

Abstract
The treatment of cancer patients with autologous T cells expressing a chimeric antigen receptor (CAR) is one of the most promising adoptive cellular therapy approaches. Reproducible manufacturing of high-quality, clinical-grade CAR-T cell products is a prerequisite for the wide application of this technology. Product quality needs to be built-in within every step of the manufacturing process. We summarize herein the requirements and logistics to be considered, as well as the state of the art manufacturing platforms available. CAR-T cell therapy may be on the verge of becoming standard of care for a few clinical indications. Yet, many challenges pertaining to manufacturing standardization and product characterization remain to be overcome in order to achieve broad usage and eventual commercialization of this therapeutic modality.
Adoptive cell therapy using naturally occurring endogenous tumor-infiltrating lymphocytes or T cells genetically engineered to express either T-cell receptors1 or chimeric antigen receptors (CAR)2 have emerged as promising cancer immunotherapy strategies. Adoptive cell therapy using CD19-targeted CAR-T cells has resulted in remarkable responses in patients with acute lymphoblastic leukemia.3–6 Promising clinical outcomes in phase 1/2 clinical trial studies have triggered active support and investment from pharmaceutical and biotechnology companies.7,8 The manufacturing of clinical-grade CAR-T cells under current good manufacturing procedure (cGMP) is a critical step and in its current state a bottleneck for the wide implementation of this promising therapeutic modality.
Adoptive cellular therapy involves the ex vivo enrichment and expansion of T lymphocytes. For therapies using T cells expressing transduced CARs or T-cell receptors, cGMP grade ancillary genetic modification reagents, such as retroviral and lentiviral vectors, are also required. One of the challenges of this largely personalized medicine is the development of efficient technologies and cost-effective clinical manufacturing platforms to support the later clinical trial phases and ultimately commercialization. In this review, we highlight the cGMP manufacturing platforms and the quality control requirements for clinical-grade CAR-T cells in early phase clinical trials.
Manufacturing of CAR-T Cells
The initial success of CD19-targeted CAR-T cells in early phase clinical trials for the treatment of hematologic malignancies has triggered a genuine interest for CAR-T cell-based therapies.4,6,9–12 The targeting of other types of cancers focusing on additional tumor-associated antigens, such as PSMA, mesothelin, GD2, HER2, and epidermal growth factor receptors, is currently an active field of research and clinical trials as well.13 Hence, the manufacturing of CAR-T cells under cGMP is a focal point for this promising therapeutic modality.
Despite the various designs and distinctive tumor-specific scFvs, the manufacturing procedure for CAR-T cells remains consistent. The procedure encompasses T-cell source collection and processing followed by CAR-T cell preparation; the latter involves T-cell selection and/or activation, genetic modification with a CAR cDNA followed by large-scale expansion, and end-of-process formulation. In-process and quality control release testing are intimately coupled to the manufacturing process to ensure the integrity of the product.
T-cell source
As a mostly autologous cell-based therapy, the CAR-T cell-manufacturing process starts from the collection of peripheral blood mononuclear cell from the patient, commonly achieved by a leukapheresis process. Consenting physicians choose the appropriate window for collection based on treatment regimens to ensure the presence of sufficient numbers of T lymphocytes. Collected apheresis products can be processed in various ways depending on the downstream procedures. Devices such as Haemonetics Cell Saver 5+, COBE2991, and Fresenius Kabi LOVO have the ability to remove gross red blood cells and platelet contaminants. Terumo Elutra and Biosafe Sepax systems provide size-based cell fractionation for the depletion of monocytes and the isolation of lymphocytes. Instruments such as CliniMACS Plus and Prodigy systems allow the enrichment of specific subsets of T cells, such as CD4+, CD8+, CD25+, or CD62L+ T cells using Miltenyi beads post-cell washing as shown in Figure 1.
Figure 1.
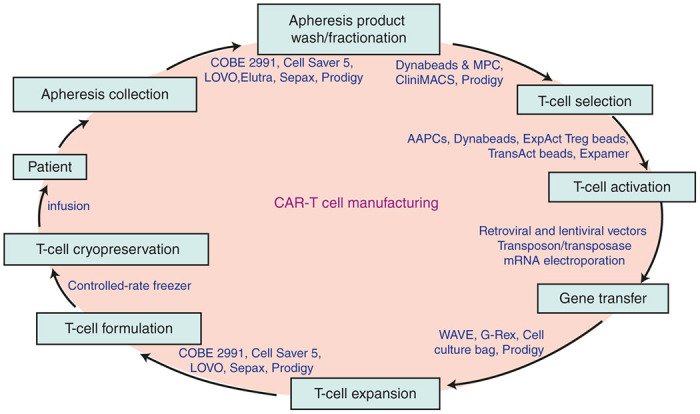
Major steps in chimeric antigen receptor-T-cell manufacturing process and examples of available technologies and devices. AAPC, artificial antigen-presenting cells; MPC, magnetic particle concentrator.
CAR-T cells generated from CD3+ population are widely used in clinical trials.3–6 However, studies from different laboratories have demonstrated that certain subsets of T cells such as naive,14 central memory,15 or memory stem cells16 may display functional advantages. Clinical-scale selection, transduction, and expansion processes have also been developed for these T-cell subsets.17,18 Although generation of CAR-T cell products initiated with T-cell populations of defined composition is an appealing strategy, T-cell subsets that provide the optimal therapeutic effect and minimal toxicity while outliving a robust and reproducible manufacturing process remain to be identified.19
The processed T-cell source material can either be used directly for downstream procedure or cryopreserved for future use. There are pros and cons for either practice. Nevertheless, cryopreserving the processed T cells allows time for product release testing and more flexibility for downstream process planning.
