboration Risk of Bias Tool and the Effective Public Health Practice Project Quality Assessment Tool: methodological research. J Eval Clin Pract 2012;18:12–18. 10.1111/j.1365-2753.2010.01516.x [DOI] [PubMed] [Google Scholar]
- 17.Graham N, Haines T, Goldsmith CH et al. Reliability of 3 assessment tools used to evaluate randomized controlled trials for treatment of neck pain. Spine (Phila Pa 1976) 2012;37:515–22. 10.1097/BRS.0b013e31822671eb [DOI] [PubMed] [Google Scholar]
- 18.Streiner DL, Norman GR. Health measurement scales: a practical guide to their development and use [Paperback]. 4th edn Toronto: Oxford University Press, 2008. http://www.amazon.ca/Health-Measurement-Scales-practical-development/dp/0199231885 (accessed 23 Apr 2014). [Google Scholar]
- 19.Berkey CS, Hoaglin DC, Mosteller F et al. A random-effects regression model for meta-analysis. Stat Med 1995;14:395–411. http://www.ncbi.nlm.nih.gov/pubmed/7746979 (accessed 8 Sep 2015). [DOI] [PubMed] [Google Scholar]
- 20.Huedo-Medina TB, Sánchez-Meca J, Marín-Martínez F et al. Assessing heterogeneity in meta-analysis: Q statistic or I2 index? Psychol Methods 2006;11:193–206. 10.1037/1082-9nn89X.11.2.193 [DOI] [PubMed] [Google Scholar]
- 21.Han B, Eskin E. Random-effects model aimed at discovering associations in meta-analysis of genome-wide association studies. Am J Hum Genet 2011;88:586–98. 10.1016/j.ajhg.2011.04.014 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Steyerberg EW, Harrell FE, Borsboom GJ et al. Internal validation of predictive models: efficiency of some procedures for logistic regression analysis. J Clin Epidemiol 2001;54:774–81. http://www.ncbi.nlm.nih.gov/pubmed/11470385 (accessed 5 Mar 2014). [DOI] [PubMed] [Google Scholar]
- 23.Riley RD, Hayden JA, Steyerberg EW et al. Prognosis Research Strategy (PROGRESS) 2: prognostic factor research. PLoS Med 2013;10:e1001380 10.1371/journal.pmed.1001380 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Ioannidis JPA, Boffetta P, Little J et al. Assessment of cumulative evidence on genetic associations: interim guidelines. Int J Epidemiol 2008;37:120–32. 10.1093/ije/dym159 [DOI] [PubMed] [Google Scholar]
- 25.Hirschhorn JN. Genomewide association studies—illuminating biologic pathways. N Engl J Med 2009;360:1699–701. 10.1056/NEJMp0808934 [DOI] [PubMed] [Google Scholar]
- 26.Visscher PM, Brown MA, McCarthy MI et al. Five years of GWAS discovery. Am J Hum Genet 2012;90:7–24. 10.1016/j.ajhg.2011.11.029 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Li A, Meyre D. Jumping on the train of personalized medicine: a primer for non-geneticist clinicians: Part 3. Clinical applications in the personalized medicine area. Curr Psychiatry Rev 2014;10:118–32. 10.2174/1573400510666140630170549 [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
bmjopen-2015-010403supp_table.pdf (57.3KB, pdf)