T-cell activation
The ex vivo expansion of T cells requires sustained and adequate activation. T-cell activation needs a primary specific signal via the T-cell receptor (Signal 1) and costimulatory signals such as CD28, 4-1BB, or OX40 (Signal 2). T-cell activation is also required for the transduction of the CAR cDNA via retroviral vectors.
Cell-based T-cell activation.
Antigen-presenting cells, such as dendritic cells (DCs), are the endogenous activators of T-cell responses. While therapeutic applications of DCs continue to be investigated,20 DC potency varies from patient to patient. Such limitation hampers the usage of DCs as a reliable source for T-cell activation. Another cell-based T-cell activation approach is through artificial antigen-presenting cells (AAPCs).21 Irradiated K562-derived AAPCs have been used to stimulate the expansion of CAR-T cells. The generation and selection of GMP-grade HLA-matched AAPC lines is complex and requires additional resources.22
Beads-based T-cell activation:
Several biotech companies have generated off-the-shelf clinical-grade T-cell activation reagents including the Invitrogen CTS Dynabeads CD3/28, the Miltenyi MACS GMP ExpAct Treg beads, Miltenyi MACS GMP TransAct CD3/28 beads, and the Juno Stage Expamer technology. These reagents have largely simplified the ex vivo T-cell activation procedure.
Antibody-coated magnetic beads.
Dynabeads CD3/28 are uniform super-paramagnetic beads covalently coupled to CD3 and CD28 antibodies. The added value of this reagent is that it enables the selection and activation of T cells in a single step when used in conjunction with the Dynal ClinExVivo MPC magnet. As the first-generation off-the-shelf clinical-grade reagent for CD3+ T-cell selection and activation, Dynabeads are widely used by different laboratories in early clinical trials although their availability is somewhat restricted.12,23 Miltenyi ExpAct Treg beads are paramagnetic beads conjugated to CD3-biotin, CD28 and anti-biotin monoclonal antibodies. By using various beads to T-cell ratios, ExpAct Treg beads can be used to expand both regulatory T cells and conventional lineage T cells.24 Removal of the magnetic beads is required at the end of the manufacturing process upon using either Dynabeads CD3/28 or Miltenyi MACS GMP ExpAct Treg beads.25
Antibody-coated nanobeads.
Miltenyi MACS GMP TransAct CD3/28 beads are polymeric nanomatrix conjugated to CD3 or to CD28 monoclonal antibodies. The advantage of the TransAct CD3/CD28 beads is that they are biodegradable, and therefore do not require removal prior to formulation, although upstream T-cell purification is needed prior to activation. Studies from our laboratory and others have shown that both ExpAct Treg beads and TransAct CD3/28 beads are comparable to Dynabeads CD3/28 for CAR-T cell manufacturing.18,24
Expamer technology.
The most recent development in T-cell activation reagent is the Expamer from Juno Therapeutics. Its unique core Streptamer technology has been used to isolate viral-specific lymphocytes.26,27 It has been reported recently that as a soluble and dissociable T-cell stimulation reagent, Expamer efficiently induces T cell receptor (TCR) signaling and efficiently activates T cells to support retroviral transduction and expansion.26,28 Although Expamer-activated T cells remain to be functionally assessed in vivo, this soluble reagent is very attractive for large-scale clinical manufacturing as it can be easily added and removed from cell suspensions and provides consistent product purity to enable automation.
Activation with anti-CD3 antibodies.
The engagement of T-cell surface CD3 molecules with soluble anti-CD3 monoclonal antibodies also supports T-cell activation in the presence of IL-2. Adequate activation and expansion of patient peripheral blood mononuclear cells with anti-CD3 monoclonal antibody OKT3 for the production of autologous and allogeneic CD19-CAR T cells has been reported.6,29
Genetic modification of T cells
Current CAR-T cell therapies largely rely on stable CAR expression upon delivery by viral and nonviral gene transfer systems. There are three major types of stable gene expression vectors used for clinical applications: γ-retroviral vectors, lentiviral vectors, and the transposon/transposase system. Messenger RNA transfer-mediated gene expression is another method to introduce CARs into cells while avoiding long-term expression.
γ-retroviral vectors were the first viral vectors used to provide stable CD19 CAR expression.30 They are presently used in approximately a fifth of all clinical trials requiring gene transfer delivery.31 In addition to high gene expression, another attractive feature of retroviral vectors is the availability of multiple stable packaging cell lines with wide tropism.32,33 Long-term patient follow-up studies have shown the safety profile of retroviral vectors in the context of adoptive T-cell therapy.34–38 Moreover, studies from our laboratory established that it is feasible to generate enough cGMP grade vector stocks to likely support phase 3 studies and beyond, by expanding retroviral stable packaging producer cell lines in the scalable Pall iCELLis bioreactors.39
Lentiviral vectors are widely used as they can transduce nondividing cells—albeit not cells in G0 phase—and display a safer genomic integration profile at least in the context of genetically modified hematopoietic stem cells.40,41 Similar to γ-retroviral vectors, lentiviral vectors mediate high gene transfer efficiency and drive stable level of CAR expression. The obstacles for scaling up the lentiviral vector production platform are multiple. They include the lack of widely available stable vector packaging systems due to the intrinsic fusogenic property of the commonly used VSV-G envelop, lot size limitation, and lot-to-lot variability imparted by the current multi-plasmids transient transfection procedure.42,43
Both retroviral and lentiviral vectors are complicated complex biological reagents that require intensive and expensive biosafety testing. A relatively new plasmid based expression system, the transposon/transposase system, has been used to introduce anti-CD19 CAR into T cells by electroporation. The advantages of this system are its simple manufacturing procedure, relatively low cost, and straightforward release testing. Integration is random, posing a potential oncogenic risk secondary to mutagenesis. Ongoing anti-CD19 CAR-T cell trial using the sleeping beauty transposon/transposase system shows low T-cell toxicity.22 However, the efficacy of CAR-T cells generated by this approach remains to be demonstrated.
Messenger RNA (mRNA) transfer provides a cytoplasmic expression system that enables transient expression of the transgene. Unlike stable and permanent expression of the transgene introduced by viral transduction or plasmid DNA transfection, in vitro transcribed mRNA can be introduced into cells by electroporation or by endocytosis. No genomic integration events occur in this process and therefore the concerns of genotoxicity, and potential generation of replication-competent retrovirus are eliminated. RNA transfection allows the expression of the transgene for about 1 week, and has been used to deliver mRNA for TCR/CAR, chemokine receptors, and cytokines.44–46 This approach may be advantageous to screen potentially toxic CAR molecules that could cross-react with normal tissues. Beatty and colleagues found that repetitive infusions of mRNA-transduced CAR T cells targeting mesothelin transiently persisted in peripheral blood of patients and elicited an antitumor effect.47
More recently, a proof-of-concept study reported that the electroporation of transcription activator-like effector nucleases specific for disrupting TCRα and CD52 molecules allows the generation of “off-the-shelf” CAR-T cells from third-party healthy donors.48
Expansion of CAR-T cells
Depending on the CAR-T cell modification strategy, there are several expansion platforms readily available to generate therapeutic doses of CAR-T cells (Figure 1).
Expansion of CAR-T cells using GE bioreactors.
The GE WAVE bioreactor system is a widely used device for expansion. This scalable system consists of a single use Cellbag Bioreactor, a temperature-enabling electric rocking base, and a range of optional controllers, pumps and probes. The Cellbag Bioreactor is placed on a rocking base that is equipped to maintain bag inflation and gently rocks the cell bag for rapid gas transfer and mixing. The perfusion functionality of the WAVE allows for automatic feeding and waste removal. Cells can rapidly expand to more than 107 cells/mL and this system can support up to 25-L cell culture in a single bioreactor. This platform is widely used by academic centers and biotech companies for cell expansion to support phase 1/2 clinical trials.25,49
Expansion of CAR-T cells using G-Rex bioreactors.
G-Rex is a relatively new platform. It is a cell culture flask with a gas-permeable membrane at the base that allows cells to grow to a high density without compromising gas exchange. The advantages of this system include its low seeding density, the one time upfront feeding regimen, the ease of growing the cells in an incubator, and the volume reduction feature at the time of harvest.50 However, cell expansion kinetics is largely affected if the cells are disturbed while in culture; therefore one drawback of the current configuration is that in-process cell sampling is not recommended.
Expansion of CAR-T cells using Prodigy.
One of the newest technologies being explored for CAR-T cell expansion is the Miltenyi CliniMACS Prodigy system. This system is currently used for stem cell enrichment and virus-reactive T-cell preparation.51,52 The CliniMACS Prodigy system is a combination of a cell washer, the CliniMACS magnetic cell separation system, and a cell cultivation device. NK cells have been successfully isolated from leukapheresis products and expanded to clinically relevant dose.53 It has also been reported that Prodigy supports lentiviral transduction of T cells with CARs.54 Equipped with a flexible programming suite, the CliniMACS Prodigy system aims to fully integrate and automate the complex multi-step CAR-T-cell processing and manufacturing procedures.
Expansion of CAR-T cells through recursive AAPC stimulation.
The expansion of CAR-T cells generated by the transposon/transposase system relies on the selective propagation upon recursive stimulation with γ-irradiated AAPCs in presence of IL-2 and IL-21. K562, a human leukemic cell line that does not express HLA class I A, HLA class I B nor HLA class II alleles, has been genetically modified to express a wide array of costimulatory molecules such as CD40, CD40L, CD70, CD80, CD83, CD86, CD137L, ICOSL, GITRL, and CD134L to facilitate T-cell expansion.55 Clinical-grade K562 cells genetically modified to express CD32, CD64, CD86, CD137, and membrane bound IL15 are currently used to support CD19-specific CAR-T cells clinical trials.22
Clinical CAR-T Cell Manufacturing Quality Checkpoints
The delivery of personalized medicine such as CAR-T cells depends on the release of complex biological products. Achieving sufficient numbers of cells displaying consistent quality at relatively low cost is challenging.56–58 The quality of CAR-T cell products is subject to donor-to-donor variation but is also largely dependent on the manufacturing environment as well as the quality and availability of ancillary raw materials and reagents. The quality of CAR-T cells needs to be carefully monitored and integrated into the manufacturing process.
Qualification of manufacturing facilities
CAR-T cell manufacturing currently requires GMP facilities with ISO5 cell processing clean rooms. The manufacturing facilities include complex and costly infrastructure and systems to support compliance with cGMP regulations.59 The facilities must be properly equipped with (i) Facilities systems (e.g., air-handlers, 24/7 alarm monitoring systems); (ii) Environmental monitoring equipment (e.g., viable and nonviable particle counters); (iii) Manufacturing process equipment (e.g., cell washers, bioreactors); and (iv) Analytical equipment (e.g., automatic cell counters, flow cytometers). The manufacturing facilities and equipment need to be properly maintained, and must have the capability to support the manufacturing process, as well as to perform adequate quality control testing. Another pivotal component for GMP facilities is the workforce. Highly skilled and dedicated personnel with extensive knowledge of GMP manufacturing, quality control, and quality assurance is the key to maintain a compliant GMP manufacturing environment.
Qualification of ancillary components
CAR-T cell manufacturing involves the use of a variety of ancillary components such as one-time use disposables, culture medium, reagents for genetic modification, cytokines, formulation medium, and cryopreservation reagents (Table 1). Clinical manufacturing requires raw materials and components qualified or approved for human use. The certificate of analysis from qualified vendors must meet established acceptance criteria and be reviewed for each lot. Routine testing of raw materials by the quality control laboratory is needed to guarantee product integrity. Establishment of backup vendors for critical materials is highly recommended to mitigate the risk of supply chain interruptions. Viral vectors are themselves complex biological materials and engender their own set of release tests and regulatory path. The detailed requirements have been summarized in a recent review.60
Table 1. Example of ancillary components for clinical CAR-T manufacturing.
| Manufacturing phases | Ancillary components |
|---|---|
| T-cell selection and activation | ○ Selection accessory set, such as the CliniMACS set |
| ○ Components for selection medium, such as X-VIVO 15, OpTimizer, human serum albumin | |
| ○ Selection reagents, such as Dynabeads, TransAct beads, ExpAct beads, Expamers | |
| Genetic modification | ○ Viral vector, such as retroviral vector and lentiviral vector |
| ○ Nonviral gene modification reagents, such as DNA plasmids and mRNA | |
| T-cell expansion | ○ Components for the expansion medium, such as X-VIVO 15, IL2, cytokines |
| Formulation | ○ Cell washing accessory sets |
| ○ Components in cell washing medium, such as phosphate buffered saline | |
| ○ Components in formulation medium, such as plasmalyte buffer | |
| Cryopreservation | ○ Components used in cryopreservation medium, such as dimethyl sulfoxide |
Qualification of manufacturing process
A controlled, robust, and reproducible manufacturing platform is essential for the success of cellular therapies. It integrates the combined readiness of (i) facilities, utilities, equipment qualification, and environmental monitoring plan, (ii) raw materials selection, (iii) standard operation procedures and batch production-controlled records, (iv) in-process and end-of-process sampling plans; (v) trained manufacturing personnel; and (vi) quality control and analytical assays.61 Although each of the constituents should be tested individually, the qualification process allows the identification of remaining challenges not previously anticipated during the evaluation and qualification of each component. Thorough documentation and timely analysis of the manufacturing processes are essential for the successful execution of the complex CAR-T cell manufacturing procedure. Data generated from the manufacturing process qualification runs allow manufacturers to gain deeper understanding of the process challenges and of the process-to-process product quality attributes variability. Robustness of product quality control is demonstrated through process performance, product quality, preventive/corrective action, change control management and review.
In-process testing and release testing of cellular products
According to cGMP regulations, quality is built into the design of the process and in every manufacturing steps.61,62 Due to the complex nature of CAR-T cell products, a cautiously devised list of in-process and release tests is required to provide adequate evidence of identity, safety, purity, and potency. The identity of CAR-T cell products is commonly characterized by CAR surface expression. Safety requires the lack of harmful contaminations, such as endotoxin, mycoplasma, replication competent retrovirus or lentivirus, and prevention of genotoxicity by appropriately limiting the level of transgene integration. Purity of the product relies in part on specified levels of CD3+ and CAR+ T cells. The impurities introduced by patient starting material and ancillary components, such as undesired cell types, tumor burden, and residual beads, have to be below certain specific levels approved by the Food and Drug Administration. Up to now, the potency of CAR-T cells is often determined by in vitro cytotoxic T lymphocyte assay or interferon-γ secretion. Table 2 summarizes examples of release assays for CAR-T cells using different genetic modification methods. A number of release assays used in on-going phase 1 clinical trials are summarized in a recent review article as well.60
Table 2. Examples of CAR-T cell release tests.
| Parameter | Release testing for CAR-T introduced by retroviral and lentiviral vector | Release testing for CAR-T introduced by transposon/transposase | Release testing for CAR-T introduced by mRNA electroporation |
|---|---|---|---|
| Safety | • Gram stain/sterility | • Gram stain/sterility | • Gram stain/sterility |
| • Mycoplasma | • Mycoplasma | • Mycoplasma | |
| • Endotoxin level | • Endotoxin level | • Endotoxin level | |
| • Copies of transgene insertion | |||
| • RCR/RCL | |||
| Purity | • % CD3+ T cells | • % CD3+ T cells | • % CD3+ T cells |
| • %CAR-T cells | • %CAR-T cells | ||
| • Residual tumor burden | • Residual AAPCs | ||
| • Residual beads | |||
| Identity | • % CAR T cells |
||
| Potency |
In vitro CTL or IFN-γ secretion |
||
| Reference | 25 | 22 | 47 |
AAPC, artificial antigen-presenting cells; CAR, chimeric antigen receptor; CTL, cytotoxic T lymphocyte; IFN, interferon; RCL, replication-competent lentivirus; RCR, replication-competent retrovirus.
Depending on the nature of the specific product, additional testing may be required. Establishment of a complete testing panel with the guidance of the Food and Drug Administration in the early development phase is essential for the final commercialization of the product.
Manufacturing CAR-T cells toward commercialization
As personalized therapies, autologous cell-based therapies pose a distinct set of manufacturing challenges. In contrast with the allogeneic donor bulk manufacturing scale-up concept, scaling up for patient-specific CAR-T cell manufacturing requires the ability to accommodate multiple independent productions in parallel. Current CAR-T cell-manufacturing platforms are labor intensive. Issues that relate to quality control and single-lot-release add onto the complexity of the individual manufacturing and drastically increase the cost of goods. Pharmaceutical and biotechnology companies are just entering the adoptive cell therapy space; at present, the most extensive experience in CAR-T manufacturing still lies in the academic centers. Partnerships between industry and academic centers, such as JUNO Therapeutics with Memorial Sloan Kettering Cancer Center, Novartis with the University of Pennsylvania, and Kite Pharmaceuticals with the National Cancer Institute8 facilitate process transfer and improvement, accelerate development and promote future commercialization prospects for this promising therapeutic modality.
Challenges for the development of more controlled and cost-effective manufacturing process has yet to be overcome. The therapeutically active T cells or subsets of T cells remain to be identified in the manufacturing starting material; the complicated manufacturing process needs to be simplified to promote standardization and yield products of increased defined composition; implementation of fully closed systems to alleviate costly cGMP manufacturing environmental requirements—such as GMP plants and labor intensive environmental monitoring plans—and automation to avoid operator-introduced errors are also pressing issues that need to be addressed. Indeed, new manufacturing tools need to be developed and tested.63
Future Aspects for CAR-T Cell Manufacturing
CAR-T cells have been shown to be one of the most promising therapeutic modalities for treatment of refractory hematologic malignancies. The design of CARs has evolved drastically over the years.2 Features such as the coexpression of costimulatory molecules,64 cytokines,65,66 and suicide genes67,68 are incorporated to further improve efficacy and safety. The tumor targets for CAR-T cells have expanded from CD19 to a great range of new targets, including but not limited to CD20, CD22, CD30, CD33, CD138, CD171, CEA, epidermal growth factor receptor, EFGRvIII, ErbB, FAP, GD2, Glypican 3, Her 2, Mesothelin, and NKG2D.13 Adoptive CAR-T cell therapy will hopefully prove to be as effective in solid tumors as in onco-hematological indications.
The initiation of manufacturing with defined subpopulations of T cells that can be derived from a blood draw instead of a leukapheresis product would reduce the scale and therefore the cost of manufacturing. New sources of T cells that could alleviate the need to obtain autologous T cells are also being investigated.69
The intensified interest from biotech and pharmaceutical companies will surely accelerate the development of improved manufacturing platforms.7 An increasing number of tools are available for clinical CAR-T cell manufacturing and consortiums such as the CCRM and NNMI are forming (http://ccrm.ca/cell-manufacturing; http://www.manufacturing.gov/nist-nnmi-institutes.html). The optimization of appropriate quality control release testing and tracking of products will need to be drastically improved in terms of efficiency and cost effectiveness. The simplification of workflows, the increase in process robustness, and the implementation of automated closed systems should enable scalability and reduce the cost of goods while maintaining the efficiency of the CAR T-cell products.59 The participation and cooperation between the various stakeholders should accelerate the path to commercialization.
Acknowledgments
We thank Michel Sadelain for thoroughly reviewing the manuscript. This work is supported by the NCI P30 CA08748, P50 CA086438, DOD and Starr Foundation Tri-Institutional Stem Cell Initiative
I.R. is a scientific co-founder of Juno Therapeutics and a consultant for Juno Therapeutics. X.W. has no conflict.
References
- Yee, C (2014). The use of endogenous T cells for adoptive transfer. Immunol Rev 257: 250–263. [DOI] [PubMed] [Google Scholar]
- Sadelain, M, Brentjens, R and Rivière, I (2013). The basic principles of chimeric antigen receptor design. Cancer Discov 3: 388–398. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Brentjens, RJ, Davila, ML, Riviere, I, Park, J, Wang, X, Cowell, LG, et al. (2013). CD19-targeted T cells rapidly induce molecular remissions in adults with chemotherapy-refractory acute lymphoblastic leukemia. Sci Transl Med 5:177ra138. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Grupp, SA, Kalos, M, Barrett, D, Aplenc, R, Porter, DL, Rheingold, SR et al. (2013). Chimeric antigen receptor-modified T cells for acute lymphoid leukemia. N Engl J Med 368: 1509–1518. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Davila, ML, Riviere, I, Wang, X, Bartido, S, Park, J, Curran, K, et al. (2014). Efficacy and toxicity management of 19-28z CAR T cell therapy in B cell acute lymphoblastic leukemia. Sci Transl Med 6:224ra225. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kochenderfer, JN, Dudley, ME, Kassim, SH, Somerville, RP, Carpenter, RO, Stetler-Stevenson, M et al. (2015). Chemotherapy-refractory diffuse large B-cell lymphoma and indolent B-cell malignancies can be effectively treated with autologous T cells expressing an anti-CD19 chimeric antigen receptor. J Clin Oncol 33: 540–549. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Flemming, A (2014). Deal watch: Pfizer and GSK join race for T cell cancer therapies. Nat Rev Drug Discov 13: 568–569. [DOI] [PubMed] [Google Scholar]
- June, CH, Riddell, SR and Schumacher, TN (2015). Adoptive cellular therapy: a race to the finish line. Sci Transl Med 7:280ps287. [DOI] [PubMed] [Google Scholar]
- Davila, ML, Riviere, I, Wang, X, Bartido, S, Park, J, Curran, K, et al. (2014). Efficacy and toxicity management of 19-28z CAR T cell therapy in B cell acute lymphoblastic leukemia. Sci Transl Med 6:224ra225. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Brentjens, RJ, Davila, ML, Riviere, I, Park, J, Wang, X, Cowell, LG, et al. (2013). CD19-targeted T cells rapidly induce molecular remissions in adults with chemotherapy-refractory acute lymphoblastic leukemia. Sci Transl Med 5:177ra138. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Porter, DL, Levine, BL, Kalos, M, Bagg, A and June, CH (2011). Chimeric antigen receptor-modified T cells in chronic lymphoid leukemia. N Engl J Med 365: 725–733. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lee, DW, Kochenderfer, JN, Stetler-Stevenson, M, Cui, YK, Delbrook, C, Feldman, SA et al. (2015). T cells expressing CD19 chimeric antigen receptors for acute lymphoblastic leukaemia in children and young adults: a phase 1 dose-escalation trial. Lancet 385: 517–528. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gill, S., M. V. Maus, and D. L. Porter. 2015. Chimeric antigen receptor T cell therapy: 25 years in the making. Blood Rev. [DOI] [PubMed]
- Hinrichs, CS, Borman, ZA, Gattinoni, L, Yu, Z, Burns, WR, Huang, J et al. (2011). Human effector CD8+ T cells derived from naive rather than memory subsets possess superior traits for adoptive immunotherapy. Blood 117: 808–814. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Berger, C, Jensen, MC, Lansdorp, PM, Gough, M, Elliott, C and Riddell, SR (2008). Adoptive transfer of effector CD8+ T cells derived from central memory cells establishes persistent T cell memory in primates. J Clin Invest 118: 294–305. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gattinoni, L, Lugli, E, Ji, Y, Pos, Z, Paulos, CM, Quigley, MF et al. (2011). A human memory T cell subset with stem cell-like properties. Nat Med 17: 1290–1297. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wang, X, Naranjo, A, Brown, CE, Bautista, C, Wong, CW, Chang, WC et al. (2012). Phenotypic and functional attributes of lentivirus-modified CD19-specific human CD8+ central memory T cells manufactured at clinical scale. J Immunother 35: 689–701. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Casati, A, Varghaei-Nahvi, A, Feldman, SA, Assenmacher, M, Rosenberg, SA, Dudley, ME et al. (2013). Clinical-scale selection and viral transduction of human naïve and central memory CD8+ T cells for adoptive cell therapy of cancer patients. Cancer Immunol Immunother 62: 1563–1573. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sommermeyer, D, Hudecek, M, Kosasih, PL, Gogishvili, T, Maloney, DG, Turtle, CJ et al. (2016). Chimeric antigen receptor-modified T cells derived from defined CD8(+) and CD4(+) subsets confer superior antitumor reactivity in vivo. Leukemia 30: 492–500. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Vacchelli, E, Vitale, I, Eggermont, A, Fridman, WH, Fučíková, J, Cremer, I et al. (2013). Trial watch: Dendritic cell-based interventions for cancer therapy. Oncoimmunology 2: e25771. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kim, JV, Latouche, JB, Rivière, I and Sadelain, M (2004). The ABCs of artificial antigen presentation. Nat Biotechnol 22: 403–410. [DOI] [PubMed] [Google Scholar]
- Singh, H, Huls, H, Kebriaei, P and Cooper, LJ (2014). A new approach to gene therapy using Sleeping Beauty to genetically modify clinical-grade T cells to target CD19. Immunol Rev 257: 181–190. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Brentjens, RJ, Rivière, I, Park, JH, Davila, ML, Wang, X, Stefanski, J et al. (2011). Safety and persistence of adoptively transferred autologous CD19-targeted T cells in patients with relapsed or chemotherapy refractory B-cell leukemias. Blood 118: 4817–4828. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wang, X, Stefanski, J, Borquez-Ojeda, O, Qu, J, Hack, A, He, Q, et al. (2015). Comparison of CTS Dynabeadsc CD3/CD28, Miltenyi TransAct CD3/28 and ExpAct beads for large-scale CAR T cell manufacturing. European Society of Gene and Cell Therapy Collaborative Congress 2015, Helsinki, Finland. A31. [Google Scholar]
- Hollyman, D, Stefanski, J, Przybylowski, M, Bartido, S, Borquez-Ojeda, O, Taylor, C et al. (2009). Manufacturing validation of biologically functional T cells targeted to CD19 antigen for autologous adoptive cell therapy. J Immunother 32: 169–180. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Odendahl, M, Grigoleit, GU, Bönig, H, Neuenhahn, M, Albrecht, J, Anderl, F et al. (2014). Clinical-scale isolation of ‘minimally manipulated’ cytomegalovirus-specific donor lymphocytes for the treatment of refractory cytomegalovirus disease. Cytotherapy 16: 1245–1256. [DOI] [PubMed] [Google Scholar]
- Freimüller, C, Stemberger, J, Artwohl, M, Germeroth, L, Witt, V, Fischer, G et al. (2015). Selection of adenovirus-specific and Epstein-Barr virus-specific T cells with major histocompatibility class I streptamers under Good Manufacturing Practice (GMP)-compliant conditions. Cytotherapy 17: 989–1007. [DOI] [PubMed] [Google Scholar]
- Bashour KT, Graef P, Stemberger C, Lothar G, Odegard V and Ramsborg CG (2015). Functional characterization of a T cell stimulation reagent for the production of therapeutic chimeric antigen receptor T cells. ASH 57th Annual Meeting & Exposition, Orlando, FL. [Google Scholar]
- Brudno, JN, Somerville, RP, Shi, V, Rose, JJ, Halverson, DC, Fowler, DH, et al. (2016). Allogeneic T cells that express an anti-CD19 chimeric antigen receptor induce remissions of B-cell malignancies that progress after allogeneic hematopoietic stem-cell transplantation without causing graft-versus-host disease. J Clin Oncol 34(10):1112–1121. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Brentjens, RJ, Latouche, JB, Santos, E, Marti, F, Gong, MC, Lyddane, C, et al. (2003). Eradication of systemic B-cell tumors by genetically targeted human T lymphocytes co-stimulated by CD80 and interleukin-15. Nat Med 9:279–286. [DOI] [PubMed] [Google Scholar]
- Deichmann, A and Schmidt, M (2013). Biosafety considerations using gamma-retroviral vectors in gene therapy. Curr Gene Ther 13: 469–477. [DOI] [PubMed] [Google Scholar]
- Ghani, K, Wang, X, de Campos-Lima, PO, Olszewska, M, Kamen, A, Rivière, I et al. (2009). Efficient human hematopoietic cell transduction using RD114- and GALV-pseudotyped retroviral vectors produced in suspension and serum-free media. Hum Gene Ther 20: 966–974. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Miller, AD, Garcia, JV, von Suhr, N, Lynch, CM, Wilson, C and Eiden, MV (1991). Construction and properties of retrovirus packaging cells based on gibbon ape leukemia virus. J Virol 65: 2220–2224. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bonini, C, Grez, M, Traversari, C, Ciceri, F, Marktel, S, Ferrari, G et al. (2003). Safety of retroviral gene marking with a truncated NGF receptor. Nat Med 9: 367–369. [DOI] [PubMed] [Google Scholar]
- Brenner, MK and Heslop, HE (2003). Is retroviral gene marking too dangerous to use? Cytotherapy 5: 190–193. [DOI] [PubMed] [Google Scholar]
- Macpherson, JL, Boyd, MP, Arndt, AJ, Todd, AV, Fanning, GC, Ely, JA et al. (2005). Long-term survival and concomitant gene expression of ribozyme-transduced CD4+ T-lymphocytes in HIV-infected patients. J Gene Med 7: 552–564. [DOI] [PubMed] [Google Scholar]
- Muul, LM, Tuschong, LM, Soenen, SL, Jagadeesh, GJ, Ramsey, WJ, Long, Z, et al. (2003). Persistence and expression of the adenosine deaminase gene for 12 years and immune reaction to gene transfer components: long-term results of the first clinical gene therapy trial. Blood 101:2563–2569. [DOI] [PubMed] [Google Scholar]
- Scholler, J, Brady, TL, Binder-Scholl, G, Hwang, WT, Plesa, G, Hege, KM, et al. (2012). Decade-long safety and function of retroviral-modified chimeric antigen receptor T cells. Sci Transl Med 4:132ra153. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wang, X, Olszewska, M, Qu, J, Wasielewska, T, Bartido, S, Hermetet, G et al. (2015). Large-scale clinical-grade retroviral vector production in a fixed-bed bioreactor. J Immunother 38: 127–135. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Naldini, L, Blömer, U, Gallay, P, Ory, D, Mulligan, R, Gage, FH et al. (1996). In vivo gene delivery and stable transduction of nondividing cells by a lentiviral vector. Science 272: 263–267. [DOI] [PubMed] [Google Scholar]
- Vannucci, L, Lai, M, Chiuppesi, F, Ceccherini-Nelli, L and Pistello, M (2013). Viral vectors: a look back and ahead on gene transfer technology. New Microbiol 36:1–22. [PubMed] [Google Scholar]
- Ni, Y, Sun, S, Oparaocha, I, Humeau, L, Davis, B, Cohen, R et al. (2005). Generation of a packaging cell line for prolonged large-scale production of high-titer HIV-1-based lentiviral vector. J Gene Med 7: 818–834. [DOI] [PubMed] [Google Scholar]
- Throm, RE, Ouma, AA, Zhou, S, Chandrasekaran, A, Lockey, T, Greene, M et al. (2009). Efficient construction of producer cell lines for a SIN lentiviral vector for SCID-X1 gene therapy by concatemeric array transfection. Blood 113: 5104–5110. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhao, Y, Zheng, Z, Cohen, CJ, Gattinoni, L, Palmer, DC, Restifo, NP, et al. (2006). High-efficiency transfection of primary human and mouse T lymphocytes using RNA electroporation. Mol Ther 13:151–159. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yoon, SH, Lee, JM, Cho, HI, Kim, EK, Kim, HS, Park, MY et al. (2009). Adoptive immunotherapy using human peripheral blood lymphocytes transferred with RNA encoding Her-2/neu-specific chimeric immune receptor in ovarian cancer xenograft model. Cancer Gene Ther 16: 489–497. [DOI] [PubMed] [Google Scholar]
- Rowley, J, Monie, A, Hung, CF and Wu, TC (2009). Expression of IL-15RA or an IL-15/IL-15RA fusion on CD8+ T cells modifies adoptively transferred T-cell function in cis. Eur J Immunol 39: 491–506. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Beatty, GL, Haas, AR, Maus, MV, Torigian, DA, Soulen, MC, Plesa, G et al. (2014). Mesothelin-specific chimeric antigen receptor mRNA-engineered T cells induce anti-tumor activity in solid malignancies. Cancer Immunol Res 2: 112–120. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Poirot, L, Philip, B, Schiffer-Mannioui, C, Le Clerre, D, Chion-Sotinel, I, Derniame, S et al. (2015). Multiplex genome-edited T-cell manufacturing platform for “Off-the-Shelf” adoptive T-cell immunotherapies. Cancer Res 75: 3853–3864. [DOI] [PubMed] [Google Scholar]
- Levine, BL (2015). Performance-enhancing drugs: design and production of redirected chimeric antigen receptor (CAR) T cells. Cancer Gene Ther 22: 79–84. [DOI] [PubMed] [Google Scholar]
- Bajgain, P, Mucharla, R, Wilson, J, Welch, D, Anurathapan, U, Liang, B et al. (2014). Optimizing the production of suspension cells using the G-Rex “M” series. Mol Ther Methods Clin Dev 1: 14015. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kumaresan, P, Figliola, M, Moyes, JS, Huls, MH, Tewari, P, Shpall, EJ, et al. (2015). Automated cell enrichment of cytomegalovirus-specific T cells for clinical applications using the cytokine-capture system. JoVE 104–e52808: 1–10. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Stroncek, DF, Tran, M, Frodigh, SE, David-Ocampo, V, Ren, J, Larochelle, A, et al. (2015). Preliminary evaluation of a highly automated instrument for the selection of CD34+ cells from mobilized peripheral blood stem cell concentrates. Transfusion 56(2): 511–517. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Granzin, M, Soltenborn, S, Müller, S, Kollet, J, Berg, M, Cerwenka, A et al. (2015). Fully automated expansion and activation of clinical-grade natural killer cells for adoptive immunotherapy. Cytotherapy 17: 621–632. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nickolay, LE, Cheung, GWC, Pule, M, Thrasher, A, Johnston, I, Kaiser, A et al. (2015). Automated lentiviral transduction of T cells with CARs using the CliniMACS Prodigy. European Society of Gene and Cell Therapy Collaborative Congress 2015, Helsinki, Finland. A26. [Google Scholar]
- Suhoski, MM, Golovina, TN, Aqui, NA, Tai, VC, Varela-Rohena, A, Milone, MC et al. (2007). Engineering artificial antigen-presenting cells to express a diverse array of co-stimulatory molecules. Mol Ther 15: 981–988. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Heathman, TR, Nienow, AW, McCall, MJ, Coopman, K, Kara, B and Hewitt, CJ (2015). The translation of cell-based therapies: clinical landscape and manufacturing challenges. Regen Med 10:49–64. [DOI] [PubMed] [Google Scholar]
- Digiusto, DL and Kiem, HP (2012). Current translational and clinical practices in hematopoietic cell and gene therapy. Cytotherapy 14: 775–790. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gee, AP (2015). Manufacturing genetically modified T cells for clinical trials. Cancer Gene Ther 22: 67–71. [DOI] [PubMed] [Google Scholar]
- Hourd, P, Chandra, A, Alvey, D, Ginty, P, McCall, M, Ratcliffe, E et al. (2014). Qualification of academic facilities for small-scale automated manufacture of autologous cell-based products. Regen Med 9: 799–815. [DOI] [PubMed] [Google Scholar]
- Wang, X and Rivière, I (2015). Manufacture of tumor- and virus-specific T lymphocytes for adoptive cell therapies. Cancer Gene Ther 22: 85–94. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Campbell, A, Brieva, T, Raviv, L, Rowley, J, Niss, K, Brandwein, H et al. (2015). Concise review: process development considerations for cell therapy. Stem Cells Transl Med 4: 1155–1163. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rathore, AS and Winkle, H (2009). Quality by design for biopharmaceuticals. Nat Biotechnol 27: 26–34. [DOI] [PubMed] [Google Scholar]
- Kaiser, AD, Assenmacher, M, Schröder, B, Meyer, M, Orentas, R, Bethke, U et al. (2015). Towards a commercial process for the manufacture of genetically modified T cells for therapy. Cancer Gene Ther 22: 72–78. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Stephan, MT, Ponomarev, V, Brentjens, RJ, Chang, AH, Dobrenkov, KV, Heller, G et al. (2007). T cell-encoded CD80 and 4-1BBL induce auto- and transcostimulation, resulting in potent tumor rejection. Nat Med 13: 1440–1449. [DOI] [PubMed] [Google Scholar]
- Pegram, HJ, Purdon, TJ, van Leeuwen, DG, Curran, KJ, Giralt, SA, Barker, JN, et al. (2015). IL-12-secreting CD19-targeted cord blood-derived T cells for the immunotherapy of B-cell acute lymphoblastic leukemia. Leukemia 29:415–422. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Koneru, M, Purdon, TJ, Spriggs, D, Koneru, S and Brentjens, RJ (2015). IL-12 secreting tumor-targeted chimeric antigen receptor T cells eradicate ovarian tumors. Oncoimmunology 4:e994446. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wang, X, Chang, WC, Wong, CW, Colcher, D, Sherman, M, Ostberg, JR et al. (2011). A transgene-encoded cell surface polypeptide for selection, in vivo tracking, and ablation of engineered cells. Blood 118: 1255–1263. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Di Stasi, A, Tey, SK, Dotti, G, Fujita, Y, Kennedy-Nasser, A, Martinez, C et al. (2011). Inducible apoptosis as a safety switch for adoptive cell therapy. N Engl J Med 365: 1673–1683. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Themeli, M, Rivière, I and Sadelain, M (2015). New cell sources for T cell engineering and adoptive immunotherapy. Cell Stem Cell 16: 357–366. [DOI] [PMC free article] [PubMed] [Google Scholar]